
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a Brønsted acid Al–MIL-53 metal–organic framework catalyst and its application in [4 + 2] cycloadditions†
Ruilian Lia,
Yi Jiangb,
Jian Zhaob,
Daniele Ramellac,
Yu Peng *a and
Yi Luan*b
*a and
Yi Luan*b
aHunan Agricultural University, Hunan 410128, P. R. China. E-mail: pengy7505@hunau.net; Tel: +86-731-84617022
bSchool of Materials Science and Engineering, University of Science and Technology Beijing, 30 Xueyuan Road, Haidian District, Beijing 100083, P. R. China. E-mail: yiluan@ustb.edu.cn
cTemple University-Beury Hall, 1901, N. 13th Street, Philadelphia, PA 19122, USA
First published on 11th July 2017
Abstract
Two Al–MIL-53 derived metal–organic frameworks (MOFs) were developed to serve as efficient heterogeneous Brønsted acid catalysts. Sulfonic acid functional groups were successfully incorporated into the framework by post-synthetic modification (PSM) using commercially available reagents. The synthesized Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H Brønsted acid–MOF catalysts were fully characterized by SEM, powder XRD, FTIR, TGA and N2 adsorption/desorption isotherms. An efficient [4 + 2] cycloaddition of 2-vinyl-substituted phenols was evaluated using the newly synthesized Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H catalysts. The Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H catalysts were found to be compatible with a variety of substituted substrates; finally, they can be recycled five times without compromising the yield or selectivity of the reaction.
1 Introduction
Although homogeneous Brønsted acids, such as H2SO4 and HCl, are widely used as cheap catalysts, they are impractical to recycle, and they cause severe equipment corrosion as well as environmental pollution.1 As a result, heterogeneous Brønsted acid catalysts are highly desirable.2 Solid supports, such as organic polymers3 and porous materials,4 have been commonly utilized as heterogeneous Brønsted acidic catalyst carriers.5 Among several porous materials reported in the literature, metal–organic frameworks (MOFs) demonstrated several advantages in their highly tailorable nature, porous structure and large surface area.6 MOF materials have been widely utilized as catalytic materials for a wide variety of organic transformations.7 In general, Brønsted acid/MOF systems have been obtained using organic ligand bearing a pre-installed acid functional group.8 However, this approach limits the variety of Brønsted acid MOFs that can be prepared and increases the cost of Brønsted acid MOFs.9 More synthetic approaches to access Brønsted acid MOFs are therefore necessary for the preparation of low pKa catalytic materials.10Post-synthetic modification (PSM) of porous MOFs is a rapid approach for the synthesis of functionalized MOF scaffolds through stable covalent bonds.11 Brønsted acidic moieties can be introduced easily and rapidly taking advantage of PSM methods.12 Yaghi and co-workers13 developed a PSM procedure that employs 1,3-propanesultone to synthesize IRMOF-3 derived Brønsted acid MOF; Wang and coworkers instead14 applied a similar PSM strategy to the synthesis of Brønsted acid MOF catalysts. More recently, Cohen and co-workers have developed an anhydride approach for the modification of amino-functionalized MOFs using the modified MOFs for organocatalytic reactions.15 For the installation of a highly acidic sulfonic group, the choice of the PSM reagent is highly important. For example, the use of chlorosulfonic acid as sulfonating agent could lead to the complete collapse of the crystalline structure.16 For this reason, a commercially available reagent for the introduction of a sulfonyl group under mild reaction condition would be highly desirable.17
Hetero-Diels–Alder (HDA) reactions are among the most useful organic transformations in heterocyclic synthesis as they can afford a broad range of structural complexity.18 ortho-Quinone methide (o-QM)19 is composed of a cyclohexadiene motif in conjugation with a carbonyl and a methylene group. ortho-Quinone methide intermediates can undergo 1,4-conjugate addition reactions with nucleophiles and [4 + 2] cycloaddition reactions, including HDA reaction, with various dienophiles.20 Chromanes have been prepared by HDA reactions with an o-quinone methide used as dienophile.21 However, only very limited examples of such reactivity22 have been reported applied to the synthesis of 4H-chromene derivatives, which are useful in medicinal chemistry.23 An unreactive alkyne is generally required to access 4H-chromenes via an HDA reaction. Alternatively, a tandem [4 + 2]/elimination reaction sequence may result in the construction of 4H-chromenes from the corresponding 2π partners. The elimination of a stable molecule, such as a benzylic substituent, is the common method for o-QM generation. It has been proposed that the mild protonation of 2-ethynylphenol might result in a benzylic carbocation intermediate. The generated benzylic carbocation would subsequently tautomerize to the desired o-QM intermediate.
In this work, we wish to report two Al–MIL-53 derived Brønsted acid MOFs for [4 + 2] cycloadditions/elimination catalysis tandem reaction.24 The newly synthesized Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H MOF catalysts bear sulfonic acid groups, and were both obtained from commercially available reagent. A variety of substituted 4H-chromenes was generated from different 2-vinyl-substituted phenols. Furthermore, the synthesized Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H catalysts can be readily filtered and separated from the reaction solution, allowing five-time recycle and reuse of the catalyst without compromising the yield and selectivity of the reaction.
2 Experimental section
2.1 Preparations of MOF support
Al–MIL-53–NH2 was prepared from 2-aminoterephthalic acid, per literature procedure.252.2 Preparations of Al–MIL-53–RSO3H
1.85 g of Al–MIL-53–NH2 (0.85 mmol based on a MW of 2181.28 g mol−1) was suspended in 20 mL of CHCl3; then, 2.0 equiv. of 1,3-propanesultone (207 mg, 1.7 mmol) were added. The mixture was stirred slowly at 25 °C for 12 h, after which the solvent was decanted. Fresh DMF (10 mL) was added once a day for three days; CHCl3 was then used to rinse the crystals once a day for two days. The crystals were dried under vacuum at 40 °C before use.2.3 Preparations of Al–MIL-53–ArSO3H
1.85 g of Al–MIL-53–NH2 (0.85 mmol based on a MW of 2181.28 g mol−1) were suspended in 20 mL of CHCl3; 2.0 equiv. of o-sulfobenzoic acid anhydride (313 mg, 1.7 mmol) were then added. The mixture was stirred slowly at 25 °C for 12 h, after which the solvent was decanted. Fresh DMF (10 mL) was added once a day for three days; CHCl3 was then used to rinse the crystals once a day for two days. The crystals were dried under vacuum at 40 °C before use.2.4 [4 + 2] cycloaddition reaction conditions
In a general catalytic reaction, 1 mol% of the Al–MIL-53–NH2 derived MOF acid was added to 1.00 mL of chlorobenzene in an oven-dried round-bottom reaction vessel. 2.0 mmol of organic substrate was added then. The solution was stirred at 40 °C for 3 h. The reaction mixture was passed through a silica gel plug and eluted with 5 mL of dichloromethane. Then, the filtrate was concentrated under reduced pressure and the residue was purified by flash chromatography over silica gel to afford the dimeric product. The yield was calculated based on the isolated product.3 Results and discussion
Al–MIL-53–NH2 was synthesized using 2-amino-1,4-benzenedicarboxylate (NH2–H2BDC) as the organic linker.Subsequently, Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H were modified post-synthetically using the corresponding reagent in chloroform solvent (Scheme 1).
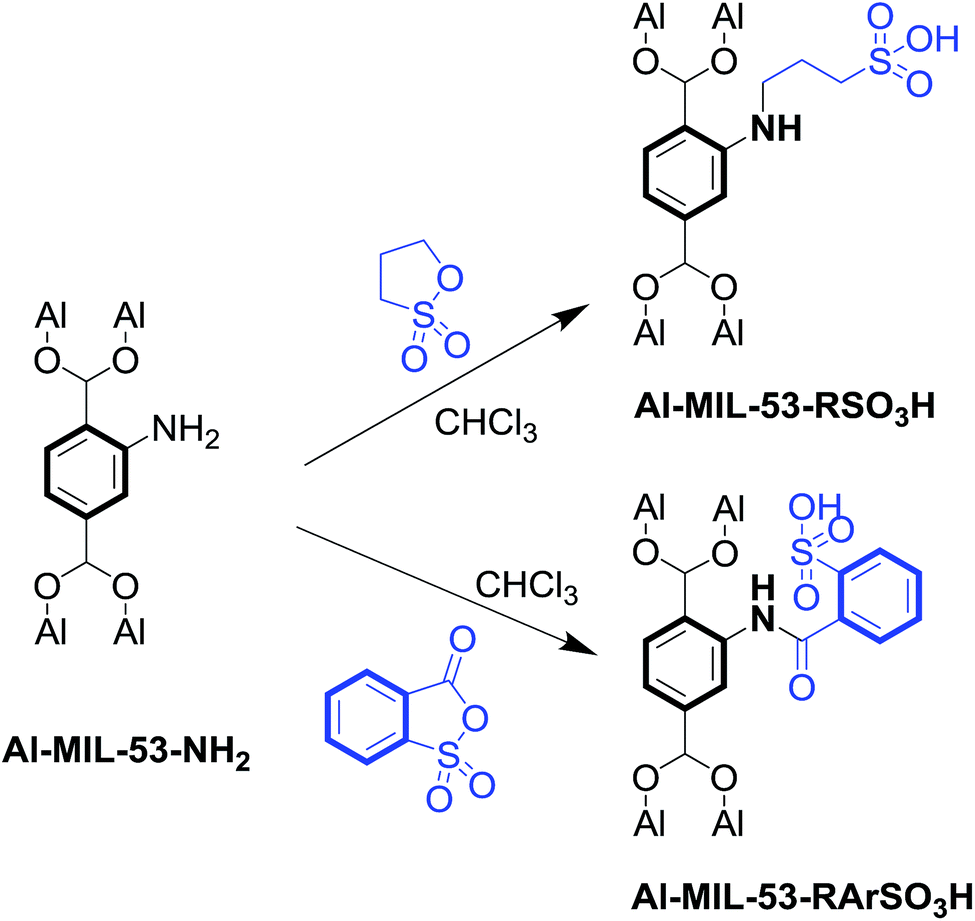 | ||
| Scheme 1 Schematic illustration of the post-synthetic modification of Al–MIL-53–NH2. | ||
The Al–MIL-53–NH2 crystals appeared to be uniform nanorods 35 nm wide and 210 nm long (Fig. 1a). The synthesis of the Al–MIL-53–RSO3H catalyst was achieved by treating Al–MIL-53–NH2 crystals with 1,3-propanesultone in chloroform solution (Scheme 1). As evidenced by scanning electron microscopy, the structural morphology was maintained during the post-synthetic modification of the Al–MIL-53–NH2 crystals (Fig. 1b). Similarly, the structural morphology of Al–MIL-53–ArSO3H was also maintained (Fig. 1c).
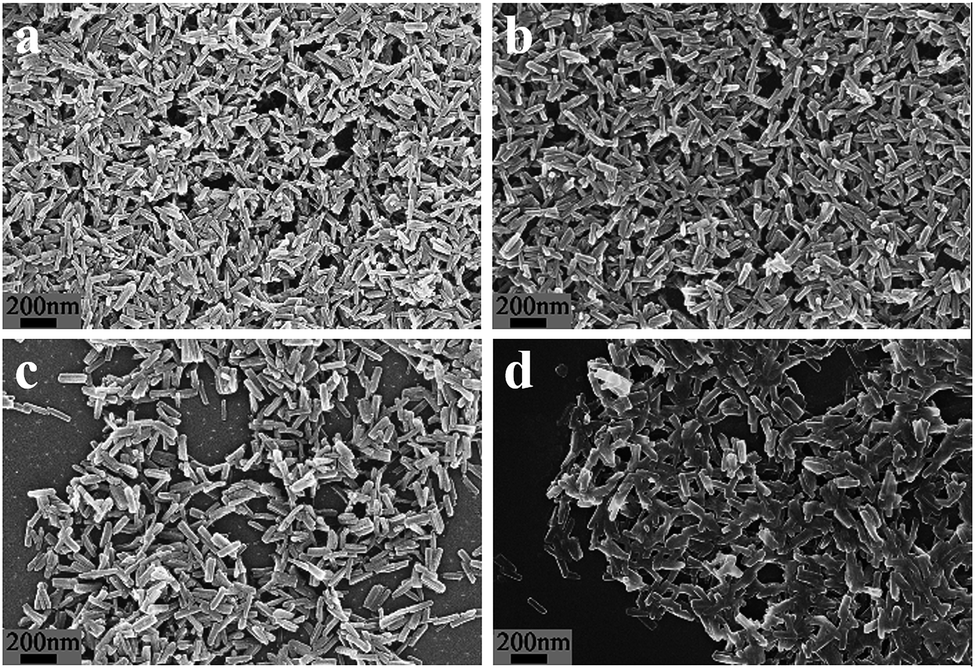 | ||
| Fig. 1 SEM of (a) Al–MIL-53–NH2, (b) Al–MIL-53–RSO3H, (c) Al–MIL-53–ArSO3H and (d) recycled Al–MIL-53–ArSO3H. | ||
Powder X-ray diffraction (PXRD) studies were performed to confirm the successful synthesis of Al–MIL-53–NH2 MOF and its sulfonated derivative.17 Post-synthetic modification with 1,3-propanesultone did not cause the decomposition of the Al–MIL-53–NH2 crystalline structure, underlining the stability of the Al–MIL-53–NH2 derived Brønsted acid (Fig. 2b). Moreover, treatment with o-sulfobenzoic acid anhydride also maintained the crystalline structure of Al–MIL-53–NH2 (Fig. 2c).
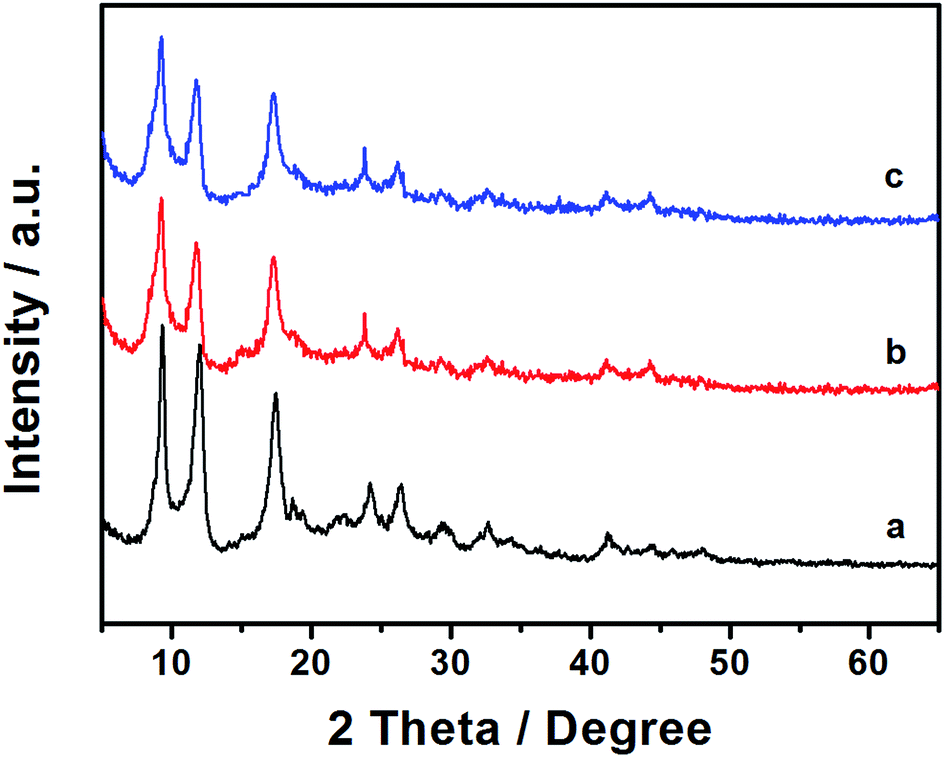 | ||
| Fig. 2 Powder XRD of (a) Al–MIL-53–NH2, (b) Al–MIL-53–RSO3H and (c) Al–MIL-53–ArSO3H. | ||
The modification ratio of aromatic sulfonic moiety on Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H was measured by 1H NMR, by comparing the spectra of the modified materials with that of Al–MIL-53–NH2 (Fig. 3). Al–MIL-53–NH2 digested in the presence of HF clearly showed three peaks. The singlet proton represents the aromatic proton adjacent to the amino functional group. The two doublets instead belong to the protons in the para and meta positions with respect to the amino group. Proton nuclear magnetic resonance studies of the digested sample indicated that 20% of Al–MIL-53–RSO3H amino groups were post-synthetically functionalized (Fig. 3b). This observation was similar to results previously reported in literature; similarly, partial NH2 group conversion has also been reported.26 In a similar fashion, Al–MIL-53–ArSO3H showed a modification ratio of 21% (Fig. 3c). These results clearly demonstrated the successful post-synthetic modification of amino functional groups, as well as the new installation of new acidic moieties. Additional evidence for sulfonic acid modification was obtained via FT-IR spectra of Al–MIL-53–NH2 and Al–MIL-53–RSO3H (Fig. S2†). The 1254 cm−1 peak corresponding to the free amine group was significantly reduced, which suggests the conversion of free amino groups to alkyl sulfonic groups (Fig. S2b and c†). However, FT-IR provided limited information in terms of detailed structure, since the peak of SO3− is not significant on FT-IR spectra.
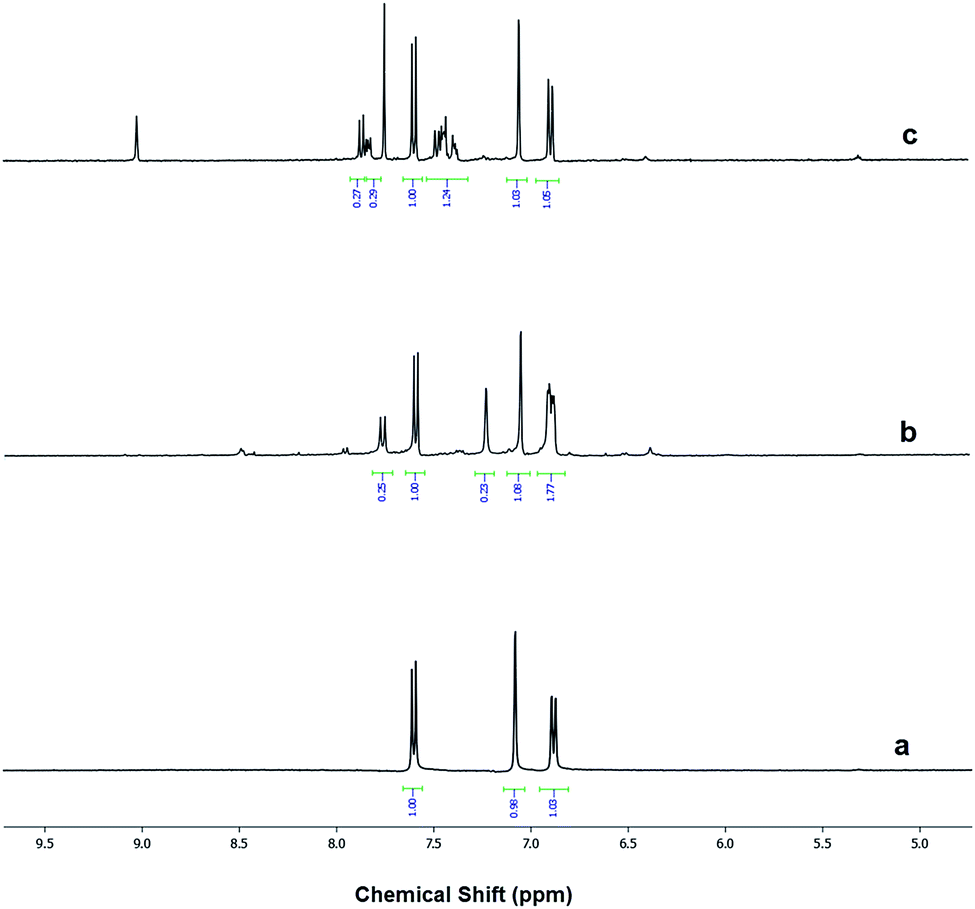 | ||
| Fig. 3 1H NMR spectra of (a) digested Al–MIL-53–NH2, (b) digested Al–MIL-53–RSO3H and (c) digested Al–MIL-53–ArSO3H. | ||
The specific surface areas of the products were analyzed by N2 adsorption–desorption measurements at 77 K. As shown in Fig. 4, the trace of the Al–MIL-53–NH2 was a type-I isotherm. Al–MIL-53–NH2 is a highly porous MOF material with a BET surface area of 540 m2 g−1, calculated by collected nitrogen adsorption isotherms (Fig. 4a). The surface area was reduced to 351 m2 g−1 for Al–MIL-53–RSO3H upon post-synthetic modification, presumably due to the pore structure filling by aromatic sulfonic groups (Fig. 4b). Similarly, the BET surface area for Al–MIL-53–ArSO3H was calculated to be 312 m2 g−1 (Fig. 4c). However, both Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H with reduced BET surface area performed well in our catalysis study, likely because the highly porous structure and the flexibility of the newly installed organic functional group allows the free entry and exit of the reaction substrate. Such a reduction in surface area was also observed in several post-synthetic modifications of MOFs reported in literature.27
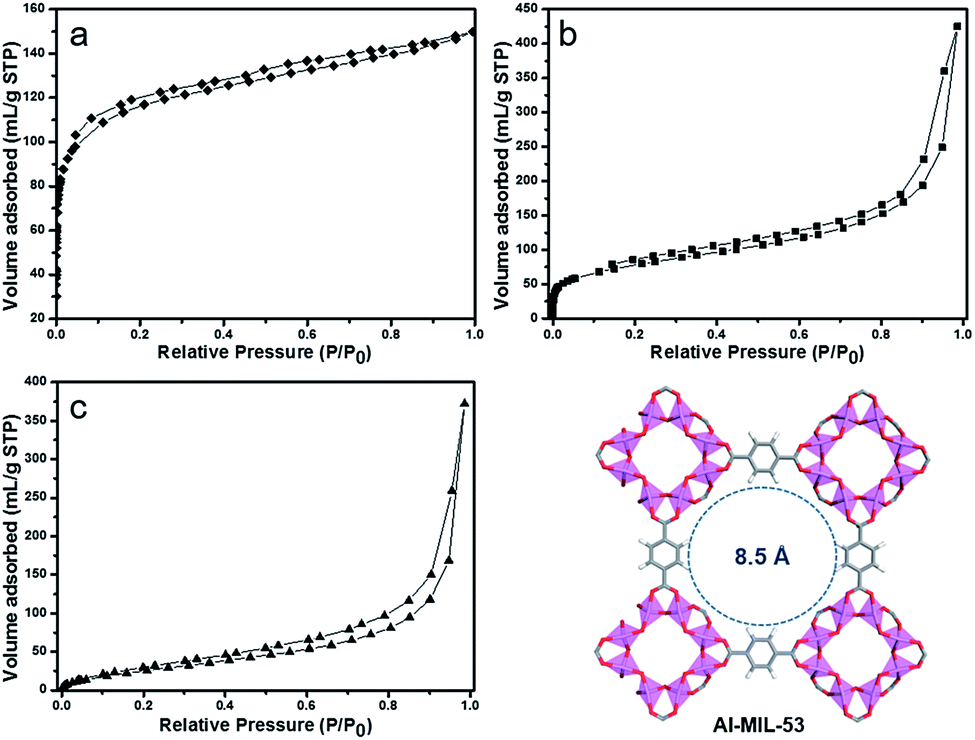 | ||
| Fig. 4 Nitrogen adsorption/desorption isotherms of (a) Al–MIL-53–NH2, (b) Al–MIL-53–RSO3H and (c) Al–MIL-53–ArSO3H. | ||
Additionally, thermal and structural stabilities of the parent Al–MIL-53–NH2, as well as of the modified Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H catalysts were examined by thermal gravimetric analysis (TGA). The weight loss of Al–MIL-53 began around 200 °C, proving its high thermal stability during organic catalysis. The weight loss of the modified Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H sample began around almost identical temperatures (Fig. 5). The TGA results proved the high thermal stability of the Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H materials, which ensures their stability in the reaction as a heterogeneous sulfonic acid catalyst. It is important to note that the stability of the material is high enough to prevent the loss of SO3 and other sulfur anhydrides, even in relatively harsh conditions.
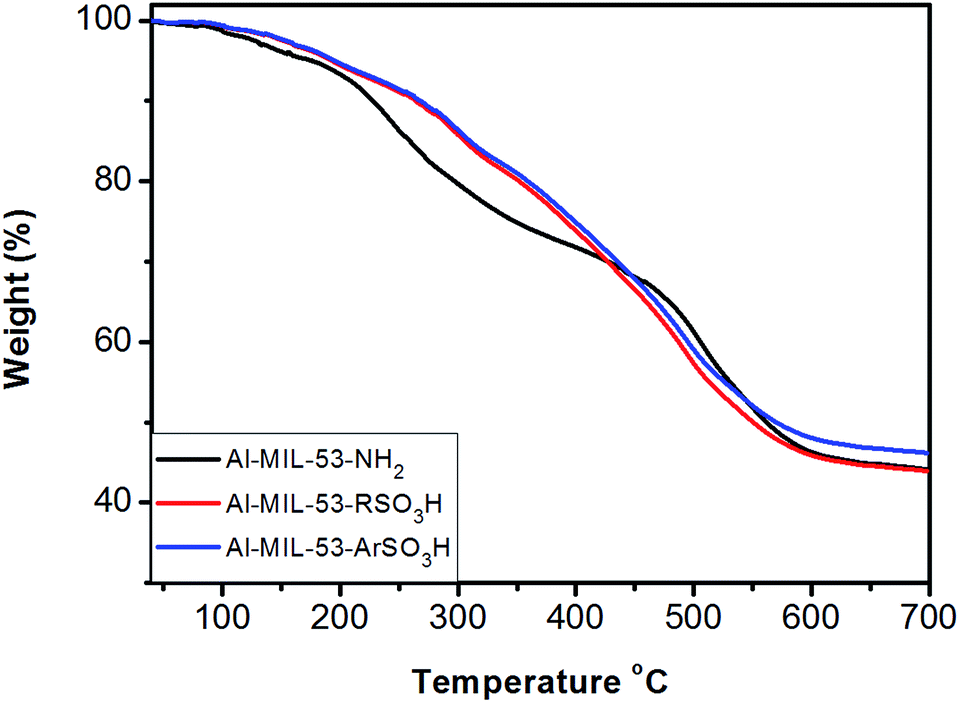 | ||
| Fig. 5 TGA of (a) Al–MIL-53–NH2, (b) Al–MIL-53–RSO3H and (c) Al–MIL-53–ArSO3H. | ||
Compound 1a was prepared following a previously reported literature procedure; the o-quinone methide intermediate was obtained through tautomerization of the benzylic carbocation.28 As reported in Table 1, cycloaddition reactions were run in the absence of catalyst (Table 1, entry 1) and with the basic Al–MIL-53–NH2 (Table 1, entry 2) as control reactions, affording no yield of 2a. Homogeneous acids were evaluated for the synthesis of 4H-chromene 2a (Table 1, entries 3–5) also affording no or poor yields and selectivity. Furthermore, hydrochloric acid and acetic acid only showed minimal reactivity at 40 °C after 3 h, presumably because of the low acidity in terms of their pKa (Table 1, entries 3–4). Only strong Brønsted acid p-TsOH functioned as an efficient catalyst in the [4 + 2] cycloaddition reaction, which gave 4H-chromene 2a a yield of 56% (Table 1, entry 5). The unsatisfactory yield was due to the incompletion of the chromene formation reaction pathway, which left significant amount of reaction intermediate 3. The turnover-number (TON) was calculated based on the total conversion of 4H-chromene 2a. 2a was then subjected to the reaction in presence of 1 mol% Al–MIL-53–RSO3H, yielding 89% of product (Table 1, entry 6). Remarkably, Al–MIL-53–ArSO3H gave the desired 4H-chromene product 2a in almost quantitative yield after 3 h in toluene (Table 1, entry 7). The TON of Al–MIL-53–ArSO3H under the optimized reaction conditions was calculated to be 98. This observation strongly demonstrated the unique property of the Al–MIL-53 derived Brønsted acid catalyst. Only the Al–MIL-53 derived acid catalyst achieved the desired cycloaddition product in high yield and high selectivity. Further screening indicated that aprotic solvents (e.g. PhCH3, CH2Cl2, THF, was calculated based on the amount of isolated product CH3CN) appeared to be more compatible (Table 1, entries 7–10), while protic solvents such as ethanol only gave very poor yields of the desired product 2a (Table 1, entry 11). The optimized reaction condition was set to use 1 mol% solid acid catalyst. Chapman reported a homodimerization of carpanone and only 46% yield was achieved using Pd2+ catalyst.29 Wan has developed a photo initiated dimerization of phenol type compound and 88% yield was achieved after 3 h.30 Schaus also reported an iron-catalyst dimerization of 2H-chromenes; 85% yield and 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereoselectivity were achieved after 12 h.31 Our strategy effectively assemble two oQM precursor, furnished a dimeric structure in high yield and selectivity, while the catalyst loading was low. Our methodology demonstrated high industrial practical use in terms of low catalyst usage, high yield and high selectivity, when compared with literature reports.
:1 diastereoselectivity were achieved after 12 h.31 Our strategy effectively assemble two oQM precursor, furnished a dimeric structure in high yield and selectivity, while the catalyst loading was low. Our methodology demonstrated high industrial practical use in terms of low catalyst usage, high yield and high selectivity, when compared with literature reports.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Yielda of 2a | Yieldb of 3 | Conv. of 1 |
| a Reaction conditions: 1a (2.0 mmol), 1 mol% catalyst based on acid group, solvent (5 mL) at 40 °C for 3 h.b Isolated yield. | |||||
| 1 | — | PhCH3 | 0% | 0% | — |
| 2 | Al–MIL-53–NH2 | PhCH3 | 0% | 0% | — |
| 3 | HCl | PhCH3 | <5% | <5% | — |
| 4 | AcOH | PhCH3 | <5% | <5% | — |
| 5 | p-TsOH | PhCH3 | 56% | 42% | 98% |
| 6 | Al–MIL-53–RSO3H | PhCH3 | 85% | 11% | 96% |
| 7 | Al–MIL-53–ArSO3H | PhCH3 | 98% | <1% | 99% |
| 8 | Al–MIL-53–ArSO3H | CH2Cl2 | 92% | <1% | 93% |
| 9 | Al–MIL-53–ArSO3H | THF | 47% | 9% | 56% |
| 10 | Al–MIL-53–ArSO3H | CH3CN | 90% | <1% | 91% |
| 11 | Al–MIL-53–ArSO3H | EtOH | 36% | 6% | 42% |
Under the optimized conditions, a broad range of 1,1-diphenylethylenes (1a–1h) was tested. The reaction showed good tolerance toward electron-rich and electron neutral functional groups. A wide range of functional groups, such as methoxy, methyl, and dioxo, performed well (Table 2, entries 1–6). The non-substituted ethylene (1b) and methyl substituted ethylene (1c) also afforded the desired products 2b and 2c in good yield (Table 2, entries 2–3). meta-Substitution patterns were also tested using methoxy groups, which provided the desired product in good yield (2d, 85% yield). In addition, very electron-rich aromatic rings, such 3,4-benzodioxole also afforded good yields (2e, 76% yield). Generally, more electron-rich functional groups provide increased yields in only 3 hours. The decreasing yield of 2e is likely due to the instability of the methylenedioxy moiety under acidic condition (Table 1, entry 5). Highly electron-rich substrate 1f provided almost quantitative yield of cycloaddition product 2f (Table 1, entry 6). Furthermore, an electron-deficient aromatic group also worked well (Table 2, entry 7). A sterically more hindering substrate was also tested affording a compromised yield 2h (Table 2, entry 8).
|
||||
|---|---|---|---|---|
| Entry | R | Ar | Product | Yield |
| a 2.0 mmol of substrate 1 and 0.02 mmol of Al–MIL-53–ArSO3H were dissolved in 2.0 mL of toluene and stirred at 40 °C for 3 h. The yield was calculated based on the amount of isolated product. | ||||
| 1 | 5-OMe |  |
2a | 98% |
| 2 | 5-OMe | 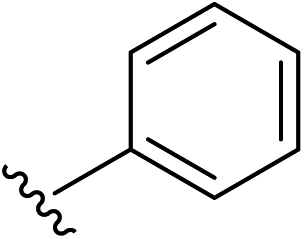 |
2b | 94% |
| 3 | 5-OMe | 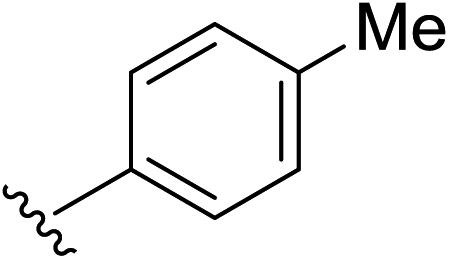 |
2c | 86% |
| 4 | 5-OMe | 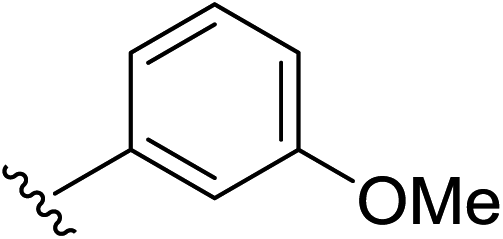 |
2d | 85% |
| 5 | 5-OMe | 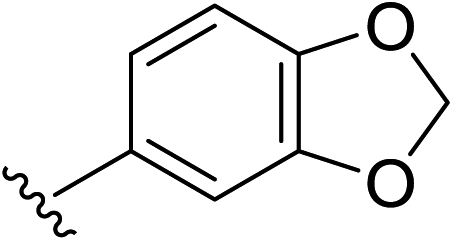 |
2e | 76% |
| 6 | 4,5-(OCH2O) |  |
2f | 95% |
| 7 | 5-OMe |  |
2g | 72% |
| 8 | 5-OMe |  |
2h | 70% |
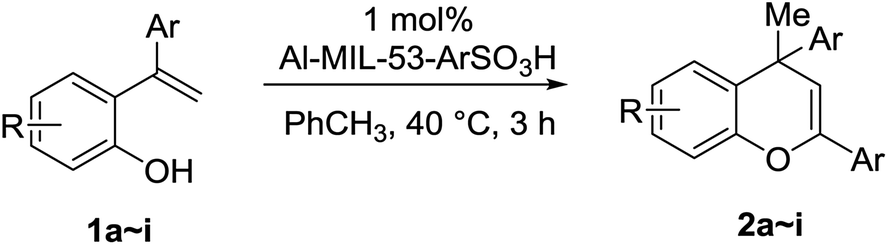
A deuterium exchange experiment was performed to further study the quinone methide mechanism (Scheme 2). Deuterated phenol 4 was synthesized and subjected to 60 °C thermal condition. However, there was no deuterium migration to the vinyl position in 7. This observation allows us to exclude the possibility of a 1,5-hydride shift mechanism for the generation of the quinone methide species.
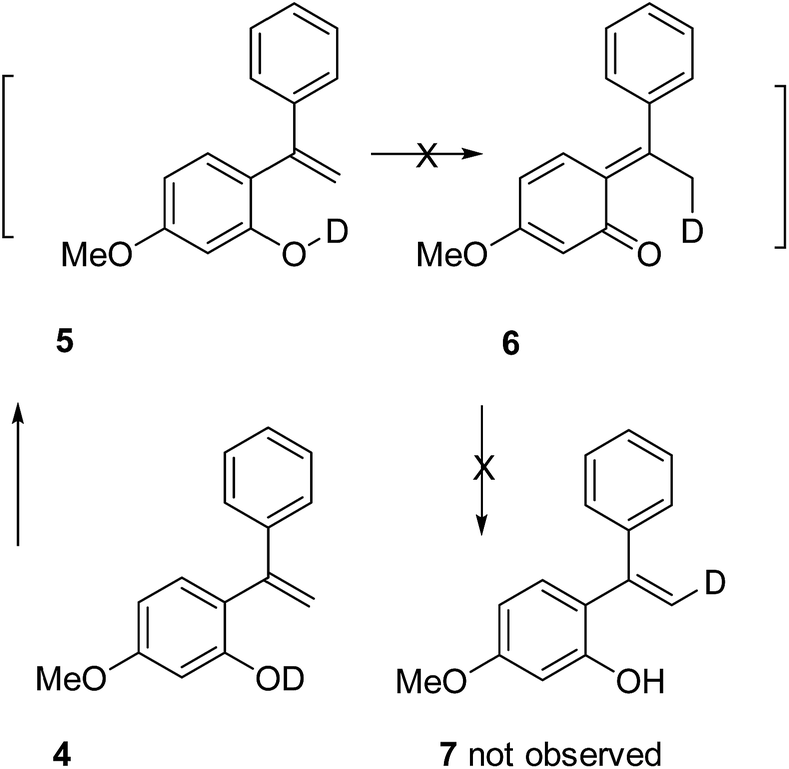 | ||
| Scheme 2 A deuterium exchange test. | ||
In order to examine its recyclability, the Al–MIL-53–ArSO3H catalyst was isolated and reused in five reaction cycles. The catalyst was centrifuged from the reaction solution and washed with chloroform. The strong covalent bond between the aromatic sulfonic acid moiety and amino group on Al–MIL-53–ArSO3H ensures the chemical stability of the active sites, which remains good, affording up to 98% yield after five reuses of the same batch of catalyst (Fig. 6). Furthermore, the supernatant liquid of the toluene suspension showed no catalytic reactivity toward the vinyl phenol substrate, which is evidence for no leakage of Al–MIL-53–ArSO3H catalyst. The SEM spectra and X-ray powder diffraction pattern of the Al–MIL-53–ArSO3H catalyst were characterized after five reuses.
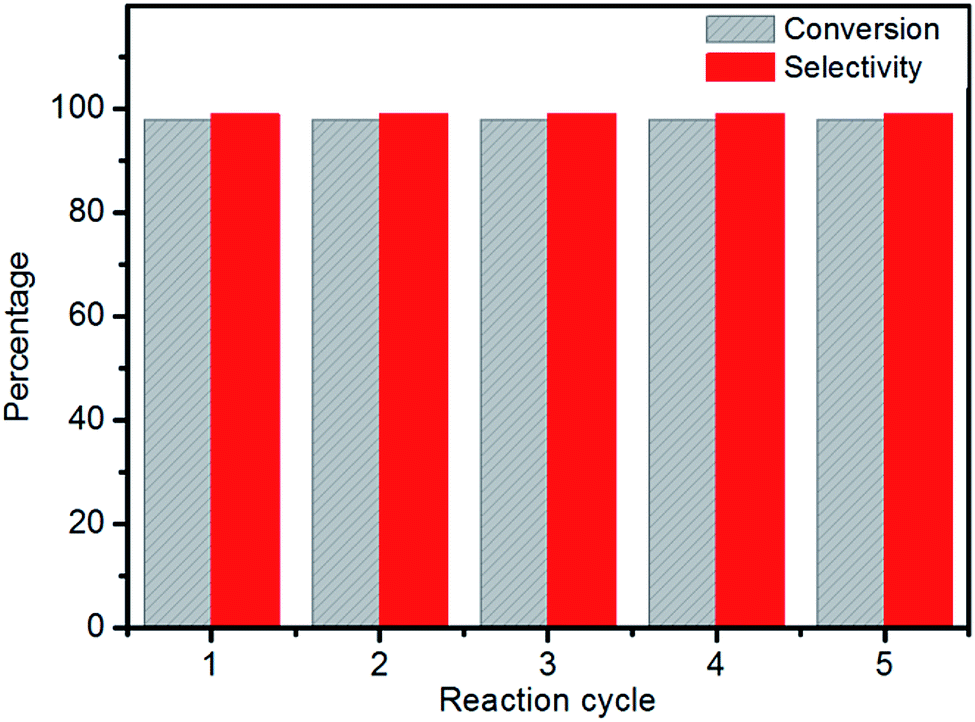 | ||
| Fig. 6 The catalyst recycling of Al–MIL-53–ArSO3H in cycloaddition reaction of 2a. | ||
These data were indistinguishable from those collected from a batch of fresh catalyst (Fig. 1d and S1†). The catalytic reaction solution was analyzed by ICP-AES and showed no detectable amounts of alumina ions. These facts clearly demonstrate that there was no catalyst decomposition or leakage over the cycloaddition process.
On the basis of the aforementioned experimental results and mechanistic findings, a possible mechanism is proposed in Scheme 2. 1,1-Diphenylethylene 1a equilibriums to form quinone methide 4 (Scheme 3). Then quinone methide 4 reacts with another equivalent of 1a through a concerted hetero-Diels–Alder reaction, to afford dimeric compound 3. Driven by steric effects, the fully substituted C-2 eliminates to generate the desired product 2a under acidic catalytic condition. The conversion of 3 to 2a was also tested using the synthesized Al–MIL-53–ArSO3H and quantitative yield was obtained for the aromatic group elimination.
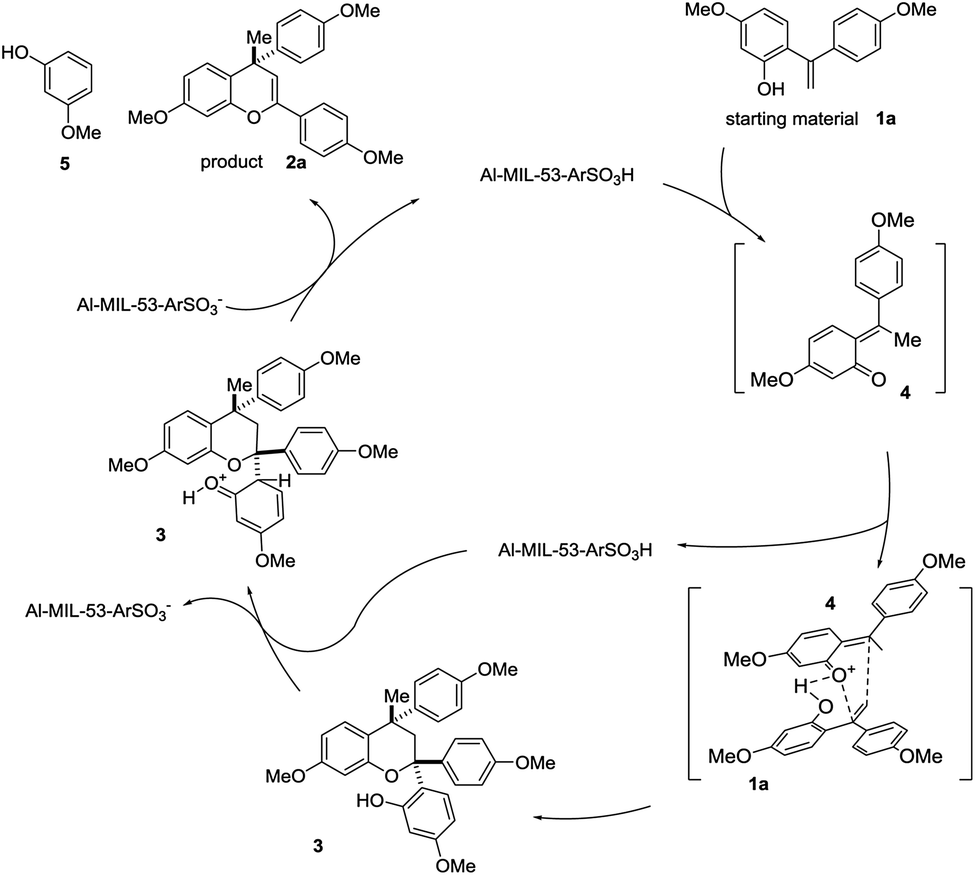 | ||
| Scheme 3 Proposed mechanism for the Al–MIL-53–ArSO3H promoted cycloaddition reaction. | ||
4 Conclusions
In conclusion, heterogeneous Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H catalysts bearing an aromatic sulfonic acid group were synthesized. The structural morphology of both Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H were well retained after post-synthetic modification using commercial available reagents. Al–MIL-53–RSO3H and Al–MIL-53–ArSO3H showed good to high activity and selectivity in the [4 + 2] cycloaddition at only 1 mol% catalyst loadings. Our methodology demonstrated higher reaction efficiency and higher selectivity than other literature reported dimerization system, while only low catalyst loading was utilized. Al–MIL-53–ArSO3H demonstrated superior catalytic activity over its non-aromatic partner Al–MIL-53–RSO3H. The Al–MIL-53–ArSO3H catalyst was chemically stable, possibly due to its strong covalent bonds and it did not show any leaching during cycloaddition catalysis. Further studies involving new applications of the Al–MIL-53 derived catalysts are in progress.Acknowledgements
This work is supported by Beijing Natural Science Foundation (2172037) and National Natural Science Foundation of China (No. 51503016). Y. P. thanks the State Key Laboratory of Chemical Resource Engineering and Y. L. thanks the Fundamental Research Funds for the Central Universities (Grant No. FRF-TP-16-004A3) for funding support.Notes and references
- (a) C. H. Cheona and H. Yamamoto, Chem. Commun., 2011, 47, 3043–3056 RSC; (b) S. Liang, G. B. Hammond and B. Xu, Chem. Commun., 2015, 51, 903–906 RSC; (c) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999–1010 CrossRef CAS.
- (a) S. M. George, Chem. Rev., 1995, 95, 475–476 CrossRef CAS; (b) Y. Chang and C. Bae, Curr. Org. Synth., 2011, 8, 208–236 CrossRef CAS; (c) O. Kwon, S. Park and G. Seo, Chem. Commun., 2007, 40, 4113–4115 RSC; (d) S. Liang, J. Jasinski, G. B. Hammond and B. Xu, Org. Lett., 2015, 17, 162–165 CrossRef CAS PubMed; (e) X. X. Huang, A. Goswami, X. X. Zou, S. Hayes, V. Shah, T. Minko, Z. M. Tao and T. Asefa, BioNanoScience, 2014, 4, 27–36 CrossRef; (f) H. Zhang, X. R. Lin, S. Chin and M. W. Grinstaff, J. Am. Chem. Soc., 2015, 137, 12660–12666 CrossRef CAS PubMed.
- B. J. Gao, D. L. Kong and Y. Zhang, J. Mol. Catal. A: Chem., 2008, 286, 143–148 CrossRef CAS.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- (a) Y. Luan, Y. Qi, J. Yu, H. Gao and S. E. Schaus, RSC Adv., 2014, 4, 34199–34203 RSC; (b) X. Du, C. X. Zhao, X. Y. Li, H. W. Huang, Y. Q. Wen, X. J. Zhang and J. Q. Li, J. Alloys Compd., 2017, 700, 83–91 CrossRef CAS; (c) X. Du, C. X. Zhao, X. Y. Li, H. W. Huang, J. H. He, Y. Q. Wen and X. J. Zhang, Eur. J. Inorg. Chem., 2017, 2017, 2517–2524 CrossRef CAS; (d) X. Du, C. X. Zhao, M. Y. Zhou, T. Y. Ma, H. W. Huang, M. Jaroniec, X. J. Zhang and S. Z. Qiao, Small, 2017, 13, 1602592 CrossRef PubMed.
- (a) A. Corma, H. Carcia and F. X. L. I. Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed; (b) J. Gascon, A. Corma, F. Kapteijn and F. X. L. I. Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkovc and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- (a) J. Juan-Alcaniz, R. Gielisse, A. B. Lago, E. V. Ramos-Fernandez, P. Serra-Crespo, T. Devic, N. Guillou, C. Serre, F. Kapteijn and J. Gascon, Catal. Sci. Technol., 2013, 3, 2311–2318 RSC; (b) Y. D. Zang, J. Shi, F. M. Zhang, Y. J. Zhong and W. D. Zhu, Catal. Sci. Technol., 2013, 3, 2044–2049 RSC; (c) Y. X. Zhou, Y. Z. Chen, Y. Hu, G. Huang, S. H. Yu and H. L. Jiang, Chem.–Eur. J., 2014, 20, 14976–14980 CrossRef CAS PubMed; (d) M. Zheng, Y. Liu, C. Wang, S. Liu and W. Lin, Chem. Sci., 2012, 3, 2623–2627 RSC; (e) Y. Liu, X. B. Xi, C. C. Ye, T. F. Gong, Z. W. Yang and Y. Cui, Angew. Chem., Int. Ed., 2014, 53, 13821–13825 CrossRef CAS PubMed; (f) B. Y. Li, K. Y. Leng, Y. M. Zhang, J. J. Dynes, J. Wang, Y. F. Hu, D. X. Ma, Z. Shi, L. K. Zhu, D. L. Zhang, Y. Y. Sun, M. Chrzanowski and S. Q. Ma, J. Am. Chem. Soc., 2015, 137, 4243–4248 CrossRef CAS PubMed.
- J. Jiang and O. M. Yaghi, Chem. Rev., 2015, 115, 6966–6997 CrossRef CAS PubMed.
- (a) M. B. Gawande, A. K. Rathi, I. D. Nogueira, R. S. Varma and P. S. Branco, Green Chem., 2013, 15, 1895–1899 RSC; (b) M. G. Goesten, J. Juan-Alcaniz, E. V. Ramos-Fernandez, K. B. S. S. Gupta, E. Stavitski, H. van Bekkum, J. Gascon and F. Kapteijn, J. Catal., 2011, 281, 177–187 CrossRef CAS.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC.
- D. Britt, C. Lee, F. J. Uribe-Romo, H. Furukawa and O. M. Yaghi, Inorg. Chem., 2010, 49, 6387–6389 CrossRef CAS PubMed.
- Y. Luan, N. N. Zheng, Y. Qi, J. Yu and G. Wang, Eur. J. Inorg. Chem., 2014, 2014, 4268–4272 CrossRef CAS.
- S. J. Garibay, Z. Q. Wang and S. M. Cohen, Inorg. Chem., 2010, 49, 8086–8091 CrossRef CAS PubMed.
- C. G. Piscopo, A. Polyzoidis, M. Schwarzer and S. Loebbecke, Microporous Mesoporous Mater., 2015, 208, 30–35 CrossRef CAS.
- (a) M. G. Goesten, J. Juan-Alcañiz, E. V. Ramos-Fernandez, K. B. S. S. Gupta, E. Stavitski, H. van Bekkum, J. Gascon and F. Kapteijn, J. Catal., 2011, 281, 177–187 CrossRef CAS; (b) C. Qi, D. Ramella, A. M. Wensley and Y. Luan, Adv. Synth. Catal., 2016, 358, 2604–2611 CrossRef CAS.
- (a) V. Eschenbrener-Lux, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2014, 53, 11146–11157 CrossRef PubMed; (b) X. Liu, D. Wang, H. Gao, Z. Yang, Y. Xing, H. Cao, W. L. He, H. H. Wang, J. M. Gu and H. Y. Hu, Dyes Pigm., 2016, 134, 155–163 CrossRef CAS.
- T. P. Pathak and M. S. Sigman, J. Org. Chem., 2011, 76, 9210–9215 CrossRef CAS PubMed.
- (a) M. S. Singh, A. Nagaraju, N. Anand and S. Chowdhury, RSC Adv., 2014, 4, 55924–55959 RSC; (b) X. Liu, D. Wang, H. Gao, Z. Yang, Y. Xing, H. Cao, W. L. He, H. H. Wang, J. M. Gu and H. Y. Hu, Phys. Chem. Chem. Phys., 2016, 18, 7341–7348 RSC.
- (a) S. B. Ferreira, F. C. Silva, A. C. Pinto, D. T. G. Gonzaga and V. F. Ferreira, J. Heterocycl. Chem., 2009, 46, 1080–1097 CrossRef CAS; (b) N. Willis and C. D. Bray, Chem.–Eur. J., 2012, 18, 9160–9173 CrossRef CAS PubMed.
- (a) M. Fujiwara, M. Sakamoto, K. Komeyama, H. Yoshida and K. Takaki, J. Heterocycl. Chem., 2015, 52, 59–66 CrossRef CAS; (b) D. Wang, Q. S. Guo, H. Gao, Z. Yang, Y. Xing, H. Cao, W. L. He, H. H. Wang, J. M. Gu and H. Y. Hu, Polym. Chem., 2016, 7, 3714–3721 RSC.
- (a) M. Costa, T. A. Dias, A. Brito and F. Proença, Eur. J. Med. Chem., 2016, 123, 487–507 CrossRef CAS PubMed; (b) C. Bingi, N. R. Emmadi, M. Chennapuram, Y. Poornachandra, C. G. Kumar, J. B. Nanubolu and K. Atmakur, Bioorg. Med. Chem. Lett., 2015, 25, 1915–1919 CrossRef CAS PubMed; (c) M. N. Semenova, D. V. Tsyganov, O. R. Malyshev, O. V. Ershov, I. N. Bardasov, R. V. Semenov, A. S. Kiselyov and V. V. Semenov, Bioorg. Med. Chem. Lett., 2014, 24, 3914–3918 CrossRef CAS PubMed.
- (a) D. M. Zhang, Y. J. Guan, E. J. M. Hensen, T. Xue and Y. M. Wang, Catal. Sci. Technol., 2014, 4, 795–802 RSC; (b) J. L. Yan, S. Jiang, S. F. Ji, D. Shi and H. F. Cheng, Sci. China: Chem., 2015, 58, 1–8 CrossRef.
- Z. Y. Wang and A. Stein, Chem. Mater., 2008, 20, 1029–1040 CrossRef CAS.
- S. J. Garibay, Z. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 7341–7349 CrossRef CAS PubMed.
- Y. Luan, Y. Qi, H. Y. Gao, R. S. Andriamitantsoa, N. N. Zheng and G. Wang, J. Mater. Chem. A, 2015, 3, 17320–17331 CAS.
- W. X. Zhao, Z. B. Wang, B. Y. Chu and J. W. Sun, Angew. Chem., Int. Ed., 2015, 54, 1910–1913 CrossRef CAS PubMed.
- O. L. Chapman, M. R. Engel, J. P. Springer and J. C. Clardy, J. Am. Chem. Soc., 1971, 93, 6696–6698 CrossRef CAS.
- M. K. Nayak and P. Wan, Photochem. Photobiol. Sci., 2008, 7, 1544–1554 CAS.
- Y. Luan, H. Sun and S. E. Schaus, Org. Lett., 2011, 13, 6480–6483 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06201j |
| This journal is © The Royal Society of Chemistry 2017 |