
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethane combustion over Pd/CoAl2O4/Al2O3 catalysts prepared by galvanic deposition†
Yuji Mahara a,
Takumi Tojoa,
Kazumasa Murataa,
Junya Ohyamaab and
Atsushi Satsuma*ab
a,
Takumi Tojoa,
Kazumasa Murataa,
Junya Ohyamaab and
Atsushi Satsuma*ab
aGraduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan. E-mail: satsuma@chembio.nagoya-u.ac.jp
bUnit of Elements Strategy Initiative for Catalysts & Batteries, Kyoto University, Kyoto 615-8530, Japan
First published on 11th July 2017
Abstract
Pd/CoAl2O4/Al2O3 methane combustion catalysts were synthesized using a galvanic deposition (GD) method (PdCoAl-GD). This PdCoAl-GD catalyst showed higher activities and turnover frequencies (TOFs) than conventional Pd/Al2O3. According to X-ray diffraction (XRD), X-ray absorption fine structure (XAFS) and scanning transmission electron microscopy (STEM) measurements, PdCoAl-GD was composed of a CoAl2O4 phase supported on γ-Al2O3 and dispersed Pd nanoparticles of 2–7 nm in size on CoAl2O4. Operando Pd K-edge XAFS measurements indicated that the Pd in PdCoAl-GD was oxidized to highly active methane combustion PdO species at low temperatures. PdCoAl-GD also showed high activity (light-off tests) when PdO was initially present on the catalysts. Methane temperature-programmed reaction (CH4-TPR) measurements on PdCoAl-GD revealed that PdO was reduced by CH4 at low temperatures. The GD method used herein achieved PdO species that were effective for C–H activation.
Introduction
Natural gas vehicles are becoming widely used because of their cleaner exhaust gas characteristics compared to conventional fossil fuel-based automobiles. However, unburned methane emissions are hazardous and have a high greenhouse effect coefficient that is 21 times higher than carbon dioxide. Complete oxidation of methane using supported heterogeneous catalysts has become increasingly important for emission reduction of unburned methane.1–3 Supported Pd catalysts have been used because of their high activity in methane combustion,4–37 and alumina-supported Pd catalysts (Pd/Al2O3) are used, especially, due to their high activity and thermal stability. However, methane combustion activity at low temperatures is still insufficient and the development of a highly active catalyst at low temperatures is strongly desired.For the aim of designing catalysts with higher activity, it is important to control the Pd–metal oxide interaction. Cargnello et al. synthesized Pd@CeO2 catalysts that have attracted attention because of their exceptional methane combustion activities,36 and the influence of the Pd–CeO2 interfaces on high methane combustion activity has been experimentally and theoretically studied.7,38,39 Zou et al. prepared alumina-supported core–shell NiO@PdO catalysts with higher Pd utilization efficiencies than Pd/CeO2. However, these catalysts were prepared by a complex method (i.e., multi-step reactions and protecting agents); thus, a simpler preparation method is desired. The addition of transition metal (e.g., Ni or Co) oxides to Pd/Al2O3 catalysts has been reported to improve their methane combustion activity.4,6,13,23,25,30,32 However, to improve the activity, Pd is required to be in good contact with the additional element species. In this sense, we used a galvanic deposition (GD) method to enhance the interaction between Pd and CoOx species.21 The GD method allowed the support of a metal over a second metal with a higher ionization tendency. Thus, different metals were deposited upon contact between the two metals, thereby enhancing their interfacial interaction. Pd–Co/Al2O3 catalysts prepared by a GD method showed significantly higher methane combustion activities compared to Pd/Al2O3 and Pd–Co/Al2O3 prepared by a conventional impregnation method. However, this improvement in activity was insufficient because of the poor contact between Pd and the relatively large Co particles. The interfacial contact between Pd and Co is expected to improve by precipitating Pd with a GD method after dispersion of Co in a solid solution in Al2O3.
In this study, we synthesized highly active Pd/CoAl2O4/Al2O3 methane combustion catalysts with enhanced interfacial contact between Pd and CoAl2O4 using a GD method. The oxidation state of Pd during the reaction was observed in situ by operando X-ray absorption fine structure (XAFS) spectroscopy.
Experimental section
Catalyst preparation
Co(NO3)2·6H2O (98%) was purchased from Kishida Chemical. A 4.5 wt% Pd(NO3)2 aqueous solution and high purity Al2O3 (99%) were supplied from Cataler Corporation and SASOL, respectively.CoAl2O4/Al2O3 (5 wt% Co loading) was prepared by an impregnation method. An aqueous suspension containing boehmite and Co(NO3)2·2H2O was stirred for 1 h. After evaporation and overnight drying at 80 °C, the resulting solid was calcined at 900 °C for 3 h. The solid was denoted as CoAl900.
Pd/CoAl2O4/Al2O3 was prepared with CoAl900 and Pd(NO3)2 by a GD method as described in previous reports (Scheme 1).21,40–42 2.5 g of CoAl900 was added to a two-neck round-bottom flask that was capped with a septum. CoAl900 was reduced under flowing H2 at 600 °C for 30 min. After switching to N2, the two-neck round-bottom flask containing CoAl900 was kept at 60 °C. 40 mL of degassed H2O was added to a two-neck round-bottom flask under stirring and 0.478 mmol of aqueous Pd(NO3)2 was subsequently injected. The slurry was stirred for 1 h, centrifuged, and dried overnight. The resulting material was denoted as PdCoAl-GD. Inductively coupled plasma analysis revealed a Pd loading of 1.9 wt% on PdCoAl-GD, which was equal to the nominal loading. The Co loading was lower than that of CoAl900 (4.7 vs. 5.0 wt%), thereby revealing that a fraction of Co was dissolved into water during deposition of Pd.
 | ||
| Scheme 1 Synthetic procedure for Pd/CoAl2O4/Al2O3 using galvanic deposition method. | ||
As a reference catalyst, Pd/Al2O3 was prepared by a conventional impregnation method using a Pd(NO3)2 solution and Al2O3. The mixture was dried and calcined in air at 500 °C for 3 h (denoted as PdAl-I). A Pd/CoAl2O4/Al2O3 catalyst was also prepared by a sequential impregnation method using a Pd(NO3)2 solution and CoAl900. The mixture was dried and calcined in air at 500 °C for 3 h (denoted as PdCoAl-SI). The Pd and Co loadings of the catalysts were adjusted to 2 and 4.9 wt%, respectively.
Catalytic tests
Light-off methane combustion tests were conducted using a conventional fixed-bed flow reactor at atmospheric pressure with 20 mg of catalyst loaded in a Pyrex glass tube with an internal diameter of 4 mm. In this study, we performed catalytic tests under two pretreatment conditions: (i) O2 at 500 °C for 10 min, followed by a H2 treatment at 500 °C for 10 min (initial state of Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd0); and (ii) O2 treatment at 500 °C for 10 min (initial state of Pd:PdO). The catalytic tests were performed under flowing (100 mL min−1) 0.4% CH4/10% O2/He balance. The reaction temperature ranged from 200 to 600 °C, and the heating rate was 5 °C min−1. The gas hourly space velocity was 300000 mL (h gcat)−1. The effluent gas was analyzed by a nondispersive infrared (NDIR) CO/CO2 analyzer (Horiba VIA510) while stepwise increasing the reaction temperature.
:Pd0); and (ii) O2 treatment at 500 °C for 10 min (initial state of Pd:PdO). The catalytic tests were performed under flowing (100 mL min−1) 0.4% CH4/10% O2/He balance. The reaction temperature ranged from 200 to 600 °C, and the heating rate was 5 °C min−1. The gas hourly space velocity was 300000 mL (h gcat)−1. The effluent gas was analyzed by a nondispersive infrared (NDIR) CO/CO2 analyzer (Horiba VIA510) while stepwise increasing the reaction temperature.
Catalyst characterization
The prepared catalysts were characterized by X-ray diffraction (XRD), XAFS, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), X-ray photoelectron spectroscopy (XPS), CO adsorption, as well as methane temperature-programmed reductions (CH4-TPR).The XRD patterns of the catalysts were recorded on a Rigaku MiniFlex II/AP diffractometer with a Cu Kα radiation. All samples were measured after preparation without performing any pretreatment.
The Co K-edge XAFS spectra were obtained on the BL01B1 beamline at the SPring-8 synchrotron radiation facility (8 GeV, 100 mA) of the Japan Synchrotron Radiation Research Institute (JASRI) in Hyogo, Japan. The XAFS measurements (transmission mode) were performed ex situ. The samples were pelletized (7 mm in diameter). PdCoAl-GD and PdCoAl-SI were ex situ measured after methane combustion tests at 600 °C, while CoAl900 was measured after no pretreatment. The data analysis was performed using the Athena and Artemis softwares including in the Demeter package. The curve-fitting analysis of the EXAFS spectra was performed for the inverse Fourier transforms assuming single scattering using the theoretical parameters calculated by FEFF6. The Co K-edge EXAFS data were fitted between 3–12 Å in k space and 1–3 Å in R space.
HAADF-STEM and energy dispersive X-ray (EDX) images were collected using a JEOL JEM-2100F microscope operating at 200 kV. For TEM analysis, the spent catalysts were dispersed in methanol and dropped onto a Cu mesh provided with a carbon microgrid.
XPS measurements were performed on a JPS-9000MC system (JEOL Ltd.) with Mg Kα radiation to understand the electronic properties of the Pd surface after methane combustion. Because the peak position of the Pd2+ 3d5/2 derived from PdO bulk was 337.3 eV, this value was fit to Pdn+ without constraint conditions. The Pd2+ 3d3/2 and Pdn+ 3d3/2 peaks were analyzed considering that the area of this peak was two-thirds of that of 3d5/2 and four analysis peaks were obtained.
CO chemisorption was carried out on a BELCAT (Bel Japan Inc.) apparatus, while CH4-TPR was carried out using a NDIR CO/CO2 analyzer (Horiba VIA510). CO chemisorption was performed after oxidation (O2 at 500 °C) and reduction (H2 at 50 °C) pretreatments. For the CH4-TPR measurements, 50 mg of the sample were heated from 200 to 600 °C at a rate of 5 °C min−1 under flowing 0.4% CH4 in N2 (50 mL min−1) after a pretreatment with flowing O2 at 500 °C.
Operando Pd K-edge XAFS spectroscopy
Operando Pd K-edge XAFS measurements were performed at BL01B1, SPring-8, Japan. The samples were pelletized (7 mm in diameter) and set in a quartz glass XAFS cell. The measurement sequence was carried out at the same conditions employed for the methane combustion catalytic tests. Thus, after a pretreatment with flowing O2 (500 °C) and H2 (500 °C), the catalyst was contacted with flowing 0.4% CH4/10% O2/He (reaction mixture) and heated at a rate of 5 °C min−1 from 200 to 500 °C. During this heating, quick Pd K-edge XAFS spectra were obtained in a transmission mode. In addition, the composition (i.e., CH4, CO, CO2, H2O, O2, etc.) of the outlet gas was analyzed by mass spectrometry (MS).Results and discussion
Catalytic activity
We performed methane combustion tests on the prepared catalysts. The catalysts were H2 pretreated at 500 °C to reduce the Pd species to a metallic phase. Fig. 1(a) shows the light-off methane combustion curves for PdCoAl-GD, PdCoAl-SI, PdAl-I, and CoAl900. T10 was defined as the temperature at which methane conversion reached 10%. As shown in Table 1, PdCoAl-GD showed the best low-temperature combustion activity because its T10 (266 °C) was ca. 20 °C lower than those of PdAl-I and PdCoAl-SI. T10 of PdCoAl-SI was slightly lower than that of PdAl-I. Compared with the catalysts previously reported, PdCoAl-GD showed comparable high activity (Table 2 and Fig. S1†).4,5,7,11,22,24,33,36,43 The apparent activity followed a different trend PdAl-I > PdCoAl-SI at high temperatures. CoAl900 showed no catalytic activity below 400 °C, thereby suggesting that Pd species were the active sites in this reaction. The turnover frequencies (TOFs) for the prepared catalysts were estimated by the following equation:
 | ||
| Fig. 1 (a) Methane combustion light-off curves of PdCoAl-GD, PdCoAl-SI, PdAl-I and CoAl900. Pretreatment: flowing O2 at 500 °C followed by flowing H2 at 500 °C. (b) Methane combustion TOF at 300 °C over PdCoAl-GD, PdCoAl-SI and PdAl-I. | ||
| Catalysts | T10 (°C) | CO uptakea (10−6 mol g−1) | dPd–COb (nm) | dPd-TEM (nm) | TOF at 300 °C (h−1) |
|---|---|---|---|---|---|
| a CO uptake was measured by CO chemisorption after an oxidation pretreatment with O2 at 500 °C and reduction with H2 at 50 °C.b The Pd particle size was estimated by assuming a CO to surface metal atom ratio of 1:1. |
|||||
| PdCoAl-GD | 266 | 48.5 | 4.3 | 4.2 | 590 |
| PdCoAl-SI | 285 | 33.5 | 6.3 | 6.2 | 304 |
| PdAl-I | 288 | 57.6 | 3.8 | 4.2 | 160 |
| Catalysts | Reaction ratea (μmolCH4 gcat−1 s−1) | Pd mass activitya (10 × μmolCH4 gPd−1 s−1) | Ref. |
|---|---|---|---|
| a All value is estimated from the results of methane combustion at 300 °C. | |||
| PdCoAl-GD (this work) | 8.3 | 42 | — |
| 3% Pd/CeO2-os | 7.4 | 25 | 24 |
| Pd@CeO2/H–Al2O3 | 5.1 | 51 | 36 |
| Co@C | 1.8 | — | 43 |
| 1.09 wt% Pd/CeO2 | 1.5 | 14 | 7 |
| Pd/H-ZMS-5(8.0) | 1.5 | 24 | 5 |
| Pd/Ni–Al2O3-GD | 1.3 | 2.6 | 33 |
| Pd/Co3O4–P | 1.3 | 12 | 11 |
| Pd/Co3O4–F | 1.2 | 11 | 11 |
| 1.93AuPd1.95/3DOM CoCr2O4 | 0.6 | — | 22 |
| NiO@PdO/Al2O3(2:1) |
0.3 | 14 | 4 |
The number of surface Pd atoms was estimated from the CO chemisorption data. The diameter of the Pd particles obtained by CO chemisorption was also confirmed by the particle size distribution obtained from the HAADF-STEM images. The CO uptake, Pd particle size and TOF values at 300 °C for the catalysts are summarized in Fig. 1(b) and Table 1. The TOF at 300 °C for methane combustion followed the trend PdCoAl-GD > PdCoAl-SI > PdAl-I. The TOF of PdCoAl-GD (590 h−1) was twice that of PdCoAl-SI (304 h−1), and four times higher than that of PdAl-I (160 h−1). PdCoAl-GD, PdCoAl-SI and PdAl-I were estimated to have average Pd particle sizes of 4.3, 6.3 and 3.8 nm, respectively. According to a previous work, catalysts having large sized Pd particles exhibited higher TOFs for methane combustion.37 Therefore, we prepared several Pd/Al2O3 and Pd/Co/Al2O3 catalysts with varying Pd particle sizes, and we measured the TOF values of these catalysts (Table S1†). PdCoAl-GD showed a noticeably high TOF even considering the Pd size effects (Fig. S2†). Considering the above data, PdCoAl-GD was particularly active toward the methane combustion reaction in the low-temperature range.
Catalyst structure
The catalyst structure was characterized by XRD, XAFS spectroscopy and HAADF-STEM observations.Fig. 2 shows the XRD patterns of the prepared catalysts. The diffraction patterns confirmed the presence of a γ-Al2O3 phase in all cases. Since the diffraction lines originated from Pd were hardly observed, we assumed that Pd was in the form of highly dispersed nanoparticles. The samples containing Co showed XRD patterns characteristics of Co3O4 or CoAl2O4 phases, although we could not discriminate between these structures. Therefore, to investigate the surrounding structure of Co in more detail, XAFS measurements were carried out. Fig. 3 shows the normalized Co K-edge X-ray absorption near-edge spectroscopy (XANES) spectra for PdCoAl-GD, PdCoAl-SI, CoAl900, and reference materials. All the prepared catalysts showed XANES spectra similar to that of CoAl2O4. Fig. 4 shows the k3-weighted Co K-edge EXAFS spectra of CoAl900, PdCoAl-GD, PdCoAl-SI, and reference materials. CoAl900, PdCoAl-GD and PdCoAl-SI showed EXAFS spectra similar to that of CoAl2O4 (and not Co3O4). The curve-fitting results of PdCoAl-GD are shown in Table 3. The spectra of PdCoAl-GD showed a first peak at 1.6 Å originated by the Co–O shell and a second peak at 3.0 Å ascribed to the Co–Al, Co–O and Co–Co shells of CoAl2O4 (Table 3). CoAl900 and PdCoAl-SI also showed spectra similar to that of CoAl2O4. These results indicated that Co in CoAl900 was almost completely dissolved into the γ-Al2O3 phase upon calcination at 900 °C forming a CoAl2O4 phase. Additionally, we obtained the Co 2p X-ray photoelectron spectroscopy (XPS) spectra of all the samples (Fig. S3†). Therefore, CoAl2O4 species were considered to be present near the surface of γ-Al2O3.
 | ||
| Fig. 2 X-ray diffraction patterns of: PdCoAl-GD, PdCoAl-SI, PdAl-I and CoAl900. The bottom peaks indicate the position of the reference materials. | ||
 | ||
| Fig. 3 Normalized absorption spectra at the Co K-edge for PdCoAl-GD (red), PdCoAl-SI (green), CoAl900 (pink) and references (Co foil: royal blue, CoO: orange, Co3O4: gray and CoAl2O4: black). | ||
 | ||
| Fig. 4 (a) k3-weighted Co K-edge EXAFS spectra and (b) Fourier transform of PdCoAl-GD (red), PdCoAl-SI (green), CoAl900 (pink) and references (black). Dotted lines: raw data; solid line: fitting data; marked: imaginary part of the Fourier transform. | ||
Fig. 5 shows HAADF-STEM and EDX elemental mapping images of PdCoAl-GD, PdCoAl-SI and PdAl-I. The white contrast in the HAADF-STEM images was produced by the Pd nanoparticles, while the green, yellow and red regions in the EDX elemental mapping were ascribed to Al, Co and Pd, respectively. In the case of PdCoAl-GD and PdCoAl-SI, the Co species were mainly overlapped with those of Al. According to the Co K-edge XAFS spectra, PdCoAl-GD and PdCoAl-SI possessed a CoAl2O4 phase dispersed over γ-Al2O3. In the case of PdCoAl-GD, Pd species with a size of 2–7 nm were detected overlapping with Al and Co species. Since GD proceeds upon contact between a Pd precursor and reduced Co species, Pd nanoparticles were deposited onto CoAl2O4 species. The Pd size distributions were obtained by analyzing the HAADF-STEM images (Fig. 5). The average Pd particle size calculated by STEM was in rough agreement with that estimated by CO chemisorption (Table 1) However, the enhanced activity cannot be explained only by characterizing the catalyst before methane combustion. A detailed analysis of the state of Pd during reaction is necessary.
 | ||
| Fig. 5 HAADF-STEM images, EDX element mapping, and Pd particle size histograms for PdCoAl-GD, PdCoAl-SI, and PdAl-I. | ||
Operando Pd K-edge XAFS measurements
We performed operando Pd K-edge XAFS measurements to clarify the Pd oxidation state during methane combustion. The samples were analyzed at the same conditions used for the catalytic tests including a H2 pretreatment (see Fig. 1). Fig. 6 shows a series of normalized Pd K-edge XANES spectra of PdCoAl-GD. First, after the oxidation pretreatment, Pd was in form of PdO (Fig. 6(b)). Pd was subsequently reduced to Pd0 after the H2 pretreatment (Fig. 6(c)). Pd was slightly oxidized when contacted with flowing reaction gases at 200 °C (Fig. 6(d)). The fraction of PdO gradually increased with temperature until complete oxidation of Pd. Similar operando XAFS measurements were conducted for PdCoAl-SI and PdAl-I (Fig. S4 and S5†). All Pd K-edge XANES spectra were described with a linear combination fitting (LCF) of two XANES spectra i.e., the one obtained after oxidation at 500 °C (Fig. 6(b)), and that obtained after reduction at 500 °C (Fig. 6(c)). The amount of PdO (PdO ratio) in the catalysts was estimated using LCF. During the XAFS measurements, the outlet gases were analyzed by MS, and the CH4 conversion was calculated with the reaction temperature. Fig. 7(a)–(c) show the PdO ratio and CH4 conversion values under reaction for PdCoAl-GD, PdCoAl-SI and PdAl-I. In the case of PdCoAl-GD, the CH4 conversion increased with the PdO ratio (900 s, reaction temperature: 275 °C). A similar trend was found for PdCoAl-SI and PdAl-I, although CH4 conversion and PdO formation were observed at higher temperatures. Moreover, we performed operando XAFS studies for several Pd/Al2O3 catalysts having different catalytic activities (Table S2 and Fig. S6†). As shown in Fig. 7(d), the relationship between T10 and the PdO ratio at 310 °C was roughly linear for all the catalysts. The catalysts with higher PdO contents showed lower T10 values. Considering the operando XAFS measurements above described, PdO was the main active species. Thus, the low light-off temperature of PdCoAl-GD can be ascribed to the oxidation of Pd0 to PdO at low temperatures.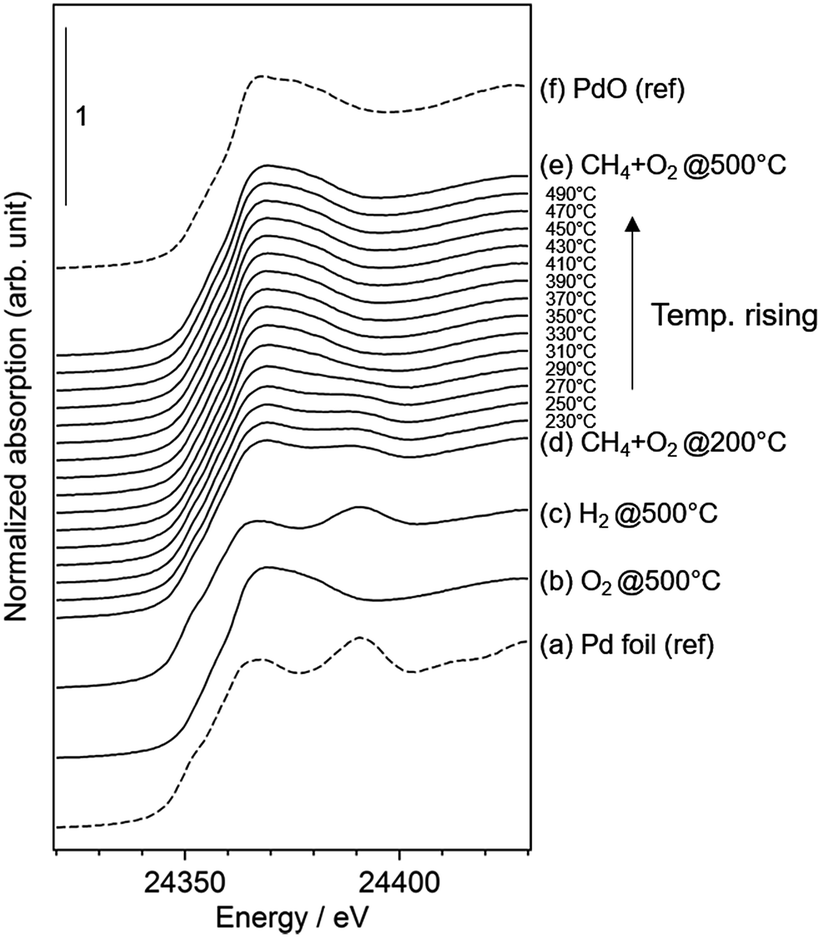 | ||
| Fig. 6 Normalized Pd K-edge XANES spectra of: (a) Pd foil as a reference; (b–e) results for PdCoAl-GD under operando XAFS measurements; (f) PdO as a reference. | ||
 | ||
| Fig. 7 PdO ratio and CH4 conversion as a function of time for: (a) PdCoAl-GD, (b) PdCoAl-SI and (c) PdAl-I during operando XAFS measurements. (d) Correlation between T10 and the PdO ratio at 310 °C over Pd-based catalysts. Red circle: PdCoAl-GD; green triangle: PdCoAl-SI; blue square: PdAl-I; black down-pointing triangle; series of Pd/Al2O3 (see Fig. S6†). | ||
Methane combustion over PdO state catalysts
As indicated in the previous section, the main active species for this reaction is PdO. This result led us to make the following question: What would happen if Pd was initially in form of PdO? Would the catalysts show similar catalytic activities?Fig. 8 shows the methane combustion results of the catalysts previously treated with O2 with the aim to oxidized Pd to PdO. Although Pd was initially as PdO, PdCoAl-GD showed the highest activity among all the catalysts tested. When Pd was initially as Pd0, PdCoAl-SI and PdAl-I showed different trends above 300 °C (Fig. 1) in virtue of their Pd particle size (6.3 vs. 3.8 nm, respectively). Pd was gradually oxidized during the catalytic tests such that PdO was main active species in this reaction. Since the Pd surface area of PdCoAl-SI was lower than that of PdAl-I, the former material was considered to have lower Pd oxidation rates. As indicated in Fig. 8, the activity of the catalysts having PdO as the initial phase followed the trend PdCoAl-GD > PdCoAl-SI > PdAl-I in all the temperature ranges.
 | ||
| Fig. 8 Methane combustion light-off curves of: PdCoAl-GD, PdCoAl-SI, PdAl-I and CoAl900. Pretreatment: flowing O2 at 500 °C. | ||
The rate-limiting step for methane combustion on PdO is the activation of the C–H bond in CH4 that takes place at the beginning of the reaction.44,45 In this step, the PdO surface is partially reduced by the CH4 molecule. Thus, to investigate the reducibility of PdO, CH4-TPR measurement was conducted shown as Fig. 9. The PdO nanoparticles of PdCoAl-GD were reduced by CH4 at a lower temperature as compared to PdCoAl-SI and PdAl-I (by 14 and 25 °C, respectively). This trend was similar to that of the catalytic activities (Fig. 8). The CH4-TPR results indicated the activity of PdO toward the activation of the C–H bond was improved in PdCoAl-GD as compared to PdCoAl-SI and PdAl-I. This enhancement can be explained as follows. According to the Pd 3d XPS results, the Pd 3d5/2 XPS peak of PdCoAl-GD was different for the catalysts prepared herein (Fig. 10). In the case of PdAl-I, the Pd 3d5/2 XPS peak was fitted with a peak almost derived from Pd2+. In the case of PdCoAl-SI and PdCoAl-GD, a good fit was not obtained with only one peak derived from Pd2+, and the presence of a peak derived from Pdn+ (0 < n < 2) was confirmed. In the case of PdCoAl-GD, which showed the highest catalytic activity, the binding energy of Pdn+ shifted to the lower energy side, and the ratio of Pdn+ was the largest among the catalysts tested. These results suggest that the electronic state of PdO changed as a result of the interaction with CoAl2O4, becoming electron enriched. Thus, the contact of PdO and CoAl2O4 can alter the electron states of PdO, resulting in enhanced C–H activation.
 | ||
| Fig. 9 CH4-TPR results for: PdCoAl-GD (red), PdCoAl-SI (green) and PdAl-I (blue). | ||
 | ||
| Fig. 10 Pd 3d XPS spectra of PdCoAl-GD, PdCoAl-SI, and PdAl-I. Circle, raw data; blue, Pd2+ 3d5/2; cyan, Pd2+ 3d3/2; pink, Pdn+ 3d5/2; yellow, Pdn+ 3d3/2; red, sum of fitting; black, background. | ||
Conclusion
Pd/CoAl2O4/Al2O3 catalysts prepared by a GD method (PdCoAl-GD) showed high activity toward the methane combustion reaction. PdCoAl-GD was comprised of a CoAl2O4 phase on γ-Al2O3 and dispersed Pd nanoparticles of 2–7 nm in size on a CoAl2O4 phase. Operando XAFS measurements revealed that the light-off temperature decreased for the PdO phase (i.e., the main active species) generated at lower temperatures. Furthermore, PdCoAl-GD showed the highest methane combustion activity among the catalysts tested herein even when PdO was initially present on the catalysts. As revealed by CH4-TPR, PdO in PdCoAl-GD was reduced at low temperatures by CH4. PdO species of the catalyst prepared by GD method were effective in activating the C–H bond of CH4. GD is a simple preparation method comparable to conventional impregnation approaches. Thus, further research on the industrial applications of GD catalysts and mechanism elucidation should be carried out.Acknowledgements
This work was supported by Grant-in-Aids from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, “Elements Strategy Initiative to Form Core Research Center” program, and Grants-in-Aid for Scientific Research KAKENHI (B) (15H04186). The XAFS measurements at the SPring-8 were carried out through the approval (proposal no. 2015B1482, 2016A1662) of Japan Synchrotron Radiation Research Institute (JASRI).References
- R. J. Farrauto, Science, 2012, 337, 659–660 CrossRef CAS PubMed.
- P. Gélin and M. Primet, Appl. Catal., B, 2002, 39, 1–37 CrossRef.
- T. V. Choudhary, S. Banerjee and V. R. Choudhary, Appl. Catal., A, 2002, 234, 1–23 CrossRef CAS.
- X. Zou, Z. Rui and H. Ji, ACS Catal., 2017, 7, 1615–1625 CrossRef CAS.
- Y. Lou, J. Ma, W. Hu, Q. Dai, L. Wang, W. Zhan, Y. Guo, X.-M. Cao, Y. Guo, P. Hu and G. Lu, ACS Catal., 2016, 6, 8127–8139 CrossRef CAS.
- J. Shen, R. E. Hayes, X. Wu and N. Semagina, ACS Catal., 2015, 5, 2916–2920 CrossRef CAS.
- S. Colussi, A. Gayen, M. F. Camellone, M. Boaro, J. Llorca, S. Fabris and A. Trovarelli, Angew. Chem., Int. Ed., 2009, 48, 8481–8484 CrossRef CAS PubMed.
- K. Sekizawa, H. Widjaja, S. Maeda, Y. Ozawa and K. Eguchi, Appl. Catal., A, 2000, 200, 211–217 CrossRef CAS.
- R. J. Farrauto, M. C. Hobson, T. Kennelly and E. M. Waterman, Appl. Catal., A, 1992, 81, 227–237 CrossRef CAS.
- M. Lyubovsky and L. Pfefferle, Appl. Catal., A, 1998, 173, 107–119 CrossRef CAS.
- Z. Chen, S. Wang, Y. Ding, L. Zhang, L. Lv, M. Wang and S. Wang, Appl. Catal., A, 2017, 532, 95–104 CrossRef CAS.
- C. L. Pieck, C. R. Vera, E. M. Peirotti and J. C. Yori, Appl. Catal., A, 2002, 226, 281–291 CrossRef CAS.
- Y. Liu, S. S. Wang, T. Sun, D. Gao, C. Zhang and S. S. Wang, Appl. Catal., B, 2012, 119–120, 321–328 CrossRef CAS.
- L. M. T. Simplício, S. T. Brandão, E. A. Sales, L. Lietti and F. Bozon-Verduraz, Appl. Catal., B, 2006, 63, 9–14 CrossRef.
- M. Monai, T. Montini, M. Melchionna, T. Duchoň, P. Kúš, C. Chen, N. Tsud, L. Nasi, K. C. Prince, K. Veltruská, V. Matolín, M. M. Khader, R. J. Gorte and P. Fornasiero, Appl. Catal., B, 2017, 202, 72–83 CrossRef CAS.
- H. Yoshida, T. Nakajima, Y. Yazawa and T. Hattori, Appl. Catal., B, 2007, 71, 70–79 CrossRef CAS.
- R. Burch and P. K. Loader, Appl. Catal., B, 1994, 5, 149–164 CrossRef CAS.
- B. Yue, R. Zhou, Y. Wang and X. Zheng, Appl. Surf. Sci., 2006, 252, 5820–5828 CrossRef CAS.
- H. Liao, M. Liu and P. Zuo, Catal. Commun., 2016, 76, 62–66 CrossRef CAS.
- J. H. Park, J. H. Ahn, H. I. Sim, G. Seo, H. S. Han and C. H. Shin, Catal. Commun., 2014, 56, 157–163 CrossRef CAS.
- Y. Mahara, J. Ohyama, T. Tojo, K. Murata, H. Ishikawa and A. Satsuma, Catal. Sci. Technol., 2016, 6, 1–10 Search PubMed.
- Z. Wang, J. Deng, Y. Liu, H. Yang, S. Xie, Z. Wu and H. Dai, Catal. Today, 2017, 281, 467–476 CrossRef CAS.
- A. Satsuma, T. Tojo, K. Okuda, Y. Yamamoto, S. Arai and J. Oyama, Catal. Today, 2015, 242, 308–314 CrossRef CAS.
- G. Ercolino, G. Grzybek, P. Stelmachowski, S. Specchia, A. Kotarba and V. Specchia, Catal. Today, 2015, 257, 66–71 CrossRef CAS.
- H. Widjaja, K. Sekizawa, K. Eguchi and H. Arai, Catal. Today, 1997, 35, 197–202 CrossRef CAS.
- G. Guo, K. Lian, F. Gu, D. Han and Z. Wang, Chem. Commun., 2014, 50, 13575–13577 RSC.
- G. Di Carlo, G. Melaet, N. Kruse, L. F. Liotta, G. Pantaleo and A. M. Venezia, Chem. Commun., 2010, 46, 6317–6319 RSC.
- P. Stefanov, S. Todorova, A. Naydenov, B. Tzaneva, H. Kolev, G. Atanasova, D. Stoyanova, Y. Karakirova and K. Aleksieva, Chem. Eng. J., 2015, 266, 329–338 CrossRef CAS.
- H. Nassiri, K.-E. Lee, Y. Hu, R. E. Hayes, R. W. J. Scott and N. Semagina, ChemPhysChem, 2017, 18, 238–244 CrossRef CAS PubMed.
- Y. Liu, S. Wang, D. Gao, T. Sun, C. Zhang and S. Wang, Fuel Process. Technol., 2013, 111, 55–61 CrossRef CAS.
- Z. Wu, J. Deng, Y. Liu, S. Xie, Y. Jiang, X. Zhao, J. Yang, H. Arandiyan, G. Guo and H. Dai, J. Catal., 2015, 332, 13–24 CrossRef CAS.
- X. Zou, Z. Rui, S. Song and H. Ji, J. Catal., 2016, 338, 192–201 CrossRef CAS.
- X. Pan, Y. Zhang, Z. Miao and X. Yang, J. Energy Chem., 2013, 22, 610–616 CrossRef CAS.
- N. M. Kinnunen, J. T. Hirvi, M. Suvanto and T. A. Pakkanen, J. Mol. Catal. A: Chem., 2012, 356, 20–28 CrossRef CAS.
- G. Zhu, J. Han, D. Y. Zemlyanov and F. H. Ribeiro, J. Am. Chem. Soc., 2004, 126, 9896–9897 CrossRef CAS PubMed.
- M. Cargnello, J. J. D. Jaen, J. C. H. Garrido, K. Bakhmutsky, T. Montini, J. J. C. Gamez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef CAS PubMed.
- A. Y. Stakheev, A. M. Batkin, N. S. Teleguina, G. O. Bragina, V. I. Zaikovsky, I. P. Prosvirin, A. K. Khudorozhkov and V. I. Bukhtiyarov, Top. Catal., 2013, 56, 306–310 CrossRef CAS.
- M. Monai, T. Montini, M. Melchionna, T. Duchoň, P. Kúš, N. Tsud, K. Prince, V. Matolin, R. J. Gorte and P. Fornasiero, Appl. Catal., B, 2015, 197, 271–279 CrossRef.
- T. P. Senftle, A. C. T. van Duin and M. J. Janik, ACS Catal., 2017, 7, 327–332 CrossRef CAS.
- Y. Mahara, H. Ishikawa, J. Ohyama, K. Sawabe and A. Satsuma, Catal. Today, 2016, 265, 2–6 CrossRef CAS.
- Y. Mahara, H. Ishikawa, J. Ohyama, K. Sawabe, Y. Yamamoto, S. Arai and A. Satsuma, Chem. Lett., 2014, 43, 910–912 CrossRef CAS.
- J. Ohyama, H. Ishikawa, Y. Mahara, T. Nishiyama and A. Satsuma, Bull. Chem. Soc. Jpn., 2016, 89, 914–921 CrossRef CAS.
- H. Wang, C. Chen, Y. Zhang, L. Peng, S. Ma, T. Yang, H. Guo, Z. Zhang, D. S. Su and J. Zhang, Nat. Commun., 2015, 6, 7181 CrossRef CAS PubMed.
- Y. C. Chin, M. García-Diéguez and E. Iglesia, J. Phys. Chem. C, 2016, 120, 1446–1460 CAS.
- Y. H. Chin, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2013, 135, 15425–15442 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06150a |
| This journal is © The Royal Society of Chemistry 2017 |