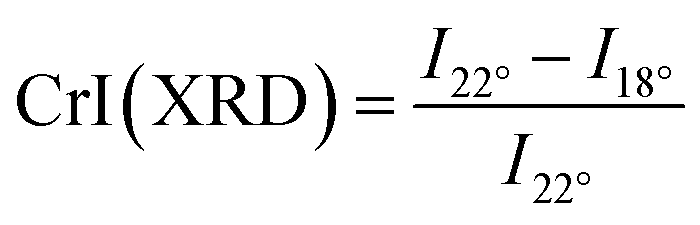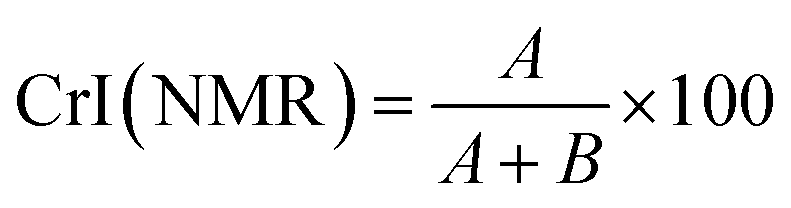DOI:
10.1039/C7RA06141B
(Paper)
RSC Adv., 2017,
7, 40600-40607
Favored surface-limited oxidation of cellulose with Oxone® in water
Received
1st June 2017
, Accepted 11th August 2017
First published on 21st August 2017
Abstract
A novel method for favored primary alcohol oxidation of cellulose was developed. Cellulose pulp and Cladophora nanocellulose were oxidized in a one-pot procedure by Oxone® (2KHSO5·KHSO4·K2SO4) and efficient reaction conditions were identified. The effects of the reaction on the morphology, viscosity and chemical structure of the products obtained were studied. The primary alcohol groups were oxidized to carboxyl groups and the content of carboxyl groups was determined by conductometric titration. SEM, capillary-type viscometry and XRD were applied to characterize the products and to investigate the influence of oxidation. For the first time, low-cost and stable Oxone® was used as a single oxidant to oxidize cellulose into carboxyl cellulose. The oxidation is an inexpensive and convenient process to produce carboxylic groups on the surface of the cellulose fibers and to make the cellulose fibers charged. Particularly, this method can avoid the use of halogens and potentially toxic radicals and constitute a green route to access carboxylated cellulose. Further, sodium bromide could be used as a co-oxidant to the Oxone® and increase the carboxylic acid content by 10–20%. The Oxone® oxidation is a promising method for oxidation of cellulose and might facilitate the production of CNC.
Introduction
Cellulose is a renewable, sustainable and highly abundant polymer. It is biodegradable, non-petroleum based, has little environmental impact, and is considered safe to animal and human health.1,2 Oxidation of alcohol groups present in the cellulose to ketones, aldehydes and carboxylic acid derivatives is one of the most important modifications of cellulose and has been studied for more than half a century.3 Oxidized cellulose exhibits interesting properties, such as gelation, complexation, antiflocculation, adhesion, as well as a number of biological effects,4 which enrich the applications of cellulose manifold, including medical applications, adhesion-prevention5 and wound-healing.6 The selective oxidation of the primary alcohol groups of cellulose and its applications are some of the most active research areas in recent years, with numerous articles published.7 In particular, the preparation of nanofibrillated cellulose from various sources of cellulose by introduction of surface charges from carboxyl groups, which are commonly introduced by either carboxymethylation or 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) mediated oxidation, has led to a large number of applications.8–11
In 1994, TEMPO-mediated oxidation was firstly used to convert primary alcohol groups in polysaccharides to the corresponding polyuronic analogues by De Nooy et al.12 and has been further developed foremost by Isogai and co-workers.7,13 In recent years, homologous compounds of TEMPO have also been developed for use as oxidants of cellulose, e.g. 2-azaadamantane N-oxyl (AZADO),14 4-acetamido-TEMPO,15 or phthalimide-N-oxyl (PINO).16 Individual nanocellulose fibers are produced by TEMPO-mediated oxidation and successive mild disintegration in water and during the process C6 alcohol groups are selectively oxidized. As useful as the TEMPO-mediated oxidation is, causing mild oxidation, low depolymerization, being mainly surface limited and charge introducing, there are still some possible drawbacks associated with the reaction. TEMPO might require stoichiometric amounts of halogen-containing co-oxidants (TEMPO/NaBr/NaClO and TEMPO/NaClO/NaClO2 oxidation systems) to mediate the oxidation reaction,17 which should be avoided in industrial processes.18 Halogen free TEMPO-based methods relying on electrochemical oxidation of TEMPO have been developed, which can obviate the use of halogen-containing co-oxidants. Another possible drawback of the TEMPO-mediated process is the risk of having trace amounts of reactive radicals in the product, which might limit the usefulness of the material in e.g. bioapplications.19 Accessible, safe, green and low-cost reagents should be employed in the oxidation of cellulose. To meet the requirements of efficient and selective alcohol oxidation of cellulose, while avoiding the production of toxic side products and contamination of final products, development of alternative selective oxidation methods to complement TEMPO-oxidation is desirable.
Oxone® is a commercial trade name for a stable triple salt (2KHSO5·KHSO4·K2SO4) containing potassium peroxymonosulfate (KHSO5), a salt of Caro's acid (H2SO5), as the active component. It is regarded as non-toxic with an acute oral toxicity LD50 of 2000 mg kg−1 in rat compared to 1050 mg kg−1 for TEMPO. Furthermore, Oxone® is readily soluble in water, widely commercially available and inexpensive.20 Oxone® can be used as sole oxidant for the oxidation of aldehydes,21 ketones to esters via Baeyer–Villiger oxidations,22 olefins23 and alcohols,24 which illustrates Oxone's potency as oxidant.20 Oxone® has also been used for bleaching and delignification25–27 of cellulose. To the best of our knowledge, there is no report regarding oxidation of cellulose employing Oxone® as the pivotal oxidant whereas the related oxidation using ammonium persulfate has been reported by Luong and co-workers.28 However, the yields of CNCs produced in Luong's work are low, i.e. only 36% for wood pulp, which would influence the method's usefulness in large-scale applications.
In this work, we investigated suitable reaction conditions for Oxone® oxidation of cellulose pulp and the effect of the oxidation on cellulose pulp and highly crystalline Cladophora nanocellulose.29 The morphology, viscosity, degree of crystallinity, chemical structure and the content of carboxyl groups of oxidized products were studied with SEM, capillary-type viscometry, XRD, FTIR and 13C NMR. Results of oxidations using Oxone® as the sole oxidant as well as Oxone® in combination with sodium bromide as co-oxidant are described.
Experimental
Materials
Never-dried cellulose pulp (Iβ-rich cellulose) was supplied by Domsjö Fabrikerna AB, Örnsköldsvik, Sweden. Nanocellulose from Cladophora algae (Iα-rich cellulose)30 was provided by FMC Biopolymer. Oxone® (monopersulfate compound, 2KHSO5·KHSO4·K2SO4), sodium bromide (NaBr) and other chemicals were of reagent or analytical grade and were used as received from commercial suppliers. Deionized water was used throughout all experiments.
Oxone® oxidation
Cellulose sample was dispersed in 100 mL of distilled water through high-energy ultrasonication for 12 min using a 30/30 s pulse and output power of 500 W (VibraCell 750 W, Sonics, U.S.A). The dispersion was transferred to a round-bottom flask and heated to specified temperatures under magnetic stirring in an oil bath. Oxone® and sodium bromide (when sodium bromide was used), were then added to the reaction flask, which was sealed by a stopper. At the specified time the reaction mixture was cooled in air and purified by three cycles of centrifugation/washing/redispersion with 10 mM hydrochloric acid solution, after which samples were dried at 45 °C in vacuum oven for >24 hours.
Summarizing, the investigated materials were unmodified cellulose pulp (u-CP), unmodified Cladophora cellulose (u-CC), cellulose pulp oxidized without sodium bromide (o-CP) and with it (o-CP-Br) as well as Cladophora cellulose oxidized without sodium bromide (o-CC) and with it (o-CC-Br). The yields of o-CP, o-CP-Br, o-CC and o-CC-Br were 79%, 85%, 70% and 83%, respectively.
Conductometric titration
The carboxylic group content was determined by conductometric titration18 using a Mettler Toledo T90 titrator. The cellulose sample (100 mg) was dispersed in 60 mL 1 mM NaCl(aq) through high-energy ultrasonication and then purged with N2 for at least 20 min to remove dissolved gases. Before the titration, the pH of the dispersion was adjusted to <2.8 with concentrated hydrochloric acid under nitrogen atmosphere. NaOH (50 mM, aq) was used as titrant and added at a rate of 0.01 mL min−1 throughout the titration process. The content of carboxylic acid was calculated by making linear fits to the linear regions (corresponding to strong acid and base) and the plateau region (corresponding to weak acid, i.e., carboxylic acid) in the conductivity vs. titrant volume curves.
Oxime formation and determination of total aldehyde and ketone content by CHN elemental analysis
According to a previously published procedure,31 samples were subjected to Schiff base reactions with hydroxylamine, converting any aldehydes (C6 position) and/or ketones (C2 and C3 position) to oximes. In short, dried sample (50 mg) was added to 20 mL 0.01 M acetate buffer solution (pH 4.5), after which 1.65 mL aqueous hydroxylamine solution (50 wt%) was added and the reaction was performed under magnetic stirring at room temperature for 24 h. The product was dialyzed for 7 days in water and then dried through freeze-drying. The product recovery was >91 wt% for all samples. CHN analysis was performed by Analytische Laboratorien (Lindlar, Germany).
Chlorite oxidation and determination of aldehyde content by conductometric titration
Aldehydes were oxidized to carboxylic acids according to a previously published method,31 with minor modifications. Briefly, dried sample (500 mg) and 3.6 g sodium chlorite was added to 100 mL 0.01 M acetate buffer solution (pH 4.5), and the reaction was conducted under magnetic stirring for 24 h at 40 °C. The product was dialyzed for 7 days in water and then dried through freeze-drying. The product recovery was >95 wt% for all samples. The carboxylic group content of samples was determined by conductometric titration according to the protocol above and the aldehyde content was calculated by subtracting the amount of carboxylic acid obtained before chlorite oxidation from the amount obtained after chlorite oxidation.
Scanning electron microscopy (SEM)
SEM micrographs of all samples were performed with a LEO 1550 field-emission SEM instrument (Zeiss, Germany). Samples were mounted on aluminum stubs by means of double-sided adhesive carbon tape and sputtered with Au/Pd to reduce charging effects.
X-ray diffraction
X-ray diffraction (XRD) diffractograms were recorded with a diffractometer having Bragg-Bretano geometry (Cu Kα radiation; λ = 1.54 Å) (D5000, Siemens/Bruker, Germany). A crystallinity index CrI(XRD) of the cellulose and its derivatives was calculated using Segal's empirical equation.32| |
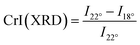 | (1) |
where CrI stands for crystallinity index of cellulose. I22° represents the total peak diffraction intensity at 22° and I18° is the peak diffraction intensity corresponding to the amorphous background at 18°.
Intrinsic viscosity
The intrinsic viscosity was measured for the samples according to the ASTM standard method D1795-96,33 using an Ubbelohde viscometer (SI Analytics GmbH, Germany). Measurements were repeated five times for each sample and all measurements were performed at 25.0 °C. The experiments were recorded with a video camera and a digital timer for accurate timing. Intrinsic viscosities of the samples were calculated according to the ASTM standard method D1795-96.
Fourier transform infrared (FT-IR) spectroscopy
FT-IR spectra were recorded on a Tensor 27 FTIR spectrometer (Bruker, Germany). The resolution was set to 4 cm−1 with 128 scans over a range of 4000–400 cm−1. Samples were ground with KBr (1 wt% sample in KBr) and compressed into pellets for FT-IR measurements.
Solid-state CP/MAS 13C NMR spectroscopy
Solid state NMR experiments were performed on a Varian Inova-600 operating at 14.7 T and equipped with a 3.2 mm solid-state probe. Measurements were conducted at 301 K with a MAS spinning rate of 15 kHz. The CP/MAS 13C NMR spectra were recorded using a cross-polarization pulse sequence followed by proton decoupling during acquisition. Acquisition parameters included a 2.9 μs 1H 90° pulse, 1200 μs contact time, 25 ms acquisition time, and 54 s recycle delay to allow for complete thermal equilibrium. Two crystallinity indices C4 CrI(NMR) and C6 CrI(NMR) were calculated according to a previously published method,34 by integrating the signals at 86–92 (A) and 80–86 ppm (B) for C4 CrI(NMR) and for 68–63 (A) and 63–58 ppm (B) for C6 CrI(NMR) and applying eqn (2).| |
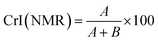 | (2) |
Results and discussion
Effect of reaction conditions
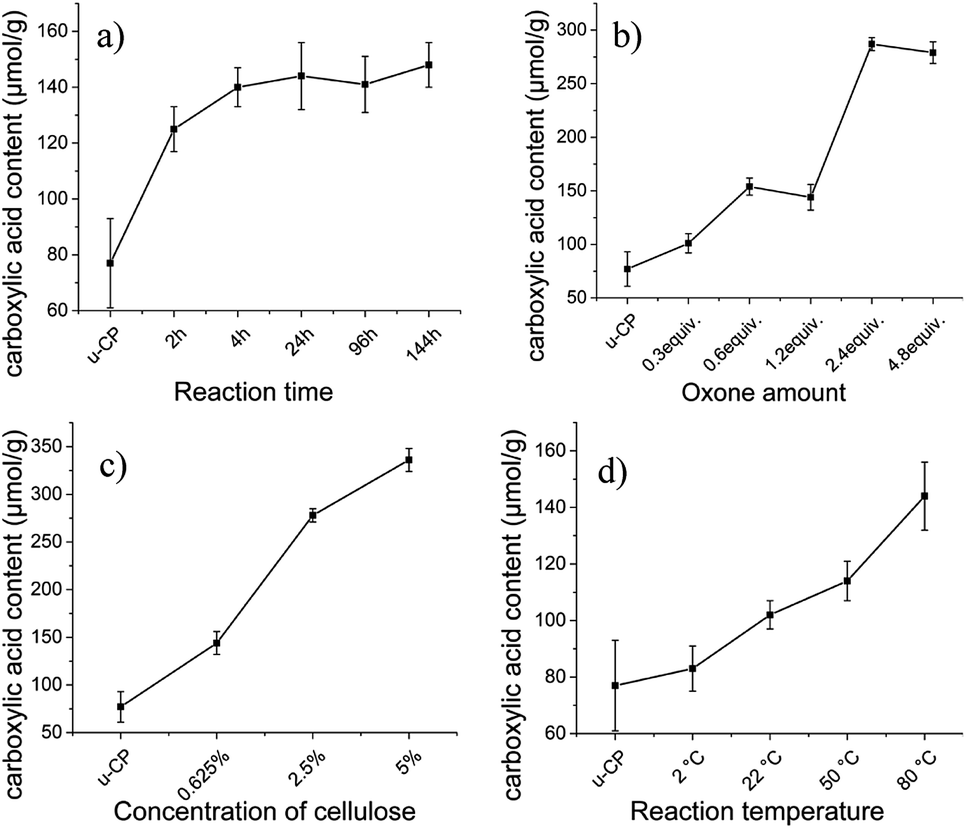
In order to find suitable oxidation conditions for achieving high carboxyl content and to improve the oxidation efficiency for cellulose pulp, several reaction parameters were screened including reaction time, amount of oxidant (Oxone®), cellulose concentration and temperature. Suitable oxidation conditions obtained for cellulose pulp were also applied to Cladophora nanocellulose.
During the screening of oxidation conditions, the following conditions were chosen as starting points based on a rough first screen: the reaction time was 24 hours, amount of Oxone® was 1.2 equiv., concentration of cellulose was 0.625 wt% in the reaction solution and the temperature was 80 °C. When one parameter was altered, the other parameters were fixed. The effects of these reaction parameters on the carboxylic acid content of cellulose are presented in Fig. 1. Fig. 1a and d show that carboxylic acid content of cellulose was moderately affected by prolonging the reaction time or increasing the reaction temperature under these conditions. The small influence of oxidation time and temperature on the carboxylic acid content might be due to the low oxidation efficiency under these conditions. The lowest carboxylic acid content (83 μmol g−1, which was close to the value of original cellulose pulp, 77 μmol g−1) was obtained at the lowest investigated reaction temperature (2 °C), which indicates that heating was beneficial for the oxidation reaction. After increasing the amount of Oxone® and the concentration of cellulose pulp, significant increase of the carboxylic acid content was obtained (see Fig. 1b and c). However, employing amounts of Oxone® exceeding 2.4 equivalents provided little effect on the carboxylic acid content. The highest value of carboxylic acid content (336 μmol g−1) of cellulose pulp was observed when the concentration of cellulose pulp was increased to 5 wt%. As the viscosity of 5 wt% Cladophora cellulose was too high to achieve efficient stirring during the reaction, 2 wt% of cellulose was selected as concentration for the following experiments to make a reasonable comparison for different sources of cellulose. Based on the results presented in Fig. 1 and the former experiments, the following conditions were selected for further studies: a reaction time of 24 hours, 2.4 equiv. of Oxone®, 2 wt% of cellulose and a reaction temperature of 80 °C.
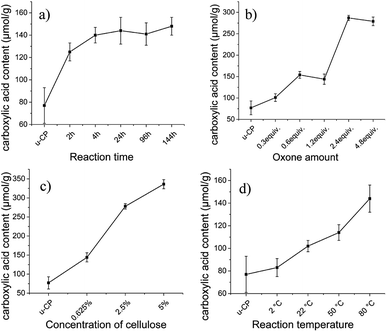 |
| | Fig. 1 Effect of (a) reaction time, (b) amount of Oxone®, (c) concentration of cellulose and (d) reaction temperature, on the carboxylic acid content of oxidized cellulose pulp. u-CP represents unmodified pulp cellulose and is used as a reference. Error bars represent standard deviation from the mean (n = 3). | |
Effect of pH
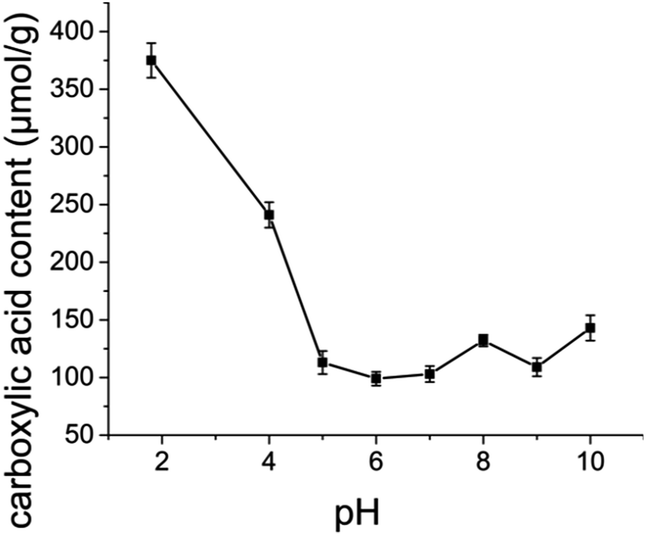
Oxone® oxidation of cellulose pulp was conducted in buffered solutions of different pH and the obtained carboxylic acid content of cellulose pulp produced is presented in Fig. 2. For experiments at pH 4, 5 and 6, acetate buffer solutions were used, while phosphate buffer solutions were employed in experiments at pH 7 and 8 and bicarbonate buffer solutions at pH 9 and 10. For the oxidation performed in distilled water, where a final pH of 1.8 was detected, a significantly higher carboxylic acid content was obtained, which demonstrated that the oxidation efficiency was higher when the reaction was carried out at lower pH in the absence of buffer.
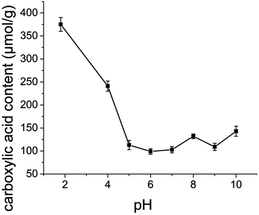 |
| | Fig. 2 Effect of solution pH on the carboxylic acid content of oxidized cellulose pulp. Error bars represent standard deviation from the mean (n = 3). | |
Physical–chemical characterization
To determine the aldehyde content, aldehydes were oxidized to carboxylic acids by employing sodium chlorite. The aldehyde content was calculated as the difference between the carboxylic acid content before and after the oxidation with sodium chlorite. As seen in Table 1, small amounts of aldehyde were present in samples o-CP and o-CC. In order to improve the oxidation efficiency for the transformation of alcohols to carboxylic acids, sodium bromide was introduced into the Oxone® oxidation system.24 The data in Table 1 shows that the samples produced in the presence of sodium bromide provide a 10–20% increase in carboxylic acid content. As can be seen from the results presented in Table 1 Oxone® seem to partly also oxidize the C2 and/or C3 hydroxyl groups providing ketones.
Table 1 Summary of the physicochemical characterization of Oxone®-oxidized samples
| |
u-CP |
o-CP |
o-CP-Br |
u-CC |
o-CC |
o-CC-Br |
| Average carboxylic acid content based on analysis of three different samples. From CHN analysis of oximes. The difference in carboxylic acid content before and after chlorite oxidation. The difference between the total aldehyde and ketone content and the aldehyde content. The sum of the carboxylic acid, aldehyde and ketone content. Average of five measurements. After sonication. |
| Carboxylic acid contenta (μmol g−1) |
77 ± 16 |
356 ± 17 |
450 ± 8 |
109 ± 4 |
252 ± 7 |
279 ± 11 |
| Total aldehyde and ketone contentb (μmol g−1) |
94 |
210 |
179 |
76 |
141 |
112 |
| Aldehyde contentc (μmol g−1) |
15 |
42 ± 11 |
27 ± 5 |
19 |
19 ± 6 |
15 ± 1 |
| Ketone contentd (μmol g−1) |
79 |
168 |
152 |
57 |
122 |
97 |
| D.O.e (%) |
1.49 |
6.48 |
7.78 |
2.08 |
4.41 |
4.78 |
| [η]f (dL g−1) |
3.20 ± 0.04 |
0.36 ± 0.01 |
0.37 ± 0.01 |
3.29 ± 0.01 |
1.79 ± 0.02 |
1.80 ± 0.01 |
| C4 CrI(NMR) (%) |
52 |
55 |
54 |
82 |
84 |
88 |
| C6 CrI(NMR) (%) |
35 |
43 |
45 |
80 |
87 |
89 |
| CrI(XRD) (%) |
79 (79)g |
78 |
76 |
92 (91)g |
92 |
92 |
SEM
Oxone® oxidation not only transformed the primary hydroxyl groups to carboxylate groups, but also influenced the morphology of the materials, which may be caused by the acidic conditions during oxidation. The morphology of cellulose pulp and Cladophora nanocellulose and their corresponding oxidized products are shown in Fig. 3 and 4. Morphological changes induced by the oxidation process could be observed: Cladophora cellulose was shaped into sheets of cellulose, and cellulose pulp was shaped into irregular small pieces. The addition of the co-oxidant NaBr did not affect the morphology as compared to the use of Oxone® alone according to SEM images. Particularly, after oxidation, Cladophora cellulose became more compact, referring to Fig. 4e and f compared to Fig. 4d. Interestingly, some small pores could be found in Fig. 3c and 4c (cellulose pulp oxidized by Oxone® with NaBr), which might be caused by evolving bromine gas produced during the oxidation.
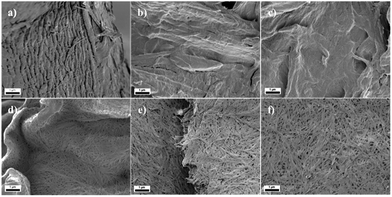 |
| | Fig. 3 SEM micrographs of (a) u-CP, (b) o-CP, (c) o-CP-Br, (d) u-CC, (e) o-CC and (f) o-CC-Br. Scale bar: 1 μm. | |
 |
| | Fig. 4 SEM micrographs of (a) u-CP, (b) o-CP, (c) o-CP-Br, (d) u-CC, (e) o-CC and (f) o-CC-Br. Scale bar: 100 nm. | |
Hua et al. observed that the surface area and pore sizes decreased and the compactness of Cladophora increased during TEMPO-mediated oxidation as the degree of carboxylation increased.35 The SEM micrographs presented here indicate that oxidation proceeded here too.
X-ray diffraction
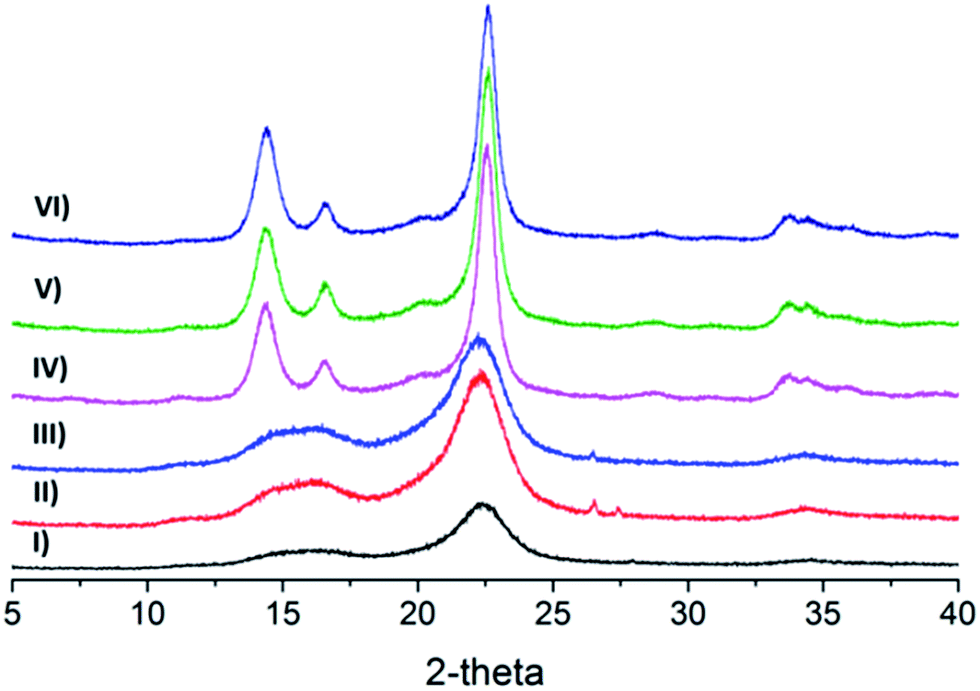
XRD was used to characterize the effects of oxidation on the crystallinity of the cellulose samples. The XRD diffractograms of samples presented in Fig. 5 and Table 1 provides the crystallinity index (CrI) of the samples. The diffractograms of samples for unmodified cellulose and oxidized cellulose appear to be almost identical, which demonstrates that the oxidative process has little influence on the crystallinity. The unaffected crystallinity is a strong indication that the Oxone® oxidation did not affect the ring structure of the glucose units as e.g. periodate oxidation does.36–38 The CrI for o-CP decreases marginally after oxidation as compared to the sonicated unoxidized samples, and the products oxidized with sodium bromide (o-CP-Br) show some marginal decrease compared to o-CP, which might be a result of the participation of bromine and its damage on the structure of cellulose during the reaction. The CrI for Cladophora cellulose samples was not affected at all; neither by pure Oxone® oxidation nor by Oxone®/NaBr oxidation. The unaffected crystallinity of the cellulose indicates that the crystalline core of the fibers is not influenced by the Oxone® oxidation and that it is limited to the surface of fibers.
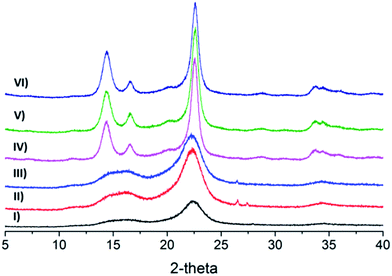 |
| | Fig. 5 X-ray diffractograms of celluloses, (I) u-CP, (II) o-CP, (III) o-CP-Br, (IV) u-CC, (V) o-CC and (VI) o-CC-Br. | |
Intrinsic viscosity
To evaluate whether any depolymerization occurred during oxidation, the intrinsic viscosity of the samples was determined. Measurements were conducted before and after oxidation with and without sodium bromide. The alkaline bis(ethylenediamine) copper(II) hydroxide solvent which was used during the process of sample dissolution and measurement may cause degradation of the cellulose through β-elimination if aldehydes and/or ketones, are present.39 However, in the work by Carlsson et al.,18 it was reported that the effect of β-elimination was negligible during the course of dissolution and measurements, for low content of aldehydes and/or ketones in the cellulose.
The intrinsic viscosity results are presented in Table 1. It was obvious that Oxone® oxidation drastically decreased the viscosity of cellulose pulp, while the viscosity of oxidized Cladophora cellulose decreased somewhat more moderately. The lower decrease for Cladophora cellulose corresponding to a lower extent of depolymerization may be due to the higher crystallinity of original Cladophora cellulose hindering the contact between the oxidant and the cellulose. After completion of the oxidation reaction, the pH of the reaction solution was below 2, where hydrolysis and depolymerization of cellulose might be induced and result in samples with low viscosity. On the course of the oxidation reactions, it was noticed that the viscosity of the reaction mixtures decreased.
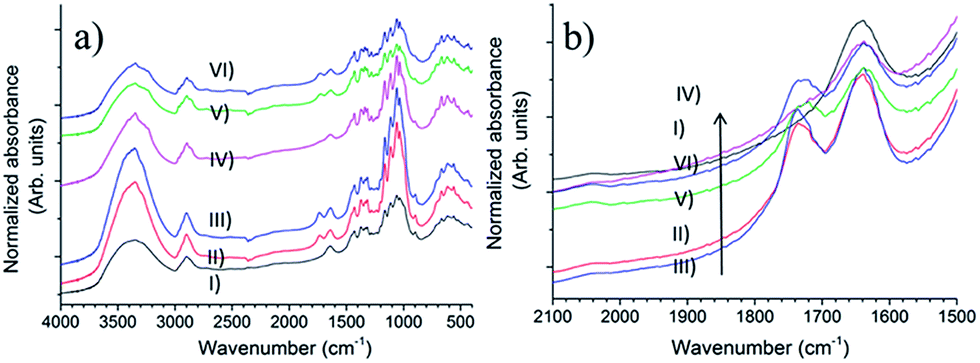
FTIR spectroscopy
The FTIR spectra of samples are shown in Fig. 6. Some effects of the Oxone® oxidation can be observed. The most obvious effect is the appearance of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band at 1732 cm−1 assigned to the carbonyl group,4 indicating the oxidation reaction occurred successfully. In Fig. 6b, after normalization with respect to the C–H stretching vibration at 2897 cm−1,40 a stronger absorbance peak could be found when the cellulose samples were oxidized with the help of sodium bromide (comparing curve II to III and V to VI, respectively), which demonstrates that sodium bromide improves the efficiency of Oxone® oxidation.
O stretching band at 1732 cm−1 assigned to the carbonyl group,4 indicating the oxidation reaction occurred successfully. In Fig. 6b, after normalization with respect to the C–H stretching vibration at 2897 cm−1,40 a stronger absorbance peak could be found when the cellulose samples were oxidized with the help of sodium bromide (comparing curve II to III and V to VI, respectively), which demonstrates that sodium bromide improves the efficiency of Oxone® oxidation.
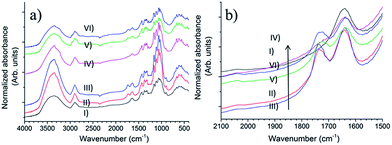 |
| | Fig. 6 (a) Full FTIR spectra and (b) the region of 2100–1500 cm−1 of samples (I) u-CP, (II) o-CP, (III) o-CP-Br, (IV) u-CC, (V) o-CC and (VI) o-CC-Br. | |
Selectivity of Oxone® oxidation according to solid-state CP/MAS 13C NMR spectroscopy
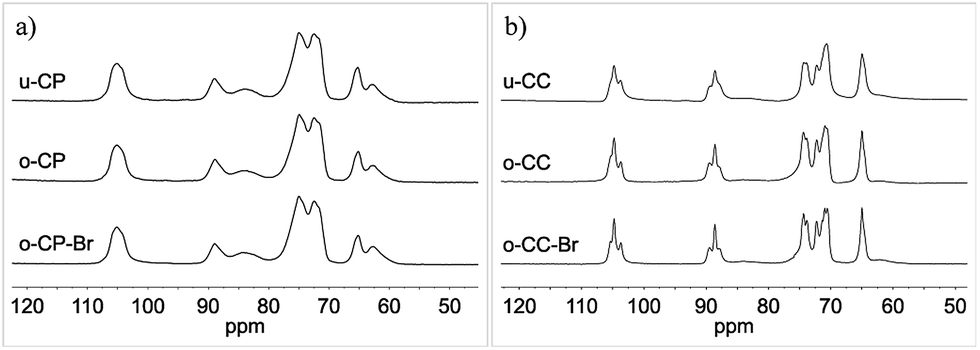
The CP/MAS 13C NMR spectra of cellulose pulp and of Cladophora cellulose and their Oxone® oxidized derivatives are presented in Fig. 7a and b. As can be seen in these spectra there seemed to be very small differences between the different samples and the Oxone® did not appear to affect the bulk material. These observations were further supported by the calculated CrI(NMR)-values34 presented in Table 1. According to the CrI(NMR) the crystallinity of the materials was only slightly affected by the Oxone® oxidation and increased moderately after Oxone® oxidation. The results are in sharp contrast to the results obtained during periodate oxidation of cellulose, which induce drastic lowering of the CrI(NMR) as the ring opening disrupts the crystallinity of the cellulose.36 The CrI(NMR) data presented indicated that the Oxone® oxidation was primarily affecting the amorphous parts of the cellulose pulp and that it was predominantly confined to the surface of the cellulose fibers for Cladophora cellulose.
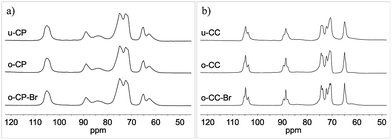 |
| | Fig. 7 Solid-state CP/MAS 13C NMR full spectra of (a) u-CP, o-CP and o-CP-Br; (b) u-CC, o-CC and o-CC-Br. | |
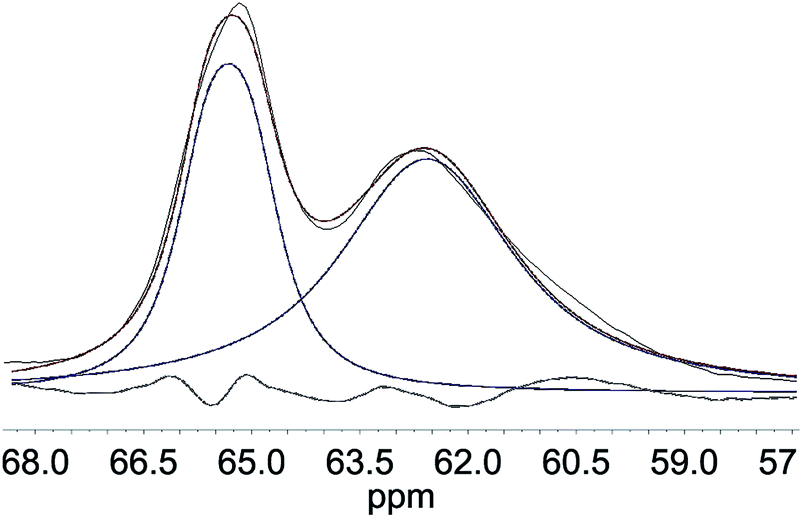
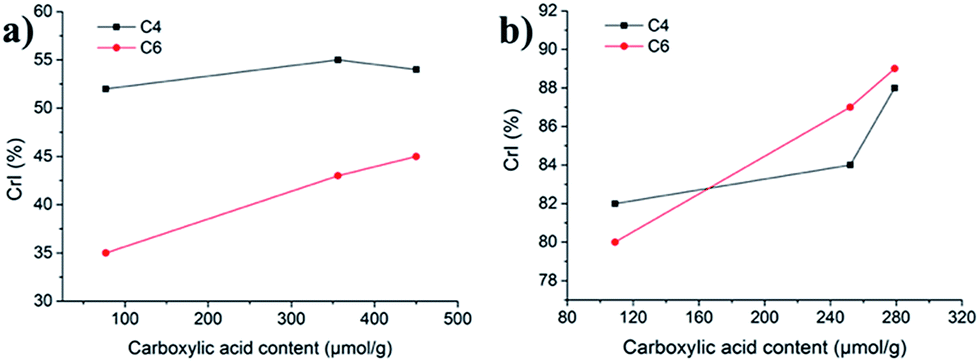
Deconvolution of the C6-region of the pulp derivatives is presented in Fig. 8 and shows that the crystallinity of the C6 (C6 CrI(NMR)) increased at increasing levels of oxidation, from 35% for u-CP to 43% for o-CP and further to 45% for the o-CP-Br (Table 1 and Fig. 9a). These results indicate that the C6 was particularly affected by the Oxone® oxidation whereas the cellulose as a whole was not affected based on the C4 CrI(NMR) and CrI(XRD) results presented in Table 1. These results are in accordance with the results published by Vignon and co-workers for TEMPO-mediated oxidation of different celluloses.41 Thus, there are strong indications that the Oxone® primarily oxidized the accessible C6 hydroxyl groups of the cellulose pulp. It is also possible that the C1 reducing end groups, generated as a result of cellulose degradation and detected in the viscosity measurements as lower degree of polymerization, can be oxidized resulting in C1 carboxyl groups. For the highly crystalline Cladophora the effect was not as pronounced as for the pulp and both the crystallinity of C6 and C4 increased during the oxidation of the material (Fig. 9b). Interestingly, the same trend was observed when Cladophora cellulose was subjected to TEMPO-mediated oxidation, where the C6 CrI(NMR) increased from 82% for u-CC to 86% for TEMPO-oxidized Cladophora cellulose with a carboxylic acid content of 0.6 mmol per g of cellulose with a concomitant increase in C4 CrI(NMR) from 82% to 89%.30
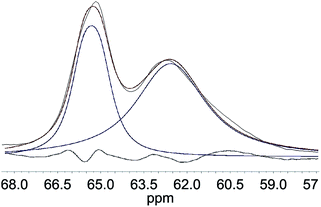 |
| | Fig. 8 Deconvolution of the C6 region of u-CP, where the peak at 65.3 ppm represents crystalline and the peak at 62.5 ppm represents amorphous cellulose (black line = original, blue line = fitted, red line = sum of fittings). | |
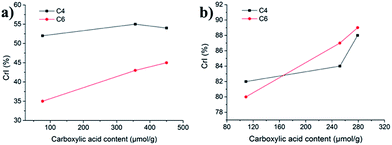 |
| | Fig. 9 Crystallinity indices of the C4 and C6 regions versus carboxylic acid content for (a) pulp derivatives and (b) Cladophora derivatives. | |
Selective methods for oxidation of cellulose are rare. Periodate oxidation is selective to oxidation of the vicinal C2 and C3 hydroxyl groups and provides 2,3-dialdehyde cellulose (DAC) with concomitant glucose ring opening via cleavage of C2–C3 bond. TEMPO-mediated oxidation is selective towards oxidation of accessible C6 primary alcohols and introduces charge to the surface of the cellulose fibers via formation of carboxylic acids. Ammonium persulfate was shown to preferentially oxidize the primary C6 alcohol.28 Oxone® also seems to demonstrate similar selectivity to ammonium persulfate and TEMPO-mediated oxidations and provides a viable alternative for C6 oxidation and possibly to some extent also C1 oxidation where CNC is the desired product.
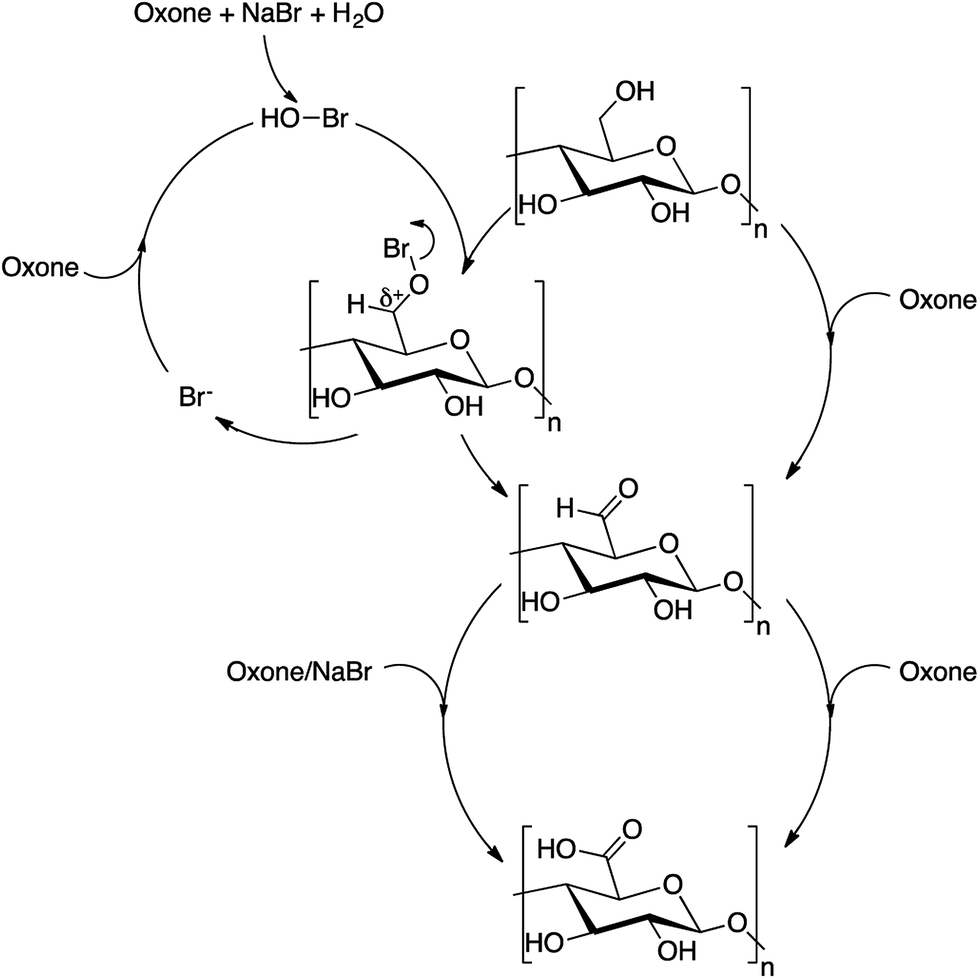
Mechanism of oxidation
A plausible reaction mechanism for the Oxone® cellulose oxidation is depicted in Scheme 1. In the scheme, we propose two reaction routes, one with NaBr and the other without NaBr as co-oxidant. The results of determination of aldehydes and ketones supported the formation of aldehydes during the oxidation. Further, the reddish-brown color of the reaction mixture, when sodium bromide was used as co-oxidant, indicates the formation of bromine during the oxidation. Moriyama et al.42 reported a similar plausible reaction mechanism for the oxidation of alcohols with Oxone® using potassium bromide as co-oxidant. Koo et al.24 and Raghavan et al.43 have reported formation of hypobromous acid as product of peroxymonosulfate oxidation of bromide ions.
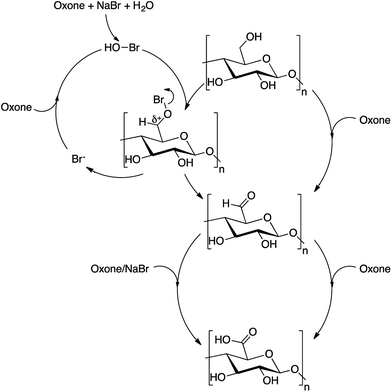 |
| | Scheme 1 Plausible reaction mechanism for Oxone® oxidation for cellulose. | |
The Oxone® oxidation described in this work provides a novel route to access C6-carboxylate cellulose. The method is convenient and relies on a non-toxic and low-cost bulk chemical, which is widely commercially available and easy to handle. A possible drawback of the method is the depolymerization of the cellulose material during oxidation, which is more profound for celluloses of lower crystallinity e.g. cellulose pulp. Hence, the Oxone® oxidation method described herein is probably best suited for production of CNC.
Conclusions
A novel route for the oxidation of cellulose with Oxone® as the single oxidant has been reported for the first time. Efficient one-pot Oxone® oxidation conditions were obtained by screening experiments conducted on cellulose pulp (Iβ-rich cellulose) and the final selected conditions were also applied to Cladophora cellulose (Iα-rich cellulose). SEM, XRD and capillary-type viscometry were applied to characterize the process of the oxidation. Further, FTIR and solid-state CP/MAS 13C NMR indicate that the C6-hydroxyl group was preferentially oxidized. The content of carboxylic acid groups was measured by conductometric titration. The oxidation method is a green, convenient and low-cost process to obtain carboxylate cellulose and charged cellulose fibers. The effect of sodium bromide on the Oxone® oxidation was also investigated. The Oxone® oxidation might have some limitations such as inducing depolymerization, but it might on the other hand be desirable for production of CNC. Oxone can be suitable for oxidizing highly crystalline cellulose like Cladophora cellulose, which is not as prone to depolymerization.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The Swedish NMR Centre is acknowledged for spectrometer time and Dr Diana Bernin is acknowledged for sample preparation and for performing the NMR experiments. Ollie and Elof Ericsson's Foundation are gratefully acknowledged for their financial support. Chang-Qing Ruan thanks the China Scholarship Council (CSC) for financial support. One of the authors (A. M.) is Wallenberg Academy Fellow and thanks the Knut and Alice Wallenberg Foundation for their support.
References
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS PubMed.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- E. C. Yackel and W. O. Kenyon, J. Am. Chem. Soc., 1942, 64, 121–127 CrossRef CAS.
- D. da Silva Perez, S. Montanari and M. R. Vignon, Biomacromolecules, 2003, 4, 1417–1425 CrossRef CAS PubMed.
- D. M. Wiseman, L. Saferstein and S. Wolf, US pat., 6500777, 2002.
- M. D. Finn, S. R. Schow and E. D. Schneiderman, J. Oral Maxillofac. Surg., 1992, 50, 608–612 CrossRef CAS PubMed.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- H. Fukuzumi, T. Saito, T. Wata, Y. Kumamoto and A. Isogai, Biomacromolecules, 2009, 10, 162–165 CrossRef CAS PubMed.
- T. Saito, M. Hirota, N. Tamura, S. Kimura, H. Fukuzumi, L. Heux and A. Isogai, Biomacromolecules, 2009, 10, 1992–1996 CrossRef CAS PubMed.
- T. Saito, Y. Nishiyama, J.-L. Putaux, M. Vignon and A. Isogai, Biomacromolecules, 2006, 7, 1687–1691 CrossRef CAS PubMed.
- H. Yano, J. Sugiyama, A. N. Nakagaito, M. Nogi, T. Matsuura, M. Hikita and K. Handa, Adv. Mater., 2005, 17, 153–155 CrossRef CAS.
- A. E. J. De Nooy, A. C. Besemer and H. van Bekkum, Carbohydr. Res., 1995, 269, 89–98 CrossRef CAS.
- A. Isogai and Y. Kato, Cellulose, 1998, 5, 153–164 CrossRef CAS.
- S. Takaichi and A. Isogai, Cellulose, 2013, 20, 1979–1988 CrossRef CAS.
- T. Isogai, T. Saito and A. Isogai, Cellulose, 2011, 18, 421–431 CrossRef CAS.
- G. Biliuta, L. Fras, M. Drobota, Z. Persin, T. Kreze, K. Stana-Kleinschek, V. Ribitsch, V. Harabagiu and S. Coseri, Carbohydr. Polym., 2013, 91, 502–507 CrossRef CAS PubMed.
- S. Coseri, G. Biliuta, B. C. Simionescu, K. Stana-Kleinschek, V. Ribitsch and V. Harabagiu, Carbohydr. Polym., 2013, 93, 207–215 CrossRef CAS PubMed.
- D. O. Carlsson, J. Lindh, L. Nyholm, M. Strømme and A. Mihranyan, RSC Adv., 2014, 4, 52289–52298 RSC.
- S. L. Kachkarova-Sorokina, P. Gallezot and A. B. Sorokin, Chem. Commun., 2004, 2844–2845 RSC.
- H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329–3371 CrossRef CAS PubMed.
- K. S. Webb and S. J. Ruszkay, Tetrahedron, 1998, 54, 401–410 CrossRef CAS.
- J. O. Edwards, R. H. Pater, R. Curclf and F. D. Furia, Photochem. Photobiol., 1979, 30, 63–70 CrossRef CAS.
- S. Rani and Y. D. Vankar, Tetrahedron Lett., 2003, 44, 907–909 CrossRef CAS.
- B.-S. Koo, C. K. Lee and K.-J. Lee, Synth. Commun., 2002, 32, 2115–2123 CrossRef CAS.
- V. Jafari, H. Sixta and A. van Heiningen, Holzforschung, 2014, 68, 497–504 CrossRef CAS.
- E. L. Springer, Tappi J., 1990, 73, 175–178 CAS.
- D. Stewart, Cellul. Chem. Technol., 2000, 34, 287–298 CAS.
- A. C. W. Leung, S. Hrapovic, E. Lam, Y. Liu, K. B. Male, K. A. Mahmoud and J. H. T. Luong, Small, 2011, 7, 302–305 CrossRef CAS PubMed.
- A. Mihranyan, J. Appl. Polym. Sci., 2011, 119, 2449–2460 CrossRef CAS.
- D. O. Carlsson, J. Lindh, M. Strømme and A. Mihranyan, Biomacromolecules, 2015, 16, 1643–1649 CrossRef CAS PubMed.
- U. J. Kim, S. Kuga, M. Wada, T. Okano and T. Kondo, Biomacromolecules, 2000, 1, 488–492 CrossRef CAS PubMed.
- L. Segal, J. Creely, A. Martin and C. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef CAS.
- ASTM, Standard D1795–96(2007)e1, Standard Test Method For Intrinsic Viscosity of Cellulose, ASTM International, 1996 Search PubMed.
- R. Teeäär, R. Serimaa and T. Paakkarl, Polym. Bull., 1987, 17, 231–237 CrossRef.
- K. Hua, I. Rocha, P. Zhang, S. Gustafsson, Y. Ning, M. Strømme, A. Mihranyan and N. Ferraz, Biomacromolecules, 2016, 17, 1224–1233 CrossRef CAS PubMed.
- J. Lindh, D. O. Carlsson, M. Strømme and A. Mihranyan, Biomacromolecules, 2014, 15, 1928–1932 CrossRef CAS PubMed.
- J. Lindh, C. Ruan, M. Strømme and A. Mihranyan, Langmuir, 2016, 32, 5600–5607 CrossRef CAS PubMed.
- C. Q. Ruan, M. Strømme and J. Lindh, Cellulose, 2016, 23, 2627–2638 CrossRef CAS.
- A. Potthast, T. Rosenau and P. Kosma, in Polysaccharides ii, Springer, 2006, pp. 1–48 Search PubMed.
- M. Åkerholm, B. Hinterstoisser and L. Salmén, Carbohydr. Res., 2004, 339, 569–578 CrossRef PubMed.
- S. Montanari, M. Roumani, L. Heux and M. R. Vignon, Macromolecules, 2005, 38, 1665–1671 CrossRef CAS.
- K. Moriyama, M. Takemura and H. Togo, J. Org. Chem., 2014, 79, 6094–6104 CrossRef CAS PubMed.
- N. Narender, P. Srinivasu, M. Ramakrishna Prasad, S. Kulkarni and K. Raghavan, Synth. Commun., 2002, 32, 2313–2318 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Jonas Lindh
and
Jonas Lindh