
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A micro-solid phase extraction in glass pipette packed with amino-functionalized silica for rapid analysis of petroleum acids in crude oils†
Gang-Tian Zhu *a,
Sheng Hea,
Xiao-Mei Heb,
Shu-Kui Zhuc and
Yu-Qi Fengb
*a,
Sheng Hea,
Xiao-Mei Heb,
Shu-Kui Zhuc and
Yu-Qi Fengb
aKey Laboratory of Tectonics and Petroleum Resources (Ministry of Education), China University of Geosciences, Wuhan 430074, P. R. China. E-mail: zhugangtian@163.com
bKey Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), Department of Chemistry, Wuhan University, Wuhan 430072, P. R. China
cState Key Laboratory of Biogeology and Environmental Geology, China University of Geosciences, Wuhan 430074, P. R. China
First published on 21st August 2017
Abstract
In this work, a micro-solid phase extraction (micro-SPE) method was developed for the analysis of petroleum acids in crude oils. Plastic vessels were found to be unsuitable as the micro-SPE vessel, because of the plasticizer interferences and the serious adhesion of oil samples on vessel surfaces. To reduce or eliminate these influences, a glass pipette was selected as the micro-SPE vessel. With the glass pipette micro-SPE format, amino-functionalized silica was chosen as the sorbent for extraction of petroleum acids based on mix-mode hydrogen bonding/ion-exchange interactions. The conditions of the micro-SPE were carefully optimized. Under the optimized conditions, no evaporation was needed, which can save time and reduce losses of analytes. By coupling with gas chromatography-mass spectrometry (GC-MS), the proposed micro-SPE method for analysis of petroleum acids was proved to be easily operated, fast (the micro-SPE can be accomplished within 5 min), solvent-saving (3 mL of solvent), reproducible and sensitive (the limits of detection range from 2 ng g−1 to 6 ng g−1).
1. Introduction
Petroleum acids in crude oils are well-known as the main corrosion agents in petroleum refining devices.1–3 Besides, because of the surfactant property and high biotoxicity, the contents of petroleum acids are important values in the risk assessment of petroleum pollution.4–7 On the other hand, to geochemists, petroleum acids are significant molecular markers that can provide valuable information on origin, biodegradation and maturation process of petroleum.8–10 Gas chromatography-mass spectrometry (GC-MS) is an important tool for the detection of individual petroleum acids because of its powerful qualitative capacity.11–14 Due to the extremely complex components of crude oils, selective separation of acids from oils is indispensable prior to GC-MS analysis.15,16 Base extraction is a typical method to separate petroleum acids from oils, but it is laborious and needs large amounts of oil sample and solvent.17 For instance, in Rowland and co-workers' recent work, the base extraction involved a complex multistep process with consumptions of 15 g of crude oil and more than 190 mL of solvents.18 Nonaqueous ion exchange chromatography-based methods have also been applied to the separation of petroleum acids for many years.19 In early works, large scale chromatographic column was used, and the consumptions of ion exchange resin, oil sample and solvent were extremely high.20 The commercial solid phase extraction (SPE) cartridge can reduce these consumptions greatly.21 However, the existing SPE methods still have some drawbacks. In Freitas and co-workers’ work, 10 mL of sampling solution and 55 mL of organic solvent were used in the whole SPE procedure, and 20 mL of desorption solution needed to be slowly evaporated under flowing N2.22 The consumptions of sample and solvents were still on the high side, and the evaporation of large amount of desorption solution was time-consuming and might cause losses of analytes with low boiling points.Micro-SPE apparatuses,23–25 such as in-pipette-tip26–28 and in-syringe formats,29–31 are frequently utilized in bioanalysis. The unique properties of these formats, including rapid, convenient and low consumptions of sample and solvent,32 inspire us to develop an extraction method to meet the challenges in the analysis of petroleum acids.
In this work, for the first time, attempts were made to develop a micro-SPE based method for the analysis of petroleum acids in crude oils. The types of vessel and sorbent are two key factors to a micro-SPE. Firstly, the influences of different plastic and glass vessels to the extraction were investigated. Because of the hydrocarbon matrix of crude oil, the use of plastic vessels would cause plasticizer interferences and serious adhesion of oil samples on vessel surfaces, while the use of glass vessels could reduce or eliminate these influences. Here, glass pipette, a common glass vessel in lab, was selected as the micro-SPE vessel. With this micro-SPE format, the extraction efficiencies of three different sorbents were compared. It was found that amino-functionalized silica possessed superior efficiency, due to its mix-mode hydrogen bonding/ion-exchange interactions. To the best of our knowledge, this is the first time that hydrogen bonding interaction was proposed and validated in extraction of petroleum acids. In addition, under the optimized conditions, no evaporation was needed, which can save time and reduce losses of analytes. Therefore, by coupling with GC-MS, a simple, rapid, solvent-saving and effective micro-SPE method for the analysis of petroleum acids in crude oils was established.
2. Experimental section
2.1 Chemicals and materials
HPLC grade n-hexane, dichloromethane (DCM), ethly acetate (EtAc), methanol (MeOH) and acetone were obtained from Fisher Scientific (PA, USA). Cyclohexanecarboxylic acid (CHA), decanoic acid (DA), lauric acid (LA), 1-adamantanecarboxylic acid (ACA), 1-adamantaneacetic acid (AAA), 1-naphthaleneacetic acid (NAA), hexadecanoic acid, octadecanoic acid, N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA), and trifluoroacetic acid (TFA) were purchased from Aladdin Chemical Reagent (Shanghai, China). Bicyclo[2,2,1]heptane-2-carboxylic acid (BCHCA) and 3-noradamantanecarboxylic acid (NACA) were supplied by Thermo Fisher Scientific (MA, USA). trans-4-Ethylcyclohexanecarboxylic acid (t-ECHA), trans-4-isopropylcyclohexanecarboxylic acid (t-iPCHA), trans-4-butylcyclohexanecarboxylic acid (t-BCHA) and trans-4-pentylcyclohexanecarboxylic acid (t-PCHA) were purchased from Adamas-beta (Shanghai, China). Cotton wool was supplied by Xuzhou Hygiene of Material Factory (Xuzhou, China). Amino-functionalized silica, carboxylic acid-functionalized silica and quaternary amine-functionalized silica were obtained from Weltech (Wuhan, China). Crude oil samples were supported from our cooperative oil company.2.2 Study of the interferences from different vessels in nonaqueous extraction
Various glass vessels (glass vial and glass pipette) and plastic vessels (plastic centrifuge tubes, medical syringes and plastic pipette tips) were washed with 1 mL of hexane or EtAc individually. The washing solutions were evaporated to dryness and redissolved in 100 μL of hexane, and then derivatized with 10 μL of MTBSTFA under vortex at room temperature for 5 min prior to analysis by GC-MS. Washing glass vial with n-hexane was selected as the control group.2.3 Preparation of the glass pipette micro-SPE device
Glass pipettes with a length of 15 cm, an inner diameter from 2 mm to 8 mm and a capacity of 1.5 mL were purchased from Aopu Huabo (Wuhan, China). Amino-functionalized silica was packed in the glass pipette with cottons (1 mg) on both ends. As shown in Fig. S1 (ESI†), the sorbent bed is in the middle of the glass pipette (the top of the thinner tube).2.4 Sample preparation with glass pipette micro-SPE
The glass pipette micro-SPE devices were directly used for extraction experiments. A schematic of the micro-SPE approach is shown in Scheme 1. Crude oils were diluted ten times with hexane. 1-Naphthaleneacetic acid (NAA) was added at a concentration of 50 ng mL−1 to the diluted samples to serve as the internal standard (IS) for quantification. The diluted oil sample (1 mL) was aspirated and dispensed 10 cycles to allow petroleum acids to be adsorbed on the sorbent. After washing twice with 1 mL of hexane/DCM (1/1, v/v), the trapped petroleum acids were eluted with 100 μL of TFA/EtAc (1/99, v/v). The eluate was derivatized with 10 μL of MTBSTFA under vortex at room temperature for 5 min prior to GC-MS analysis. | ||
| Scheme 1 Schematic of the procedure for the glass pipette micro-SPE based method for analysis of petroleum acids. | ||
In the experiments for optimization of extraction conditions, diluted oil sample was spiked with eleven petroleum acid standards (Table S1, ESI†) at a certain concentration (100 ng mL−1), and the IS (NAA) was added in the eluate rather than the sampling solution.
To obtain a blank sample for calibration and validation purposes, a crude oil was fractionated into saturates, aromatics, resins and asphaltenes (SARA fractionation method),33 and then the saturates, aromatics and asphaltenes were remixed together as the matrix free of petroleum acids.
2.5 GC-MS analysis
Analysis of petroleum acids was carried out on a GC-MS system consisting of an Agilent 7890 GC, a 7683B autosampler and a 5975C mass spectrometer (CA, USA). The GC separation was performed on a DB-5MS capillary column, 30 m (length) × 0.25 mm (i.d.) × 0.25 μm (film thickness), purchased from Agilent. High purity helium (99.9995%) was used as the carrier gas at a flow rate of 1.0 mL min−1. The injector temperature was 300 °C. The injection volume was 1.0 μL in splitless mode. The oven temperature was programmed from 100 °C to 210 °C at 6 °C min−1, then to 300 °C at 12 °C min−1 and held for 5 min. The solvent delay was 8.5 min. The mass spectrometer was operated in the electron impact mode (70 eV). The temperatures of ion source and transfer-line were set at 230 °C and 300 °C, respectively. The temperature of quadrupole was held at 150 °C. A full scan ranging from m/z 50 to 550 was performed.3. Results and discussion
3.1 Influences of different vessels in nonaqueous extraction
To investigate the potential interferences from vessels in nonaqueous extraction, different glass vessels and plastic vessels were washed by nonpolar (hexane) or low polar solvent (EtAc), then the washing solutions were derivatized with MTBSTFA and analyzed by GC-MS. The results are shown in Fig. S2 (ESI†). It can be seen that when plastic vessels, including plastic centrifuge tubes, medical syringes and plastic pipette tips, were washed by hexane or EtAc, the signals of some compounds dissolved from plastic would dominate the GC-MS chromatograms (Fig. S2, ESI†). On the contrary, the GC-MS chromatograms of the washing solutions with glass vials and glass pipettes display much lower intensity of signals (Fig. S2, ESI†). To obtain quantifiable results for comparison, the peak areas of the signals representing hexadecanoic acid and octadecanoic acid (two common used plasticizers, also two potential petroleum acids in crude oils) were calculated and shown in Table S2 (ESI†). Washing glass vial with hexane was selected as the control group. The peak areas of the two acids in washing solutions with glass pipettes are similar to those in the control group, while the peak areas with plastic centrifuge tubes, medical syringes and plastic pipette tips are 425–2812, 398–3875 and 49–316 times higher than those in the control group. These results indicate that in nonaqueous extraction, such as extraction of petroleum acids from crude oils, using glass vessels instead of plastic vessels can reduce or eliminate the interferences from vessels.In addition, it was found that when plastic vessels were used to deal with crude oils, the strong adhesion of oils on the plastic surfaces would make the washing step in extraction process difficult. This issue could be distinctly improved by using glass vessels. Furthermore, glass vessels are more convenient to be reused after washing and high-temperature calcination, while plastic vessels are generally single-use.
Here, glass pipette, a common glass vessel in lab, was selected as the vessel of micro-SPE for extraction of petroleum acids. The glass pipette micro-SPE device was prepared by packing sorbents in a glass pipette with cottons on both ends (Fig. S1, ESI†), which is simple and low cost. The procedure of the micro-SPE is shown in Scheme 1. The SPE procedure is easy-operated, fast and suitable for in-field sample preparation without any electrical device.
3.2 Optimization of conditions for the micro-SPE method
In nonaqueous system, especially in nonpolar solvent system, hydrogen bonding is usually a key interaction in adsorption. Most of the components in crude oils are nonpolar hydrocarbon compounds. However, in the existing chromatography-based methods for extraction of petroleum acids, almost all used ion-exchange materials as the sorbents based on the ion-exchange adsorption.21,22 To the best of our knowledge, hydrogen bonding has never been mentioned in previous works. Here, as the sorbent for extraction of petroleum acids, amino-functionalized silica (AFS) was introduced and compared with quaternary amine-functionalized silica (strong anion exchange, SAX) and carboxylic acid-functionalized silica (CAFS). The results are shown in Fig. 1. It can be seen that the AFS provides superior performance in extraction of petroleum acids. SAX also exhibits adsorption capacity of the acids, but the desorption is incomplete because of the strong interaction. Increasing the volume or acidity of the desorption solution may improve the recovery, but this would also increase the difficulty of subsequent process. Interestingly, it was found that CAFS could also adsorb petroleum acids and its extraction efficiency is close to that of SAX. We propose that the adsorptions of petroleum acids on SAX and CAFS are based on ion-exchange and hydrogen bonding interaction, respectively, while the adsorption on AFS is based on mix-mode hydrogen bonding/ion-exchange interactions (Scheme 2).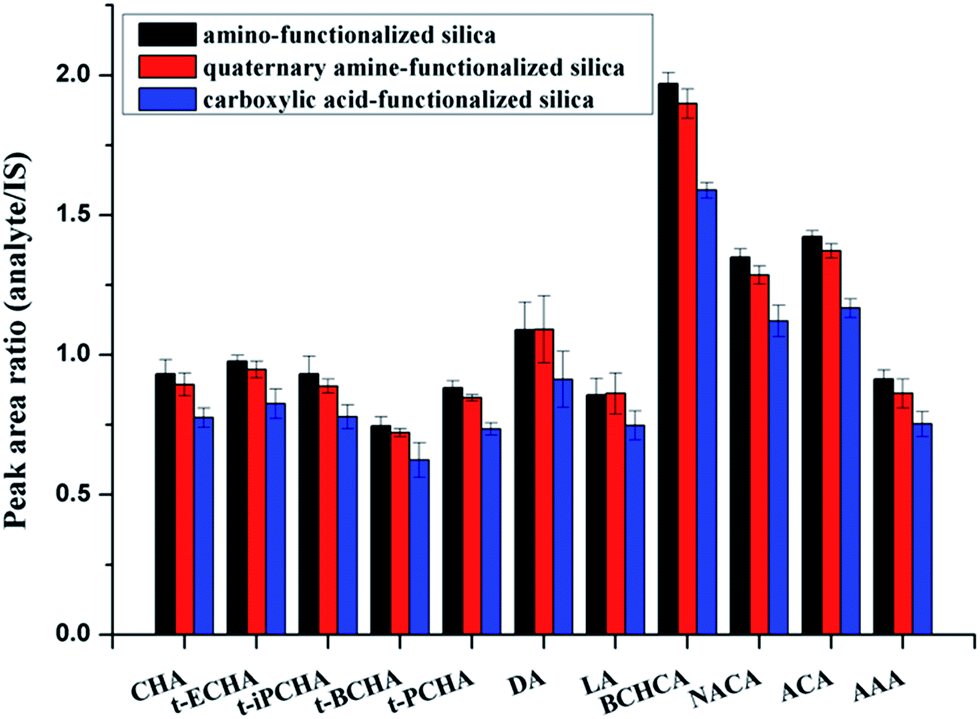 | ||
| Fig. 1 Extraction efficiencies of three different sorbents. | ||
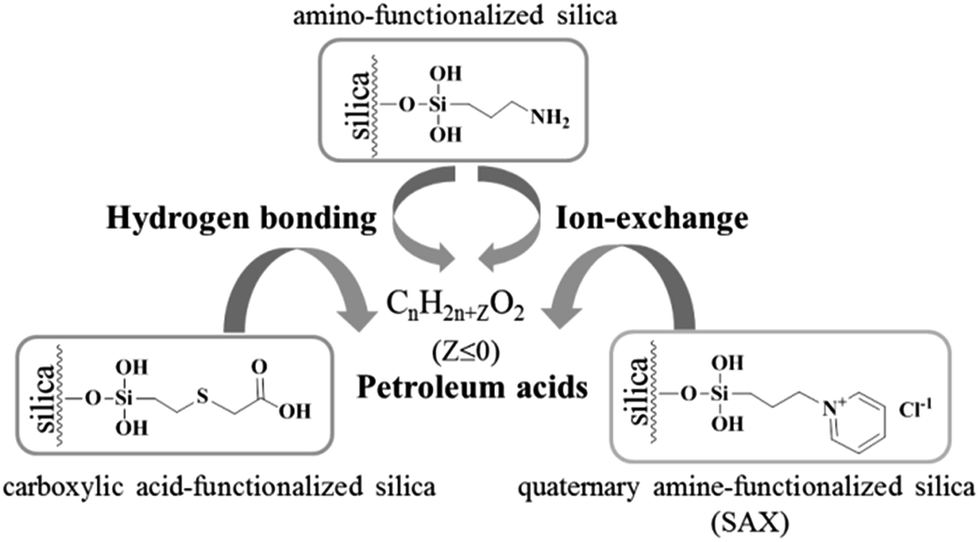 | ||
| Scheme 2 Schematic of the interactions between analytes and sorbents. | ||
AFS was selected as the sorbent for the following glass pipette micro-SPE experiments. To achieve the best performance, a series of parameters, including desorption solution, derivatization conditions, sorbent amount, sampling cycle and eluting cycle, were investigated.
Six different solutions were used to evaluate the desorption efficiency. Fig. 2 shows that when pure organic solvents without acid were used, the desorption efficiency of MeOH is much better than that of EtAc or acetone. This further indicates the existence of hydrogen bonding interaction between petroleum acids and AFS. The addition of TFA can improve the desorption efficiency obviously. And overall, the efficiency of TFA/EtAc (1/99, v/v) is slightly better than that of TFA/MeOH (1/99, v/v) or TFA/acetone (1/99, v/v). Thus, TFA/EtAc (1/99, v/v) was selected as the desorption solution for further experiments.
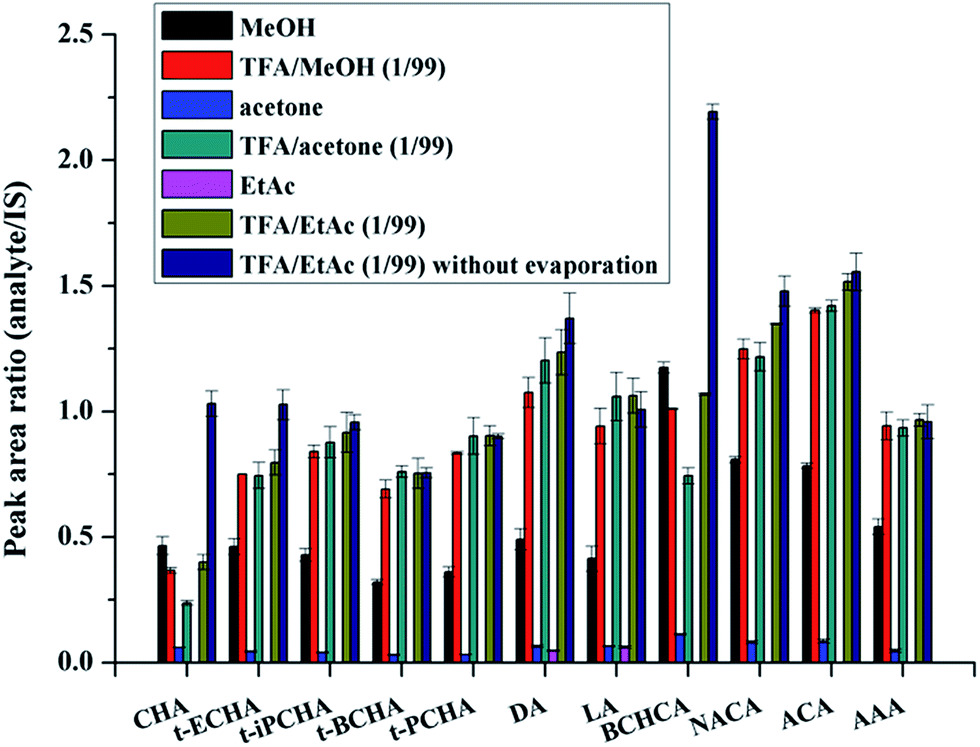 | ||
| Fig. 2 Effect of desorption solution on extraction efficiency. | ||
It should be noted that all the extraction experiments above needed an evaporation step (after eluting step) under flowing N2. The evaporation step was regarded to be used to eliminate strong polar interferents (MeOH, TFA) that could affect the subsequent derivatization or GC-MS analysis. Besides, in previous works, evaporation was also used for concentration of the large volume of desorption solution.22 We made attempts to remove the evaporation step and directly performed the derivatization of petroleum acids in the desorption solution. To reach this goal, the amount of derivatization regent should be enough to ensure the completely derivatization of both TFA and petroleum acids. Various amounts of the derivatization reagent were used to test the derivatization efficiency. Results show that the derivatization efficiency keeps almost constant when the derivatization reagent is substantial excess (Fig. S3, ESI†), and 10 μL of the derivatization reagent was chosen for further experiments. On this condition, the derivatization efficiency in TFA/EtAc (1/99, v/v) was compared with that in hexane. Fig. 3 demonstrates that the two efficiencies are comparable. Moreover, it was found that the retention time of the derivative product of TFA in GC was earlier than those of all petroleum acids, and thus a suitable solvent delay time of MS was set to skip the detection of the high concentration by-product. The comparison of the analysis results with evaporation step and without evaporation step is shown in Fig. 2. It can be seen that the evaporation step would cause the losses of analytes, especially the petroleum acids with low boiling points. For instance, the losses of the two analytes with the lowest boiling points (CHA and BCHCA) are more than 50% in the evaporation step. Therefore, the time-consuming and unprofitable evaporation step was removed from this work. This strategy may provide inspirations for other works to remove evaporation, such as extraction of fatty acids from edible oils prior to GC or GC-MS analysis,34 in which organic solvent containing TFA or formic acid is frequently used as the desorption solution.
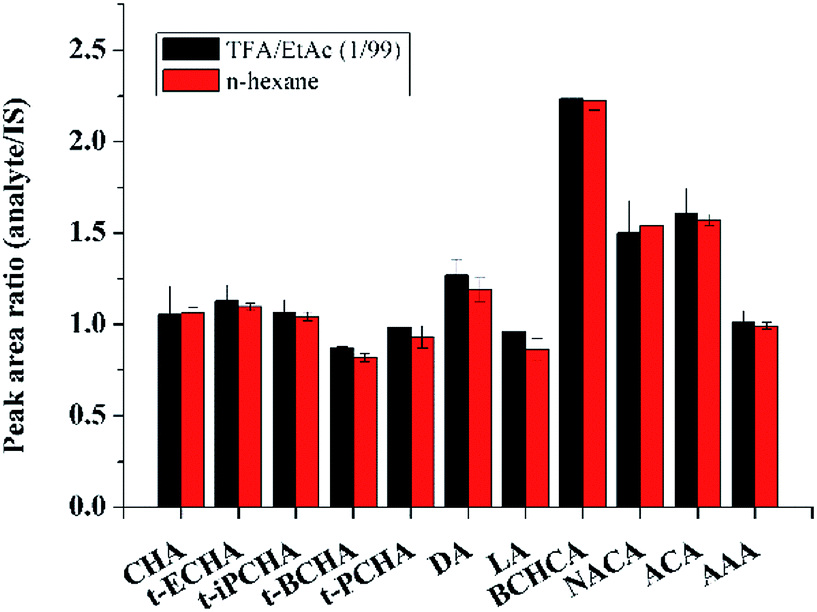 | ||
| Fig. 3 Derivatization efficiencies of petroleum acids with MTBSTFA in TFA/EtAc (1/99, v/v) and n-hexane. | ||
The effect of derivatization time was investigated from 2 min to 30 min. Fig. S4 (ESI†) indicates that high derivatization efficiency can be achieved within 5 min, because of the high silylation reactivity of carboxylic acid. Therefore, 5 min was selected as the derivatization time for further experiments.
Various amounts of sorbent ranging from 0 mg to 20 mg were packed in the glass pipette to extract petroleum acids. When no AFS was used, signals of petroleum acids could also be observed (Fig. 4), although with poor recoveries, because of the hydroxyl groups on the surface of cotton that could also provide hydrogen bonding interactions with petroleum acids. The recoveries keep almost constant with sorbent amounts from 2 mg to 20 mg (Fig. 4). Considering the stability and the back pressure, 5 mg of AFS was chosen for further experiments.
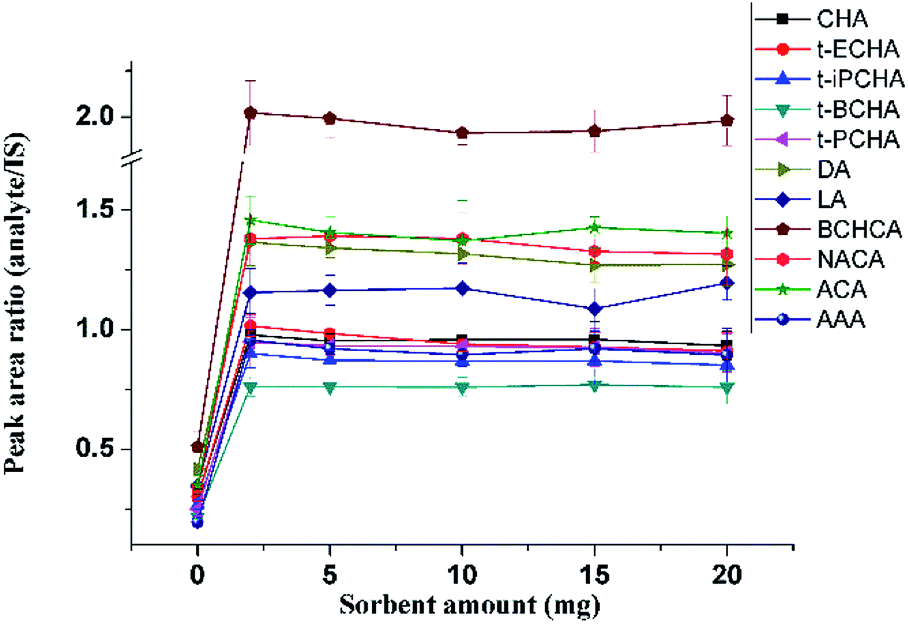 | ||
| Fig. 4 Effect of sorbent amount on extraction efficiency. | ||
As shown in Scheme 1, the extraction steps were performed by simply aspiration and dispensation of solutions. The cycles of aspiration and dispensation in the sampling and eluting steps were optimized. As shown in Fig. S5 (ESI†), with the increase of sampling cycle or eluting cycle from 5 to 40, there are no obvious differences for the recoveries of petroleum acids. Thus, 10 cycles were selected for both the sampling and eluting steps. Notably, 10 cycles of aspiration and dispensation of solutions requires only about 1 min. Thus, under the optimized conditions, the glass pipette micro-SPE procedure can be accomplished within 5 min.
The preparation reproducibility of the developed glass pipette micro-SPE was tested, and results show that the batch to batch relative standard deviations (RSDs) (n = 5) range from 4.2% to 8.5%, indicating the good preparation reproducibility. The recoveries of petroleum acids were found to be 74–92% in crude oil spiked with petroleum acid standards (Table S3, ESI†).
3.3 Validation of the method
To validate the micro-SPE approach for analysis of petroleum acids, calibration cures were constructed firstly. Good linear correlations are achieved with correlation coefficients (R) greater than 0.9983 (Table S4, ESI†). The limits of detection (LODs) and limits of quantitation (LOQs) of petroleum acids range from 2 ng g−1 to 6 ng g−1 and 6 ng g−1 to 20 ng g−1, respectively. The accuracy of the method was examined and referred to as relative recovery, and the precision of the method was assessed by measuring intra-day and inter-day RSDs. Results show that the relative recoveries are between 79% and 115%, and the intra-day and inter-day RSDs are less than 12.3% and 12.4% (Table 1), illustrating that both the accuracy and precision of the developed method are acceptable.| Analytes | Intra-day precision (RSD, %; n = 5) | Inter-day precision (RSD, %; n = 3) | Recovery (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Low 20 ng g−1 | Medium 200 ng g−1 | High 2000 ng g−1 | Low 20 ng g−1 | Medium 200 ng g−1 | High 2000 ng g−1 | Low 20 ng g−1 | Medium 200 ng g−1 | High 2000 ng g−1 | |
| CHA | 3.1 | 4.1 | 2.3 | 11.4 | 2.6 | 3.5 | 94 | 96 | 89 |
| t-ECHA | 4.2 | 2.2 | 2.5 | 3.5 | 2.5 | 2.6 | 84 | 83 | 86 |
| t-iPCHA | 2.3 | 2.7 | 2.2 | 5.1 | 2.4 | 5.6 | 86 | 83 | 86 |
| t-BCHA | 10.0 | 1.4 | 3.2 | 11.1 | 2.9 | 4.6 | 85 | 89 | 90 |
| t-PCHA | 6.1 | 2.4 | 2.6 | 4.6 | 5.4 | 4.1 | 83 | 84 | 89 |
| DA | 7.3 | 5.1 | 3.4 | 8.2 | 12.4 | 7.9 | 114 | 103 | 97 |
| LA | 9.5 | 12.3 | 2.5 | 9.2 | 6.4 | 5.6 | 115 | 112 | 104 |
| BCHCA | 2.5 | 4.3 | 2.9 | 2.1 | 2.6 | 7.7 | 86 | 89 | 86 |
| NACA | 10.3 | 2.9 | 3.5 | 3.1 | 4.7 | 5.5 | 79 | 83 | 82 |
| ACA | 4.3 | 3.3 | 1.8 | 4.7 | 2.1 | 4.1 | 81 | 88 | 84 |
| AAA | 3.7 | 2.3 | 4.0 | 5.7 | 2.0 | 2.0 | 83 | 81 | 87 |
3.4 Applications in real samples
To study the applicability, the micro-SPE method was applied to analysis of petroleum acids from three crude oils with different densities; meanwhile, a typical base extraction method18 was also utilized to deal with these samples for comparison. As listed in Table 2, the analytical results with the two methods are comparable. These results indicate the feasibility of the micro-SPE based method for analysis of petroleum acids in complex crude oils.| Crude oil | Contents (ng g−1) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CHA | t-ECHA | t-iPCHA | t-BCHA | t-PCHA | DA | LA | BCHCA | NACA | ACA | AAA | |
| a Not detected. | |||||||||||
| 1 | 515 | 33 | 22 | 27 | 43 | 202 | 298 | —a | — | 31 | — |
| (493) | (28) | (—) | (25) | (46) | (211) | (285) | (—) | (—) | (37) | (—) | |
| 2 | 162 | — | — | — | — | 1131 | 1596 | — | — | — | — |
| (150) | (—) | (—) | (—) | (—) | (1252) | (1702) | (—) | (—) | (—) | (—) | |
| 3 | 223 | 59 | 32 | — | — | 1532 | 3562 | — | — | — | — |
| (240) | (69) | (—) | (—) | (24) | (1705) | (3845) | (—) | (—) | (—) | (—) | |
3.5 Comparison of the micro-SPE method with reported methods
To evaluate the analytical performance of the proposed method, a comparison of the proposed micro-SPE method with other reported methods for the determination of petroleum acids is presented in Table 3. Compared with the existing methods for analysis of petroleum acids in crude oils, the micro-SPE method required much smaller amounts of oil sample and solvent, and shorter time. Because few previous works on analysis of petroleum acids in crude oils presented the LOD or LOQ result, two reported works on analysis of petroleum acids in water samples were selected to compare with this work. It can be seen that the LOQ in this work is lower than that in direct analysis of water sample after centrifugation, but higher than that in analysis of water sample after LLE or SPE. Considering the significant differences on enrichment factor and complexity between the crude oil and water samples, the sensitivity of the micro-SPE method is acceptable.| Sample preparation method | Consumption | Sample | Instrument | LOQ (ng g−1) | Ref. | |
|---|---|---|---|---|---|---|
| Time (min) | Solvent (mL) | |||||
| a Alumina modified with 1,4-bis-(n-propyl)diazoniabicyclo[2.2.2]octane chloride silsesquioxane.b Liquid–liquid extraction. | ||||||
| SPE (SAX) | >60 | >260 | Crude oil (10 g) | GC-MS | — | 21 |
| SPE (Dab-Al2O3)a | >30 | 55 | Crude oil | GC-MS | — | 22 |
| Base extraction | >60 | 190 | Crude oil (15 g) | GC × GC-MS | — | 18 |
| Centrifugation | >5 | 0 | River water (10 mL) | LC-MS | 100 | 35 |
| LLEb or SPE (Oasis HLB) | >40 | >35 | Tailings water (100 mL) | LC-MS | 0.3–6 | 36 |
| Micro-SPE (amino-functionalized silica) | <5 | 3 | Crude oil (0.1 g) | GC-MS | 6–20 | This work |
4. Conclusions
A glass pipette micro-SPE method was developed for extraction of petroleum acids from crude oils with low-consumptions of sample, solvent and time. Good recoveries were achieved by using amino-functionalized silica as the sorbent for extraction based on mix-mode hydrogen bonding/ion-exchange interactions. The whole sample preparation procedure is easy and fast without any evaporation step. This research may provide a promising method for analysis of petroleum acids in chloroform bitumen sample that is difficult to obtain in large quantity. We also believe that the glass pipette micro-SPE packed with other specific sorbent has great potentials in analysis of nonaqueous samples such as petroleum, edible oils and volatile oils.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial supports from the National Natural Science Foundation of China (21607137), Fundamental Research Funds for the Central Universities, China University of Geosciences (Wuhan) (CUG160606), China National Science and Technology Major Project (2016ZX05006003-001), and China Geological Survey Project Grant (12120114046901).References
- M. J. Wilde and S. J. Rowland, Anal. Chem., 2015, 87, 8457–8465 CrossRef CAS PubMed.
- K. D. Duncan, D. R. Letourneau, G. W. Vandergrift, K. Jobst, E. Reiner, C. G. Gill and E. T. Krogh, J. Mass Spectrom., 2016, 51, 44–52 CrossRef CAS PubMed.
- M. P. Barrow, L. A. McDonnell, X. D. Feng, J. Walker and P. J. Derrick, Anal. Chem., 2003, 75, 860–866 CrossRef CAS PubMed.
- M. B. Woudneh, M. C. Hamilton, J. P. Benskin, G. H. Wang, P. McEachern and J. R. Cosgrove, J. Chromatogr. A, 2013, 1293, 36–43 CrossRef CAS PubMed.
- J. P. Swigert, C. Lee, D. C. L. Wong, R. White, A. G. Scarlett, C. E. West and S. J. Rowland, Chemosphere, 2015, 124, 1–9 CrossRef CAS PubMed.
- M. P. Barrow, M. Witt, J. V. Headley and K. M. Peru, Anal. Chem., 2010, 82, 3727–3735 CrossRef CAS PubMed.
- J. Liggio, S. M. Li, K. Hayden, Y. M. Taha, C. Stroud, A. Darlington, B. D. Drollette, M. Gordon, P. Lee, P. Liu, A. Leithead, S. G. Moussa, D. Wang, J. O'Brien, R. L. Mittermeier, J. R. Brook, G. Lu, R. M. Staebler, Y. M. Han, T. W. Tokarek, H. D. Osthoff, P. A. Makar, J. H. Zhang, D. L. Plata and D. R. Gentner, Nature, 2016, 534, 91–94 CrossRef CAS PubMed.
- B. Bennett, C. M. Aitken, D. M. Jones, P. Farrimond and S. R. Larter, Org. Geochem., 2007, 38, 1977–1985 CrossRef CAS.
- J. A. Valencia-Dávila, C. Blanco-Tirado and M. Y. Combariza, Fuel, 2017, 193, 168–177 CrossRef.
- W. K. Seifert, E. J. Gallegos and R. M. Teeter, Angew. Chem., Int. Ed., 1971, 10, 747–748 CrossRef CAS.
- J. S. Clemente and P. M. Fedorak, Chemosphere, 2005, 60, 585–600 CrossRef CAS PubMed.
- J. V. Headley, K. M. Peru and M. P. Barrow, Mass Spectrom. Rev., 2016, 35, 311–328 CrossRef CAS PubMed.
- W. Jie, X. F. Cao, L. W. Chai, J. Q. Liao, Y. Huang and X. Y. Tang, Anal. Methods, 2015, 7, 2149–2154 RSC.
- J. V. Headley, K. M. Peru and M. P. Barrow, Mass Spectrom. Rev., 2009, 28, 121–134 CrossRef CAS PubMed.
- X. Pan and R. P. Philp, Org. Geochem., 2006, 37, 1085–1100 CrossRef CAS.
- F. C. Damasceno, L. D. A. Gruber, A. M. Geller, M. C. Vaz de Campos, A. O. Gomes, R. C. L. Guimarães, V. F. Péres, R. A. Jacques and E. B. Caramão, Anal. Methods, 2014, 6, 807–816 RSC.
- I. Dzidic, A. C. Somerville, J. C. Raia and H. V. Hart, Anal. Chem., 1988, 60, 1318–1323 CrossRef CAS.
- C. E. West, J. Pureveen, A. G. Scarlett, S. K. Lengger, M. J. Wilde, F. Korndorffer, E. W. Tegelaar and S. J. Rowland, Rapid Commun. Mass Spectrom., 2014, 28, 1023–1032 CrossRef CAS PubMed.
- W. K. Seifert and W. G. Howells, Anal. Chem., 1969, 41, 554–562 CrossRef CAS.
- D. M. Jewell, J. H. Weber, J. W. Bunger, H. Plancher and D. R. Latham, Anal. Chem., 1972, 44, 1391–1395 CrossRef CAS.
- D. M. Jones, J. S. Watson, W. Meredith, M. Chen and B. Bennett, Anal. Chem., 2001, 73, 703–707 CrossRef CAS PubMed.
- J. F. de Conto, S. Nascimento Jdos, D. M. de Souza, L. P. da Costa, S. M. Egues, S. Freitas Ldos and E. V. Benvenutti, J. Sep. Sci., 2012, 35, 1044–1049 CrossRef PubMed.
- J. Plotka-Wasylka, N. Szczepanska, M. de la Guardia and J. Namiesnik, TrAC, Trends Anal. Chem., 2015, 73, 19–38 CrossRef CAS.
- T. Deng, D. P. Wu, C. F. Duan and Y. F. Guan, J. Chromatogr. A, 2016, 1456, 105–112 CrossRef CAS PubMed.
- C. J. Zhang, G. K. Li and Z. M. Zhang, J. Chromatogr. A, 2015, 1419, 1–9 CrossRef CAS PubMed.
- G. T. Zhu, X. M. He, S. He, X. Chen, S. K. Zhu and Y. Q. Feng, ACS Appl. Mater. Interfaces, 2016, 8, 32182–32188 CAS.
- J. Rappsilber, M. Mann and Y. Ishihama, Nat. Protoc., 2007, 2, 1896–1906 CrossRef CAS PubMed.
- M. Atakay, Ö. Çelikbıçak and B. Salih, Anal. Chem., 2012, 84, 2713–2720 CrossRef CAS PubMed.
- G. T. Zhu, X. S. Li, X. M. Fu, J. Y. Wu, B. F. Yuan and Y. Q. Feng, Chem. Commun., 2012, 48, 9980–9982 RSC.
- G. T. Zhu, X. Chen, X. M. He, H. Wang, Z. Zhang and Y. Q. Feng, Chem.–Eur. J., 2015, 21, 4450–4456 CrossRef CAS PubMed.
- G. T. Zhu, X. Chen, X. M. He, Z. Zhang, X. S. Li, B. F. Yuan and Y. Q. Feng, RSC Adv., 2015, 5, 75341–75347 RSC.
- M. Abdel-Rehim, J. Chromatogr. A, 2010, 1217, 2569–2580 CrossRef CAS PubMed.
- W. Zhang, S. Zhu, S. He and Y. Wang, J. Chromatogr. A, 2015, 1380, 162–170 CrossRef CAS PubMed.
- F. Wei, Q. Zhao, X. Lv, X. Y. Dong, Y. Q. Feng and H. Chen, J. Agric. Food Chem., 2013, 61, 76–83 CrossRef CAS PubMed.
- D. Shang, M. Kim, M. Haberl and A. Legzdins, J. Chromatogr. A, 2013, 1278, 98–107 CrossRef CAS PubMed.
- M. Bataineh, A. C. Scott, P. M. Fedorak and J. W. Martin, Anal. Chem., 2006, 78, 8354–8361 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06763a |
| This journal is © The Royal Society of Chemistry 2017 |