
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heteropolytungstostannate as a homo- and heterogeneous catalyst for Knoevenagel condensations, selective oxidation of sulfides and oxidative amination of aldehydes
Roushan Khoshnavazi *,
Leila Bahrami and
Manuchehr Rezaei
*,
Leila Bahrami and
Manuchehr Rezaei
Department of Chemistry, University of Kurdistan, P.O. Box 66135-416, Sanandaj, Iran. E-mail: r.khoshnavazi@uok.ac.ir; khoshnavazi@yahoo.com; Fax: +98 8733 624 133; Tel: +98 8733 624 133
First published on 25th September 2017
Abstract
Heteropolytungstostannate K11H[P2W18O68(HOSnIVOH)3]·20H2O (1) is used as a homo- and heterogeneous catalyst for Knoevenagel condensations. New Fe3O4@SiO2@CH2(CH2)2NH3+[P2W18O68(HOSnIVOH)3]11− nanocatalyst was fabricated through electrostatic grafting of (1) on amine-modified silica-coated magnetic nanoparticles. The new magnetically recoverable nanocatalyst was characterized using infrared spectroscopy (IR), scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray powder diffractometry (XRD), energy-dispersive X-ray analysis (EDX) and alternating gradient force magnetometry (AGFM). The results indicate that the particles are mostly spherical in shape and have an average size of approximately <30 nm. The catalytic activity of the catalyst was investigated in the selective oxidation of sulfides to sulfoxides and oxidative amination of aldehydes in an aqueous medium. The catalyst was reused at least five times without significant decrease in catalytic activities, and the structure of the catalyst remained unchanged after recycling.
Introduction
Polyoxometalates (POMs) are a large class of molecular metal-oxide clusters with unmatched structural variety and versatility properties. POMs due to diversity in structure and composition have been used in many fields, including catalysis, medicine and material science.1–4 Owing to their redox and acidic properties, POMs exhibit catalytic activity in both homogeneous and heterogeneous catalysis.5,6 The application of POMs as catalysts is limited because they are solids with a low surface area (less than 10 m2 g−1) and also have high solubility in polar solvents. To overcome the limitations, POMs have been supported on inert and high-surface-area materials. Self-assembly techniques are being developed for designing catalysts based on polyoxometalates such as self-assembly of polyoxometalate with cations and positively charged nanoparticles that could facilitate catalyst recycling via filtration or centrifugation.7–11 Recently, magnetic nanoparticles (MNPs) have become promising candidates as high-surface-area materials for immobilization of homogeneous catalysts.12,13 Owing to their exceptionally high surface areas, MNPs exhibit high activity under mild conditions. Most examples of immobilization of POMs on surfaces have been limited to non-covalent interactions between Keggin heteropolyacids through electrostatic interaction of external oxygen atoms of the POMs with protonated hydroxyl groups of the surface.In comparison with the Keggin and Dawson-type POMs, the catalytic properties of sandwich-type POMs as heterogeneous catalysts have not been extensively studied.14–17 In this research, we investigate the catalytic properties of tin-containing sandwich-type polyoxometalates [P2W18O68(HOSnIVOH)3]12−. The structure of [P2W18O68(HOSnIVOH)3]12− (1) consists of two A-type α-PW9O34 anions linked by three tin(IV) cations leading to A-type sandwich POM18 (Fig. 1). Tin-containing sandwich-type polyoxometalates are more hydrolytically stable than transition-metal-substituted sandwich-type polyoxometalates in aqueous solution18–20 and thus are more suitable for oxidation reactions. Encouraged by these results, we evaluated the interaction of 1 with already prepared amine-modified silica-coated magnetite nanoparticles. The catalytic activity of the tin-containing sandwich-type polyoxometalate and the prepared nanocomposite was examined in reactions such as Knoevenagel condensations, oxidation of sulfides with H2O2 and oxidative amination of aldehydes in a green medium.
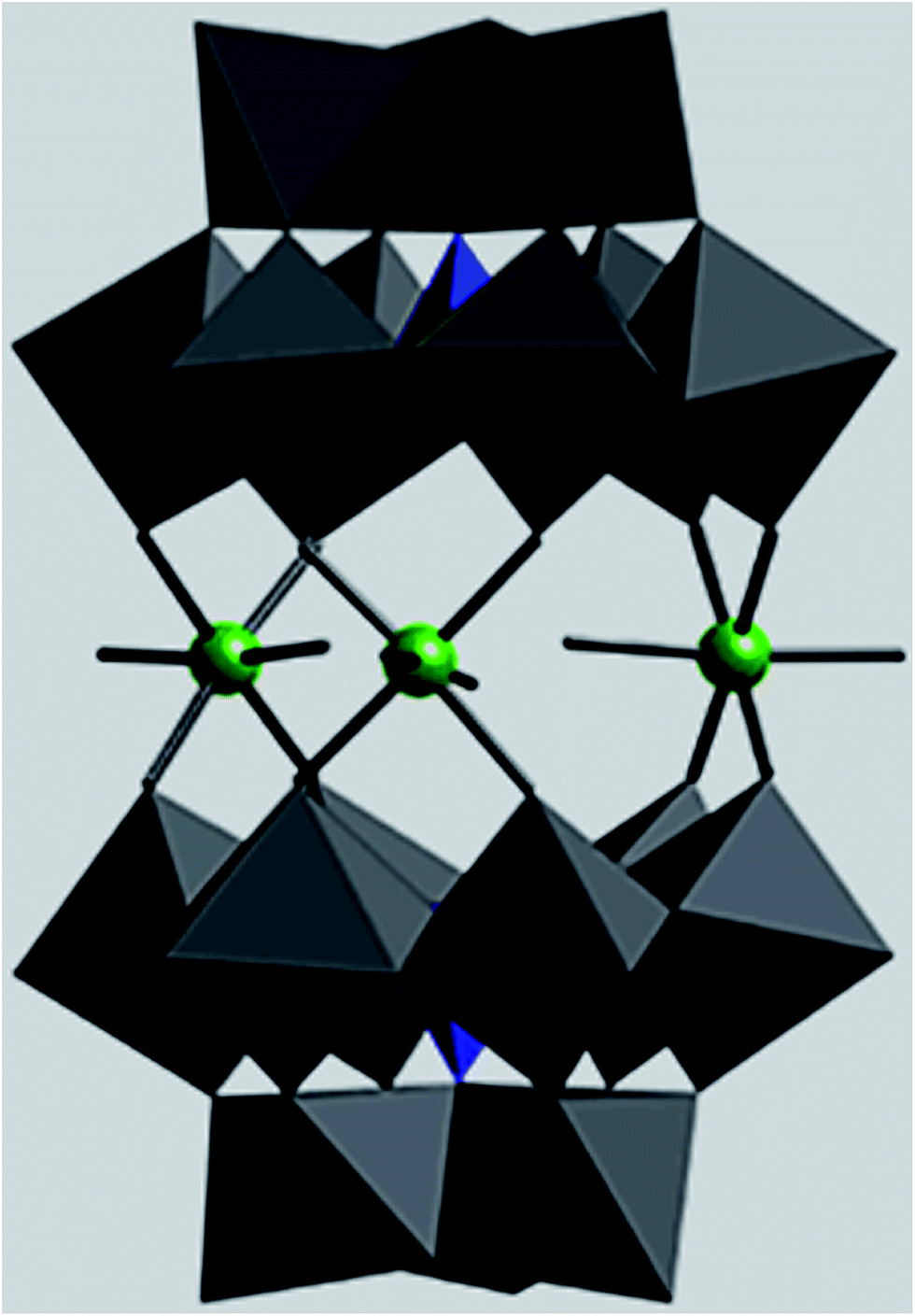 | ||
| Fig. 1 Polyhedral representation of [P2W18O68(HOSnIVOH)3]12−. | ||
Experimental section
Materials
All reagents were commercially obtained from Merck chemical company, Acrōs Organics and Sigma-Aldrich and used without further purification. Heteropolytungstostannate K11H[P2W18O68(HOSnIVOH)3]·20H2O was prepared according to the literature.18 The Fe3O4 nanoparticles, silica-coated magnetite nanoparticles (Fe3O4@SiO2) and Fe3O4@SiO2CH2(CH2)2NH2 were prepared according to the literature.21–23Characterization
The crystal structure of the synthesized nanocomposite was characterized by a Philips (X'Pert MPD) diffractometer in 40 kV and 30 mA with a CuKα radiation (λ = 1.5418 Å). Infrared spectra were recorded on a Bruker Vector 22 FT-IR using KBr pellet. The morphology of nanocomposite was revealed using a scanning electron microscope (FESEM-TESCAN MIRA3 from Czech Republic) and a transmission electron microscope (TEM, Zeiss-EM10C 80 KV).The elements in the nanocomposite sample were probed using energy-dispersive X-ray (EDX) spectroscopy accessory to the FESEM-TESCAN MIRA3 scanning electron microscopy. The magnetic properties were investigated using a home-made alternative gradient force magnetometer (AGFM) in the magnetic field range of −5000 to 5000 Oe at room temperature. Textural characteristics of the outgassed samples were obtained from nitrogen physisorption at 77 K using a Brunauer, Emmett and Teller surface area and porosity analyzer instrument.
Synthesis
Fe3O4@SiO2@CH2(CH2)2NH3+[P2W18O68(HOSnIVOH)3]11− (1a). Fe3O4@SiO2CH2(CH2)2NH2 (0.1 g) was dispersed in 20 mL of deionized water and ultrasonicated for 30 min. Then, a solution of 1 (0.1 g in 5 mL deionized water) was added dropwise into the solution, ultrasonicated for 30 min and the mixture was stirred under reflux condition for 24 h. The obtained solid was then magnetically collected from the solution and washed abundantly with water/ethanol and dried at 60 °C.
The Knoevenagel condensations. In a typical experiment, aldehyde (1 mmol) and malononitrile (1.2 mmol), 0.010 g of 1 as catalyst and water or ethanol (1 mL) were placed in a 20 mL glass bottle at 25 °C. The reaction mixture was constantly stirred vigorously. The progress was monitored through TLC (eluent: n-hexane/EtOAc, 2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). After completion of the reaction, the reaction mixture was transferred to a separating funnel and EtOAc (3 × 5 mL) was added and then the products were separated. After evaporation of EtOAc, the obtained products were recrystallized from EtOH and pure products were obtained in 85–95% yield.
:1). After completion of the reaction, the reaction mixture was transferred to a separating funnel and EtOAc (3 × 5 mL) was added and then the products were separated. After evaporation of EtOAc, the obtained products were recrystallized from EtOH and pure products were obtained in 85–95% yield.
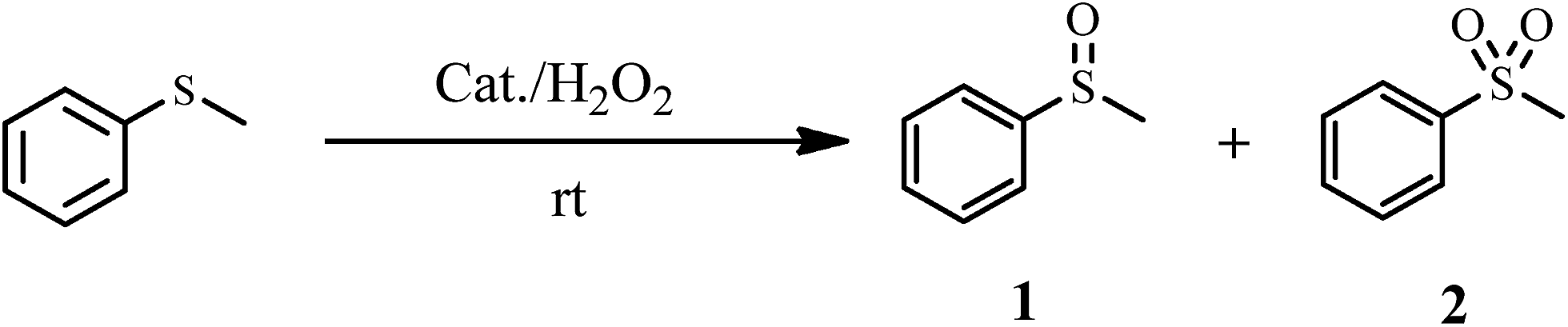
The oxidation of sulfides to sulfoxides. The catalytic activity of the catalyst was investigated in the selective oxidation of sulfides to sulfoxides using 30% H2O2 as a green oxidant. A mixture containing a sulfide (1 mmol), H2O (1.25 mL), 30% H2O2 (6 mmol) and catalyst 1a (0.030 g) was stirred at room temperature for the time specified. The progress of the reaction was monitored through TLC (eluent: n-hexane/EtOAc, 2
:1). After completion of the reaction, the catalyst was separated from the product using an external magnet. The product was extracted with CH2Cl2 (3 × 5 mL) and the combined organics were washed with brine (10 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to afford the corresponding pure sulfoxide in most cases.
The oxidative amination of aldehydes. While stirring aldehyde (1 mmol), hydroxylamine hydrochloride (1.2 mmol) and NaHCO3 (1.2 mmol) in water (1 mL), 1a nanocatalyst (0.020 g) was added to the mixture at a temperature of 90 °C. After completion of the reaction and separation of the magnetic nanocatalyst using an external magnet, the reaction mixture was extracted with ethyl acetate and washed with brine and then dried with Na2SO4. For further purification, the products were recrystallized from hexane and dichloromethane mixture to afford the desired products. The obtained products were identified on comparison of the physical properties and spectral data with pure samples and those reported in the literature.
Results and discussion
Synthesis and characterization
Magnetic Fe3O4 nanoparticles were prepared by chemical co-precipitation of Fe2+ and Fe3+ ions in the basic solution and silica-coated magnetite nanoparticles were prepared by the Stöber method.21 The Fe3O4@SiO2CH2(CH2)2NH2 nanoparticle was prepared by covalent grafting of silanol groups of Fe3O4@SiO2 surface and 3-aminopropyltriethoxysilane (APTS).Magnetic nanoparticles of 1a was prepared by simple direct addition of heteropolytungstostannate of [P2W18O68(HOSnIVOH)3]12− aqueous solution to Fe3O4@SiO2CH2(CH2)2NH2 nanoparticle. The magnetic nanoparticle of Fe3O4@SiO2@CH2(CH2)2NH2 was protonated by the acidic hydrogen and the hydrogen of hydroxide groups in the structure of K11H[P2W18O68(OHSnIVOH)3]·20H2O. The anionic [P2W18O68(OHSnIVOH)3]12− was attached to positively charged magnetic nanoparticles, Fe3O4@SiO2@CH2(CH2)2NH3+ nanoparticle through electrostatic interactions. The nanocatalyst synthesis pathway is shown in Scheme 1.
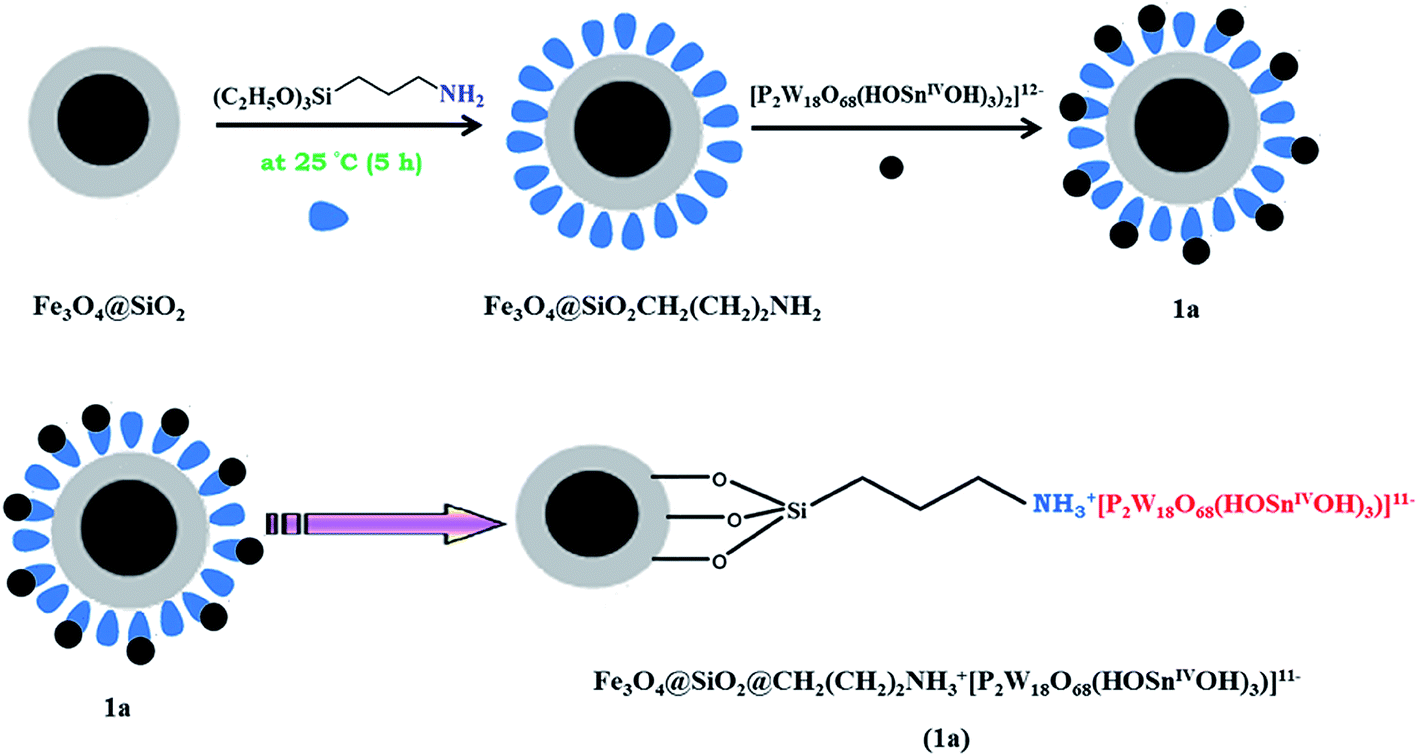 | ||
| Scheme 1 The synthesis pathway for preparation of 1a nanocomposite. | ||
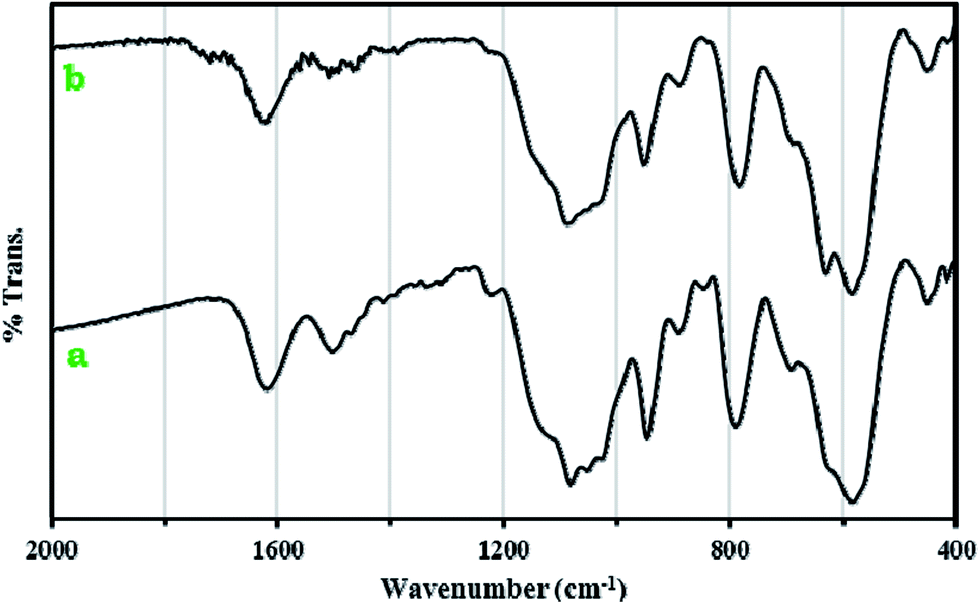
FT-IR spectra of 1a, Fe3O4@SiO2CH2(CH2)2NH2 and 1 are shown in Fig. 2. The peak at 573 cm−1 in Fe3O4@SiO2CH2(CH2)2NH2 and 1a spectra could be attributed to Fe–O stretching vibration.24 Two bands at 1070 and 800 cm−1 are ascribed to the symmetrical and asymmetrical vibrations of the Si–O–Si bonds.25 The very strong bands appearing in the 700–1090 cm−1 in spectrum of 1a are clearly assigned to W–O–W, W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and P–O stretching modes of [P2W18O68(HOSnIVOH)3]12−. The results show that small shifts to the higher wavenumbers were observed compared to 1. The C–H stretching vibration bands of the anchored propyl group appeared at 2925 and 2850 cm−1.26 These results prove the formation of nanocomposite 1a.
O and P–O stretching modes of [P2W18O68(HOSnIVOH)3]12−. The results show that small shifts to the higher wavenumbers were observed compared to 1. The C–H stretching vibration bands of the anchored propyl group appeared at 2925 and 2850 cm−1.26 These results prove the formation of nanocomposite 1a.
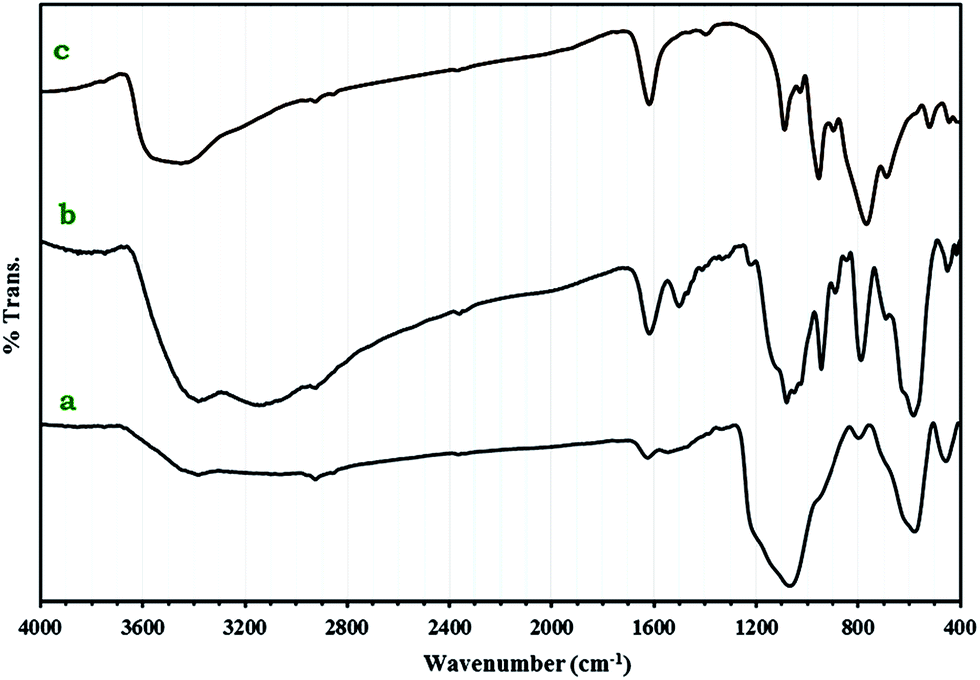 | ||
| Fig. 2 IR spectra of Fe3O4@SiO2CH2(CH2)2NH2 (a), 1a nanocomposite (b) and 1 (c). | ||
Fig. 3 shows the XRD results of Fe3O4 (a), 1a (b), and 1 (c). Diffraction peaks with 2θ = 18.2°, 30.2°, 35.6°, 43.2°,53.7°, 57.2°, 62.7° and 74.4° are attributed to Fe3O4, which indicates a cubic structure of the magnetite.27 The peaks corresponding to 1 are also observed in 1a, which is a confirmation of the successful synthesis of nanocomposite. The particle diameters that were calculated from the Scherrer formula are in the range of 18–30 nm.
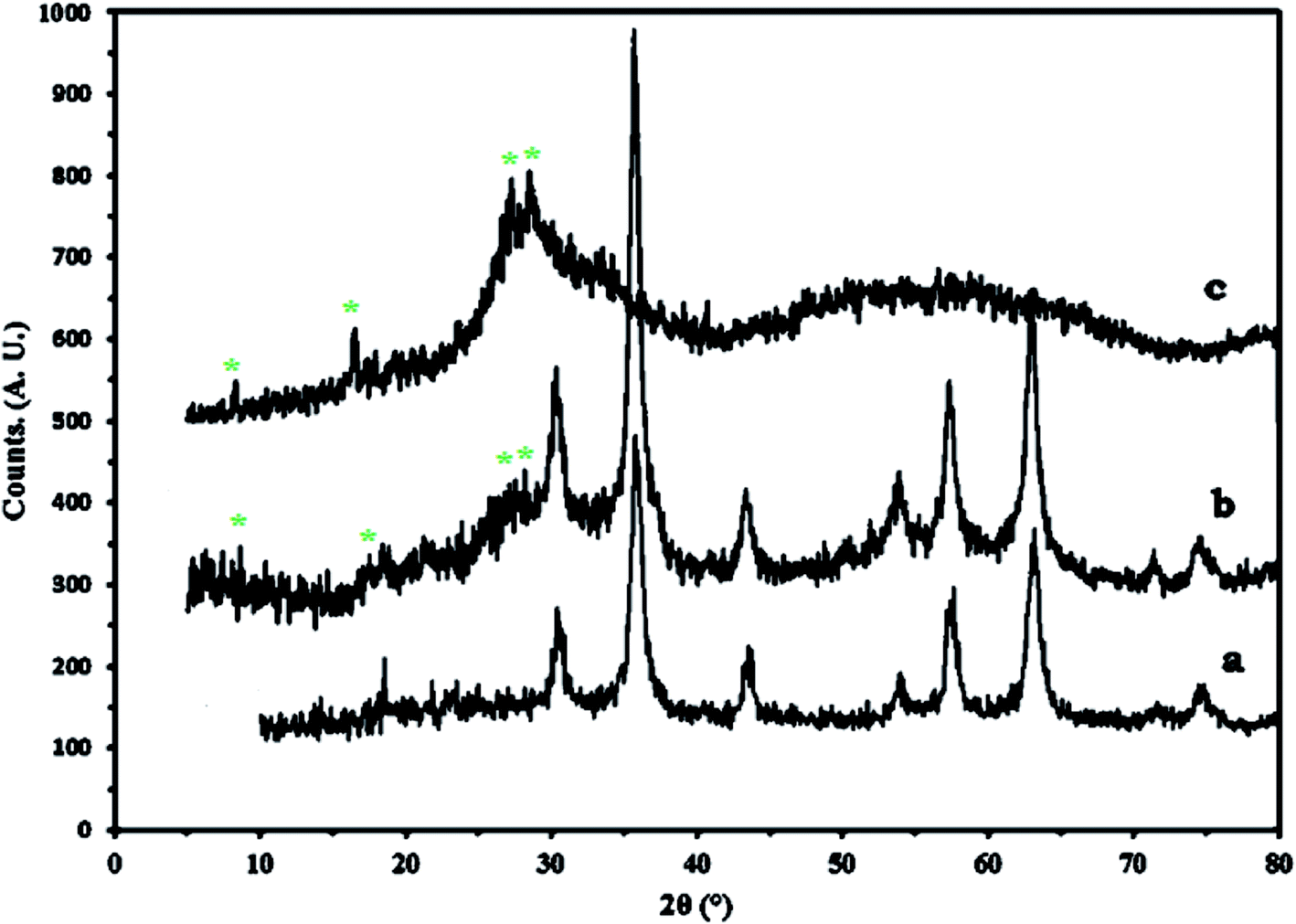 | ||
| Fig. 3 The XRD diffraction patterns of Fe3O4 (a) 1a nanocomposite (b) and 1 (c). | ||
The EDX spectrum obtained for 1a (Fig. 4) revealed that Fe, O, Si, C, N, Sn, P and W peaks are associated with Fe3O4@SiO2@CH2(CH2)2NH3+[P2W18O68(HOSnIVOH)3]11−. These results demonstrate that the mentioned polyoxometalate was immobilized onto the surface of the functionalized support.
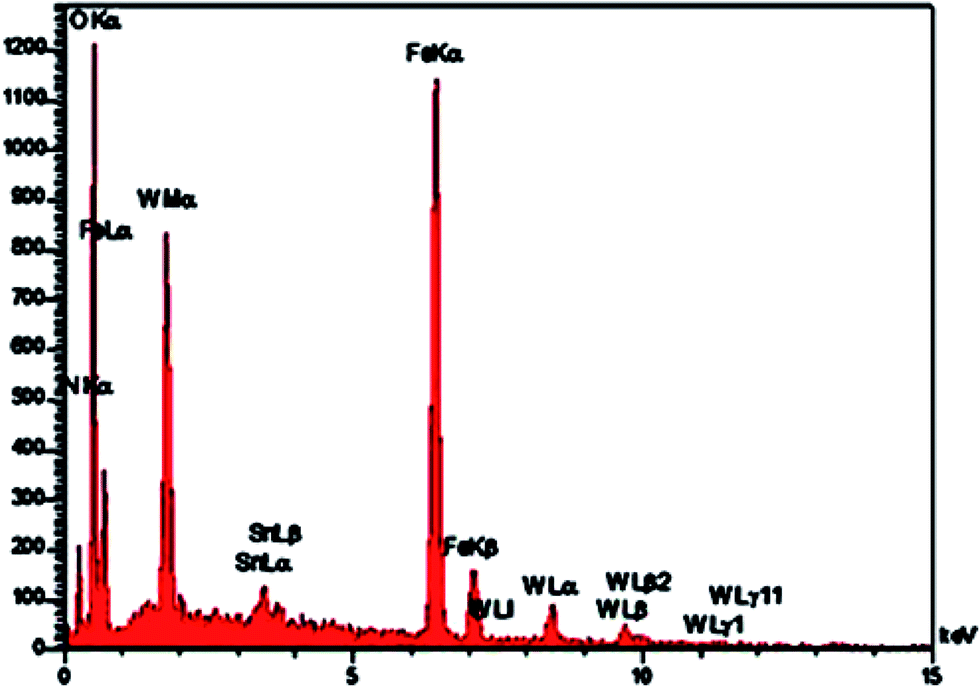 | ||
| Fig. 4 EDX analysis of 1a nanocomposite. | ||
Magnetic properties of Fe3O4@SiO2CH2(CH2)2NH2 and 1a nanocomposite were analyzed using an alternating gradient force magnetometer (AGFM) at room temperature (Fig. 5). A decrease in saturation magnetization was observed for 1a, which could be attributed to the increased mass of [P2W18O68(HOSnIVOH)3]12−. Even with this reduction in the saturation magnetization, complete magnetic separation of 1a was achieved in <20 s by placing a magnet near the vessels containing the aqueous dispersion of the nanoparticles. These results indicate the polyoxometalate coating on the magnetic support.
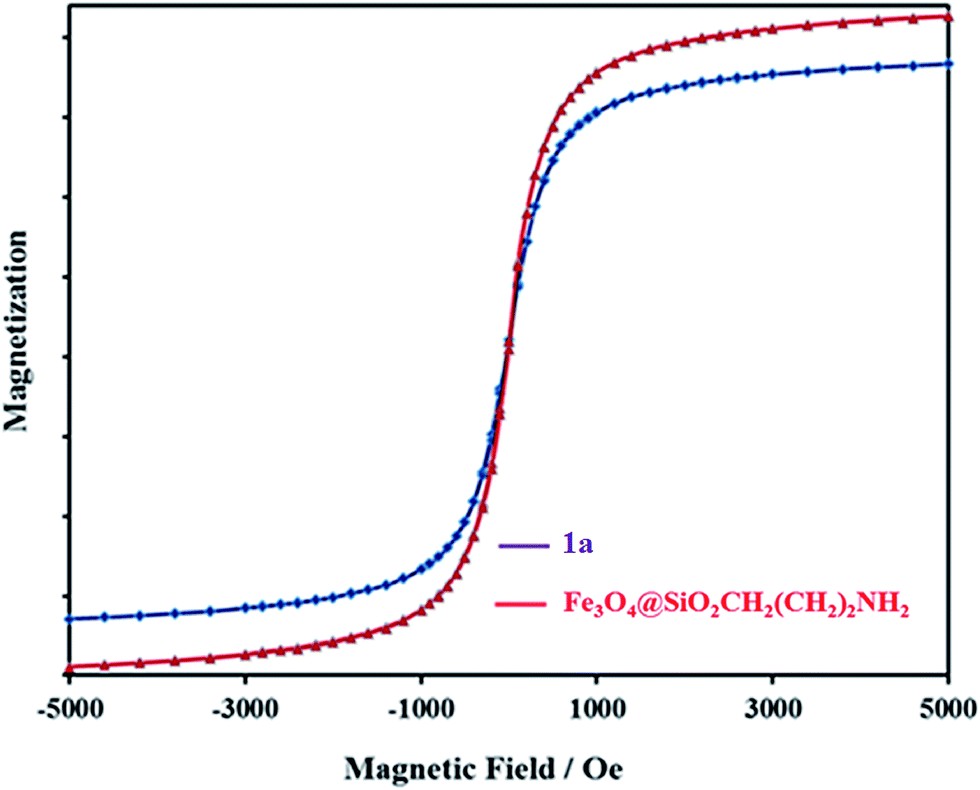 | ||
| Fig. 5 Alternating gradient force magnetometer (AGFM) of Fe3O4@SiO2CH2(CH2)2NH2 and 1a nanocomposite at room temperature. | ||
The SEM images show the morphological features of 1a, which confirm that the nanoparticles were almost spherical (Fig. 6). The nanocomposite shows uniform shape with a mean size less than 30 nm, which is consistent with the XRD results. Magnetic interactions occur between the nanoparticles, which leads to some particle aggregations as shown in the SEM images.
 | ||
| Fig. 6 SEM images of 1a nanoparticles. | ||
Nitrogen adsorption–desorption isotherms of 1a nanocomposite show a type IV isotherm with a type H1 hysteresis loop in the high range of relative pressure (Fig. 7A(a)). For the values of high relative pressure, condensation takes place giving a sharp adsorption volume increase, which confirms a mesoporous character for 1a (specific BET surface area, 56.99 m2 g−1).
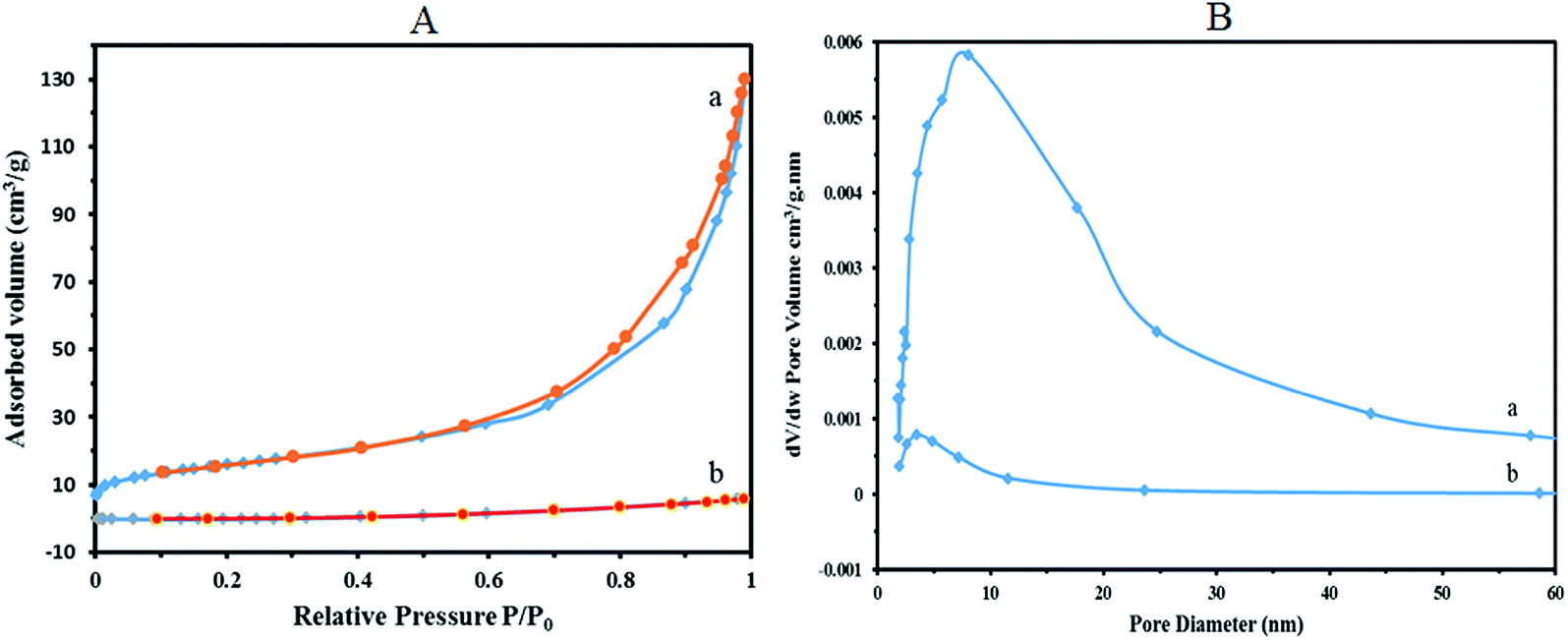 | ||
| Fig. 7 Nitrogen adsorption–desorption isotherms (A) and pore size distribution (B) of 1a and 1. | ||
The pore size distributions were calculated using the Barret–Joyner–Halenda (BJH) method applied to the adsorption branches of the isotherms. Fig. 7B(a) shows the pore size distribution for 1a nanocomposite. As shown in the figure a hierarchical set of pores centered near 50 Å and 80 Å (the average pore diameter 96.99 Å) could be observed in the nanocomposite. As expected, the surface area of the crystallite K11H[P2W18O68(HOSnIVOH)3]·20H2O (1) is very small (BJH adsorption cumulative surface area of pores, 5.318 m2 g−1). The BET analysis illustrates that surface area of 1 markedly increased by immobilization on silica-coated magnetite nanoparticles of Fe3O4@SiO2CH2(CH2)2NH2. The nitrogen adsorption–desorption isotherms and the pore size distribution of the bulk K11H[P2W18O68(HOSnIVOH)3]·20H2O (1) are shown in Fig. 7A(b) and B(b), respectively.
The catalytic applications
|
|||||
|---|---|---|---|---|---|
| Entry | Cat (X g) | Solvent | Time | Conversion | Yieldb (%) |
| a Reaction conditions: benzaldehyde (1 mmol), malononitrile (1.2 mmol), solvent (1 ml), rt, air.b Isolated yield. | |||||
| 1 | 1 (0.020) | H2O | 1 h | 100 | 85 |
| 2 | 1 (0.010) | H2O | 1 h | 100 | 90 |
| 3 | 1 (0.0050) | H2O | 2 h | 100 | 85 |
| 4 | 1 (0.020) | EtOH | 1 h | 100 | 85 |
| 5 | 1 (0.010) | EtOH | 1 h | 100 | 87 |
| 6 | 1 (0.0050) | EtOH | 2 h | 100 | 80 |
| 7 | Catalyst free | H2O/EtOH | 24 h | 40 | — |
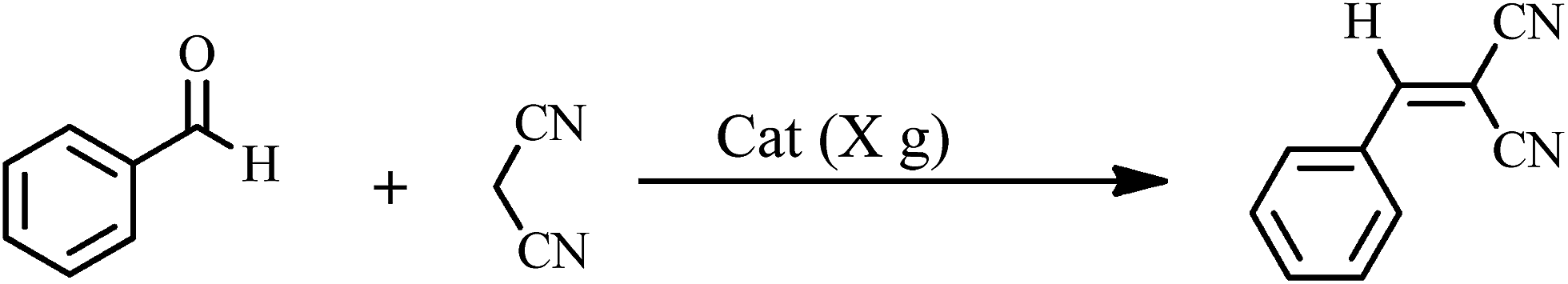
The yields of reaction show that 1 is much more effective than the Keggin POMs of [PW12O40]3− (acid or sodium salt) and the mono-lacunary POM of [PW11O39]7− (potassium salt) under similar conditions.40 The high activity of 1 compared with its similar compounds is attributed to the stannous atoms in 1. The Knoevenagel condensation catalyzed by 1 in water was carried out with good yields of 85–95% (Table 2). Reaction in absolute ethanol by POM of 1 as a heterogeneous catalyst was also carried out and we found that the products were formed with good yields of 85–95% (Table 3). The catalyst was reused at least five times without any significant decrease in catalytic activities, and the structure of the catalyst remained unchanged after recycling.
|
||||
|---|---|---|---|---|
| Entry | R | Time (min) cat. 1 | Conversion | Yieldb (%) cat. 1 |
| a Reaction conditions: aromatic aldehyde (1 mmol), malononitrile (1.2 mmol), H2O (1 ml), rt, air.b Isolated yield. | ||||
| 1 | 4-CH3C6H4 | 80 | 100 | 95 |
| 2 | 4-CH3OC6H4 | 10 | 100 | 95 |
| 3 | 4-NO2C6H4 | 20 | 100 | 95 |
| 4 | 3-NO2C6H4 | 10 | 100 | 95 |
| 5 | 4-ClC6H4 | 60 | 100 | 92 |
| 6 | 2-ClC6H4 | 60 | 100 | 87 |
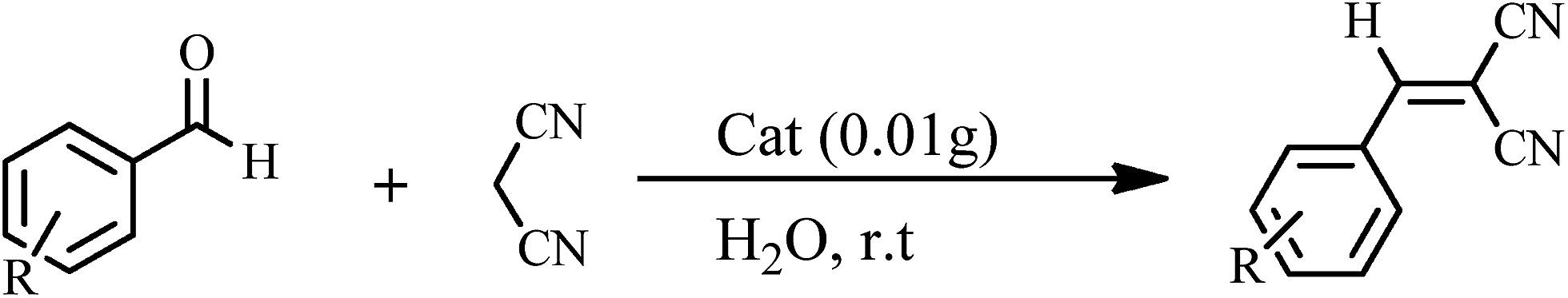
|
||||
|---|---|---|---|---|
| Entry | R | Time (min) cat. 1 | Conversion | Yieldb (%) cat. 1 |
| a Reaction conditions: aromatic aldehyde (1 mmol), malononitrile (1.2 mmol), EtOH (1 ml), rt, air.b Isolated yield. | ||||
| 1 | 4-CH3C6H4 | 80 | 100 | 95 |
| 2 | 4-CH3OC6H4 | 60 | 100 | 95 |
| 3 | 4-NO2C6H4 | 20 | 100 | 95 |
| 4 | 3-NO2C6H4 | 20 | 100 | 95 |
| 5 | 4-ClC6H4 | 60 | 100 | 90 |
| 6 | 2-ClC6H4 | 30 | 100 | 85 |
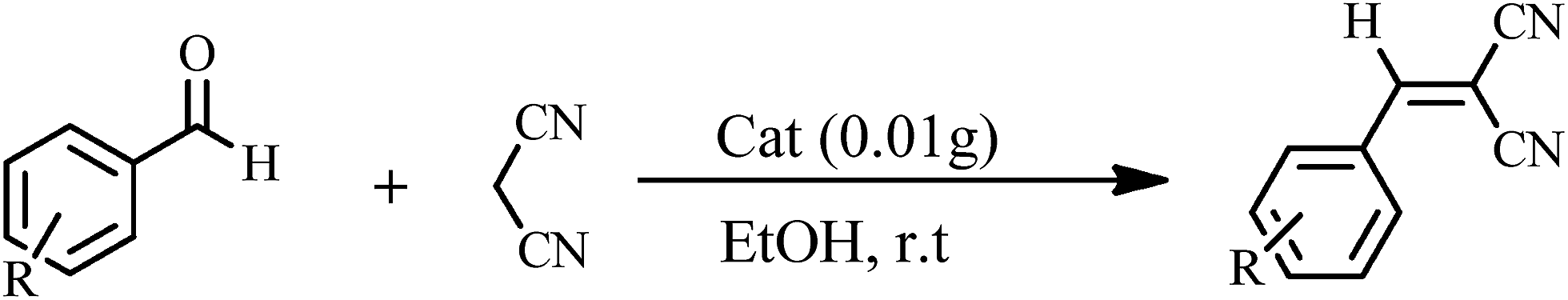
The catalytic activity of 1a was investigated in the selective oxidation of sulfides to sulfoxides using 30% H2O2 as a green oxidant at room temperature. To optimize the reaction condition, methyl phenyl sulfide was used as a model compound. The amount of catalyst and the effect of solvent type were studied on the rate of the reaction (Table 4). The appropriate amount of catalysts needed to carry out the reaction was 0.030 g. As shown in Table 4, solvents H2O and CH3CN were examined. The results show that by changing of water to acetonitrile, sulfone was the reaction product indeed of sulfoxide. Owing to the toxicity of acetonitrile, water was selected as a suitable solvent for this purpose. Investigations showed that the oxidation reaction remained incomplete without catalysts under optimized conditions (Table 4, entry 5).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat (mg) | Solvent | Time (min) | Y 1b (%) | Y 2b (%) | Conversion |
| a Reaction conditions: methyl phenyl sulfide (1 mmol), solvent (1.25 ml), H2O2 (6 mmol, 30%), rt, air.b Isolated yield. | ||||||
| 1 | 1a (10) | MeCN | 3 h | — | 90 | 100 |
| 2 | 1a (30) | MeCN | 60 | — | 92 | 100 |
| 3 | 1a (30) | H2O | 5 | 90 | — | 100 |
| 4 | Catalyst free | H2O | 24 h | 30 | — | — |
| 5 | 1 (30) | H2O | 10 h | 30 | 40 | — |
In order to generalize the scope of the reaction, a series of various sulfides such as benzylic, linear and cyclic sulfides were subjected to the experiment under optimized conditions (Table 5). According to the data listed in Table 5, the reaction was performed in a very short time with high yields. To show the chemoselectivity of this method, the sulfides that contain functional groups such as OH and COOCH3 were subjected to the reaction. The functional groups remained unchanged in the oxidation reaction (Table 5, entries 4 and 5).
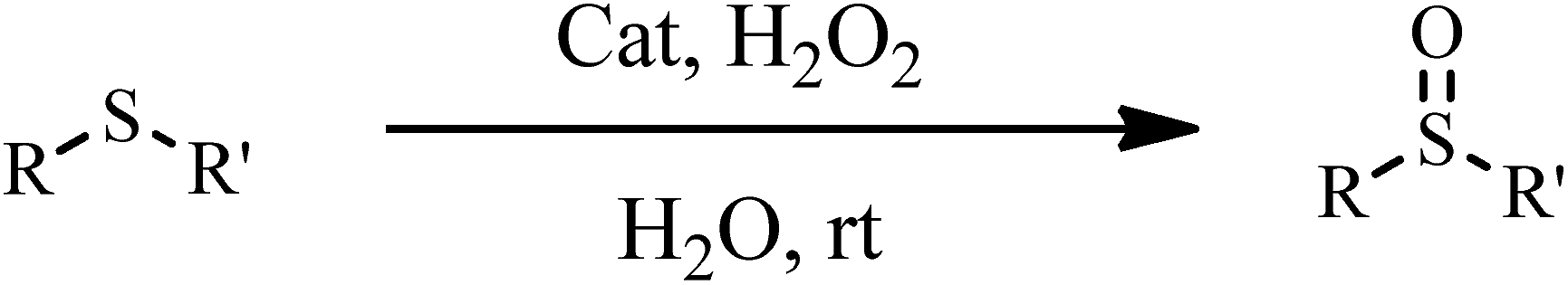
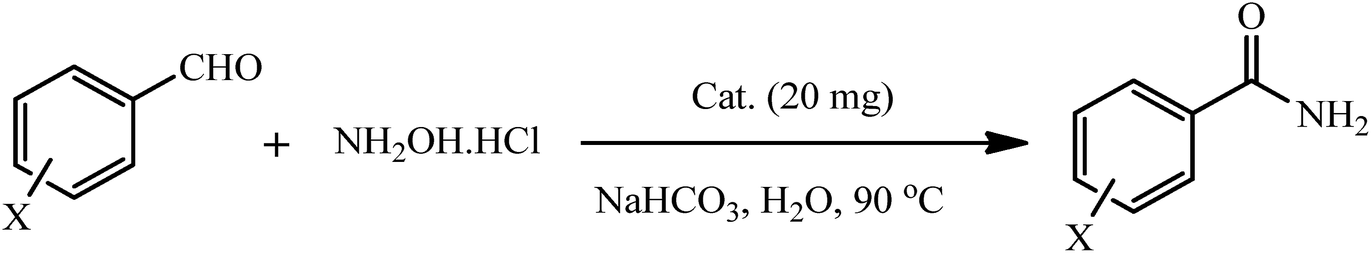
Thus, [P2W18O68(HOSnIVOH)32]12− was supported on amine-modified silica-coated magnetite nanoparticles and their catalytic performance was evaluated in the preparation of amides from aldehydes and hydroxylamine hydrochloride in an aqueous medium (Table 6). The products were confirmed by comparison with the literature. As the results show, the catalytic system displayed high performance in the synthesis of active and inactive amides. The reaction time of aldehydes was lower in the presence of electron-withdrawing groups (Table 6, entries 2, 4, 5, 6) than in the presence of electron-donating groups (Table 6, entry 3). Furthermore, the reaction time was affected by the substituted position. For example, the reaction of the ortho-derivatives (Table 6, entries 5 and 6) despite having an electron-withdrawing group has a longer reaction time. High product yield, short reaction time, solvent green conditions, facile preparation and reusability of the catalyst are the noteworthy advantages that make the present catalytic protocol superior to those reported previously.
|
||||
|---|---|---|---|---|
| Entry | X | Time (h) cat. 1a | Conversion | Yieldb (%) |
| a Reaction conditions: aldehyde (1 mmol), hydroxyl amine hydrochloride (1.2 mmol), sodium bicarbonate (1.2 mmol), catalyst (20 mg), and H2O (1 ml) at 90 °C.b Isolated yield. | ||||
| 1 | H | 3.5 | 100 | 94 |
| 2 | 4-Cl | 3 | 100 | 91 |
| 3 | 4-Me | 4 | 100 | 90 |
| 4 | 4-NO2 | 3 | 100 | 96 |
| 5 | 2-NO2 | 3.5 | 100 | 89 |
| 6 | 2,4-Cl2 | 4 | 100 | 86 |
Catalysts recovery and reuse
The recycling and stability of the catalyst were investigated. The catalyst could be easily recovered using an external magnet and further recycled. The recovered catalyst after the first use was washed thoroughly with ether and reused in a subsequent reaction. The strong coordination of POM with functionalized Fe3O4 magnetite nanoparticles makes the catalyst stable, nearly free from leaching, and reusable for a greater number of cycles (five times, (Fig. 8)). The FT-IR spectra showed no significant structural changes for the catalyst after five consecutive runs (Fig. 9).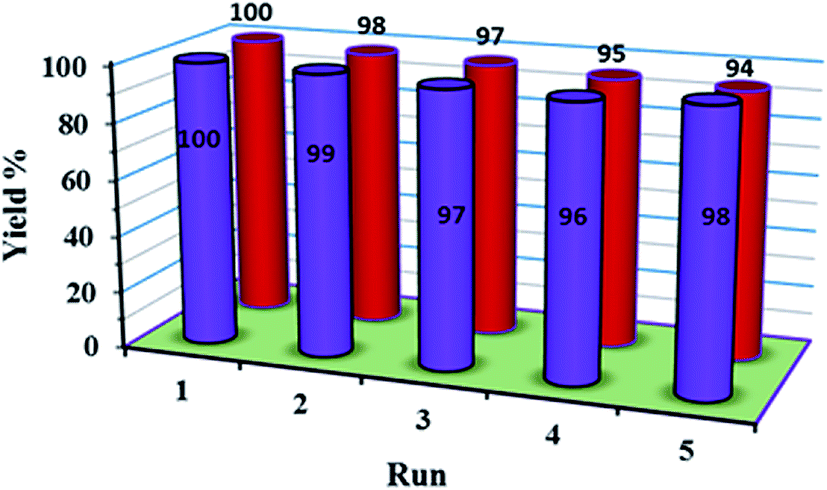 | ||
| Fig. 8 The recycling experiment of 1a for the oxidation of methyl phenyl sulfide (blue) and the recycling experiment of 1a for the oxidation oxidative amination of aldehyde (red). | ||
 | ||
| Fig. 9 The FT-IR spectra of 1a nanoparticles (a) and 1a nanoparticles after five consecutive runs (b). | ||
Conclusion
In summary, heteropolytungstostannate of K11H[P2W18O68(HOSnIVOH)3]·H2O (1) was used as a heterogeneous and homogeneous catalyst in the high selectivity and efficient Knoevenagel condensation and for fabrication of magnetically recoverable catalyst via electrostatic interaction. The new type of magnetically recoverable nanocomposite was characterized using IR, SEM, XRD, EDX and AGFM. The magnetically recoverable nanocatalyst is utilized as a high-selectivity and efficient catalyst in the oxidation of sulfides to sulfoxides and the oxidative amination of aldehydes. The catalyst could be easily separated from the reaction mixture and used for the next run over five times without any obvious decrease in the catalytic activity.Conflicts of interest
The authors declare that they have no competing interests.References
- M. T. Pope, Heteropoly and Isopoly Oxometalate, Springer-Verlag, Berlin, 1983, p. 31 Search PubMed.
- U. Kortz, M. G. Savelieff, B. S. Bassil and M. H. Dickman, Angew. Chem., Int. Ed., 2001, 40, 3384 CrossRef CAS PubMed.
- V. W. Day and W. G. Klemperer, Science, 1985, 228, 533 CAS.
- C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407 CrossRef CAS.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171 CrossRef CAS PubMed.
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199 CrossRef CAS PubMed.
- X. M. Yan, J. H. Lei, D. Liu, Y. C. Wu and W. Liu, Mater. Res. Bull., 2007, 42, 1905 CrossRef CAS.
- N. Dubey, S. S. Rayalu, N. K. Labhsetwar and S. Devotta, Int. J. Hydrogen Energy, 2008, 33, 5958 CrossRef CAS.
- M. A. Alibeik, Z. Zaghaghi and I. M. Baltork, J. Chin. Chem. Soc., 2008, 55, 1 CrossRef.
- S. Uchida and N. Mizuno, J. Am. Chem. Soc., 2004, 126, 1602 CrossRef CAS PubMed.
- N. M. Okun, M. D. Ritorto, T. M. Anderson, R. P. Apkarian and C. L. Hill, Chem. Mater., 2004, 16, 2551 CrossRef CAS.
- C. W. Lim and I. S. Lee, Nano Today, 2010, 5, 412 CrossRef CAS.
- K. K. Senapati, C. Borgohain and P. Phukan, J. Mol. Catal. A: Chem., 2011, 339, 24 CrossRef CAS.
- M. G. Egusquiza, C. I. Cabello, I. L. Botto, H. J. Thomas, S. Casuscelli, E. Herrero and D. Gazzoli, Catal. Commun., 2012, 26, 117 CrossRef CAS.
- A. Patel and K. Patel, Inorg. Chim. Acta, 2014, 419, 130 CrossRef CAS.
- V. Nardello, J. M. Aubry, D. E. D. Vos, R. Neumann, W. Adam, R. Zhang, J. E. Elshof, P. T. Witte and P. L. Alsters, J. Mol. Catal. A: Chem., 2006, 251, 185 CrossRef CAS.
- I. C. M. S. Santos, J. A. F. Gamelas, M. S. S. Balula, M. M. Q. Simoes, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, J. Mol. Catal. A: Chem., 2007, 262, 41 CrossRef CAS.
- F. Xin and M. T. Pope, J. Am. Chem. Soc., 1996, 118, 7731 CrossRef CAS.
- R. Khshnavazi and L. Bahrami, J. Coord. Chem., 2009, 62, 2067 CrossRef.
- W. H. Knoth, P. J. Domaille and R. L. Harlow, Inorg. Chem., 1986, 25, 1577 CrossRef CAS.
- X. Liu, Z. Ma, J. Xing and H. Liu, J. Magn. Magn. Mater., 2004, 270, 1 CrossRef CAS.
- M. Yamaura, R. L. Camilo, L. C. Sampaio, M. A. Macedo, M. Nakamura and H. E. Toma, J. Magn. Magn. Mater., 2004, 279, 210 CrossRef CAS.
- F. Shahbazi and K. Amani, Catal. Commun., 2014, 55, 57 CrossRef CAS.
- A. Alizadeh, M. M. Khodaei, M. Beygzadeh, D. Kordestani and M. Feyzi, Bull. Korean Chem. Soc., 2012, 33, 2546 CrossRef CAS.
- H. Ono and T. Katsumata, Appl. Phys. Lett., 2001, 78, 1832 CrossRef CAS.
- F. An and B. Gao, J. Hazard. Mater., 2007, 145, 495 CrossRef CAS PubMed.
- Y. Lin, H. Chen, K. Lin, B. Chen and C. Chiou, J. Environ. Sci., 2011, 23, 44 CrossRef CAS.
- (a) G. Jones, Org. React., 1967, 15, 204 CAS; (b) L. F. Tietze and U. Beifuss, in Comprehensive Organic Synthesis, Pergamon, New York, 1991, vol. 2, p. 341 Search PubMed.
- F. Bigi, L. Chesini, R. Maggi and G. Sartori, J. Org. Chem., 1999, 64, 1033 CrossRef CAS PubMed.
- N. Yu, J. M. Aramini, M. W. German and Z. Huang, Tetrahedron Lett., 2000, 41, 6993 CrossRef CAS.
- (a) D. S. Bose and A. V. Narsaiah, J. Chem. Res., 2001, 36 CrossRef CAS; (b) J. L. Scott and C. L. Raston, Green Chem., 2000, 2, 245 RSC.
- (a) W. Lehnert, Tetrahedron, 1974, 30, 301 CrossRef CAS; (b) B. Green, R. I. Crane, I. S. Khaidem, R. S. Leighton, S. S. Newaz and T. E. Smyser, J. Org. Chem., 1985, 50, 640 CrossRef CAS.
- P. Shanthan Rao and R. V. Venkataratnam, Tetrahedron Lett., 1991, 32, 5821 CrossRef.
- G. Bartoli, R. Beleggia, S. Giuli, A. Giuliani, E. Marcantoni, M. Massaccesi and M. Paletti, Tetrahedron Lett., 2006, 47, 6501 CrossRef CAS.
- S. Kantevari, R. Bantu and L. Nagarapu, J. Mol. Catal. A: Chem., 2007, 269, 53 CrossRef CAS.
- (a) S. Saravanamurugan, M. Palanichamy, M. Hartmann and V. Murugesan, Appl. Catal., A, 2006, 298, 8 CrossRef CAS; (b) T. I. Reddy and R. S. Varma, Tetrahedron Lett., 1997, 38, 1721 CrossRef CAS.
- M. L. Kantam, B. M. Choudary, C. V. Reddy, K. K. Rao and F. Figueras, Chem. Commun., 1998, 1033 RSC.
- (a) C. F. H. Allen and F. W. Spangler, Org. Syn. Coll., 1955, 3, 377 Search PubMed; (b) L. Rand, J. V. Swisher and C. J. Cronin, J. Org. Chem., 1962, 27, 3505 CrossRef CAS.
- (a) D. S. Acker and W. R. Hertler, J. Am. Chem. Soc., 1962, 84, 3370 CrossRef CAS; (b) G. Cardillo, S. Fabbroni, L. Gentilucci, M. Gianotti and A. Tolomelli, Synth. Commun., 2003, 33, 1587 CrossRef CAS.
- S. Zhao, Y. Chen and Y. F. Song, Appl. Catal., A, 2014, 475, 140 CrossRef CAS.
- S. L. Jain, B. S. Rana, B. Singh, A. K. Sinha, A. Bhaumik, M. Nandi and B. Sain, Green Chem., 2010, 12, 374 RSC.
- S. Doherty, J. G. Knight, M. A. Carroll, A. R. Clemmet, J. R. Ellison, T. Backhouse, N. Holmes and R. A. Bourne, RSC Adv., 2016, 6, 73118 RSC.
- N. Salam, P. Mondal, J. Mondal, A. S. Roy, A. Bhaumik and S. M. Islam, RSC Adv., 2012, 2, 6464 RSC.
- J. M. Humphery and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243 CrossRef.
- R. K. Mylavarapu, G. C. M. Kondaiah, N. Kolla, R. Veeramalla, P. Koilkonda, A. Bhattacharya and R. Bandnewichhor, Org. Process Res. Dev., 2007, 11, 1065 CrossRef CAS.
- W. J. Yoo and C. J. Li, J. Am. Chem. Soc., 2006, 128, 13064 CrossRef CAS PubMed.
- J. Liang, J. Lv and Z. Shang, Tetrahedron, 2011, 67, 8532 CrossRef CAS.
- S. C. Ghosh, J. S. Y. Ngiam, C. L. L. Chai, A. M. Seayad, T. T. Dang and A. Chen, Adv. Synth. Catal., 2012, 354, 1407 CrossRef CAS.
- (a) A. M. Asencio, M. Yus and D. J. Ramon, Tetrahedron, 2012, 68, 3948 CrossRef; (b) M. A. Ali and T. Punniyamurthy, Adv. Synth. Catal., 2010, 352, 288 CrossRef CAS; (c) K. Yamaguchi, H. Kobayashi, Y. Wang, T. Oishi, Y. Ogasawara and N. Mizuno, Catal. Sci. Technol., 2013, 3, 318 RSC; (d) R. Vanjari, T. Guntreddi and K. N. Singh, Green Chem., 2014, 16, 351 RSC.
- M. Saikia and L. Saikia, RSC Adv., 2016, 6, 14937 RSC.
| This journal is © The Royal Society of Chemistry 2017 |