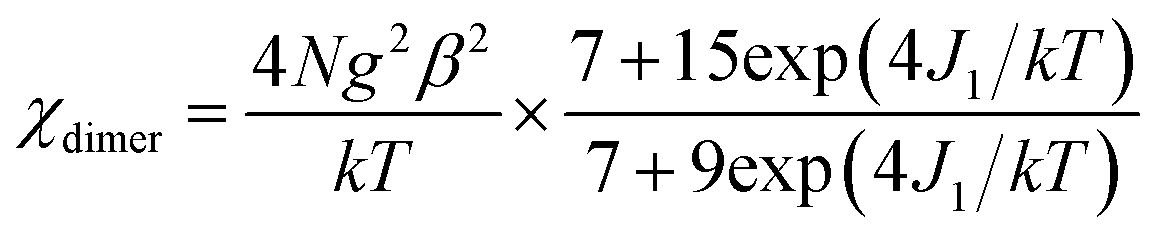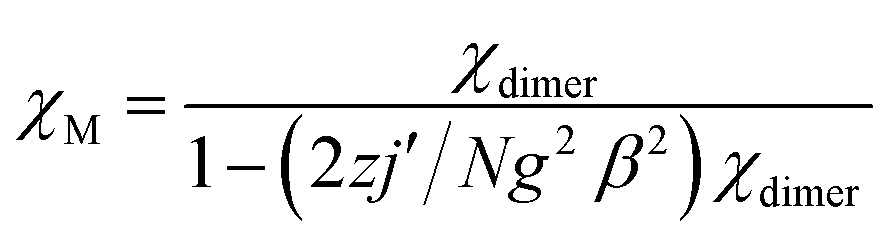DOI:
10.1039/C7RA09192C
(Paper)
RSC Adv., 2017,
7, 45504-45512
Modulating spin dynamics of binuclear LnIII–radical complexes by using different indazole radicals†
Received
20th August 2017
, Accepted 14th September 2017
First published on 25th September 2017
Abstract
The combination of LnIII ion (GdIII or DyIII) with two different indazole nitronyl nitroxide radicals results in four novel 2p–4f compounds, namely, [Ln(hfac)3(5-IndazoleNIT)]2 (Ln = Gd (1), Dy (2); hfac = hexafluoroacetylacetone; 5-IndazoleNIT = 5-(1′-oxyl-3′-oxido-4′,4′,5′,5′-tetramethyl-4,5-hydro-1H-imidazol-2-yl)-1H-Indazole) and [Ln(hfac)3(6-IndazoleNIT)]2 (Ln = Gd (3), Dy (4); 6-IndazoleNIT = 6-(1′-oxyl-3′-oxido-4′,4′,5′,5′-tetramethyl-4,5-hydro-1H-imidazol-2-yl)-1H-Indazole). Single crystal X-ray diffraction studies revealed that compounds 1–2 are binuclear isostructural complexes with local D2d symmetry, in which each 5-IndazoleNIT molecule acts as a bridging ligand linking two LnIII ions through the oxygen atom of its NO group and nitrogen atom of its indazole ring to form a cyclic four-spin system. Complexes 3–4 exhibit analogous binuclear cyclic four-spin systems, where the symmetry of central LnIII ions is D4d due to the change of location of the NO group in the indazole ring. In addition, compound 2 displays no out-of-phase alternating-current (ac) signal, whereas compound 4 exhibits obvious slow relaxation of magnetization, suggesting single-molecule magnet (SMM) behavior. The different magnetic relaxation behaviour between 2 and 4 is largely dependent on the ligand field of the central dysprosium ions.
Introduction
In recent decades, the design and synthesis of molecular nanomagnets based on anisotropic metal ions that show magnetization relaxation have aroused deep interest from the application and theoretical viewpoints.1,2 Such materials known as single-molecular magnets (SMMs) and single chain magnets (SCMs), have potential applications in high-density information storage,3 nanoscale electronics,4 and quantum computing.5 The general feature of SMMs is that the magnetic bistable state arises from overcoming the effective energy barrier (Ueff). One of the challenging problems in this field is to increase the blocking temperature at which slow relaxation of the magnetization can occur, which relies on the anisotropy barrier from the combination of a large spin multiplicity in the ground state and a significant uniaxial magnetic anisotropy.6 Considerable researches have been focused not only on designing, synthesizing and characterizing new molecular species to obtain SMMs with high Ueff, but also on deep understanding the magneto–structural correlation. Compared to 3d or 3d–4f systems, pure 4f systems, especially the heavy lanthanide systems (such as TbIII or DyIII ions), are better candidates for the construction of SMMs, owing to the large number of unpaired f-electrons and large intrinsic magnetic anisotropy of the lanthanide ions originating from strong spin–orbit coupling and crystal-field effects.7–10 For lanthanide ions, the internal 4f electrons are strongly shielded by the outer 5s and 5p electrons, and hence leads to weak magnetic exchange coupling, which always lowers the effective relaxation energy barrier and induces the loss of remnant magnetization.10b,11 Recent studies show that 4f-organic radical approach has been proved to be an efficient strategy to design new SMMs.3b,12 Nitronyl nitroxide radicals (NITs) as spin carriers can coordinated to 4f ions and transmit effective magnetic coupling interaction, on account of the direct overlap of the orbitals containing the unpaired electrons.13,14
For SMMs containing lanthanide ion, the magnetic relaxation is very sensitive to the symmetry of the ligand field around the LnIII ion, and the spin dynamic can be modified by the careful adjustment of the ligand field around the metal center. It is worth mentioning that in high symmetry crystal field, for example C∞v, D∞h, D4d, D5h and D6d, the control of quantum tunneling of magnetization (QTM) comes true by tuning the local symmetry of the metal centers.15 In order to explore how the symmetry of the local crystal field around the lanthanide center affect the spin dynamics of the complex, we decided to use two different indazole nitronyl nitroxide radicals to construct radical–lanthanide SMMs. The location of the radical in the indazole may modify the ligand field of the metal ion, thus adjusting the magnetic relaxation of the molecule. Herein we synthesized four novel compounds from two different IndazoleNIT ligands (Scheme 1), namely, [Ln(hfac)3(5-IndazoleNIT)]2 (Ln = Gd (1), Dy (2); hfac = hexafluoroacetylacetone; 5-IndazoleNIT = 5-(1′-oxyl-3′-oxido-4′,4′,5′,5′-tetramethyl-4,5-hydro-1H-imidazol-2-yl)-1H-Indazole) and [Ln(hfac)3(6-IndazoleNIT)]2 (Ln = Gd (3), Dy (4); 6-IndazoleNIT = 6-(1′-oxyl-3′-oxido-4′,4′,5′,5′-tetramethyl-4,5-hydro-1H-imidazol-2-yl)-1H-Indazole). Magnetic studies showed that complexes 4 exhibit frequency-dependent ac susceptibility at low temperature, which suggests SMMs behavior. The comparison of the magnetic property between 2 and 4 and the reported results based on 3-IndazoleNIT16 highlights that the local crystal field can play an important role in modulating the magnetic relaxation.
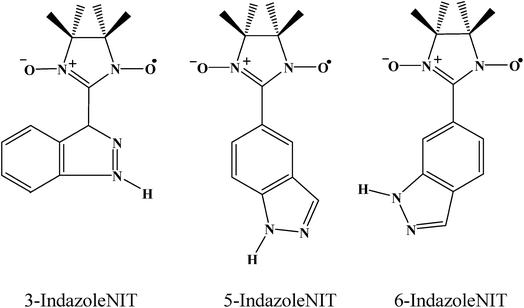 |
| | Scheme 1 Structures of the three indazole nitronyl nitroxide radicals. | |
Experimental details
Materials and physical measurements
All reagents used in the syntheses were of reagent-grade, except that the solvents were used after dried (heptane over sodium, CH2Cl2 over CaH2 and CHCl3 over P2O5) and distilled prior to use. Ln(hfac)3·2H2O was synthesized according to methods in the literature.17 The radicals (5-IndazoleNIT and 6-IndazoleNIT) were prepared by condensation of 2,3-bis(hydroxylamino)-2,3-dimethylbutane with 5- and 6-Indazolecarboxaldehyde, followed by oxidation with NaIO4 according to Ullman's procedure.18 Elemental analyses for carbon, hydrogen, and nitrogen were carried out on a PerkinElmer 240 elemental analyzer. Powder X-ray diffraction measurements were recorded on a D/Max-2500 X-ray diffractometer using Cu-Kα radiation. Infrared spectra of the compounds in KBr pellets were obtained on a Bruker TENOR 27 spectrometer in the 4000–400 cm−1 region. Direct-current (dc) magnetic susceptibilities of crystalline samples were measured on an MPMS-7 SQUID magnetometer. The data were corrected for the diamagnetism of the samples using Pascal constants. Alternating-current (ac) susceptibilities were performed on the same magnetometer under zero or 1000 Oe static field with an oscillating of 3.5 Oe at frequencies up to 1500 Hz.
Preparation of complexes of 1–4
Complexes 1–2 were synthesized following a similar procedure. A solution of Ln(hfac)3·2H2O (0.1 mmol) (Ln = Gd (1), Dy (2)) in 15 mL dry n-heptane was heated under refluxing for 2 h. After that, the solution was cooled to 60 °C, to which a solution of 5-IndazoleNIT (0.1 mmol, 27.2 mg) in CH2Cl2 (5 mL) was added. The mixture remained refluxing for 1 h, and a lot of dark purple powder produced during this period. Then the resulting mixture was cooled to room temperature and filtrated. The collected solid powder was recrystallized in CHCl3/CH3OH solution to give blue-violet crystals, which were suitable for X-ray analysis.
Complexes 3–4 were synthesized in a similar way as complex 1–2, except that 5-IndazoleNIT was replaced by 6-IndazoleNIT.
[Gd(hfac)3(5-IndazoleNIT)]2 (1). Yield 0.066 g, 63%. C58H40F36Gd2N8O16 (2103.48): calcd. C 33.01, H 1.92, N 5.33; found: C 33.22, H 1.89, N 5.42%. IR (KBr pellet): 3129 (b), 1654 (m), 1532 (w), 1401 (s), 1257 (m), 1204 (w), 1142 (s), 802 (w), 617 (m) cm−1.
[Dy(hfac)3(5-IndazoleNIT)]2 (2). Yield 0.067 g, 63%. C58H40Dy2F36N8O16 (2113.98): calcd. for C 32.95, H 1.91, N 5.30; found: C 32.81, H 1.86, N 5.08%. IR (KBr pellet): 3119 (s), 1655 (s), 1532 (w), 1498 (w), 1400 (s), 1257 (m), 1209 (m), 1149 (s), 803 (w), 618 (m) cm−1.
[Gd(hfac)3(6-IndazoleNIT)]2 (3). Yield 0.063 g, 60%. C58H40F36Gd2N8O16 (2103.48): calcd. C 33.01, H 1.92, N 5.33; found: C 33.42, H 1.77, N 5.24%. IR (KBr pellet): 3133 (vs), 1654 (s), 1531 (m), 1494 (w), 1401 (s), 1258 (s), 1207 (m), 1145 (s), 801 (w), 659 (w), 585 (w) cm−1.
[Dy(hfac)3(6-IndazoleNIT)]2 (4). Yield 0.072 g, 61%. C58H40Dy2F36N8O16 (2113.98): calcd. for C 32.95, H 1.91, N 5.30; found: C 33.11, H 1.75, N 5.44%. IR (KBr pellet): 3129 (vs), 1654 (s), 1532 (m), 1401 (s), 1258 (s), 1204 (m), 1143 (s), 801 (w), 617 (w) cm−1.
X-Ray crystallography
Diffraction intensity data were collected by using the φ–ω scan technique on an Agilent SuperNova (Dual, Cu at zero, AtlasS2, CCD) diffractometer equipped with mirror-monochromated Mo-Kα radiation (λ = 0.71073 Å) for 1–3 and Cu-Kα radiation (λ = 1.54184 Å) for complex 4, respectively. Semiempirical multiscan absorption corrections were applied by SCALE3 ABSPACK, and the programs CrysAlisPro were used for integration of the diffraction profiles.19 Structures were solved by direct methods and refined with the full-matrix least-squares technique using the ShelXT and ShelXL programs.20 There are some disordered fluorine atoms, which were refined anisotropically. Some restraints are applied, such as ISOR (anisotropic parameter) for some of the fluorine atoms, DFIX (restricting the distance between two atoms) for some of the C–F bonds. All non-hydrogen atoms were refined anisotropically, and hydrogen atoms were located and refined isotropically. Crystallographic data for the compounds 1–4 are listed in Table 1 and the powder X-ray diffraction data for all the four compounds are shown in the ESI (Fig. S1–S3†).
Table 1 Crystallographic data and structure refinement details for 1–4
| |
1 |
2 |
3 |
4 |
| Formula |
C58H40F36Gd2N8O16 |
C58H40Dy2F36N8O16 |
C58H40F36Gd2N8O16 |
C58H40Dy2F36N8O16 |
| Mr |
2103.48 |
2113.98 |
2103.48 |
2113.98 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/n |
P21/c |
P21/n |
| a (Å) |
12.0292(4) |
11.9723(4) |
11.9977(10) |
11.9689(2) |
| b (Å) |
24.1953(12) |
24.1441(9) |
25.907(2) |
25.9221(5) |
| c (Å) |
12.9431(5) |
12.9349(5) |
15.0221(10) |
12.3832(2) |
| α (°) |
90 |
90 |
90 |
90 |
| β (°) |
100.293(4) |
100.167(3) |
126.689(5) |
103.828(2) |
| γ (°) |
90 |
90 |
90 |
90 |
| V (Å3) |
3706.5(3) |
3680.3(2) |
3744.2(5) |
3730.7(2) |
| Z |
2 |
2 |
2 |
2 |
| ρcalc (mg m−3) |
1.885 |
1.908 |
1.866 |
1.882 |
| μ (mm−1) |
1.933 |
2.175 |
1.914 |
12.052 |
| F (000) |
2048 |
2056 |
2048.0 |
2056.0 |
| θ range (°) |
3.31–25.01 |
3.46–25.01 |
1.57–26.50 |
4.053–67.08 |
| GOF on F2 |
1.042 |
1.038 |
1.037 |
1.030 |
| R1, wR2 [I > 2σ(I)] |
0.0473, 0.1002 |
0.0449, 0.1024 |
0.0597, 0.1480 |
0.0570, 0.1469 |
| R1, wR2 (all data) |
0.0583, 0.1077 |
0.0559, 0.1106 |
0.0867, 0.1659 |
0.0722, 0.1597 |
Results and discussion
Crystal structure
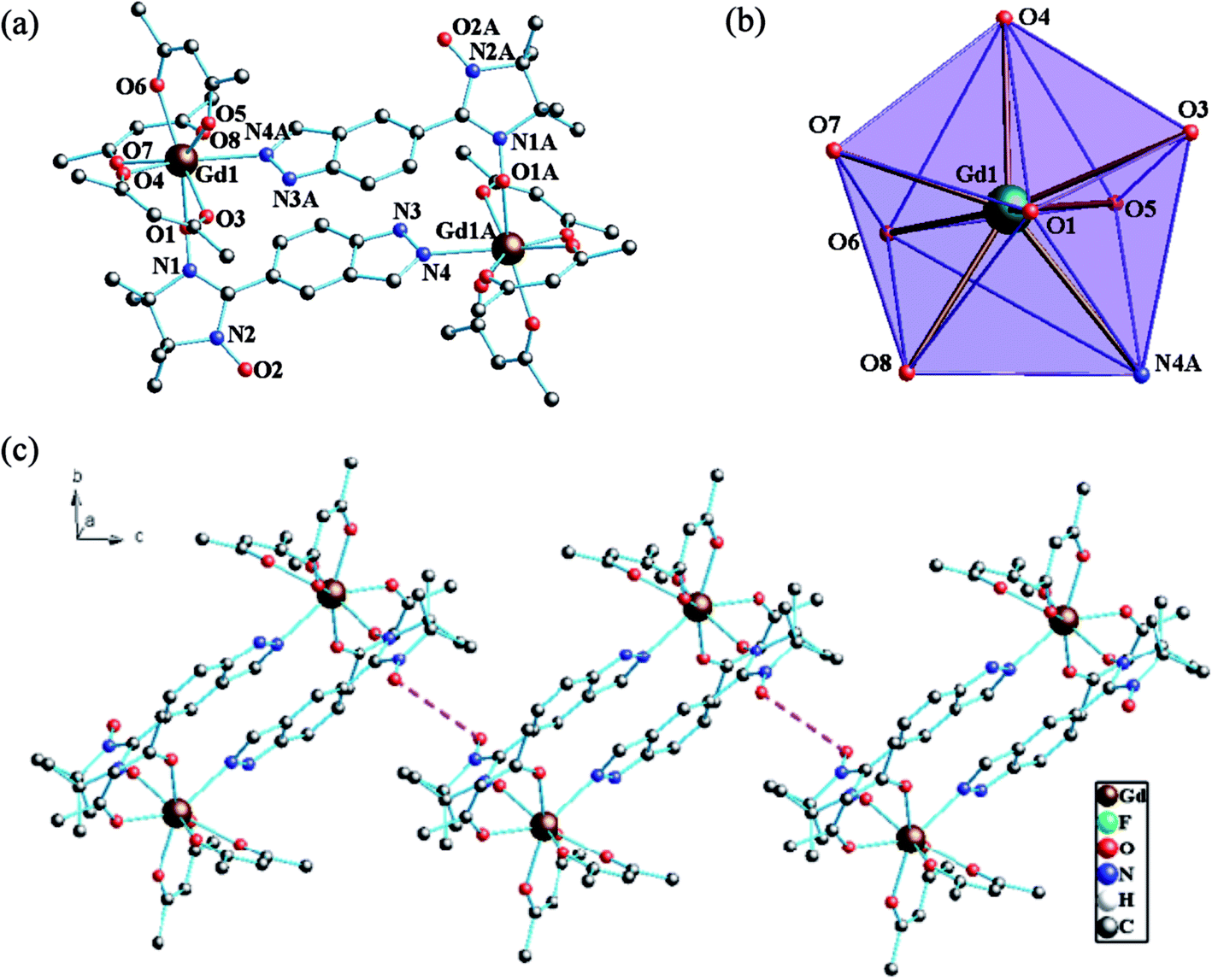
Structure of 1. Single-crystal X-ray diffraction analyses show that complexes 1–2 are isostructural and belong to monoclinic P21/n space group with Z = 2. Therefore, the structure of 1 will be described in detail as a representative example and the partially labelled crystal structure of 1 is shown in Fig. 1. Complex 1 is centrosymmetric, two indazole substituted radical ligands are coordinated to two Gd(hfac)3 units with the oxygen atoms of the nitronyl nitroxide groups and the nitrogen atoms of the indazole rings to form a cyclic dimer comprising two asymmetric units of [Ln(hfac)3(5-IndazoleNIT)]. Center ions (GdIII) are in GdO7N coordination sphere from three bischelate hfac anions and two radical ligands and in a slightly distorted triangular dodecahedron (D2d) polyhedron configuration. The Gd–O(N) (nitroxide) distance is 2.332(4) Å and the Gy–O(hfac) bond lengths are in the range of 2.339(4)–2.417(4) Å, which are comparable to those of reported Gd(hfac)3 complexes with nitronyl nitroxides. The Gd–N4A distance (2.546(4) Å) is a little longer than the normal Gd–N bonds ascribed to the bridged character of the radical ligand (Table 2). The Gd⋯Gd separation distance in every binuclear unit is 10.113(5) Å and the shortest intermolecular Gd⋯Gd separation is 12.029(5) Å. Here, the O–N–C–N–O group containing the unpair electron and the indazole ring shows average twist angles of 39.1°. However the torsion angle for Gd–O–N–C is 93.9(6)°, which is a crucial factor for Gd–rad magnetic coupling interaction.21 The shortest contacts between the uncoordinated NO groups are 4.286 Å for 1, which might lead to the weak intermolecular magnetic coupling (Fig. 1c). By employing the classic Continuous Symmetry Measures (CSM) method, the coordination sphere of Gd1 is estimated as nearly ideal D2d triangular dodecahedron with the deviation parameter S = 0.879.22
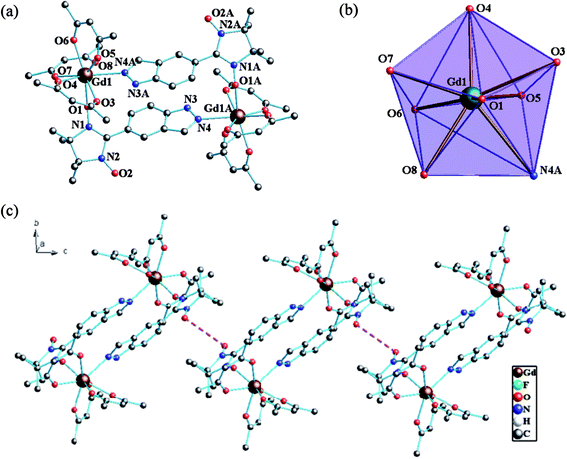 |
| | Fig. 1 (a) Simplified view of the crystal structure of 1. Fluorine and hydrogen atoms are omitted for clarity. (b) D2d-symmetry polyhedral of gadolinium atom. (c) The shortest Orad⋯Orad distance (4.286 Å) in 1 (pink dotted line). | |
Table 2 Selected bond distances (Å) and angles (°) for complexes 1–4
| |
1 Gd |
2 Dy |
3 Gd |
4 Dy |
| Ln–O(hfac) |
2.339(4)–2.417(4) |
2.317(4)–2.386(4) |
2.330(6)–2.398(5) |
2.299(6)–2.368(5) |
| Ln–O(rad) |
2.332(4) |
2.308(4) |
2.313(5) |
2.284(4) |
| Ln–N |
2.546(4) |
2.520(5) |
2.558(6) |
2.528(5) |
| N–O(rad) (coor) |
1.298(6) |
1.304(6) |
1.297(7) |
1.300(7) |
| N–O(rad) (uncoor) |
1.273(6) |
1.264(6) |
1.278(8) |
1.270(7) |
| Ln–O(rad)–N |
139.9(3) |
138.8(3) |
150.6(4) |
149.3(4) |
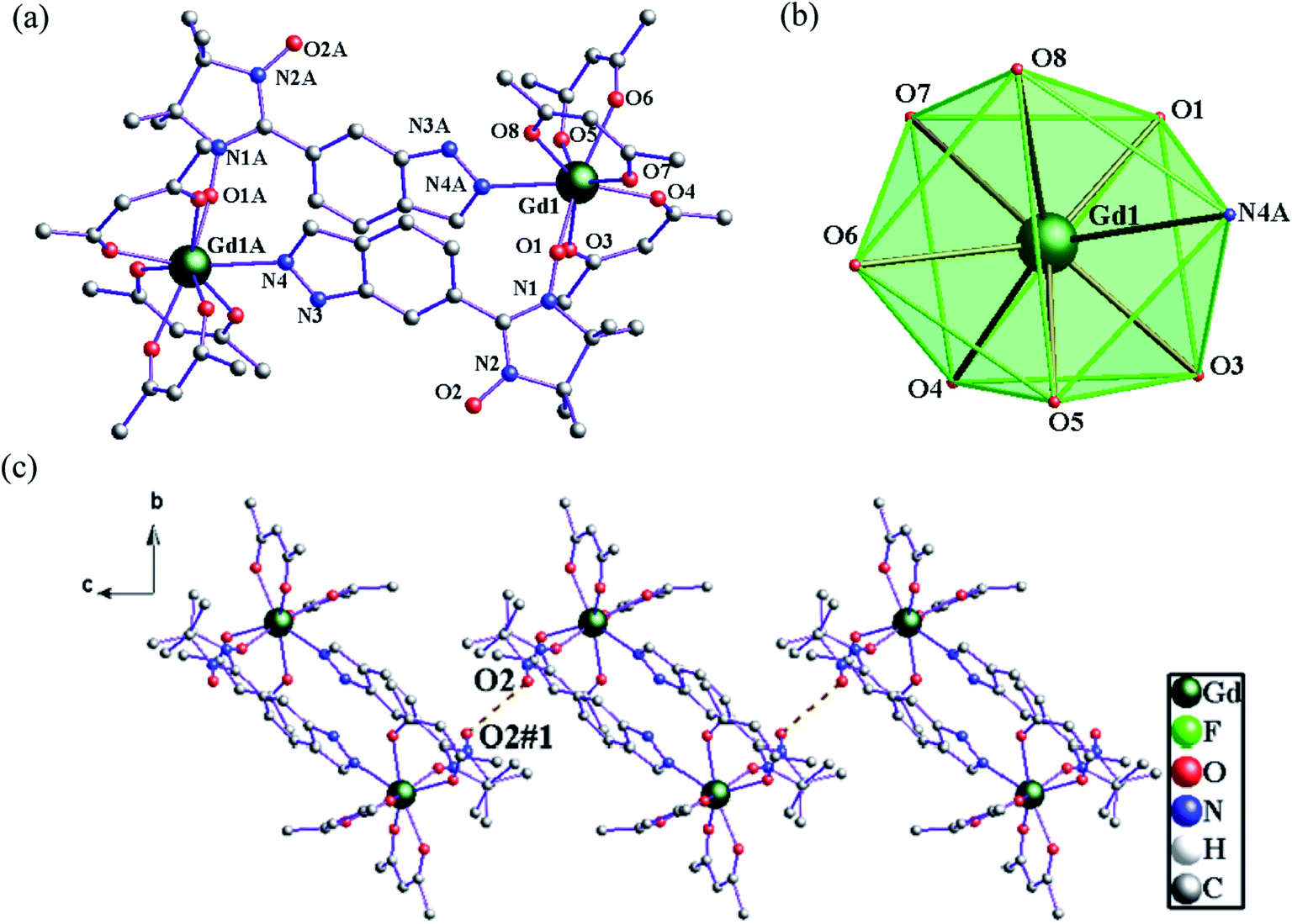
Structure of 3. Compound 3 crystallizes in space group P21/c with Z = 2. The dinuclear crystal structure of 3, shown in Fig. 2, possesses an inversion centre and comprises two bridging 6-IndazoleNIT radicals, each one coordinating two lanthanide ions via the oxygen atom from one NO group and a nitrogen atom from the indazole ring. As shown in Fig. 2a, each central GdIII ion is eight-coordinated with three bidentate β-diketonate coligands and two 6-IndazoleNIT radical ligands. When applying the D4d symmetry to the GdO7N site, CSM method gives the Gd1 the minimal value of S = 0.949. The Gd–O(hfac) distances range from 2.330(6) to 2.398(5) Å. The Gd–O(radical) and Gd–N(indazole) bond lengths for compound 3 are 2.313(5) Å and 2.558(6) Å, respectively. The torsion angle for Gd–O–N–C is 104.8(9)°, and the shortest distance between the uncoordinated NO groups are 3.729 Å for 1, which means the intermolecular radical–radical coupling is nonnegligible (Fig. 2c).
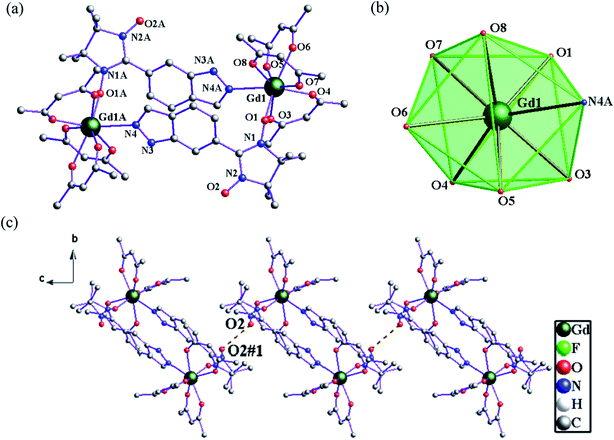 |
| | Fig. 2 (a) The crystal structure of 3. Fluorine and hydrogen atoms are omitted for clarity. (b) D4d-symmetry polyhedral of gadolinium atom. (c) The shortest Orad⋯Orad distance (3.729 Å) in 3 (brown dotted line). | |
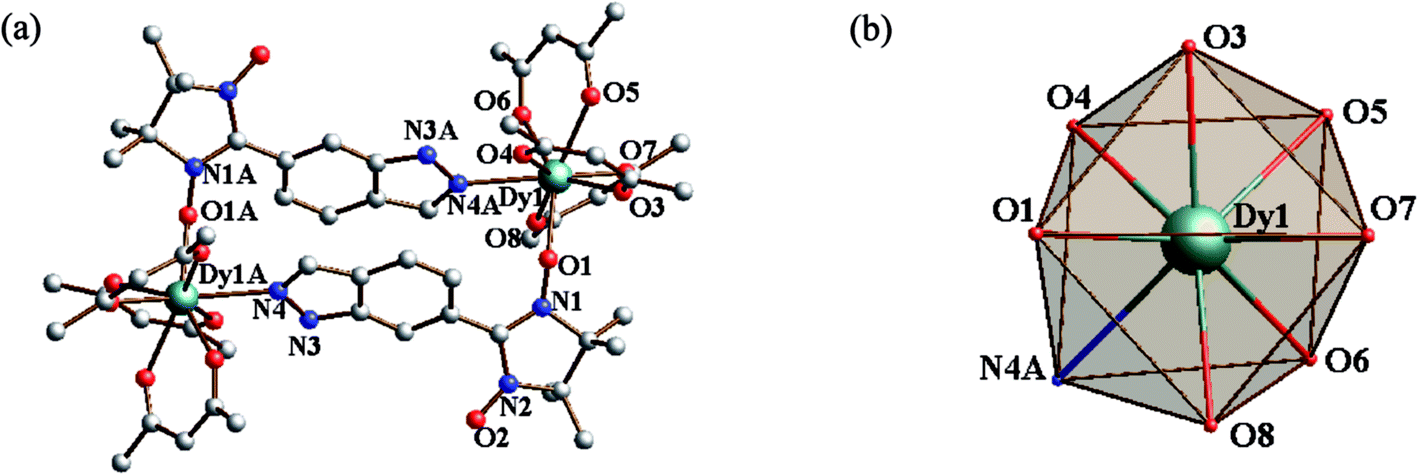
Structure of 4. The dinuclear crystal structure of 4 is shown in Fig. 3, which crystallizes in space group P21/n. The central DyIII ion has the same coordinating environment as GdIII ion in compound 3. The Dy–O(rad) (nitroxide) distance is found to be 2.284(4) Å and the Dy–O(hfac) bond lengths are in the range of 2.299(6)–2.368(5). And the central DyIII ion is also in a D4d symmetry coordination sphere with S of 0.848 (Table 3).
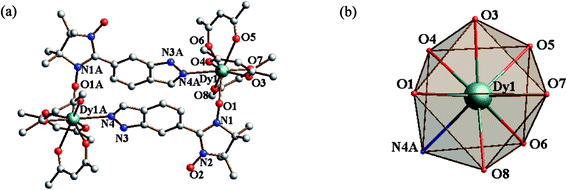 |
| | Fig. 3 (a) Simplified view of the crystal structure of 4. Fluorine and hydrogen atoms are omitted for clarity. (b) D4d-symmetry polyhedral of dysprosium atom. | |
Table 3 Lanthanide geometry analysis by SHAPE software
| Ln(III) |
D2d-DD |
C2v-TP |
D4d-AP |
| Gd1 (1) |
0.879 |
1.597 |
1.054 |
| Dy1 (2) |
0.867 |
1.682 |
1.015 |
| Gd1 (3) |
1.007 |
1.759 |
0.949 |
| Dy1 (4) |
1.093 |
1.774 |
0.848 |
Static magnetic properties
Magnetic measurements were performed on polycrystalline samples of 1–4. The phase purity of the bulk samples was confirmed by XRD analyses. Direct current (dc) magnetic susceptibilities for all the four complexes were measured in 2–300 K range under the applied magnetic field of 1 kOe.
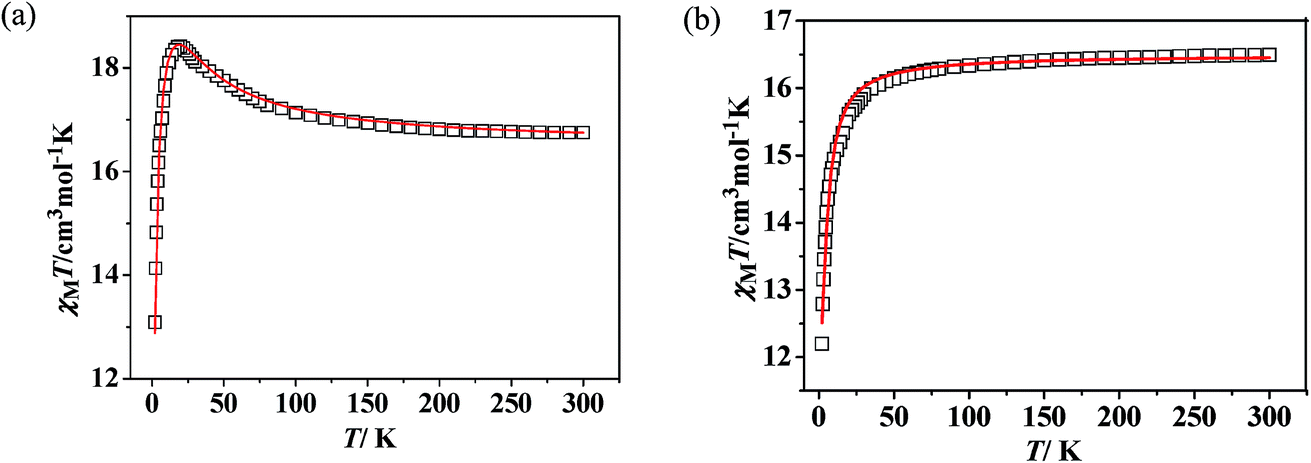
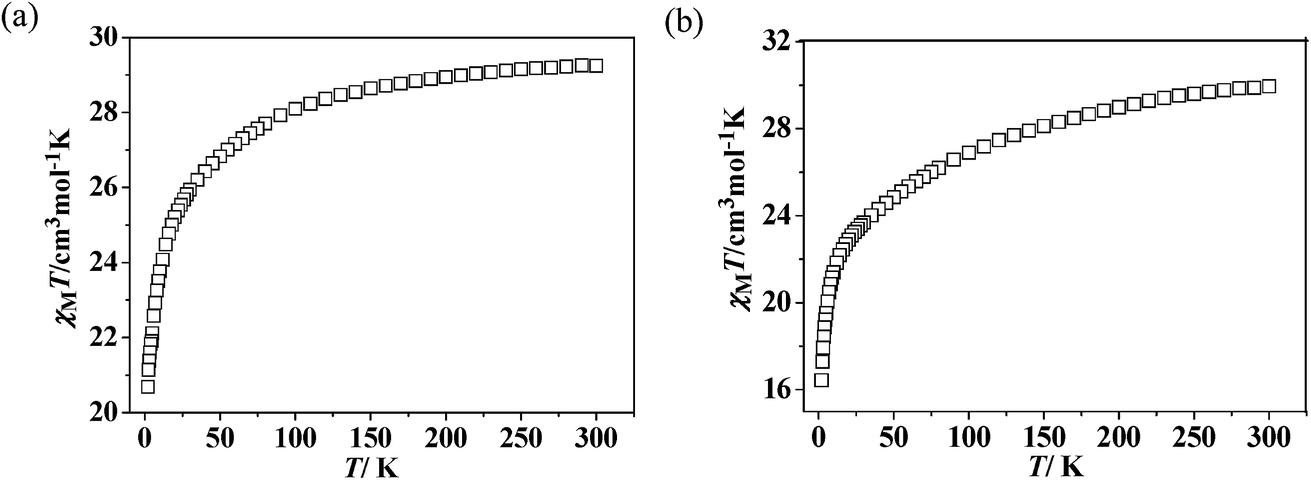
Static magnetic properties for 1 and 3. Variable temperature magnetic susceptibilities for complexes 1 and 3 are shown in Fig. 4. At room temperature, the values of χMT are 16.75 and 16.64 cm3 K mol−1 for complexes 1 and 3, respectively, in good agreement with the expected value of 16.67 cm3 K mol−1 for two uncoupled GdIII ions (8S7/2, g = 2) and two radicals (S = 1/2, 0.375 cm3 K mol−1). Upon cooling, the χMT–T plots of 1 and 3 display different behaviors. For 1, the χMT value increases slowly to reach the maximum of 18.42 cm3 K mol−1 at the temperature of 20 K. Then it begins to decrease quickly as the temperature is lowered further and reaches the least value of 13.09 cm3 K mol−1 at 2 K. The profile of the curve indicates that the interaction between GdIII ion and nitroxide radical is ferromagnetic. Based on the above structural analysis, three exchange pathways should be operative: (i) the magnetic interaction between Gd and the directly coordinated nitroxide group (J1); (ii) the magnetic interaction between two uncoordinated NO groups through space (J2); (iii) the magnetic coupling between Gd(1) and Gd(1A) through imidazoline and indazole ring (J3); (iv) the Gd(III) ion interacting with the NO group through imidazoline and indazole ring (J4). J3 and J4 are anticipated to be weak, this assumption is in accordance with previous results about the cyclic dimer GdIII-complexes with nitroxide radicals containing pyridine.23 What's more, the indazole ring is larger than the pyridine, and the distances (Gd1⋯Gd1A: 10.113(5) Å; Gd1⋯O1A (radical): 9.632 Å) between the coupled spin carries are much longer than that in the references,23 so the magnetic interactions (J3 and J4) are weaker than the reported data in structure similarly complexes. The value of J2 has relationship with the distance between the uncoordinated NO groups. For complex 1 (Fig. 1c), the Orad⋯Orad# distance is a little long (4.286 Å), so J2 is also supposed as weak interaction. Accordingly, the system was modeled as two mononuclear di-spin units (rad–Gd), and the magnetic analysis for every GdIII–rad unit was carried out by using the spin Hamiltonian Ĥ = −2JŜGd1Ŝrad1. The weak exchange interactions being considered within the mean field approximation (zJ′). Eqn (1) and (2) was introduced to analyze the magnetic coupling strength, where J1 represent the magnetic coupling for GdIII–radical.| |
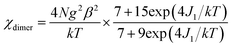 | (1) |
| |
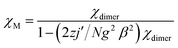 | (2) |
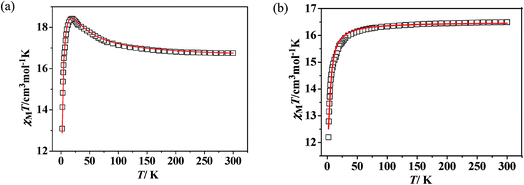 |
| | Fig. 4 Temperature dependence of DC magnetic susceptibilities for 1 (a) and 3 (b). The solid red line represents the theoretical values based on the corresponding equations. | |
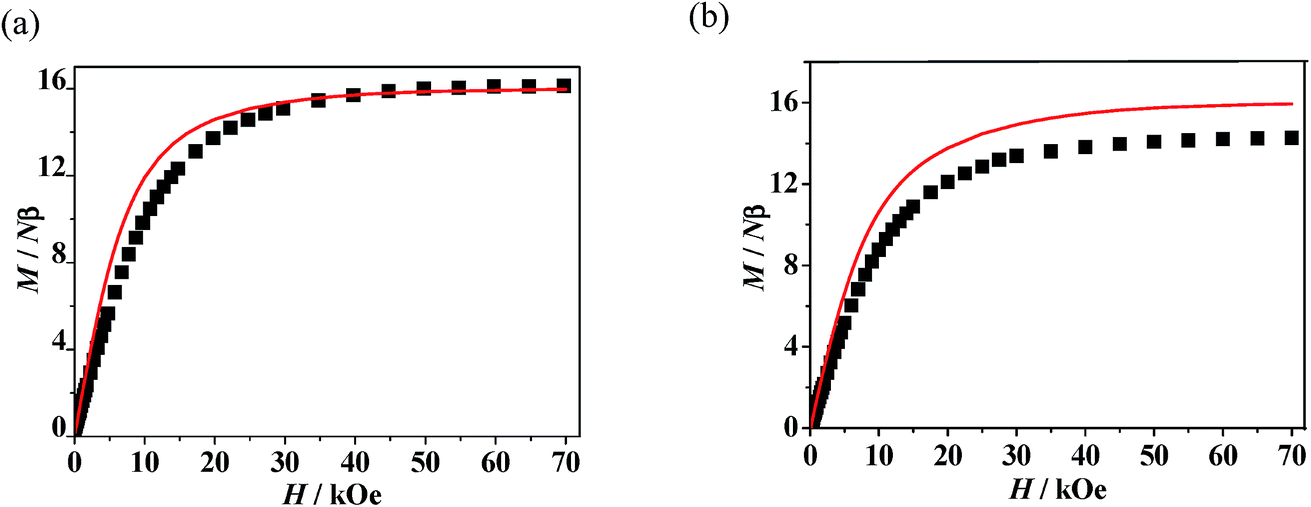
A good agreement between calculated and experimental values of the susceptibility is obtained yielding the parameters, J1 = 8.07 cm−1, zj′ = −0.028 cm−1 (g was fixed as 2). The positive values of J1 indicate the ferromagnetic interactions between GdIII ion and the direct coordinated radical, which is very common in Gd–radical systems,23,24 and the fitting results are also coordinated with the conclusion made by Ishida.21b The magnetization versus field measurements at 2 K is shown in Fig. 4a, a magnetization of 16.09 Nβ is reached at 50 kOe, which corresponds with the value for two S = 4 ferromagnetic Gd–rad units.23,24b
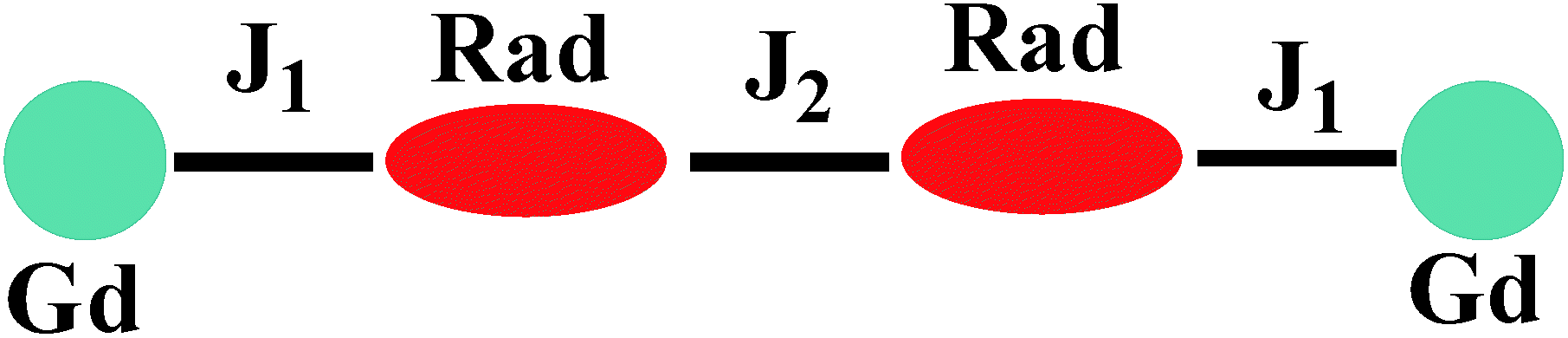
For 3, on decreasing the temperature, the χMT value remains unchanged till 80 K and then begins to decrease slowly till to reach the minimum of 12.09 cm3 K mol−1 at 2 K. Although the structure of 3 is analogical to that of 1, the Orad⋯Orad# distance (3.729) is obviously shorter than that in complex 1 (4.286 Å), the intermolecular magnetic interaction between the neighbouring uncoordinated NO groups may be strong. The bi-nuclear model for fitting the χMT value in 1 is not suit for fitting compound 3. Accordingly, the magnetic behavior of 3 can be interpreted as one linear Gd–rad–rad–Gd magnetic unit (Scheme 2). According to the structural data, the torsion angle for Gd–O–N–C is 93.9(6)° and the value of J1 should be positive.21 Consequently; the observed antiferromagnetic interaction in complex 3 is probably due to the magnetic coupling between two adjacent uncoordinated NO groups, which is stronger than the ferromagnetic interaction between Gd and the directed coordinated NO group.21 Based on the Hamiltonian equation: Ĥ = −2J1(Ŝrad1ŜGd1 + Ŝrad2ŜGd2) − 2J2Ŝrad1Ŝrad2, the software of MAGPACK25 was used to simulate the magnetic susceptibilities.
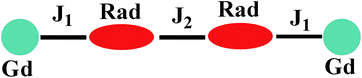 |
| | Scheme 2 The magnetic exchange pathways in complex 3. | |
The observed χMT data were well reproduced by using the approximate eqn (1) and (2), giving the best fitting parameters of g = 2.01, Jrad–Gd = 0.91 cm−1, J2 = −13.16 cm−1. The positive value of Jrad–Gd indicates the weak ferromagnetic interactions between Gd(III) and the radical, which is common in the Gd–radical system.21,23,24 In addition, the high negative J2 value indicates the strong intermolecular antiferromagnetic interactions between the uncoordinated NO groups. The magnetization versus field measurements was carried out at 2 K (Fig. 5b), and the measured magnetization is below the calculated two S = 7/2 + 1/2 units. It further confirms that the antiferromagnetic interaction between the uncoordinated NO groups overwhelms the ferromagnetic interactions between GdIII and the directed coordinated NO group.
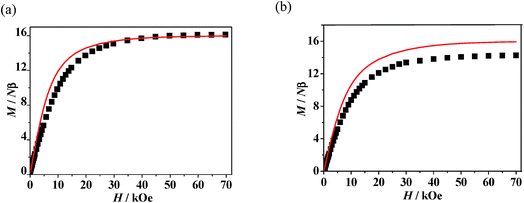 |
| | Fig. 5 (a) Field dependence of the magnetization for 1 at 2 K. The solid line represents the theoretical magnetization curve for two isolated S = 4 units. (b) Field dependence of the magnetization for 3 at 2 K. The solid lines represent the theoretical magnetization curves for two isolated S = 7/2 + 1/2 (red) units. | |
Static magnetic properties for 2 and 4. The temperature dependence of magnetic susceptibility for 2 and 4 exhibited very analogous behaviors (Fig. 6). The observed room-temperature values of χMT for 2 and 4 are 29.24 and 29.52 cm3 K mol−1, respectively, which is close to the expected value of 29.07 cm3 K mol−1 for two isolated Dy(III) ions plus two uncorrelated S = 1/2 spins. When the temperature is lowered from 300 K, the χMT values for both of the two complexes decrease slightly at first and then more quickly to the lowest values at 2 K. The decrease of χMT value upon lowering of the temperature in the high-temperature range for 2 and 4 is due to both depopulation of the LnIII stark sublevels and Ln–radical interactions through N–O. The decrease of χMT at lower temperature may be attributed to the antiferromagnetic LnIII-coordinated NO radical interaction. The field dependences of magnetization (M) for complexes 2 and 4 have been determined at 2 K in the range of 0–70 kOe (Fig. S9†). For both of the two complexes, the field-dependent magnetization value shows a rapid increase below 10 kOe. At higher fields, M increases up to 13.58 (for 2) and 15.41 Nβ (for 4) at 70 kOe, respectively, which is much lower than the expected saturation values of 22 Nβ (10 Nβ for each DyIII ion for J = 15/2 and g = 4/3, plus 2 Nβ for the two organic radicals). The large gaps can be ascribe to the presence of magnetic anisotropy and/or low-lying excited states in the systems.23,26
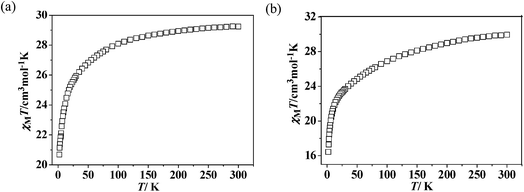 |
| | Fig. 6 Temperature dependence of χMT value for complexes 2 (a) and 4 (b). | |
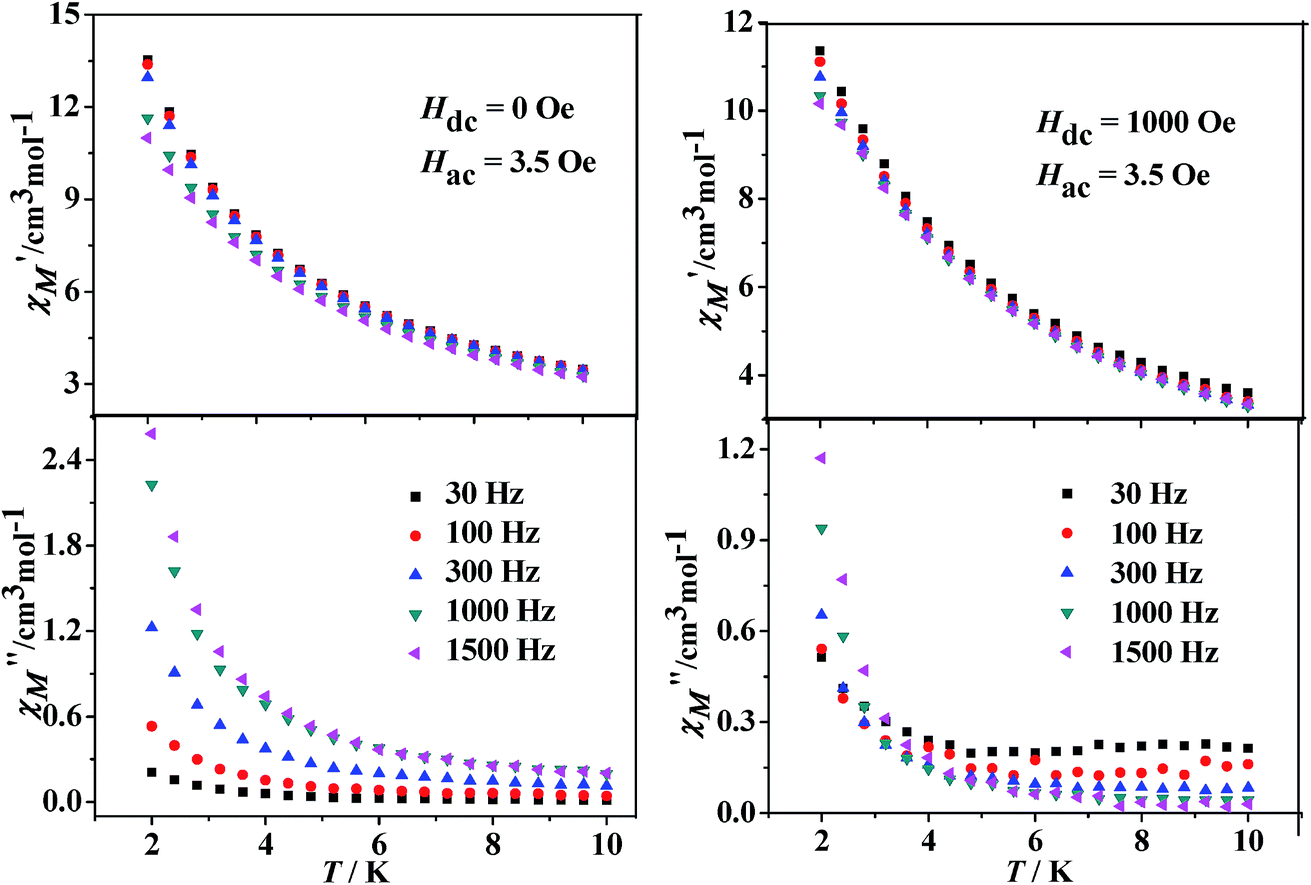
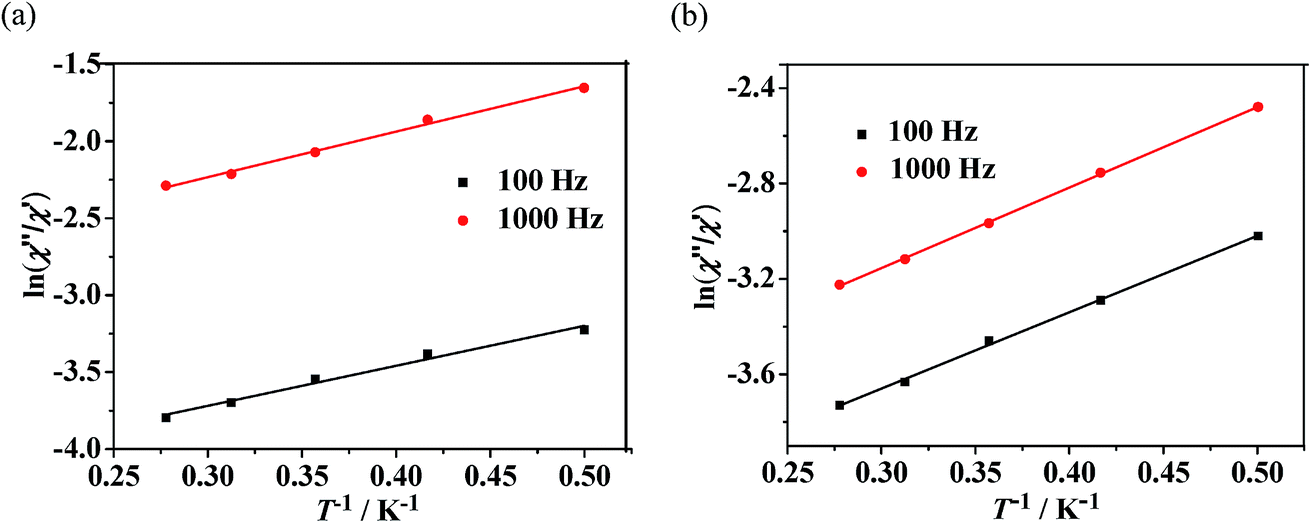
Dynamic magnetic properties for Dy(III)'s complexes (2 and 4). The dynamic magnetic susceptibility measurements of complexes 2 and 4 have been investigated in the absence or with an applied static field. The imaginary component χ′′ of the complex 2 does not show obvious frequency-dependent phenomenon (Fig. S10, see ESI†). For complex 4, frequency-dependent out-of-phase signals are observed (Fig. 7), indicating the onset of magnetization expected for single-molecule magnet (SMM) behavior. However, no peak maximum is found above 2 K even for the highest frequency investigated. The observed dynamic magnetic behavior for 4 suggests that the maxima may exist below the operating lowest temperature (2.0 K) or above the maximum frequency (1500 Hz) of the SQUID instrument.27 These behaviors show that 4 is possibly a DyIII SMM with a small relaxation barrier. Measurements performed with an applied external field (1000 Oe) did not change this situation (Fig. 7, right). For complex 4, a method proposed by Bartolomé et al.28 was used to predict the energy of the reversal barrier and the characteristic relaxation time. Supposing that there is only one characteristic relaxation process in the Debye type, a rough evaluation of the energy barrier Ea and relaxation time τ0 is given by the following equation ln(χ′′/χ′) = ln(ωτ0) + Ea/kBT. Best fit to the experimental data yielded Ea = 2.1 K and τ0 = 1.8 × 10−5 s at zero DC field or Ea = 2.3 K and τ0 = 2.5 × 10−5 s at 1 kOe static field, respectively (Fig. 8).
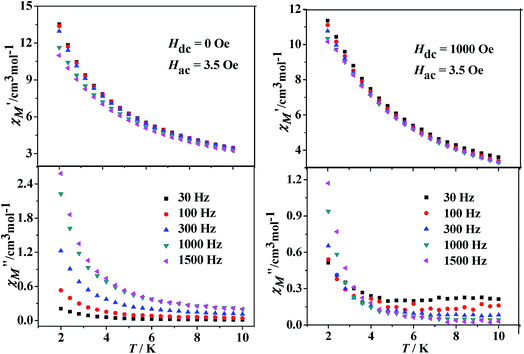 |
| | Fig. 7 Temperature dependence of the in-phase (χ′) and out-of-phase (χ′′) components of the AC magnetic susceptibility under zero (left) or 1 kOe (right) DC field for 4. | |
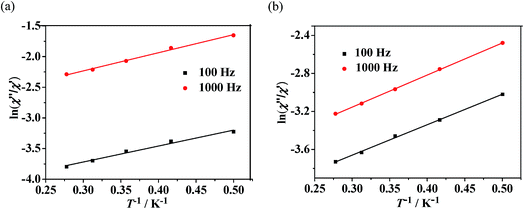 |
| | Fig. 8 Plots of the natural logarithm of χ′′/χ′ versus 1/T at 0 Oe DC field (a) or 1000 Oe DC field (b) for complex 4. The solid line represents the fitting results. | |
For Ln-based systems, the observed magnetic relaxation behavior mainly depends on the single-ion relaxation, which is extremely sensitive to strength and symmetry of the local crystal field around the center ions.29 The effect of the crystal-field can be expressed by the Hamiltonian ĤCF = ∑BqkÕqk, in which Bqk are the crystal-field parameters and Õqk are the Stephen operators.30 The Stephen operators are always considered to have relationship with the QTM. At the condition of q ≠ 0 and k = 2, 4, 6, the relaxation times can be significantly reduced. In high symmetry crystal-field, such as C∞v, D∞h, D4d, D5h and D6d, certain parameters Bqk vanish, which provides a chemical method to control QTM by tuning the local symmetry of the metal centers. As we can see, by using two indazole radical ligands with different locations of the radical, the crystal structures of complexes 2 and 4 show no drastic changes. In both of the two complexes, the central DyIII ions exhibit binuclear structure, which are all in DyO7N coordination sphere, but with different polyhedron symmetry. The D2d symmetry in complex 2 accompanied by a weaker ac signal. The D4d symmetry in complex 4 resulted in SMM behavior, in which quantum tunneling of magnetization was better suppressed.31 Zhu's research group reported an analogous binuclear Dy–radical system with D4d symmetry,21 which shows slowing the relaxation of the magnetization. The result suggests that the magnetic relaxation for the Ln–radical system is largely relied on the local coordination field.
Conclusions
In summary, four binuclear lanthanide–radical compounds have been synthesized using two different indazole nitronyl nitroxide radicals. The study on the magnetization dynamics for complexes 2 and 4 reveals that they exhibit quite distinct magnetic relaxation behaviors. Complex 4 shows obvious frequency-dependent out-of-phase signals, however, such a phenomena is not observed for 2. The difference in magnetic relaxation of these complexes is probably due to the different symmetry of local ligand field of the central Dy(III) ions. These results show that the different ligand field can drastically affect the magnetic relaxation of the magnetization.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21371133).
References
-
(a) G. Christou, D. Gatteschi, D. N. Hendrickson and R. Sessoli, MRS Bull., 2000, 25, 66 CrossRef CAS;
(b) A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., Int. Ed., 2001, 40, 1760 CrossRef CAS;
(c) R. Clérac, H. Miyasaka, M. Yamashita and C. Coulon, J. Am. Chem. Soc., 2002, 124, 12837 CrossRef.
-
(a) L. Bogani, A. Vindigni, R. Sessoli and D. Gatteschi, J. Mater. Chem., 2008, 18, 4750 RSC;
(b) J. S. Miller and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3065 RSC;
(c) Molecular Magnets Physics and Applications, ed. J. Bartolomé, F. Luis and J. F. Fernández, Springer-Verlag, Berlin, Germany, 2014 Search PubMed.
-
(a) M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M. A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8, 194 CrossRef CAS PubMed;
(b) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 10, 538 CrossRef PubMed.
-
(a) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed;
(b) S. Thiele, R. Vincent, M. Holzmann, S. Klyatskaya, M. Ruben, F. Balestro and W. Wernsdorfer, Phys. Rev. Lett., 2013, 111, 037203 CrossRef CAS PubMed;
(c) K. Hong and W. Y. Kim, Angew. Chem., Int. Ed., 2013, 52, 3389 CrossRef CAS PubMed.
-
(a) J. A. Jones, Science, 1998, 280, 229 CrossRef CAS;
(b) M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789 CrossRef CAS PubMed;
(c) M. J. Graham, J. M. Zadrozny, M. Shiddiq, J. S. Anderson, M. S. Fataah, S. Hill and D. E. Freedman, J. Am. Chem. Soc., 2014, 136, 7623 CrossRef CAS PubMed.
- D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 269 Search PubMed.
- A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., 2001, 113, 1810 CrossRef.
- G. F. Xu, Q. L. Wang, P. Gamez, Y. Ma, R. Clérac, J. K. Tang, S. P. Yan, P. Cheng and D. Z. Liao, Chem. Commun., 2010, 46, 1506 RSC.
- Y. N. Guo, G. F. Xu, P. Gamez, L. Zhao, S. Y. Lin, R. P. Deng, J. K. Tang and H. J. Zhang, J. Am. Chem. Soc., 2010, 132, 8538 CrossRef CAS PubMed.
-
(a) S. D. Jiang, B. W. Wang, H. L. Sun, Z. M. Wang and S. Gao, J. Am. Chem. Soc., 2011, 133, 4730 CrossRef CAS PubMed;
(b) R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. L. McInnes, L. F. Chibotaru and R. E. P. Winpenny, Nat. Chem., 2013, 5, 673 CrossRef CAS PubMed.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078 RSC.
-
(a) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236 CrossRef CAS PubMed;
(b) T. Rajeshkumar and G. Rajaraman, Chem. Commun., 2012, 48, 7856 RSC.
- A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., Int. Ed., 2001, 40, 1760 CrossRef CAS PubMed.
-
(a) N. Ishii, Y. Okamura, S. Chiba, T. Nogami and T. Ishida, J. Am. Chem. Soc., 2008, 130, 24 CrossRef CAS PubMed;
(b) C. Benelli and D. Gatteschi, Chem. Rev., 2002, 102, 2369 CrossRef CAS PubMed;
(c) D. Luneau and P. Rey, Coord. Chem. Rev., 2005, 249, 2591 CrossRef CAS;
(d) R. N. Liu, L. C. Li, X. L. Wang, P. P. Yang, C. Wang, D. Z. Liao and J. P. Sutter, Chem. Commun., 2010, 46, 2566 RSC.
- C. Görller-Walrand, K. Binnemans, K. A. Gschneidner and L. Eyring, Handbook on the Physics and Chemistry of Rare Earths, 1996, vol. 23 Search PubMed.
- X. Li, T. Li, X. J. Shi and L. Tian, Inorg. Chim. Acta, 2017, 456, 216 CrossRef CAS.
-
(a) Y. Z. Zheng, Y. Lan, C. E. Anson and A. K. Powell, Inorg. Chem., 2008, 47, 10813 CrossRef CAS PubMed;
(b) S. Kanegawa, M. Maeyama, M. Nakano and N. Koga, J. Am. Chem. Soc., 2008, 130, 3079 CrossRef CAS PubMed.
-
(a) E. F. Ullmann, L. Call and J. H. Osiecki, J. Org. Chem., 1970, 35, 3623 CrossRef;
(b) M. S. Davis, K. Morokum and R. N. Kreilick, J. Am. Chem. Soc., 1972, 94, 5588 CrossRef CAS.
-
(a) SCALE3 ABSPACK: Empirical absorption correction, CrysAlis-Software package, Oxford Diffraction Ltd, Oxford, 2006 Search PubMed;
(b) CrysAlisPro, Agilent Technologies, Yarnton, Oxfordshire, England, 2010 Search PubMed.
-
(a) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 2015, 71, 3 CrossRef PubMed;
(c) G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
-
(a) M. Zhu, Y. Li, L. J. Jia, L. Zhang and W. Zhang, RSC Adv., 2017, 7, 36895 RSC;
(b) T. Kanetomo and T. Ishida, Inorg. Chem., 2014, 53, 10794 CrossRef CAS PubMed.
- M. Llunell, D. Casanova, J. Cirera, J. M. Bofill, P. Alemany, S. Alvarez, M. Pinsky, D. Avnir, SHAPE 2.0, Universitat De Barcelona and the Hebrew University Of Jerusalem, Barcelona, 2003 Search PubMed.
- X. L. Mei, R. N. Liu, C. Wang, P. P. Yang, L. C. Li and D. Z. Liao, Dalton Trans., 2012, 41, 2904 RSC.
-
(a) Y. L. Wang, Y. Y. Gao, Y. Ma, Q. L. Wang, L. C. Li and D. Z. Liao, CrystEngComm, 2012, 14, 4706 RSC;
(b) P. Hu, M. Zhu, X. L. Mei, H. X. Tian, Y. Ma, L. C. Li and D. Z. Liao, Dalton Trans., 2012, 41, 14651 RSC;
(c) P. Hu, H. F. Guo, Y. Li and F. P. Xiao, Inorg. Chem. Commun., 2015, 59, 91 CrossRef CAS;
(d) S. Y. Zhou, X. Li, T. Li, L. Tian, Z. Y. Liu and X. G. Wang, RSC Adv., 2015, 5, 17131 RSC.
-
(a) J. J. Borrás-Almenar, J. M. Clemente-Juan, E. Coronado and B. S. Tsukerblat, Comput. Chem., 2001, 22, 985 CrossRef;
(b) J. J. Borrás-Almenar, J. M. Clemente-Juan, E. Coronado and B. S. Tsukerblat, Inorg. Chem., 1999, 38, 6081 CrossRef.
-
(a) K. Bernot, J. Luzon, L. Bogani, M. Etienne, C. Sangregorio, M. Shanmugam, A. Caneschi, R. Sessoli and D. Gatteschi, J. Am. Chem. Soc., 2009, 131, 5573 CrossRef CAS PubMed;
(b) P. Hu, X. F. Wang, Y. Ma, Q. L. Wang, L. C. Li and D. Z. Liao, Dalton Trans., 2014, 43, 2234 RSC;
(c) L. L. Li, S. Liu, Y. Zhang, W. Shi and P. Cheng, Dalton Trans., 2015, 44, 6118 RSC.
- Y. N. Guo, X. H. Chen, S. F. Xue and J. K. Tang, Inorg. Chem., 2008, 51, 4035 CrossRef PubMed.
-
(a) F. Luis, J. Bartolomé, J. F. Fernández, J. Tejada, J. M. Hernández, X. X. Zhang and R. Ziolo, Phys. Rev. B, 1997, 55, 11448–11456 CrossRef CAS;
(b) J. Bartolomé, G. Filoti, V. Kuncser, G. Schinteie, V. Mereacre, C. E. Anson, A. K. Powell, D. Prodius and C. Turta, Phys. Rev. B, 2009, 80, 014430 CrossRef.
- J. Long, F. Habib, P. H. Lin, I. Korobkov, G. Enright, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 5319 CrossRef CAS PubMed.
- C. Görller-Walrand, K. Binnemans, K. A. Gschneidner and L. Eyring, Handbook on the Physics and Chemistry of Rare Earths, 1996, vol. 23 Search PubMed.
-
(a) N. Ishikawa, M. Sugita, T. Ishikawa, S. Y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694 CrossRef CAS PubMed;
(b) C. R. Ganivet, B. Ballesteros, G. de la Torre, J. M. Clemente-Juan, E. Coronado and T. Torres, Chem.–Eur. J., 2013, 19, 1457 CrossRef CAS PubMed;
(c) J.-L. Liu, Y.-C. Chen, Y.-Z. Zheng, W. Q. Lin, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M.-L. Tong, Chem. Sci., 2013, 4, 3310 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1554849, 1554850, 1554950 and 1555029 contain the supplementary crystallographic data for 1–4, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra09192c |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Zhong Yi Liu*,
Feng Zhen Hua,
Si Jia Yu and
Yuan Yuan Xu
*,
Zhong Yi Liu*,
Feng Zhen Hua,
Si Jia Yu and
Yuan Yuan Xu