
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of an azo-Mn(II) complex with mild pH control using a microfluidic device
Daiki Tanaka *a,
Shunsuke Sawaia,
Dong Hyun Yoona,
Tetsushi Sekiguchia,
Takashiro Akitsub and
Shuichi Shojia
*a,
Shunsuke Sawaia,
Dong Hyun Yoona,
Tetsushi Sekiguchia,
Takashiro Akitsub and
Shuichi Shojia
aResearch Organization for Nano & Life Innovation, Waseda University, 120-5 Research Development Center, 513 Waseda-tsurumakicho, Shinjuku-ku, Tokyo 162-0041, Japan. E-mail: d.tanaka@aoni.waseda.jp
bDepartment of Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan
First published on 14th August 2017
Abstract
This study describes the synthesis of an azo-Mn(II) complex requiring an accurate pH control. The reaction conditions for the delicate azo-Mn(II) complex could be precisely controlled using a microfluidic device. The microfluidic method showed the following advantages over the conventional method: the concentration of the pH-control reagent was reduced (1/15), the reaction time was remarkably decreased from 4 h to less than 1 s, and the reaction temperature was lowered from 40 to 23 °C. Moreover, the microfluidic device suppressed the oxidation of the compound and did not require cooling to remove the heat of reaction. In addition, the conventional method uses harsh pH control, whereas the microfluidic device permits mild pH control.
1. Introduction
Azo-metal complexes have attracted attention from many researchers in recent years. Azo compounds have important uses in biofuel cell electrodes and solar power applications and as cell markers. For example, Lim et al. designed a heptamethine-azo conjugate as a near-infrared fluorescent probe to monitor mitochondrial glutathione with low background autofluorescence and serve as a cell marker.1 However, the synthesis of azo-metal complexes remains challenging and is still an active area of research. For instance, Grirrane et al. and Zhang and Jiao have reported that gold nanoparticles supported on titanium dioxide (TiO2) and nanoparticulate cerium dioxide (CeO2) catalyse the aerobic oxidation of aromatic anilines to aromatic azo compounds.2,3 Takeda, Minakata, and co-workers demonstrated the oxidative homodimerization of aniline under various conditions.4–6 Monir et al. reported the development of an efficient method for the synthesis of aromatic azo compounds in high yields through the phenyliodine(III) diacetate (PIDA)-mediated oxidative dehydrogenative coupling of anilines.7 Einaga, Akitsu, and co-workers systematically investigated organic/inorganic hybrid materials composed of metal complexes and azo compounds in poly(methyl methacrylate) films as non-crystalline solids8 that could be converted into chiral Schiff base complexes and made to regularly align.9 However, the synthesis of azo compounds via conventional methods is complicated because it requires a metal catalyst and strict control of the atmosphere and temperature.Herein, we propose a new synthetic method for a very simple azo-metal complex using a microfluidic device. Chemical processes using microfluidic devices can be highly efficient. For example, Priest et al. reported the microfluidic solvent extraction of Cu2+ ions from particle-laden aqueous solutions using 2-hydroxy-5-nonylacetophenone as an alternative to conventional solvent extraction.10 Furthermore, Kitamori and co-workers described the determination of carbamate pesticides such as carbaryl, carbofuran, propoxur, and bendiocarb using a microfluidic device developed for efficient solvent extraction.11,12 Brivio et al. and deMello et al. reviewed the effects of downscaling reaction vessels as well as the advantages of the continuous-flow microfluidic approach over conventional methods, as illustrated by a number of examples of organic reactions carried out in microfluidic devices.13–16 The Lumley group reported the use of a microfluidic device in the photochemical oxidation of cholesterol, α-terpinene, and citronellol under flow conditions, and the results were compared with those of similar batch reactions.17 Miller et al. described the rapid and high-yielding carbonylative cross-coupling reactions of aryl halides to form secondary amides using a glass-fabricated microfluidic device.18 Gunther and Jensen reviewed the flow characteristics in multiphase micro- and nanoscale systems, measurement methodology, and how these characteristics could be utilized in microfluidic applications.19
Kanai et al. described a sheath flow multi-sample injector that allowed concentration-conserving sample injection using a stepwise flow to reduce the dilution of the sample reagent.20 We have previously established the synthesis of metal complexes and protein–metal complexes using microfluidic devices.21,22 The synthesis of the metal complexes using the microfluidic device required a shorter reaction time and no temperature or atmospheric control.23,24 Furthermore, the conventional synthesis of azo compounds requires harsh pH adjustments using high concentrations of hydrochloric acid and sodium hydroxide. However, highly concentrated reagents lead to decomposition of the compounds (Fig. 1). In addition, these conventional methods require cooling to remove the heat of reaction.
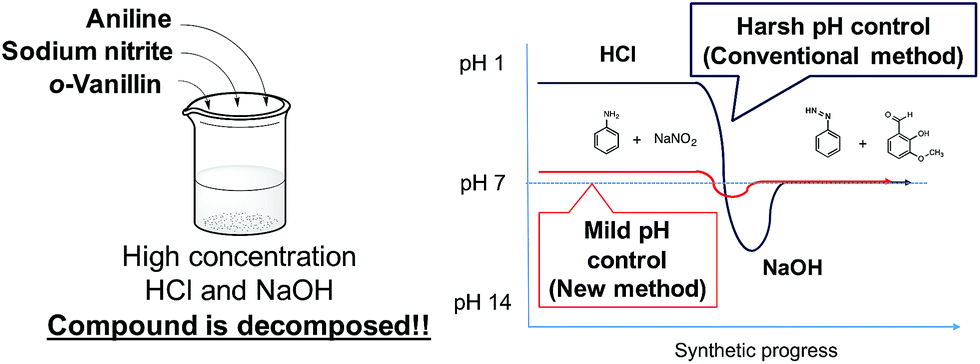 | ||
| Fig. 1 pH control in the conventional method. | ||
In this study, we demonstrate the use of a microfluidic device to synthesise azo compounds under milder conditions than those utilized in conventional methods. According to previous studies, microfluidic devices can permit a more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold increase in the rates of chemical reactions.23,24 We, therefore, focused on the high reactivity that could be achieved with a microfluidic device. The device is likely to reduce the required concentration of pH-adjustment reagents. Moreover, the microfluidic device allows reactions to be performed at the interface between reagents. The reagents are supplied one after another, and the reactants for each step are synthesised in a stepwise manner. Thus, localised excess chemical reactions do not occur. Furthermore, the conventional method requires complicated operations, thus making the synthesis difficult. However, the use of a microfluidic device eliminates the need to control these operations and allows the convenient and user-friendly synthesis of azo-Mn(II) complexes.
000-fold increase in the rates of chemical reactions.23,24 We, therefore, focused on the high reactivity that could be achieved with a microfluidic device. The device is likely to reduce the required concentration of pH-adjustment reagents. Moreover, the microfluidic device allows reactions to be performed at the interface between reagents. The reagents are supplied one after another, and the reactants for each step are synthesised in a stepwise manner. Thus, localised excess chemical reactions do not occur. Furthermore, the conventional method requires complicated operations, thus making the synthesis difficult. However, the use of a microfluidic device eliminates the need to control these operations and allows the convenient and user-friendly synthesis of azo-Mn(II) complexes.
2. Experimental
2.1 Materials and measuring equipment
The chemicals and reagents were of the highest grade commercially available (Tokyo Chemical Industry; Kanto Chemical) and used as received without further purification. The products were analysed by infrared spectroscopy (IR; FT/IR-6200, JASCO), ultraviolet-visible spectroscopy (UV-vis; U-3900, Hitachi), fast atom bombardment mass spectrometry (FAB-MS; JMS-BU25, JEOL), time-of-flight mass spectrometry (TOF-MS; JMS-T100LC, JEOL), proton NMR spectroscopy (1H-NMR; ECX500, JEOL), and X-ray diffraction (XRD; RINTUltima3, Rigaku). The conventional synthesis and the synthesis conducted using the microfluidic device were compared.2.2 Conventional synthesis
As shown in Fig. 2, the azo-Mn(II) complex was synthesised in three steps following the method reported by Akitsu et al.25 Steps I, II, and III represent the syntheses of an azo compound, azo ligand, and azo-Mn(II) complex, respectively. Steps II and III were performed under a nitrogen atmosphere.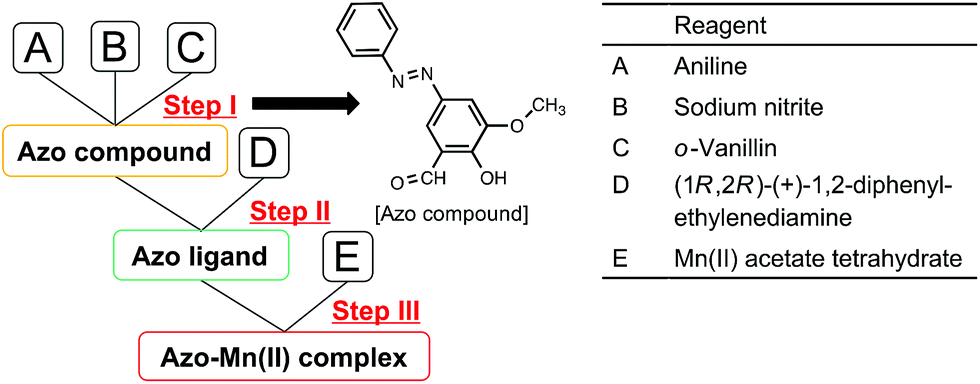 | ||
| Fig. 2 Three-step synthesis of the azo-Mn(II) complex. | ||
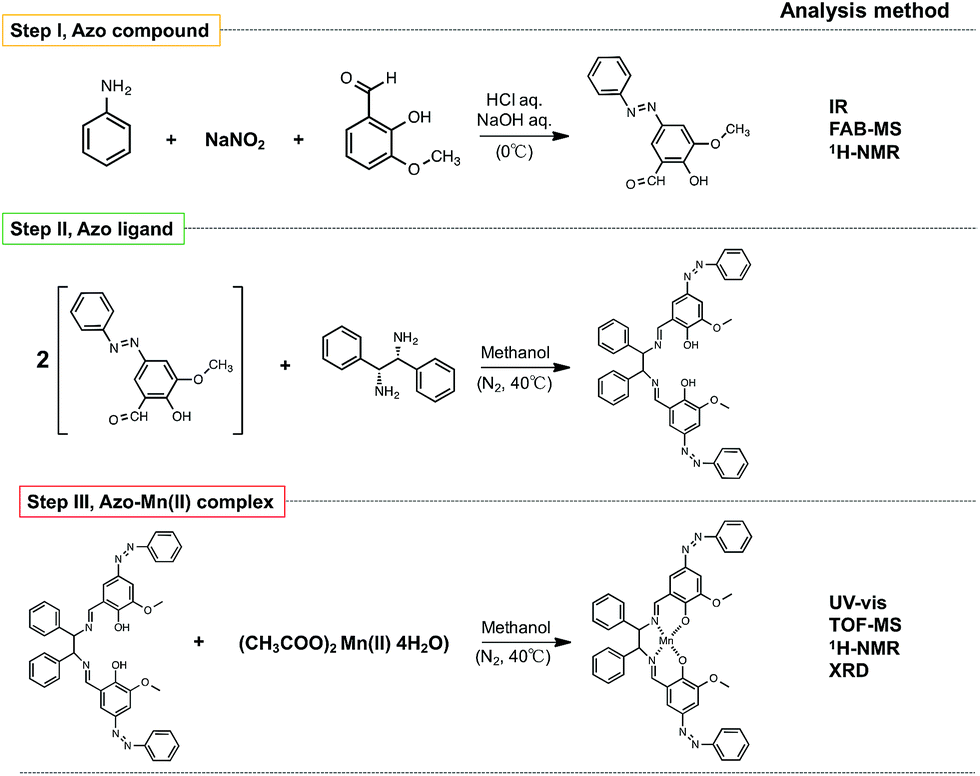 | ||
| Fig. 3 Synthetic scheme for the azo-Mn(II) complex. | ||
2.3 Synthesis with the microfluidic device
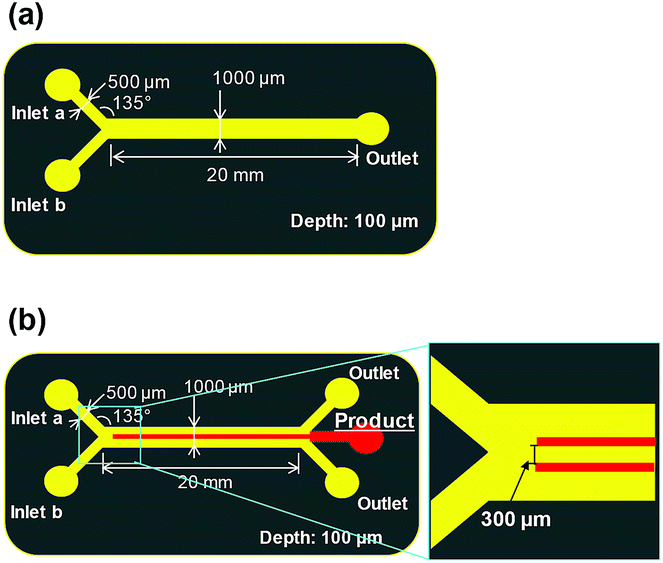 | ||
| Fig. 4 Device design and fluidic experiments. (a) Y-junction device. (b) X-junction device. | ||
| HCl concentration | Synthesis result |
|---|---|
| 1/1 (5.62 mol L−1) | Failure |
| 1/2 (2.81 mol L−1) | Failure |
| 1/5 (1.12 mol L−1) | Failure |
| 1/10 (0.56 mol L−1) | Success |
| 1/15 (0.37 mol L−1) | Success |
| 1/20 (0.28 mol L−1) | Insoluble (aniline) |
| Conventional method | Microfluidic device | |
|---|---|---|
| Reagent concentration (step I), HCl and NaOH for pH control | 5.62 mol L−1 | 0.37 mol L−1 |
| Reagent concentration (step I), azo compounds | 250 mmol L−1 | 250 mmol L−1 |
| Reagent concentration (steps II and III), azo-metal complex | 20 and 40 mmol L−1 | 20 and 40 mmol L−1 |
| Reaction temperature (step I) | 0–5 °C | 23 °C (R.T.) |
| Reaction temperature (steps II and III) | 40 °C | 23 °C (R.T.) |
| Flow rate | — | 2 and 4 μL min−1 |
| Stirring time (step I) | 1 h | — |
| Stirring time (steps II and III) | 4 h | — |
| Atmosphere | N2 | Air |
| Synthesis scale | 50 cm3 | 0.002 cm3 |
| Analytical method for the azo compound (step I) | IR, FAB-MS and 1H-NMR | |
| Analytical method for the metal complex (step III) | UV-vis, TOF-MS, 1H-NMR and XRD | |
3. Results and discussion
The FAB-MS results show the mass-to-charge ratio (m/z: [256.08 + H]+) of the products obtained after step I for both the conventional method and the microfluidic device method (Fig. 5). In addition, the 1H-NMR spectra of the products obtained from the two methods were consistent (Fig. 6). Furthermore, a peak corresponding to N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (1590 cm−1) was observed in the IR spectra (Fig. 7a). These analytical data showed that both the conventional and microfluidic device methods successfully afforded the azo compound. This result proved that the azo compound could be synthesised under mild pH control using the microfluidic device. The chemical reaction was promoted using the microfluidic device, and the reaction proceeded even at a low HCl concentration. Moreover, the synthesis in the microfluidic device did not require temperature control (Table 3) as the microfluidic device allowed the reaction to occur at the interface. The reagents were supplied one after another, and the reactants for each step were synthesised in a stepwise manner. The synthesis of difficult azo compounds requiring control of pH and temperature is also possible using microfluidic devices.
N (1590 cm−1) was observed in the IR spectra (Fig. 7a). These analytical data showed that both the conventional and microfluidic device methods successfully afforded the azo compound. This result proved that the azo compound could be synthesised under mild pH control using the microfluidic device. The chemical reaction was promoted using the microfluidic device, and the reaction proceeded even at a low HCl concentration. Moreover, the synthesis in the microfluidic device did not require temperature control (Table 3) as the microfluidic device allowed the reaction to occur at the interface. The reagents were supplied one after another, and the reactants for each step were synthesised in a stepwise manner. The synthesis of difficult azo compounds requiring control of pH and temperature is also possible using microfluidic devices.
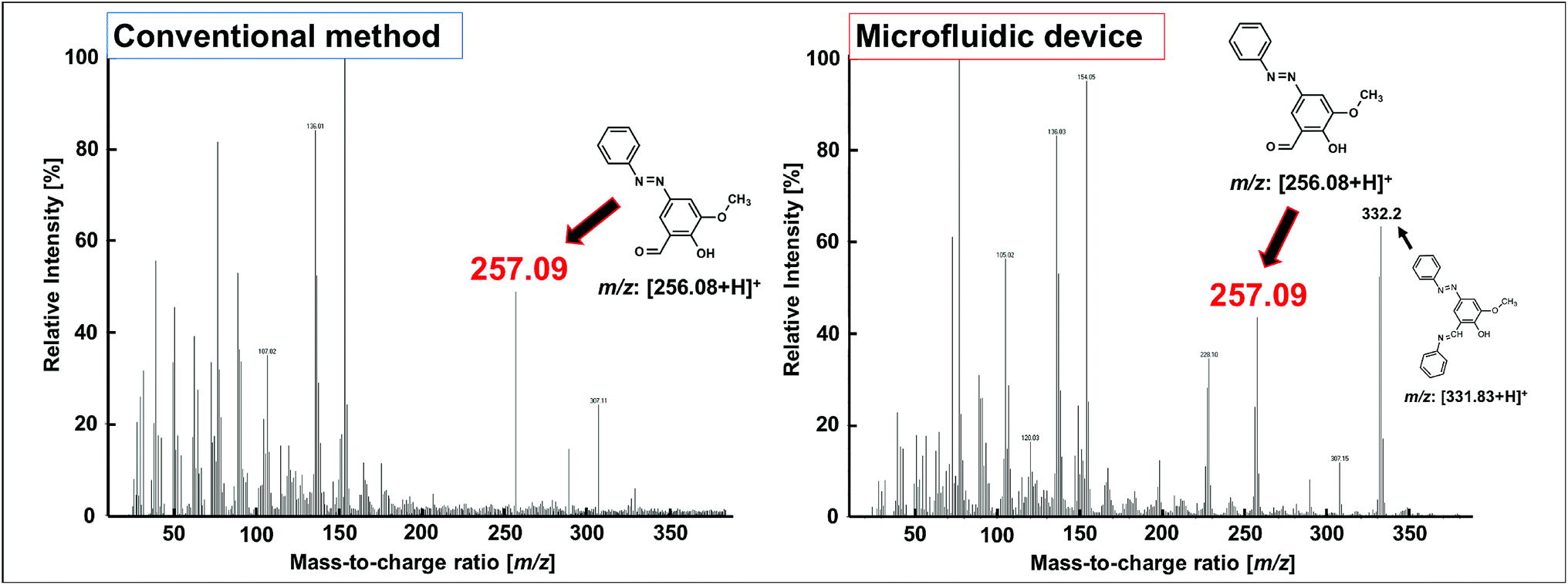 | ||
| Fig. 5 FAB-MS spectra of the azo compounds obtained in step I of the conventional method and using the microfluidic device. | ||
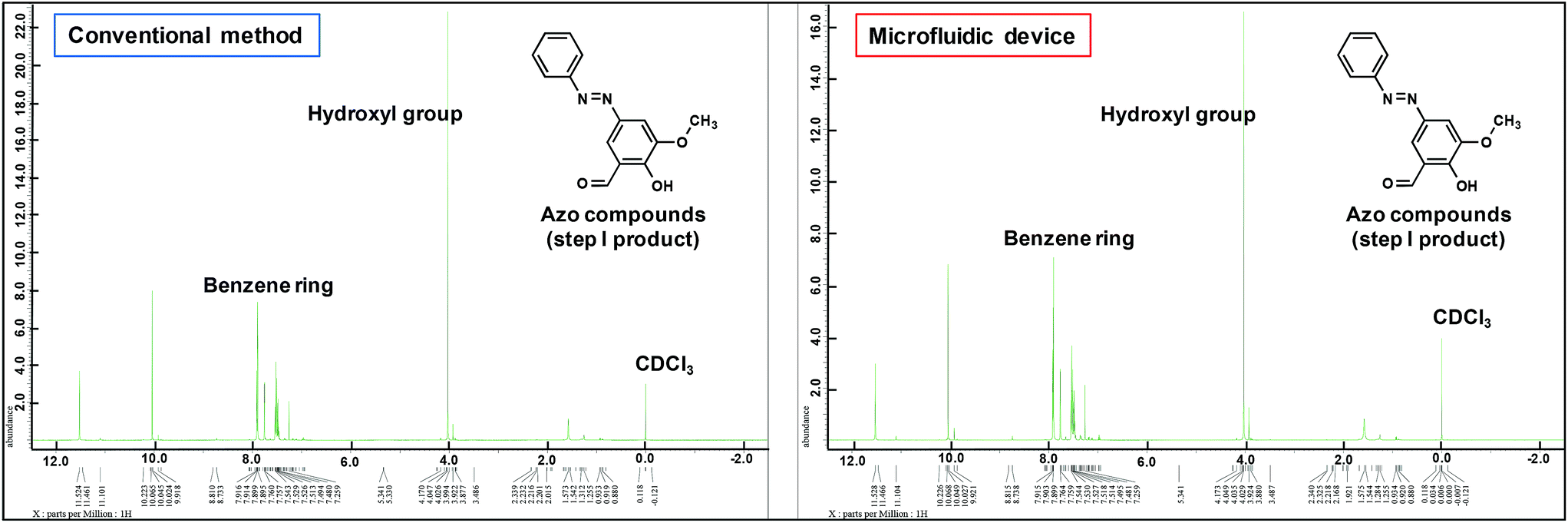 | ||
| Fig. 6 1H-NMR spectra of the azo compounds obtained in step I of the conventional method and using the microfluidic device. | ||
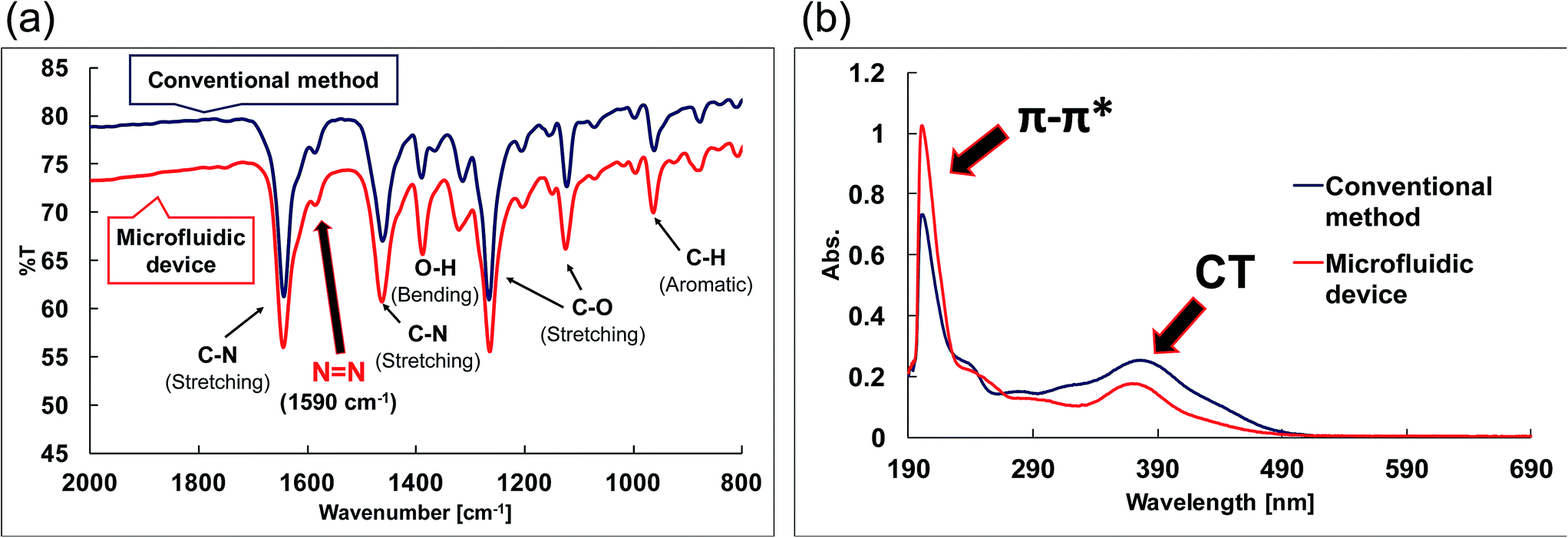 | ||
| Fig. 7 Spectroscopic measurements of the compounds. (a) IR spectra of the azo compounds (step I). (b) UV-vis spectra of the azo-Mn(II) complexes (step III). | ||
| Parameter | Conventional method | Microfluidic device |
|---|---|---|
| pH control (step I), HCl and NaOH | 5.62 mol L−1 | 0.37 mol L−1 |
| Reaction temperature (step I) | 0–5 °C | 23 °C (R.T.) |
| Reaction temperature (steps II and III) | 40 °C | 23 °C (R.T.) |
| Synthesis time (step I) | 1 h | 1 s |
| Synthesis time (steps II and III) | 4 h | 1 s |
| Atmosphere | N2 | Air |
We also tested a microfluidic device with a channel width of 200 μm. However, this device could not be used for the synthesis because the azo compound immediately clogged the channels. Therefore, in the experiments reported herein, we synthesised the azo compound using a microfluidic device with a channel width of 1 mm.
The UV-vis spectra (Fig. 7b) revealed changes in the detected π–π* and charge-transfer transitions, which indicated that the azo-Mn(II) complex had been obtained. In addition, the TOF-MS data showed peaks for the azo-Mn(II) complex (m/z: 741.2; Fig. 8). Moreover, the large peaks in the 1H-NMR spectra were consistent (Fig. 9). Overall, these results indicated that the azo-Mn(II) complex had been successfully synthesised using the microfluidic device.
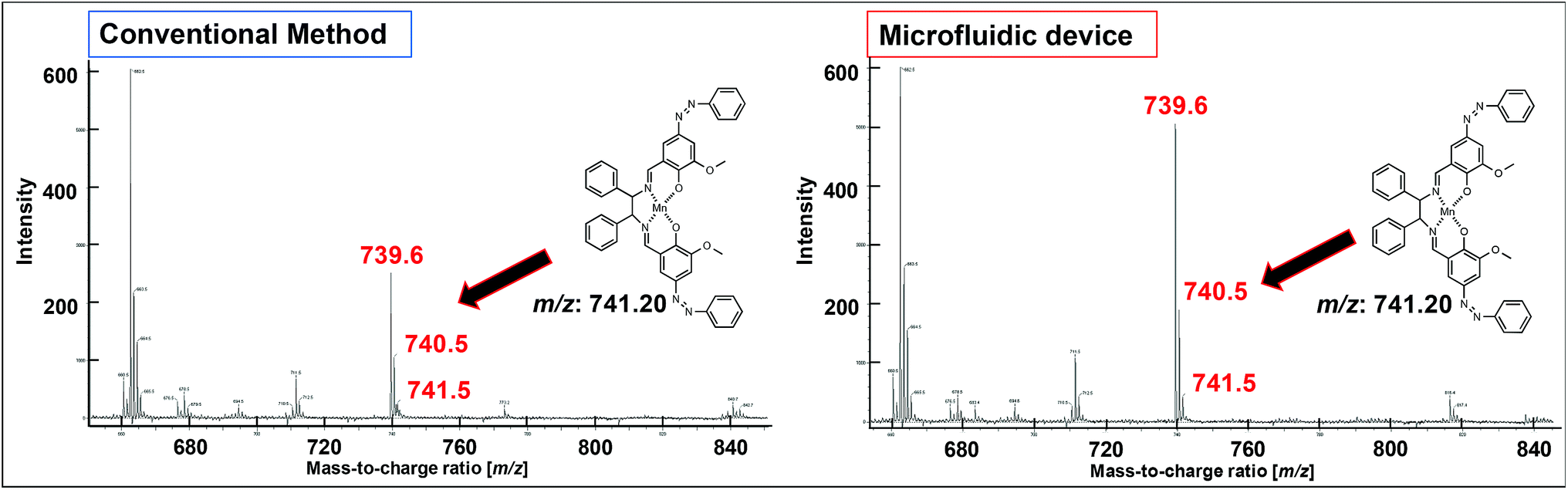 | ||
| Fig. 8 TOF-MS spectra of the azo-Mn(II) complex obtained in step III of the conventional method and using the microfluidic device. | ||
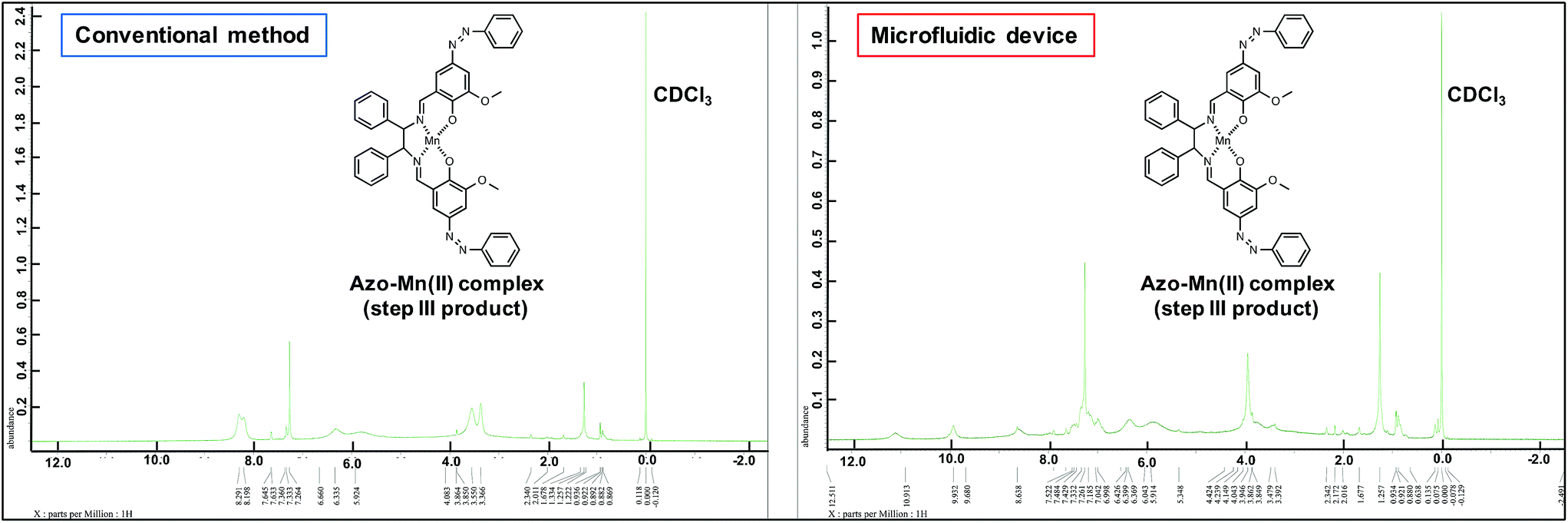 | ||
| Fig. 9 1H-NMR spectra of the azo-Mn(II) complex obtained in step III of the conventional method and using the microfluidic device. | ||
XRD measurement data could not be obtained. In the conventional method, a large amount of solvent can be gradually evaporated to promote crystal growth. Conversely, a small amount of solvent obtained using the microfluidic device method rapidly evaporated. For this reason, the product herein was obtained as a powder rather than as a crystal, and the XRD spectra could be adequately obtained.
Microfluidic devices have been suggested to lead to high reaction rates. For steps II and III, the microfluidic device was operated at room temperature and the conventional method was conducted at 40 °C. In addition, the conventional method required a reaction time of 4 h, whereas the reaction in the microfluidic device completed in less than 1 s.
Furthermore, the synthesis using the conventional method needed to be performed under a N2 atmosphere, whereas it could be performed under an air atmosphere using the microfluidic device. Zhang and Cloud reported that oxygen could easily pass through a polydimethylsiloxane (PDMS) device.26 However, Tanaka et al. have also described the synthesis of Mn(II) and Co(II) salen complexes using microfluidic devices.24 In this study, the reaction time was too short for the reagents to react with oxygen because they flowed continuously into the microfluidic device. However, in the conventional method, the reaction mixture was stirred, and metal acetate was oxidised. Furthermore, PDMS is not affected by alcohol.27 The quantities that microfluidic devices can handle are small although this problem can be solved using multiple devices in parallel. In microfluidic devices, reactions occur rapidly and are isolated from the air. Therefore, syntheses, which cannot be achieved via conventional methods, such as simultaneous organic and complex reactions, can be achieved using microfluidic devices.
One drawback of the chemical synthesis using the microfluidic device was the formation of a byproduct (m/z: [331.83 + H]+) derived from the azo compound, as shown in Fig. 5. In addition, the π–π* band in Fig. 7b indicated that the ligands were mixed with the azo-Mn(II) complex. Furthermore, Fig. 9 indicated that unreacted materials were mixed with the desired products. However, this problem could be solved using a microdroplet merging device, and we will investigate the use of this device in our future work to prevent the inclusion of unreacted materials.
4. Conclusion
An azo-Mn(II) complex was synthesised with mild pH control using a highly reactive microfluidic device. We successfully synthesised azo compounds requiring temperature control and pH control using a simple Y-junction microfluidic device. In addition, the microfluidic device formed the azo-Mn(II) complex in less than 1 s at room temperature, and the synthesis could be performed under an air atmosphere. The microfluidic device is capable of chemical synthesis similar to the conventional method although it is better suited to handling valuable and dangerous reagents. In future work, we will synthesise azo-metal complexes containing lysozyme as azo-metal complexes have optical isomerisation properties that may allow us to achieve the goal of controlling fuel cells using light. It should be noted that the amount of product generated using the microfluidic device is small. We intend to solve this problem by fabricating an integrated device that should enable practical chemical synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by a Grant-in-Aid for the Scientific Basic Research (A) (No. 16H02349) from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT), and the authors would also like to thank the MEXT Nanotechnology Platform Support Project of Waseda University.References
- S. Y. Lim, K. H. Hong, D. I. Kim, H. Kwon and H. J. Kim, J. Am. Chem. Soc., 2014, 136, 7018–7025 CrossRef CAS PubMed.
- A. Grirrane, A. Corma and H. García, Science, 2008, 322, 1661–1664 CrossRef CAS PubMed.
- C. Zhang and N. Jiao, Angew. Chem., 2010, 122, 6310–6313 CrossRef.
- S. Okumura, C. H. Lin, Y. Takeda and S. Minakata, J. Org. Chem., 2013, 781, 2090–12105 Search PubMed.
- Y. Takeda, S. Okumura and S. Minakata, Angew. Chem., Int. Ed., 2012, 51, 7804–7808 CrossRef CAS PubMed.
- J. Belmar, M. Parra, C. Zuânäiga, C. Peârez, C. Munäoz, A. Omenat and J. Serrano, Liq. Cryst., 1998, 26, 389–396 CrossRef.
- K. Monir, M. Ghosh, S. Mishra, A. Majee and A. Hajra, Eur. J. Org. Chem., 2014, 1096–1102 CrossRef CAS.
- Y. Einaga, R. Mikami, T. Akitsu and G. Li, Thin Solid Films, 2005, 493, 230–236 CrossRef CAS.
- Y. Aritake, T. Takanashi, A. Yamazaki and T. Akitsu, Polyhedron, 2011, 30, 886–894 CrossRef CAS.
- C. Priest, J. Zhou, R. Sedev, J. Ralston, A. Aota, K. Mawatari and T. Kitamori, Int. J. Miner. Process., 2011, 98, 168–173 CrossRef CAS.
- T. Ito, K. Uchiyama, S. Ohya and T. Kitamori, Jpn. J. Appl. Phys., Part 1, 2001, 40, 5469–5473 CrossRef CAS.
- A. Smirnova, K. Shimura and A. Hibara, Anal. Sci., 2007, 23, 103–107 CrossRef PubMed.
- M. Brivio, W. Verboom and D. N. Reinhoudt, Lab Chip, 2006, 6, 329–344 RSC.
- A. J. deMello, Control and detection of chemical reactions in microfluidic systems, Nature, 2006, 442/27, 394–402 CrossRef PubMed.
- M. Neumann and K. Zeitler, Org. Lett., 2012, 14/11, 2658–2661 CrossRef PubMed.
- C. A. Serra and Z. Chang, Chem. Eng. Technol., 2008, 31/8, 1099–1115 CrossRef.
- E. K. Lumley, C. E. Dyer, N. Pamme and R. W. Boyle, Org. Lett., 2012, 14/22, 5724–5727 CrossRef PubMed.
- P. W. Miller, N. J. Long, A. J. de Mello, R. Vilar, J. Passchierc and A. Geec, Chem. Commun., 2006, 546–548 RSC.
- A. Gunther and K. F. Jensen, Lab Chip, 2006, 6, 1487–1503 RSC.
- M. Kanai, S. Ikeda, J. Tanaka, J. S. Go, H. Nakanishi and S. Shoji, Sens. Actuators, A, 2004, 111, 32–36 CrossRef CAS.
- D. H. Yoon, A. Jamshaid, J. Ito, A. Nakahara, D. Tanaka, T. Akitsu, T. Sekiguchi and S. Shoji, Lab Chip, 2014, 14, 3050–3055 RSC.
- D. Tanaka, D. H. Yoon, T. Sekiguchi, S. Shoji and T. Akitsu, ISMM 2014, Singapore, 2014, pp. 243–246 Search PubMed.
- D. Tanaka, Y. Murakoshi, E. Tsuda, Y. Mitsumoto, D. H. Yoon, T. Sekiguchi, T. Akitsu and S. Shoji, Transducers 2015, Alaska, 2015, pp. 243–246 Search PubMed.
- D. Tanaka, W. Kawakubo, E. Tsuda, Y. Mitsumoto, D. H. Yoon, T. Sekiguchi, T. Akitsu and S. Shoji, RSC Adv., 2016, 6, 81862–81868 RSC.
- Y. Mitsumoto, N. Sunaga and T. Akitsu, SciFed Journal of Chemical Research, 2017, 1(1) Search PubMed , 1–12.
- H. Zhang and A. Cloud, Global Advances in Materials and Process Engineering, Coatings and Sealants Section, Dallas, 2006 Search PubMed.
- A. Mata, A. J. Fleischman and S. Roy, Biomed. Microdevices, 2005, 7(4), 28 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |