
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and insecticidal assessment of some innovative heterocycles incorporating a thiadiazole moiety against the cotton leafworm, Spodoptera littoralis†
Ahmed A. Fadda *a,
M. Abd El Salamb,
Eman H. Tawfika,
E. M. Anwarb and
H. A. Etmana
*a,
M. Abd El Salamb,
Eman H. Tawfika,
E. M. Anwarb and
H. A. Etmana
aChemistry Department, Faculty of Science, Mansoura University, 35516 Mansoura, Egypt. E-mail: afadda50@yahoo.com
bPlant Protection Research Institute, ARC, Dokki, Giza, Egypt
First published on 15th August 2017
Abstract
New 2-cyano-N-(5-ethyl-1,3,4-thiadiazol-2-yl)acetamide (1) was utilized as a versatile precursor for the synthesis of various heterocycles, such as pyrrole, pyridine, coumarin, thiazole, pyrido[2′,3′:3,4]pyrazolo[5,1-c]triazine, triazolo[5,1-c]triazine, aminopyrazole, thiophene, 1,3-dithiolane, triazolo[1,5-a]pyrimidine and benzo[d]imidazole derivatives. The newly synthesized compounds were identified by IR, MS, 1H NMR, 13C NMR, DEPT, H–H COSY, HMBC, and HSQC. Representative compounds of the synthesized products were examined and estimated as insecticidal agents against the cotton leafworm, Spodoptera littoralis.
Introduction
In recent years, the development of heterocyclic agrochemicals has become a main trend in research on pesticides because of their flexible structure, low mammalian toxicity, and high activity.1 As an important class of heterocyclic compounds, 2,5-disubstituted 1,3,4-thiadiazoles are related with many types of biological properties, probably by virtue of the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–S– group, including acaricidal,1 insecticidal,2 herbicidal,3 antioxidant,4 antibacterial,5 antidepressant,6 antidiabetic,7 antifungal,8 anticonvulsant9 and anti-inflammatory effects.10 In particular, many substituted 1,3,4-thiadiazole derivatives, including Schiff base derivatives, are of significant interest because they possess anticancer activities.11 The thiadiazole moiety acts as a “hydrogen binding domain” and “two-electron donor system”. Thiadiazoles are easily capable of crossing cellular membranes owing to their mesoionic nature and their better liposolubility, attributed to the presence of a sulfur atom. 1,3,4-Thiadiazoles are mesoionic systems,12 i.e. they are polyheteroatomic systems and contain a five-membered heterocyclic ring associated with conjugated p and π electrons and discrete regions of positive and negative charges.
C–S– group, including acaricidal,1 insecticidal,2 herbicidal,3 antioxidant,4 antibacterial,5 antidepressant,6 antidiabetic,7 antifungal,8 anticonvulsant9 and anti-inflammatory effects.10 In particular, many substituted 1,3,4-thiadiazole derivatives, including Schiff base derivatives, are of significant interest because they possess anticancer activities.11 The thiadiazole moiety acts as a “hydrogen binding domain” and “two-electron donor system”. Thiadiazoles are easily capable of crossing cellular membranes owing to their mesoionic nature and their better liposolubility, attributed to the presence of a sulfur atom. 1,3,4-Thiadiazoles are mesoionic systems,12 i.e. they are polyheteroatomic systems and contain a five-membered heterocyclic ring associated with conjugated p and π electrons and discrete regions of positive and negative charges.
The synthetic approaches adopted to obtain the newly synthesized compounds depend on regioselective attack on the cyanoacetamido moiety of the precursor 1 by different reagents, which, in one or two steps, adds a highly functionalized substituent or heterocyclic ring to the molecule.13 Cyanoacetamides are polyfunctional derivatives possessing both electrophilic and nucleophilic aspects. These chemical aspects have been used to build up diverse heterocycles with diverse ring sizes,14–26 which have wide spectrums of biological activity.
Results and discussion
Chemistry
The synthetic procedures adopted to obtain the target compounds are depicted in Schemes 1–5. The new starting compound, 2-cyano-N-(5-ethyl-1,3,4-thiadiazol-2-yl)acetamide (1), was prepared in dry benzene by treatment of 2-amino-5-ethyl-1,3,4-thiadiazole with 3-(3,5-dimethyl-1H-pyrazol-1-yl)-3-oxopropanenitrile as the cyanoacetylation reagent according to the previously reported procedure27 (Scheme 1). The structure 1 was elucidated according to its spectral data. The IR spectrum showed absorption bands at 3449 cm−1 for the NH function, stretching absorption bands at 2749–2955 cm−1 for CH aliphatic, a sharp band at 2260 cm−1 for the cyano group and a strong sharp band at 1702 cm−1 for the amidic carbonyl function. Its 1H NMR spectrum (DMSO-d6) indicated the existence of a triplet signal at δH 1.29 ppm due to protons of the methyl group, a quartet signal at δH 3.01 ppm attributed to methylene protons and two singlet signals at δH 4.06 and δH 12.73 ppm ascribed to CH2 protons and NH proton. Its 13C NMR and DEPT spectra (DMSO-d6) indicated the presence of a methyl carbon at δC 14.19 ppm and two methylene carbons at δC 23.16 and δC 26.49 ppm in addition to a cyano carbon at δC 115.57 ppm, two quaternary carbons at δC 158.44 and δC 162.69 ppm, and a carbonyl carbon at δC 166.49 ppm. The MS exhibited a molecular ion peak (M+) at m/z 196 assigned to the molecular formula C7H8N4OS.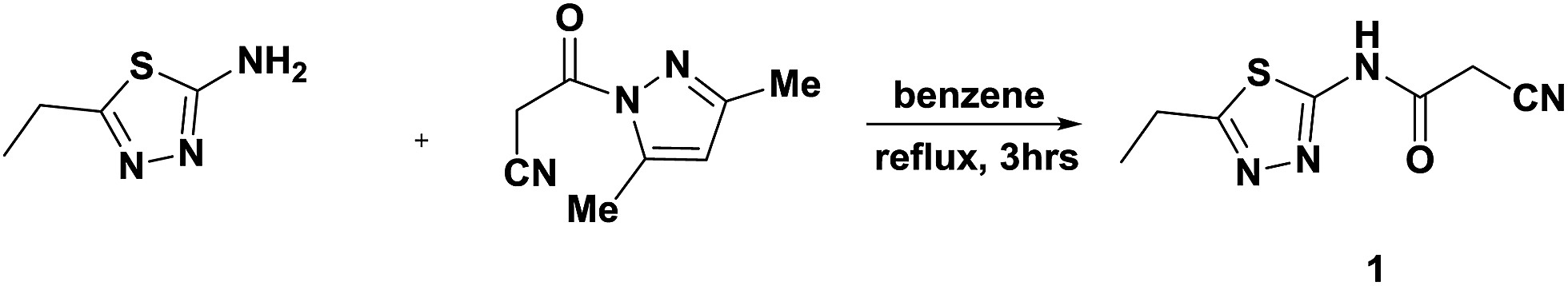 | ||
| Scheme 1 Synthesis of starting compound 1. | ||
Thus, cyclocondensation of compound 1 with 2-hydroxy-1-naphthaldehyde in hot ethanol containing piperidine as a basic catalyst furnished the coumarin derivative 2. The reaction of 1 with 1,3-dicarbonyl compounds was studied for the purpose of establishment of pyridine derivatives with effective biological activities. Therefore, it reacted with acetylacetone to afford the pyridine derivative 3. The previous product's structure was established according to its spectral data. The IR spectrum exhibited three absorption bands at 3447, 2221 and 1676 cm−1 due to OH, CN and CO function groups, respectively. The 1H NMR spectrum indicated the appearance of three new singlets at δH 2.11, 2.42 and 6.56 ppm attributed to two methyl protons and the pyridinone H-5. In addition, its 13C NMR spectrum indicated the presence of two new methyl carbons at δC 20.31 and δC 20.99 ppm and one aromatic carbon at δC 100.38 ppm. The structure of the pyridine derivative 3 was also established by 2D NMR, such as H–H COSY, HSQC and HMBC. The mass spectrum exhibited a molecular ion peak (M+) at m/z 260, which agreed with the molecular formula C12H12N4OS (Scheme 2).
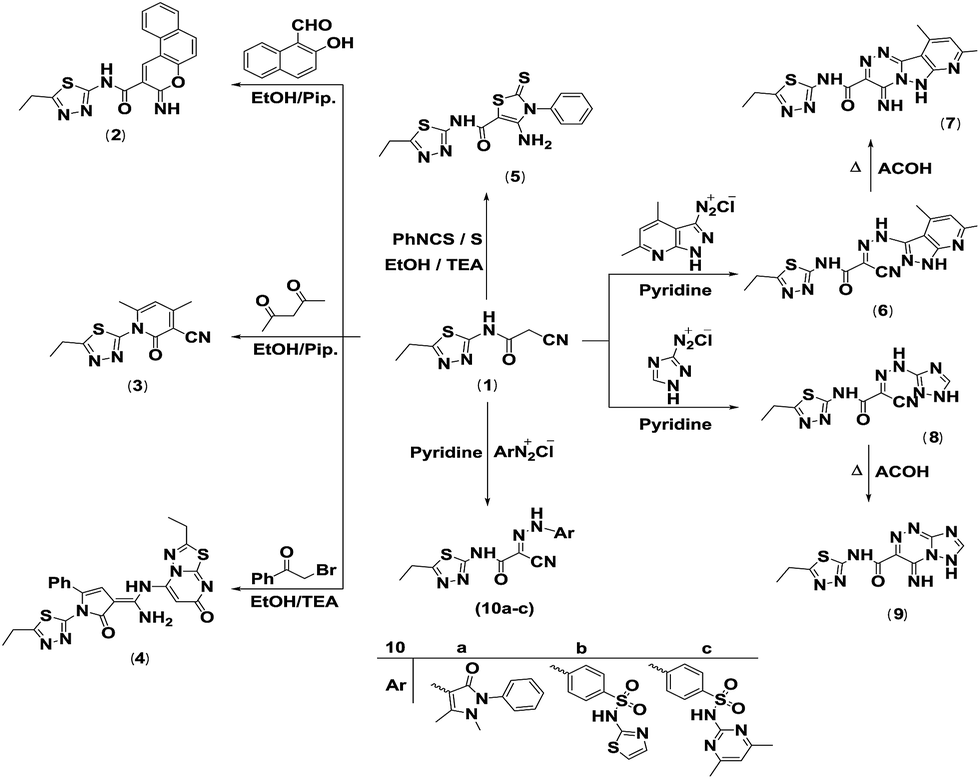 | ||
| Scheme 2 Synthesis of coumarin, pyridine, pyrrole, thiazole, triazolo[5,1-c]triazine pyrido[2′,3′:3,4]pyrazolo[5,1-c]triazine and azo compound derivatives. | ||
Recently, the reaction of α-halocarbonyl compounds with the cyanoacetamide moiety has been reported as a simple, new and effective synthetic route for the production of pyrrole derivatives.28 Hence, it was fascinating to study the reaction of 1 with phenacyl bromide. Cyclocondensation of 1 with phenacyl bromide in hot ethanol containing a catalytic amount of triethylamine furnished the pyrrole derivative 4. The proposed structure was in agreement with the analytical and spectral data. Therefore, the 1H NMR spectrum of the yielded product exhibited three singlet signals at δH 7.02, 7.06 and 7.91 ppm corresponding to the pyrrolone proton H-4, pyrimidinone H-3 and NH proton, respectively, in addition to a broad singlet signal at δH 8.63 ppm due to NH2 protons, two triplets at δH 1.26 and 1.41 ppm corresponding to two CH3, two quartets at δH 2.95 and 3.19 ppm assignable to two CH2, and a multiplet at δH 7.20–7.43 ppm assigned to aromatic protons. The IR spectrum revealed the presence of two CO groups stretching at 1666 and 1700 cm−1, an NH2 group at 3142 and 3331 cm−1 and an NH absorption band at 3408 cm−1. The mass spectrum displayed a molecular ion peak at m/z 492 ascribed to the molecular formula C22H20N8O2S2. Furthermore, Gewald reaction of compound 1 with both phenyl isothiocyanate and elemental sulfur in warm ethanol using triethylamine as a basic catalyst yielded the thiazole derivative 5. The proposed structure was in agreement with the elemental analysis, IR and MS (Scheme 2).
Recently, diazotized heterocyclic amines have been reported as a perfect building block for the synthesis of bridged-head nitrogen heterocyclic systems.29 Consequently, coupling of compound 1 with both 4,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-3-diazonium chloride and 1H-1,2,4-triazol-3-diazonium chloride30 in pyridine at 0–5 °C furnished the corresponding hydrazono compounds 6 and 8. When compounds 6 and 8 were heated under reflux in acetic acid, they cyclized to N-(5-ethyl-1,3,4-thiadiazol-2-yl)-4-imino-8,10-dimethyl-4,6-dihydropyrido[2′,3′:3,4]pyrazolo[5,1-c][1,2,4]triazine-3-carboxamide (7) and N-(5-ethyl-1,3,4-thiadiazol-2-yl)-4-imino-4,6-dihydro-[1,2,4]triazolo[5,1-c][1,2,4]triazine-3-carboxamide (9), respectively. The nucleophilic properties of the ring nitrogen enabled attack on the cyano group in order to synthesize compounds 7 and 9. The IR spectrum of 7 exhibited three absorption bands at 3449, 3433, and 3406 cm−1 due to three NH groups besides one carbonyl absorption band at 1646 cm−1. The 1H NMR spectrum displayed two D2O-exchangeable singlets at δH 12.33 and 12.82 ppm due to two NH protons, and additionally three singlets at δH 2.47, 2.79 and 6.98 ppm were characterized for two methyl protons of the pyridine ring and one aromatic proton of the pyridine ring, respectively. In addition, a triplet and a quartet were found at δH 1.31 and 3.02 ppm, characteristic of the side chain ethyl group. Its mass spectrum indicated a molecular ion peak at m/z 369 (M+), which agreed with its molecular formula of C15H15N9OS. Compound 9's structure was confirmed on the basis of spectral data. Its IR spectrum exhibited the lack of an absorption band attributed to the cyano function. Three NH absorption bands appeared at 3128, 3331 and 3440 cm−1, while a strong absorption appeared at 1661 cm−1 for the amidic carbonyl group. The 1H NMR spectrum (DMSO-d6) indicated the absence of a singlet signal assignable to methylene protons, whereas a singlet signal was found at δH 8.58 ppm due to the aromatic proton of the triazole ring, as well as two singlets at δH 11.84 and 14.37 ppm characteristic of NHCO and NH protons, while the side chain CH2CH3 protons appeared at δH 1.30 ppm as a triplet and at δH 3.02 ppm as a quartet. Moreover, the mass spectrum for the triazolo[5,1-c]triazine structure 9 displayed a molecular ion peak (M+) at m/z 291 corresponding to the molecular formula C9H9N9OS (Scheme 2).
Due to its highly biological activity, next, the reactivity of the active methylene group existing in compound 1 towards various diazonium salts was also studied. Thus, diazocoupling reaction of compound 1 with antipyrine diazonium chloride, sulfathiazole diazonium chloride and sulfamethazine diazonium chloride in pyridine at 0–5 °C afforded the hydrazone derivatives 10a–c. IR, 1H NMR and MS results were in agreement with the proposed structures. Thus, the IR spectrum of 10b exhibited an absorption band at 2231 cm−1 attributed to the cyano function, while absorption bands at 3130, 3226 and 3469 cm−1 were ascribed to three NH groups, in addition to a strong absorption band at 1665 cm−1 for the carbonyl group. Its 1H NMR spectrum (DMSO-d6) provided two olefinic protons, H-5 and H-4, of the thiazole ring, which appeared as doublets at δH 6.84 and 7.26 ppm that were coupled to each other with a coupling constant J = 4 Hz, while the aromatic protons appeared at δH 7.80 and 7.97 ppm as doublets in an AA′XX′ system, J = 8 Hz, in addition to three singlet signals at δH 12.37, 12.38 and 12.81 ppm ascribed to N–NH, NHSO2 and NHCO, respectively. Moreover, the methyl protons were appeared as triplet signals at δH 1.30 ppm, while the methylene protons appeared as quartet signal at δH 3.01 ppm. Moreover, the mass spectrum of compound 10b displayed a molecular ion peak at m/z 462 ascribed to the molecular formula C16H14N8O3S3 (Scheme 2).
The reactivity of the methylene group in the cyanoacetamide derivative 1 towards isothiocyanate was examined. Thus, treatment of 1 with phenyl isothiocyanate in DMF including potassium hydroxide at room temperature furnished the non-isolable thiocarbamoyl salt 11, which underwent heterocyclization upon reaction with α-halocarbonyl compounds chloroacetyl chloride, chloroacetone and phenacyl chloride to give the corresponding thiazole derivatives 12, 13a and 13b, respectively (Scheme 3). The structure of 12 was characterized by the appearance of a singlet signal equivalent to two protons at δH 4.02 ppm in the 1H NMR spectrum, which represent the C5 protons of the thiazolidinone moiety. In addition, a multiplet signal equivalent to five protons at δH 7.38–7.52 ppm was ascribed to the aromatic protons. The IR spectrum revealed the presence of a new absorption band at 1747 cm−1 due to the carbonyl group at C4 of the thiazole ring. According to the MS of compound 12, the m/z was 371 corresponding to its molecular formula C16H13N5O2S2. The IR spectrum of 13a exhibited stretching frequencies at 3486, 2184 and 1626 cm−1 for the NH, CN and CO frequencies, respectively. The 1H NMR spectrum exhibited two singlet signals at δH 1.88 and 7.01 ppm due to the CH3 and olefinic protons of the thiazole ring, a multiplet signal at δH 7.48–7.60 ppm related to aromatic protons, in addition to a triplet at δH 1.28 ppm and a quartet at δH 2.91 ppm assignable to the side chain ethyl group. The 13C NMR spectrum was assigned by signals at δC 13.11 and 13.98 ppm for the two methyl carbons, a signal at δC 22.71 ppm due to the methylene carbon, a signal at δC 106.89 ppm related to the methine carbon, a signal at δC 115.16 ascribed to cyano carbon, and signals at δC 128.73–130.51 ppm assigned to aromatic carbons. The structure of the thiazole derivative 13a was also established by DEPT 13C NMR and 2D NMR, such as H–H COSY, HSQC and HMBC. Its mass spectrum displayed a molecular ion peak (M+) at m/z 369, which agreed with the molecular formula C17H15N5OS2. The IR spectrum of 13b indicated an absorption band at 3446 cm−1 for the NH group, a sharp band at 2187 cm−1 for the cyano function and a strong sharp band at 1626 cm−1 for the carbonyl function. The 1H NMR spectrum (DMSO-d6) revealed the presence of two singlet signals at δH 7.19 and 14.49 ppm assignable to the olefinic proton of the thiazole ring and the NH proton, and a multiplet signal at δH 7.21–7.61 ppm assigned to aromatic protons, in addition of triplet and quartet signals at δH 1.30 and 2.96 ppm due to the ethyl group. The mass spectrum exhibited a molecular ion peak at m/z 431 attributed to the molecular formula C22H17N5OS2.
 | ||
| Scheme 3 Reaction of compound 1 with phenyl isothiocyanate. | ||
 | ||
| Scheme 4 Synthesis of arylidene, aminopyrazole and 2-pyridone derivatives. | ||
Moreover, the reaction of the intermediate 11 with chloroacetonitrile furnished (E)-2-(4-amino-3-phenylthiazol-2(3H)-ylidene)-2-cyano-N-(5-ethyl-1,3,4-thiadiazol-2-yl)acetamide (14). IR and MS results are compatible with the proposed structure (Scheme 3).
Treatment of the thiocarbamoyl salt intermediate 11 with dimethyl sulfate furnished the novel ketene N,S-acetal 15. The structure of 15 was identified by spectral data. The IR spectrum displayed absorption bands at 3448, 3313 and 2227 cm−1 due to two NH groups and the nitrile function, respectively, in addition to a carbonyl absorption band at 1638 cm−1. Its mass spectrum exhibited a molecular ion peak at m/z 345 (M+), which agrees with its molecular formula C15H15N5OS2. Its 1H NMR spectrum displayed three singlet signals at δH 2.85, 9.80 and 9.83 ppm assignable to SCH3, NHPh and NHCO, respectively, a multiplet signal at δH 7.10–7.49 ppm related to the aromatic protons, and a triplet signal at δH 1.30 ppm and a quartet signal at δH 3.04 ppm corresponding to CH2CH3 protons. The 13C NMR spectrum was identified by signals at δC 11.87 and 17.45 ppm characterized to the two methyl carbons, a signal at δC 24.38 ppm ascribed to the methylene carbon, a signal at δC 114.58 ppm assigned to the cyano carbon, signals at δC 123.6–128.41 ppm assigned to aromatic carbons, and a signal at δC 163.68 ppm attributed to the amidic carbonyl carbon atom. The structure of the ketene N,S-acetal 15 was also characterized by DEPT 13C NMR and 2D NMR, such as H–H COSY, HSQC and HMBC (Scheme 3).
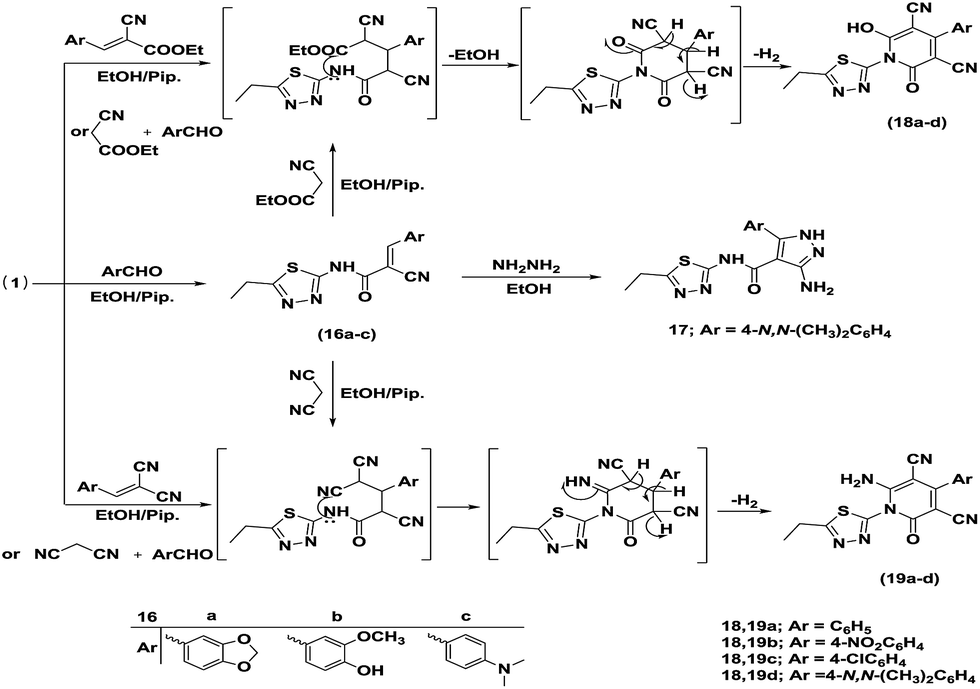
Next, the synthetic potency and applicability of cyanoacetamide derivative 1 were investigated to develop a facile and convenient route to some novel pyrazole and pyridine derivatives with an anticipated wide spectrum of bioresponses.31,32 Thus, the Knoevenagel condensation of 1 with aromatic aldehydes, namely piperonal, vanilline, and 4-N,N-dimethylbenzaldehyde, in refluxing ethanol using piperidine as a basic catalyst afforded the corresponding arylidene derivatives 16a–c. Treatment of 16c with hydrazine hydrate in boiling ethanol yielded 3-amino-5-(4-(dimethylamino)phenyl)-N-(5-ethyl-1,3,4-thiadiazol-2-yl)-1H-pyrazole-4-carboxamide (17). Michael addition of hydrazine hydrate to α,β-unsaturated nitrile 16c led to the formation of compound 17 via in situ intramolecular 1,5-dipolar cyclization through the nucleophilic addition of the amino group to the cyano function to afford dihydropyrazole, which underwent auto oxidation to furnish the target pyrazole. The assignment of structures 16a–c and 17 was supported by spectral data. The 1H NMR (DMSO-d6) spectra of structures 16a–c exhibited, in general, a singlet signal at δH 8.082–8.353 ppm attributable to vinylic protons. Furthermore, the IR spectrum of compound 17 revealed the absence of a cyano function and instead, the appearance of a new absorption band at 3280 and 3152 cm−1 assigned to an NH2 group.
One-pot reactions of the cyanoacetamide derivative 1 with ethyl cyanoacetate and different aromatic aldehydes, namely benzaldehyde, p-nitrobenzaldehyde, p-chlorobenzaldehyde and 4-N,N-dimethylbenzaldehyde (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 molar ratio), in refluxing ethanol containing a catalytic amount of piperidine yielded the 2-pyridone derivatives 18a–d, respectively. On the other hand, the 2-pyridone derivatives 18a–d, were also acquired via reaction of cyanoacetamide 1 with arylidene ethyl cyanoacetate in hot ethanol under reflux containing piperidine as the catalyst. In addition, pyridin-2-ones 18d and 19d were also obtained via the reaction of the arylidene derivative 16c with ethyl cyanoacetate and malononitrile, respectively, in ethanol in the presence of piperidine as the catalyst. Another route for the synthesis of 2-pyridone derivatives was the one-pot reaction of the cyanoacetamide derivative 1 with malononitrile and the same mentioned aromatic aldehydes (1:1:1 molar ratio) followed by refluxing in ethanol containing a few drops of piperidine to furnished 19a–d. Moreover, when arylidene malononitrile was refluxed with the cyanoacetamide derivative 1 in ethanol in the presence of piperidine, it afforded 2-pyridone derivatives 19a–d. The structures 18a–d and 19a–d were confirmed on the basis of spectral data.
:1:1 molar ratio), in refluxing ethanol containing a catalytic amount of piperidine yielded the 2-pyridone derivatives 18a–d, respectively. On the other hand, the 2-pyridone derivatives 18a–d, were also acquired via reaction of cyanoacetamide 1 with arylidene ethyl cyanoacetate in hot ethanol under reflux containing piperidine as the catalyst. In addition, pyridin-2-ones 18d and 19d were also obtained via the reaction of the arylidene derivative 16c with ethyl cyanoacetate and malononitrile, respectively, in ethanol in the presence of piperidine as the catalyst. Another route for the synthesis of 2-pyridone derivatives was the one-pot reaction of the cyanoacetamide derivative 1 with malononitrile and the same mentioned aromatic aldehydes (1:1:1 molar ratio) followed by refluxing in ethanol containing a few drops of piperidine to furnished 19a–d. Moreover, when arylidene malononitrile was refluxed with the cyanoacetamide derivative 1 in ethanol in the presence of piperidine, it afforded 2-pyridone derivatives 19a–d. The structures 18a–d and 19a–d were confirmed on the basis of spectral data.
Treatment of compound 1 with carbon disulfide in DMF, containing potassium hydroxide, at room temperature afforded the intermediate enaminonitrile 20, which underwent heterocyclization upon treatment with α-halocarbonyl compounds, such as phenacyl bromide, chloroacetyl chloride, to give the corresponding ketene S,S-dithiolane derivatives or thiophene derivatives (Scheme 5). Thus, the in situ stirring reaction of the non-isolable intermediate 20 with phenacyl bromide or chloroacetyl chloride in the presence of a protic solvent such as ethanol afforded the 1,3-dithiolane derivatives 21 and 23, respectively. On the other hand, when the intermediate enaminonitrile 20 was refluxed with phenacyl bromide in DMF only as an aprotic solvent and in the presence of a catalytic amount of triethylamine, a thiophene derivative 22 was obtained. The spectral data of the isolated products was in complete agreement with structures 21, 22 and 23. The IR spectrum of compound 22 indicated the lack of an absorption band assigned to a conjugated C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N function and showed absorption bands at 3413, 3345, 3296, 2622, 1718 and 1645 cm−1 assignable to NH, NH2, SH, PhCO, and amidic CO functions, respectively. The 1H NMR spectrum (DMSO-d6) displayed a broad singlet signal at δH 8.35 ppm assignable to NH2, and a multiplet signal in the δH 7.43–7.60 ppm region that is distinctive for aromatic protons, besides triplet and quartet signals at δH 1.31 and 2.95 ppm, respectively, corresponding to the side chain ethyl group. Its mass spectrum exhibited a molecular ion peak at m/z 390, corresponding to molecular formula C16H14N4O2S3. The 13C NMR spectrum was characterized by a signal at δC 12.37 ppm ascribed to the methyl carbons, a signal at δC 23.29 ppm ascribed to methylene carbon, a signal at δC 126.83–130.71 ppm attributed to aromatic carbons, and signals at δC 167.14 and 185.86 ppm corresponding to the two carbonyl carbon atoms. The structure of the thiophene derivative 22 was also characterized by DEPT 13C NMR and 2D NMR, such as H–H COSY, HSQC and HMBC.
N function and showed absorption bands at 3413, 3345, 3296, 2622, 1718 and 1645 cm−1 assignable to NH, NH2, SH, PhCO, and amidic CO functions, respectively. The 1H NMR spectrum (DMSO-d6) displayed a broad singlet signal at δH 8.35 ppm assignable to NH2, and a multiplet signal in the δH 7.43–7.60 ppm region that is distinctive for aromatic protons, besides triplet and quartet signals at δH 1.31 and 2.95 ppm, respectively, corresponding to the side chain ethyl group. Its mass spectrum exhibited a molecular ion peak at m/z 390, corresponding to molecular formula C16H14N4O2S3. The 13C NMR spectrum was characterized by a signal at δC 12.37 ppm ascribed to the methyl carbons, a signal at δC 23.29 ppm ascribed to methylene carbon, a signal at δC 126.83–130.71 ppm attributed to aromatic carbons, and signals at δC 167.14 and 185.86 ppm corresponding to the two carbonyl carbon atoms. The structure of the thiophene derivative 22 was also characterized by DEPT 13C NMR and 2D NMR, such as H–H COSY, HSQC and HMBC.
 | ||
| Scheme 5 Reaction of compound 1 with carbon disulfide. | ||
The ketene S,S-dithioacetal 24 was prepared by reaction of 1 with carbon disulfide in the presence of potassium hydroxide in DMF, followed by alkylation with dimethyl sulfate (Scheme 5). The structure of 24 was established on the basis of the spectral data (1H NMR, 13C NMR, DEPT 13C NMR and 2D NMR, such as H–H COSY, HSQC and HMBC).
Polarized cyanoketene S,S-acetals are utilized as a key intermediate for the synthesis of a wide-ranging variety of fused heterocycles. Thus, further reaction of 24 with 3-amino-1H-1,2,4-triazole in refluxing pyridine afforded triazolo[1,5-a]pyrimidine 25. Compound 25 was also elucidated by the spectral data. Moreover, compound 24 was also used as a versatile starting material for the synthesis of fused heterocyclic compounds by treatment with bifunctional nucleophilic reagents. Thus, heating of 24 with o-phenylenediamine in pyridine afforded benzo[d]imidazole derivative 26. The IR spectrum of 26 exhibited absorption bands at 3451, 3303, and 3130 cm−1 for three NH stretching modes, a band at 2219 cm−1 for the cyano function, and a strong absorption band for the amidic carbonyl group at 1679 cm−1. The 1H NMR spectrum revealed no signal for SCH3 protons, while a multiplet signal at δH 7.47–7.64 ppm was assigned to aromatic protons, two singlet signals at δH 7.93 ppm and δH 8.35 ppm appeared for 2NH and NHCO protons, respectively, beside a triplet at δH 1.35 ppm attributed to CH3 protons, and a quartet at δH 3.06 ppm assigned to CH2 protons. Its mass spectrum exhibited a molecular ion peak at m/z 312, attributed to the molecular formula C14H12N6OS (Scheme 5).
Insecticidal activity
| Tested compounds | LC50 (ppm) and confidence limits at 95% | LC90 (ppm) and confidence limits at 95% | Slope | Toxicity index % at LC50 value |
|---|---|---|---|---|
| a Toxicity index is defined as the ratio of the most effective compound's LC50 value to the other tested compound's LC50 value multiplied by 100. | ||||
| 10b | 64.12, 47.12, 87.01 | 259.07, 163.95, 666.16 | 2.113 ± 0.417 | 100 |
| 10c | 69.17, 53.80, 87.48 | 225.01, 164.53, 366.87 | 2.502 ± 0.351 | 92.69 |
| 7 | 75.51, 59.44, 95.05 | 235.22, 173.25 377.15 | 2.597 ± 0.355 | 84.91 |
| 10a | 91.45, 67.78, 116.76 | 288.78, 210.83 493.24 | 2.566 ± 0.429 | 70.11 |
| 9 | 101.30, 76.88, 131.68 | 422.79, 295.71, 725.63 | 2.065 ± 0.267 | 63.29 |
| 5 | 115.48, 84.07, 150.36 | 454.49, 323.96, 774.83 | 2.154 ± 0.317 | 55.52 |
| 18c | 235.41, 178.96, 309.27 | 993.61, 673.32, 1853.32 | 2.049 ± 0.288 | 27.23 |
| 18b | 240.91, 181.73, 311.86 | 1279.72, 870.94, 2322.95 | 1.767 ± 0.231 | 26.61 |
| 17 | 262.31, 194.03, 334.36 | 973.84, 719.65, 1548.13 | 2.250 ± 0.319 | 24.44 |
| 19c | 290.72, 215.83, 371.94 | 1114.06, 806.06, 1860.78 | 2.197 ± 0.322 | 22.05 |
| 22 | 294.26, 231.53, 369.04 | 1026.68, 762.93, 1571.35 | 2.362 ± 0.279 | 21.79 |
| 12 | 298.94, 224.51, 380.06 | 1098.63, 802.36, 1798.5 | 2.267 ± 0.326 | 21.44 |
| 16a | 321.34, 242.23, 408.97 | 1206.54, 872.60, 2018.38 | 2.231 ± 0.323 | 19.95 |
| 4 | 334.04, 256.08, 416.27 | 1181.66, 904.50, 1731.51 | 2.336 ± 0.291 | 19.19 |
| 19d | 344.93, 264.73, 439.91 | 1619.94, 1158.03, 2627.91 | 1.908 ± 0.222 | 18.58 |
| 18d | 349.27, 268.69, 434.98 | 1221.88, 931.20, 1810.47 | 2.357 ± 0.298 | 18.35 |
| 25 | 375.58, 294.25, 470.43 | 1504.17, 1112.55, 2304.69 | 2.127 ± 0.241 | 17.07 |
| 18a | 381.33, 296.33, 472.43 | 1310.07, 999.64, 1941.12 | 2.391 ± 0.302 | 16.81 |
| 19b | 387.34, 294.35, 485.22 | 1494.55, 1129.37, 2243.94 | 2.186 ± 0.275 | 16.55 |
| 13b | 389.93, 308.90, 482.03 | 1130.61, 855.53, 1745.80 | 2.772 ± 0.391 | 16.44 |
| 14 | 395.26, 300.87, 497.92 | 1584.55, 1162.05, 2541.15 | 2.125 ± 0.284 | 16.22 |
| 19a | 428.20, 330.22, 531.29 | 1656.80, 1261.38, 2451.76 | 2.181 ± 0.266 | 14.97 |
| 26 | 448.32, 346.01, 556.57 | 1774.75, 1340.77, 2669.08 | 2.145 ± 0.264 | 14.30 |
| 3 | 476.84, 370.61, 601.48 | 2359.00, 1688.68, 3822.99 | 1.846 ± 0.211 | 13.44 |
| 2 | 481.22, 383.88, 582.89 | 1538.40, 1215.39, 2137.24 | 2.539 ± 0.295 | 13.32 |
| 16c | 484.28, 377.30, 598.21 | 1877.49, 1418.89, 2829.16 | 2.178 ± 0.269 | 13.24 |
| 23 | 484.93, 378.15, 598.60 | 1868.67, 1414.22, 2808.55 | 2.188 ± 0.269 | 13.22 |
| 21 | 492.62, 381.52, 611.87 | 2020.45, 1503.41, 3137.98 | 2.091 ± 0.264 | 13.01 |
| 15 | 521.67, 412.29, 638.29 | 1889.44, 1445.06, 2792.62 | 2.293 ± 0.280 | 12.29 |
| 13a | 550.83, 436.27, 682.24 | 1990.66, 1462.13, 3221.37 | 2.297 ± 0.309 | 11.64 |
| 24 | 564.99, 457.82, 683.14 | 1690.21, 1306.90, 2486.15 | 2.693 ± 0.347 | 11.34 |
| 1 | 627.94, 513.81, 747.33 | 1831.85, 1459.39, 2542.52 | 2.756 ± 0.333 | 10.21 |
Experimental
Instruments
All melting points were recorded using a Gallenkamp melting point apparatus in degrees centigrade and are uncorrected. The IR spectra (KBr) were determined on a Mattson 5000 FTIR spectrophotometer at the Faculty of Science, Mansoura University. NMR spectra (1H, 13C, DEPT, H–H COSY, HSQC and HMBC) were acquired using a Bruker WP 400 MHz, 500 MHz, 300 MHz and 125 MHz at the Faculty of Pharmacy, Beni-Suef University with DMSO-d6 as the solvent, utilizing TMS as an internal standard reference, and chemical shifts are expressed as δ ppm. Mass spectra were measured using a Finnegan MAT 212 instrument at the Faculty of Science, Cairo University, and the Regional Center for Mycology & Biotechnology, Al-Azhar University. Elemental analyses were performed at the Microanalytical Unit, Faculty of Science, Cairo University. TLC (silica gel, aluminium sheets 60 F254, Merck) was carried out after all the reactions.Synthesis of 2-cyano-N-(5-ethyl-1,3,4-thiadiazol-2-yl)acetamide (1)
A solution of 2-amino-5-ethyl-1,3,4-thiadiazole (5.16 g, 0.04 mol) in dry benzene (30 mL) was added to a solution of 3-(3,5-dimethyl-1H-pyrazol-1-yl)-3-oxopropanenitrile (6.52 g, 0.04 mol) in the same solvent (15 mL) and the reaction mixture was refluxed for 3 h. After cooling, the solid precipitate was isolated by filtration and purified via recrystallization from ethanol to afford 1. White crystals; mp 230–232 °C; yield 90%; IR (KBr) ν/cm−1: 3449 (NH), 2260 (CN), 1702 (CO); 1H NMR (400 MHz, DMSO-d6): δH ppm 1.29 (t, 3H, CH3), 3.01 (q, 2H, CH2), 4.06 (s, 2H, CH2), 12.73 (s, 1H, NHCO). 13C NMR (400 MHz, DMSO-d6): δC ppm 14.19, 23.16, 26.49, 115.57, 158.44, 162.7, 166.49. MS m/z (%): 197 (M+ + 1, 83.33), 196 (M+, 69.23), 181 (89.74), 155 (94.87), 139 (100), 110 (94.87), 71 (93.59), 61 (91.03). Anal. for C7H8N4OS (196.23): calcd: C, 42.85; H, 4.11; N, 28.55%; found: C, 42.92; H, 4.05; N, 28.43%.Synthesis of N-(5-ethyl-1,3,4-thiadiazol-2-yl)-3-imino-3H-benzo[f]chromene-2-carboxamide (2)
To a solution of compound 1 (0.4 g, 0.002 mol) in absolute ethanol (25 mL) in the presence of piperidine (0.5 mL), 2-hydroxy-1-naphthaldehyde (0.35 g, 0.002 mol) was added. The reaction mixture was refluxed for 3 h and then left to cool. The formed precipitate was filtered off, purified by washing with ethanol, and then dried and recrystallized from ethanol to furnish 2. Pale yellow crystals; mp 253–255 °C; yield 85%; IR (KBr) ν/cm−1: 3435, 3322 (2NH), 1733 (CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.20 (t, 3H, CH3), 2.80 (q, 2H, CH2), 7.65–8.66 (m, 6H, Ar-H), 9.81 (s, 1H, CH). 13C NMR (400 MHz, DMSO-d6): δC ppm 13.8, 23.11, 100.79, 112.33, 115.02, 116.75, 122.55, 126.86, 128.79, 129.07, 129.38, 129.98, 137.17, 149.6, 155.03, 157.20, 159.65, 168.08. Also H–H COSY, HSQC and HMBC proved the structure. MS m/z (%): 350 (M+, 12.13), 288 (57.85), 274 (42.21), 194 (15.78), 182 (51.66), 169 (47.38), 152 (47.8), 120 (39.18), 97 (57.97), 71, (50.95), 64 (59.97), 51 (100). Anal. for C18H14N4O2S (350.4): calcd: C, 61.70; H, 4.03; N, 15.99%; found: C, 61.55; H, 3.96; N, 16.05%.
Synthesis of 1-(5-ethyl-1,3,4-thiadiazol-2-yl)-4,6-dimethyl-2-oxo-1,2-dihydropyridine-3-carbonitrile (3)
An equimolar mixture of compound 1 (0.4 g, 0.002 mol) and acetylacetone (0.21 mL, 0.002 mol) in absolute ethanol (15 mL) containing three drops of piperidine was heated under reflux for 3 h. The reaction mixture was allowed to cool and the precipitate obtained was isolated by filtration and purified through recrystallization from ethanol to afford 3. Colorless crystals; mp 175–177 °C; yield 89%; IR (KBr) ν/cm−1: 3447 (OH), 2221 (CN), 1676 (CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.37 (t, 3H, CH3), 2.11 (s, 3H, CH3), 2.42 (s, 3H, CH3), 3.19 (q, 2H, CH2), 6.56 (s, 1H, pyridine H-5). 13C NMR (400 MHz, DMSO-d6): δC ppm 13.54, 20.31, 20.99, 23.76, 100.38, 110.02, 114.97, 151.26, 158.35, 160.08, 161.93, 176.27. The H–H COSY, HSQC and HMBC results are in agreement with the proposed structure. MS m/z (%): 261 (M+ + 1, 12.78), 260 (M+, 14.02), 205 (80.89), 178 (6.97), 147 (100), 119 (14.53), 104 (6.63), 77 (10), 51 (2.45). Anal. for C12H12N4OS (260.32): calcd: C, 55.37; H, 4.65; N, 21.52%; found: C, 55.25; H, 4.55; N, 21.38%.Synthesis of (E)-5-((amino(1-(5-ethyl-1,3,4-thiadiazol-2-yl)-2-oxo-5-phenyl-1,2-dihydro-3H-pyrrol-3-ylidene)methyl)amino)-2-ethyl-7H-[1,3,4]thiadiazolo[3,2-a]pyrimidin-7-one (4)
Equimolar amounts of compound 1 (0.4 g, 0.002 mol) and phenacyl bromide (0.404 g, 0.002 mol) in absolute ethanol (25 mL) containing three drops of triethylamine was refluxed for 3 h. The solid product that formed was filtered off, purified and recrystallized from dry ethanol to furnish compound 4. Pale yellow powder; mp > 300 °C; yield 80%; IR (KBr) ν/cm−1: 3408 (NH), 3331, 3142 (NH2), 1700, 1666 (2CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.26 (t, 3H, CH3), 1.41 (t, 3H, CH3), 2.95 (q, 2H, CH2), 3.19 (q, 2H, CH2), 7.02 (s, 1H, pyrimidine H-5), 7.06 (s, 1H, pyrrole H-4), 7.2–7.43 (m, 5H, Ar-H). MS m/z (%): 492 (M+, 31.92), 368 (100), 313 (39.01), 299 (33.81), 262 (46.71), 239 (80.47), 186 (30.46), 179 (15.68), 123 (22.79). Anal. for C22H20N8O2S2 (492.58): calcd: C, 53.64; H, 4.09; N, 22.75%; found: C, 53.55; H, 4.01; N, 22.80%.Synthesis of 4-amino-N-(5-ethyl-1,3,4-thiadiazol-2-yl)-3-phenyl-2-thioxo-2,3-dihydrothiazole-5-carboxamide (5)
To an ethanolic solution (25 mL) of compound 1 (0.4 g, 0.002 mol) containing three drops of triethylamine, elemental sulfur (0.064 g, 0.002 mol) and phenyl isothiocyanate (0.23 mL, 0.002 mol) were added. The reaction mixture was continuously stirred at 60 °C for 3 h, and then poured into a beaker containing a crushed ice/water mixture acidified by a few drops of hydrochloric acid. The solid precipitate that formed was isolated by filtration, dried, and recrystallized from a mixture of DMF and ethanol (3:1) to afford compound 5. Brown powder; mp 248–250 °C; yield 75%; IR (KBr) ν/cm−1: 3446 (NH), 3373, 3244 (NH2), 1640 (CO), 1232 (CS). MS m/z (%): 364 (M+ + 1, 3.51), 363 (M+, 17.68), 330 (15.04), 298 (16.81), 266 (16.1), 235 (22.88), 208 (51), 136 (53.05), 135 (82.06), 129 (82.36), 93 (31.97), 77 (100), 74 (43.85), 60 (26.7). Anal. for C14H13N5OS3 (363.47): calcd: C, 46.26; H, 3.61; N, 19.27%; found: C, 46.22; H, 3.45; N, 19.21%.
General procedure for coupling reaction of 1 with different primary aromatic amine diazonium salts
To a cold (0–5 °C) solution of compound 1 (0.4 g, 0.002 mol) in pyridine (20 mL) was added the appropriate diazonium chloride [which was prepared by dissolving sodium nitrite (0.14 g, 0.002 mol) in cold water (3 mL) and adding to a cold solution of the appropriate aromatic amine (0.002 mol) containing an adequate amount of hydrochloric acid (1.5 mL) under continuous stirring conditions] portion-wise over a period of 25 min. The reaction mixture was kept overnight in the refrigerator, and then diluted with water. The formed solid that precipitated was filtered off, purified by washing in water, then dried and recrystallized from EtOH and DMF (2:1) to afford arylazo derivatives 6, 8 and 10a–c.
Synthesis of (E)-N-(4,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-3-yl)-2-((5-ethyl-1,3,4-thiadiazol-2-yl)amino)-2-oxoacetohydrazonoyl cyanide (6)
Black powder; mp 280–282 °C; yield 85%; IR (KBr) ν/cm−1: 3449 (NH), 2197 (CN), 1646 (CO). MS m/z (%): 371 (M+ + 2, 5.96), 370 (M+ + 1, 21.87), 369 (M+, 100), 336 (16.7), 281 (25.17), 271 (16.77), 241 (18.99), 162 (33.41), 146 (36.88), 119 (28.06), 104 (16.43), 78 (33.58). Anal. for C15H15N9OS (369.41): calcd: C, 48.77; H, 4.09; N, 34.13%; found: C, 48.32; H, 4.01; N, 34.03%.Synthesis of N-(5-ethyl-1,3,4-thiadiazol-2-yl)-4-imino-8,10-dimethyl-4,6-dihydropyrido[2′,3′:3,4]pyrazolo[5,1-c][1,2,4]triazine-3-carboxamide (7)
A solution of 6 (0.74 g, 0.002 mol) in glacial acetic acid (25 mL) was refluxed for 3 h, and then allowed to cool. The formed precipitate was filtered off, purified by washing with ethanol and recrystallized from a mixture of EtOH–DMF (1:1) to furnish compound 7. Reddish brown powder; mp > 300 °C; yield 93%; IR (KBr) ν/cm−1: 3449, 3433, 3406 (3NH), 1646 (CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.31 (t, 3H, CH3), 2.47 (s, 3H, CH3-pyridine), 2.79 (s, 3H, CH3-pyridine), 3.02 (q, 2H, CH2), 6.98 (s, 1H, pyridine H-3), 12.33 (s, 1H, NHCO), 12.82 (s, 1H, NH). Anal. for C15H15N9OS (369.41): calcd: C, 48.77; H, 4.09; N, 34.13%; found: C, 48.30; H, 3.98; N, 34.01%.
Synthesis of (E)-2-((5-ethyl-1,3,4-thiadiazol-2-yl)amino)-2-oxo-N-(1H-1,2,4-triazol-3-yl)acetohydrazonoyl cyanide (8)
Pale yellow crystals; mp 268–270 °C; yield 87%; IR (KBr) ν/cm−1: 3440 (NH), 2217 (CN), 1661 (CO). MS m/z (%): 291 (M+, 32.62), 279 (20.83), 258 (19.97), 214 (33.47), 201 (44.55), 185 (38.18), 161 (45), 156 (46.51), 143 (44.53), 129 (57.59), 116 (23.16), 100 (17.52), 78 (41.72), 69 (100), 60 (72.81). Anal. for C9H9N9OS (291.29): calcd: C, 37.11; H, 3.11; N, 43.28%; found: C, 37.05; H, 3.05; N, 43.18%.Synthesis of N-(5-ethyl-1,3,4-thiadiazol-2-yl)-4-imino-4,6-dihydro-1,2,4 triazolo[5,1-c]1,2,4 triazine-3-carboxamide (9)
A solution of 8 (0.58 g, 0.002 mol) in glacial acetic acid (20 mL) was refluxed for 3 h, and then left to cool. The formed solid precipitate was filtered off, purified by washing with ethanol and recrystallized from a mixture of EtOH–DMF (1:1) to furnish compound 9. Orange crystals; mp 290–292 °C; yield 90%; IR (KBr) ν/cm−1: 3440, 3331, 3128 (3NH), 1661 (CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.30 (t, 3H, CH3), 3.02 (q, 2H, CH2), 8.58 (s, 1H, triazole H-3), 11.84 (s, 1H, NHCO), 14.37 (s, 1H, NH). Anal. for C9H9N9OS (291.29): calcd: C, 37.11; H, 3.11; N, 43.28%; found: C, 37.00; H, 3.03; N, 43.12%.
Synthesis of (E)-N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-2-((5-ethyl-1,3,4-thiadiazol-2-yl)amino)-2-oxoacetohydrazonoyl cyanide (10a)
Deep yellow crystals; mp 218–220 °C; yield 92%; IR (KBr) ν/cm−1: 3420, 3411 (2NH), 2220 (CN), 1736, 1658 (2CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.31 (t, 3H, CH3), 2.27 (s, 3H, CH3-pyrazole), 3.02 (q, 2H, CH2), 3.18 (s, 3H, NCH3), 7.09 (m, 5H, Ar-H), 12.33 (s, 1H, NH), 12.82 (s, 1H, NH). MS m/z (%): 411 (M+ + 1, 17.75), 410 (M+, 2.21), 369 (3.31), 240 (9.15), 229 (10.51), 156 (7.79), 129 (10.99), 119 (30.09), 91 (32.82), 77 (50.67), 56 (100), 54 (27.07). Anal. for C18H18N8O2S (410.46): calcd: C, 52.67; H, 4.42; N, 27.3%; found: C, 52.59; H, 4.39; N 27.18%.
Synthesis of (E)-2-((5-ethyl-1,3,4-thiadiazol-2-yl)amino)-2-oxo-N-(4-(N-(thiazol-2-yl)sulfamoyl)phenyl)acetohydrazonoyl cyanide (10b)
Orange crystals; mp 265–267 °C; yield 95%; IR (KBr) ν/cm−1: 3469, 3226, 3165 (3NH), 2231 (CN), 1665 (CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.30 (t, 3H, CH3), 3.01 (q, 2H, CH2), 6.84 (d, 1H, thiazole H-5, J = 4 Hz), 7.26 (d, 1H, thiazole H-4, J = 4 Hz), 7.81 (d, 2H, Ar-H, J = 8 Hz), 7.97 (d, 2H, Ar-H, J = 8 Hz), 12.37 (s, 1H, N–NH), 12.38 (s, 1H, NHSO2), 12.81 (s, 1H, NHCO). MS m/z (%): 463 (M+ + 1, 1.02), 462 (M+, 4.16), 451 (2.96), 369 (9.5), 305 (7.88), 250 (10.7), 229 (4.14), 194 (6.01), 156 (17.25), 109 (21.87), 97 (49.14), 83 (51.29), 69 (100), 57 (99.58). Anal. for C16H14N8O3S3 (462.52): calcd: C, 41.55; H, 3.05; N, 24.23%; found: C, 41.23; H, 2.95; N, 23.98%.
Synthesis of (E)-N-(4-(N-(4,6-dimethylpyrimidin-2-yl)sulfamoyl)phenyl)-2-((5-ethyl-1,3,4-thiadiazol-2-yl)amino)-2-oxoacetohydrazonoyl cyanide (10c)
Orange crystals; mp 208–210 °C; yield 90%; IR (KBr) ν/cm−1: 3588, 3527, 3231 (3NH), 2232 (CN), 1666 (CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.31 (t, 3H, CH3), 2.27 (s, 6H, 2CH3-pyrimidine), 3.02 (q, 2H, CH2), 6.77 (s, 1H, pyrimidine H-5), 7.98 (m, 4H, Ar-H), 12.33 (s, 1H, N–NH), 12.79 (s, 1H, NHSO2), 12.82 (s, 1H, NHCO). MS m/z (%): 485 (M+, 1.79), 451 (1.5), 344 (5.88), 236 (4.42), 213 (41.28), 200 (6.19), 165 (15.29), 129 (19.51), 83 (45.29), 69 (46.63), 55 (61.02), 43 (100). Anal. for C19H19N9O3S2 (485.54): calcd: C, 47.00; H, 3.94; N, 25.96%; found: C, 46.92; H, 3.90; N, 25.86%.
General procedure for the synthesis of thiazole derivatives 12, 13a, b and 14
To a stirred solution of KOH (0.11 g, 0.002 mol) in DMF (25 mL), compound 1 (0.4 g, 0.002 mol) was added. The mixture was stirred for 30 min, and then phenyl isothiocyanate (0.23 mL, 0.002 mol) was added. Stirring the reaction was continued for 6 h. Then the appropriate α-halo compound [namely chloroacetyl chloride (0.16 mL, 0.002 mol), chloroacetone (0.16 mL, 0.002 mol), phenacyl chloride (0.31 g, 0.002 mol) and chloroacetonitrile (0.13 mL, 0.002 mol)] was added to the resulting mixture. The reaction mixture was stirred for an additional 3 h. Then, the reaction mixture was poured into a beaker containing a crushed ice/water mixture. The formed precipitate was filtered off, dried and purified by recrystallization from EtOH to furnish compounds 12, 13a, b and 14, respectively.Synthesis of (E)-2-cyano-N-(5-ethyl-1,3,4-thiadiazol-2-yl)-2-(5-oxo-3-phenylthiazolidin-2-ylidene)acetamide (12)
Reddish brown crystals; mp 240–242 °C; yield 92%; IR (KBr) ν/cm−1: 3461 (NH), 2211 (CN), 1747, 1656 (2CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.27 (t, 3H, CH3), 2.91 (q, 2H, CH2), 4.02 (s, 2H, CH2-thiazolidinone), 7.38–7.52 (m, 5H, Ar-H), 14.4 (s, 1H, NH). MS m/z (%): 372 (M+ + 1, 20.69), 371 (M+, 13.43), 329 (2.37), 279 (9.19), 243 (76.54), 215 (100), 169 (12.92), 141 (17.58), 132 (43.76), 124 (26.47), 93 (16.59), 77 (80.99), 73 (12.68), 51 (12.48). Anal. for C16H13N5O2S2 (371.43): calcd: C, 51.74; H, 3.53; N, 18.86%; found: C, 51.68; H, 3.48; N, 18.82%.Synthesis of (E)-2-cyano-N-(5-ethyl-1,3,4-thiadiazol-2-yl)-2-(4-methyl-3-phenylthiazol-2(3H)-ylidene)acetamide (13a)
Off white crystals; mp 265–267 °C; yield 90%; IR (KBr) ν/cm−1: 3486 (NH), 2184 (CN), 1626 (CO). 1H NMR (500 MHz, DMSO-d6): δH ppm 1.28 (t, 3H, CH3), 1.88 (s, 3H, CH3-thiazole), 2.91 (q, 2H, CH2), 7.01 (s, 1H, thiazole H-5), 7.48–7.6 (m, 5H, Ar-H). 13C NMR (125 MHz, DMSO-d6): δC ppm 13.11, 13.98, 22.71, 106.89, 115.16, 128.73, 129.42, 130.51, 136.25, 138.51, 166.51. DEPT 13C NMR, H–H COSY, HSQC and HMBC results are in agreement with the proposed structure. MS m/z (%): 370 (M+ + 1, 4.67), 369 (M+, 26.96), 351 (6.59), 324 (19), 301 (8.47), 265 (18.23), 243 (100), 241 (54.49), 212 (37.02), 186 (35.26), 154 (21.6), 126 (39.35), 77 (45.46), 68 (64), 45 (64.59). Anal. for C17H15N5OS2 (369.46): calcd: C, 55.27; H, 4.09; N, 18.96%; found: C, 55.19; H, 4.05; N, 18.91%.Synthesis of (E)-2-cyano-2-(3,4-diphenylthiazol-2(3H)-ylidene)-N-(5-ethyl-1,3,4-thiadiazol-2-yl)acetamide (13b)
Beige powder; mp 245–247 °C; yield 93%; IR (KBr) ν/cm−1: 3446 (NH), 2187 (CN), 1626 (CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.30 (t, 3H, CH3), 2.96 (q, 2H, CH2), 7.19 (s, 1H, thiazole H-5), 7.21–7.61 (m, 10H, Ar-H), 14.49 (s, 1H, NH). MS m/z (%): 432 (M+ + 1, 7.15), 431 (M+, 8.76), 408 (10.59), 390 (10.12), 373 (14.88), 366 (40.31), 316 (22.04), 292 (22.45), 284 (12.94), 250 (37.24), 243 (47.44), 241 (27.25), 197 (24.51), 192 (32.43), 152 (50.08), 132 (33.16), 102 (40.44), 90 (43.04), 83 (64.41), 53 (64.15), 51 (100), 43 (57.47). Anal. for C22H17N5OS2 (431.53): calcd: C, 61.23; H, 3.97; N, 16.23%; found: C, 61.20; H, 3.93; N, 16.19%.Synthesis of (E)-2-(4-amino-3-phenylthiazol-2(3H)-ylidene)-2-cyano-N-(5-ethyl-1,3,4-thiadiazol-2-yl)acetamide (14)
Deep green powder; mp 225–227 °C; yield 45%; IR (KBr) ν/cm−1: 3415 (NH), 3209, 3154 (NH2), 2186 (CN), 1644 (CO). MS m/z (%): 371 (M+ + 1, 2.43), 370 (M+, 1.52), 359 (5.92), 330 (2.41), 297 (9.83), 271 (100), 225 (1.46), 174 (37.98), 129 (79.78), 77 (18.66), 60 (25.27), 51 (42.58). Anal. for C16H14N6OS2 (370.45): calcd: C, 51.88; H, 3.81; N, 22.69%; found: C, 51.85; H, 3.79; N, 22.67%.
Synthesis of (E)-2-cyano-N-(5-ethyl-1,3,4-thiadiazol-2-yl)-3-(methylthio)-3-(phenylamino)acrylamide (15)
To a stirred solution of KOH (0.11 g, 0.002 mol) in DMF (25 mL), compound 1 (0.4 g, 0.002 mol) was added. The mixture was stirred for 30 min, and then phenyl isothiocyanate (0.23 mL, 0.002 mol) was added. The reaction mixture was stirred for 6 h. Then dimethyl sulfate (0.19 mL, 0.002 mol) was added to the resulting mixture. Stirring continued for an additional 3 h. Then, the reaction mixture was poured into a beaker containing a crushed ice/water mixture. The formed precipitate was filtered off, dried and purified by recrystallization from EtOH to afford 15. Colourless crystals; mp 270–272 °C; yield 70%; IR (KBr) ν/cm−1: 3448, 3313 (2NH), 2227 (CN), 1638 (CO). 1H NMR (500 MHz, DMSO-d6): δH ppm 1.30 (t, 3H, CH3), 2.85 (s, 3H, SCH3), 3.04 (q, 2H, CH2), 7.1–7.49 (m, 5H, Ar-H), 9.8 (s, 1H, NHPh), 9.83 (s, 1H, NHCO). 13C NMR (125 MHz, DMSO-d6): δC ppm 11.87, 17.45, 24.38, 114.58, 123.6, 124.38, 128.41, 139.45, 157.02, 162.71, 163.68, 164.87, 179.58. DEPT 13C NMR, H–H COSY, HSQC and HMBC results are in agreement with the proposed structure. MS m/z (%): 346 (M+ + 1, 1.74), 345 (M+, 3.78), 344 (9.39), 331 (9.35), 298 (23.07), 297 (100), 296 (45.5), 252 (93.69), 237 (9.73), 224 (7.7), 180 (12.87), 156 (12.72), 154 (17.42), 118 (5.91), 85 (16.87), 70 (12.05), 58 (4.37), 44 (10.43). Anal. for C15H15N5OS2 (345.44): calcd: C, 52.16; H, 4.38; N, 20.27%; found: C, 52.11; H, 4.32; N, 20.22%.General procedure for the synthesis of arylidenes 16a–c
An equimolar mixture of cyanoacetamide 1 (0.4 g, 0.002 mol) and the appropriate aldehyde (namely piperonal, vanilline, and 4-N,N-dimethylbenzaldehyde) (0.002 mol) in ethanol (25 mL) containing piperidine (0.5 mL) was refluxed for 3 h. The obtained product was filtered off and recrystallized from EtOH to afford compounds 16a–c.Synthesis of (E)-3-(benzo[d]1,3 dioxol-5-yl)-2-cyano-N-(5-ethyl-1,3,4-thiadiazol-2-yl)acrylamide (16a)
Pale yellow crystals; mp 285–287 °C; yield 90%; IR (KBr) ν/cm−1: 3452 (NH), 2221 (CN), 1651 (CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.30 (t, 3H, CH3), 2.98 (q, 2H, CH2), 4.1 (br s, 1H, NH), 6.2 (s, 2H, CH2-dioxolane), 7.15 (d, 1H, Ar-H, J = 8 Hz), 7.57 (d, 1H, Ar-H, J = 8.4 Hz), 7.69 (s, 1H, Ar-H), 8.35 (s, 1H, vinylic-H). MS m/z (%): 329 (M+ + 1, 23.55), 328 (M+, 100), 327 (93.83), 299 (14.72), 268 (2.95), 207 (5.52), 200 (57.9), 170 (87.75), 142 (39.55), 114 (70.73), 87 (10.52), 73 (9.88), 63 (7.81). Anal. for C15H12N4O3S (328.35): calcd: C, 54.87; H, 3.68; N, 17.06%; found: C, 54.85; H, 3.63; N, 16.95%.Synthesis of (E)-2-cyano-N-(5-ethyl-1,3,4-thiadiazol-2-yl)-3-(4-hydroxy-3-methoxyphenyl)acrylamide (16b)
Deep yellow crystals; mp 140–142 °C; yield 92%; IR (KBr) ν/cm−1: 3454 (OH), 3350 (NH), 2211 (CN), 1709 (CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.26 (t, 3H, CH3), 2.87 (q, 2H, CH2), 3.83 (s, 3H, OCH3), 6.89 (d, 1H, Ar-H, J = 8 Hz), 7.43 (d, 1H, Ar-H, J = 8.4 Hz), 7.72 (s, 1H, Ar-H), 8.08 (s, 1H, vinylic-H), 8.99 (s, 1H, OH), 9.02 (s, 1H, NHCO). MS m/z (%): 331 (M+ + 1, 34.66), 330 (M+, 100), 301 (5.1), 299 (2.73), 202 (25.53), 170 (36.67), 156 (13.85), 130 (10.67), 114 (11.17), 76 (3.28), 73 (4.35), 56 (1.54). Anal. for C15H14N4O3S (330.36): calcd: C, 54.54; H, 4.27; N, 16.96%; found: C, 54.51; H, 4.23; N, 16.95%.Synthesis of (E)-2-cyano-3-(4-(dimethylamino)phenyl)-N-(5-ethyl-1,3,4-thiadiazol-2-yl)acrylamide (16c)
Orange crystals; mp 290–292 °C; yield 93%; IR (KBr) ν/cm−1: 3455 (NH), 2214 (CN), 1651 (CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.30 (t, 3H, CH3), 2.98 (q, 2H, CH2), 3.09 (s, 6H, N(CH3)2), 6.85 (d, 2H, Ar-H, J = 8.8 Hz), 7.93 (d, 2H, Ar-H, J = 8.8 Hz), 8.27 (s, 1H, vinylic-H), 12.85 (s, 1H, NHCO). MS m/z (%): 328 (M+ + 1, 10.89), 327 (M+, 46.31), 298 (1.56), 272 (3.04), 199 (100), 171 (33.82), 156 (8.7), 128 (2.69), 101 (1.36), 85 (0.94), 73 (2.5). Anal. for C16H17N5OS (327.40): calcd: C, 58.70; H, 5.23; N, 21.39%; found: C, 58.55; H, 5.21; N, 21.33%.Synthesis of 3-amino-5-(4-(dimethylamino)phenyl)-N-(5-ethyl-1,3,4-thiadiazol-2-yl)-1H-pyrazole-4-carboxamide (17)
Equimolar amounts of arylidene derivative 16c (0.654 g, 0.002 mol) and hydrazine hydrate (80%, 0.1 mL, 0.002 mol) in 20 mL of ethanol were heated under reflux for 3 h, then allowed to cool. The obtained solid precipitate was filtered off, purified by washing with ethanol, dried, and recrystallized from EtOH to furnish compound 17. Yellow powder; mp 295–297 °C; yield 55%; IR (KBr) ν/cm−1: 3411 (2NH), 3280, 3152 (NH2), 1638 (CO). MS m/z (%): 357 (M+, 6.92), 341 (6.42), 310 (14.8), 309 (37.32), 294 (100), 278 (20.25), 266 (80.14), 251 (11.74), 214 (9.2), 199 (9.85), 174 (9.81), 159 (18.12), 157 (22.63), 147 (23.35), 130 (28.89), 118 (29.54), 104 (29.44), 96 (33.58), 77 (39.75), 70 (42.92), 55 (40.84). Anal. for C16H19N7OS (357.44): calcd: C, 53.77; H, 5.36; N, 27.43%; found: C, 53.72; H, 5.35; N, 27.39%.
General procedure for the synthesis of pyridin-2-ones 18a–d
Synthesis of 1-(5-ethyl-1,3,4-thiadiazol-2-yl)-6-hydroxy-2-oxo-4-phenyl-1,2-dihydropyridine-3,5-dicarbonitrile (18a)
Yellow crystals; mp 290–292 °C; yield 65%; IR (KBr) ν/cm−1: 3475 (OH), 2216 (2CN), 1694 (CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.25 (t, 3H, CH3), 2.94 (q, 2H, CH2), 7.21–7.41 (m, 5H, Ar-H). 13C NMR (125 MHz, DMSO-d6): δC ppm 11.96, 24.35, 44.17, 97.56, 101.15, 114.87, 117.8, 127.36, 127.51, 127.54, 138.21, 153.66, 158.87, 159.87, 160.28. MS m/z (%): 350 (M+ + 1, 21.24), 349 (M+, 80.58), 348 (100), 332 (8.96), 320 (5.88), 261 (2.92), 165 (5.7), 139 (3.44), 127 (3.16), 85 (3.88), 56 (4.57). Anal. for C17H11N5O2S (349.37): calcd: C, 58.44; H, 3.17; N, 20.05%; found: C, 58.40; H, 3.14; N, 19.99%.Synthesis of 1-(5-ethyl-1,3,4-thiadiazol-2-yl)-6-hydroxy-4-(4-nitrophenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (18b)
Deep brown powder; mp 250–252 °C; yield 85%; IR (KBr) ν/cm−1: 3446 (OH), 2199 (2CN), 1670 (CO), 1522 (NO2). MS m/z (%): 394 (M+, 5.01), 373 (5.45), 336 (5.59), 329 (28.08), 301 (16.62), 300 (62.34), 292 (1.40), 284 (23.61), 265 (32.96), 257 (34.87), 256 (100), 255 (25.28), 246 (19.31), 227 (5.23), 192 (4.91), 137 (6.49), 112 (14.78), 93 (17.79), 80 (23.63), 79 (20.13), 68 (12.58), 48 (9.77). Anal. for C17H10N6O4S (394.37): calcd: C, 51.78; H, 2.56; N, 21.31%; found: C, 51.74; H, 2.52; N, 21.30%.Synthesis of 4-(4-chlorophenyl)-1-(5-ethyl-1,3,4-thiadiazol-2-yl)-6-hydroxy-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (18c)
Yellow powder; mp 265–267 °C; yield 82%; IR (KBr) ν/cm−1: 3504 (OH), 2213 (2CN), 1692 (CO). MS m/z (%): 368 (M+ − 15, 13.47), 366 (1.08), 353 (11.22), 328 (4.88), 313 (4.31), 297 (6.47), 265 (9.98), 236 (17.53), 229 (8.88), 159 (11.13), 137 (10.87), 123 (24.83), 109 (26.98), 97 (50.06), 84 (39.46), 71 (57.77), 69 (100), 57 (93.35), 43 (91.87). Anal. for C17H10ClN5O2S (383.81): calcd: C, 53.2; H, 2.63; N, 18.25%; found: C, 53.15; H, 2.62; N, 18.19%.Synthesis of 4-(4-(dimethylamino)phenyl)-1-(5-ethyl-1,3,4-thiadiazol-2-yl)-6-hydroxy-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (18d)
Orange powder; mp 285–287 °C; yield 82%; IR (KBr) ν/cm−1: 3445 (OH), 2209 (2CN), 1705 (CO). MS m/z (%): 392 (M+, 3.00), 370 (3.08), 369 (5.40), 351 (4.94), 330 (3.73), 328 (5.18), 304 (20.99), 287 (8.14), 271 (31.05), 248 (16.77), 240 (100), 207 (45.35), 185 (23.2), 181 (48.16), 176 (11.13), 156 (62.39), 153 (31.52), 138 (20.18), 121 (18.63), 86 (82.77), 84 (51.31), 60 (45.65), 52 (49.38), 44 (98.88). Anal. for C19H16N6O2S (392.44): calcd: C, 58.15; H, 4.11; N, 21.42%; found: C, 58.12; H, 4.08; N, 21.38%.General procedure for the synthesis of pyridin-2-ones 19a–d
Synthesis of 6-amino-1-(5-ethyl-1,3,4-thiadiazol-2-yl)-2-oxo-4-phenyl-1,2-dihydropyridine-3,5-dicarbonitrile (19a)
Yellow powder; mp 240–242 °C; yield 75%; IR (KBr) ν/cm−1: 3444 (OH), 3343, 3208 (NH2), 2212, 2163 (2CN), 1638 (CO). MS m/z (%): 348 (M+, 1.38), 330 (7.03), 329 (11.78), 298 (2.47), 201 (2.9), 156 (13.48), 135 (26.66), 129 (33.61), 119 (16.51), 100 (8.53), 93 (23.43), 77 (100), 74 (75.38), 73 (43.98), 60 (82.11), 45 (72.42). Anal for C17H12N6OS (348.38): calcd: C, 58.61; H, 3.47; N, 24.12%; found: C, 58.57; H, 3.42; N, 24.05%.Synthesis of 6-amino-1-(5-ethyl-1,3,4-thiadiazol-2-yl)-4-(4-nitrophenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (19b)
Brown powder; mp 240–242 °C; yield 90%; IR (KBr) ν/cm−1: 3446 (OH), 3342, 3217 (NH2), 2210 (2CN), 1636 (CO), 1521 (NO2). MS m/z (%): 394 (M+ + 1, 1.96), 393 (M+, 6.7), 363 (2.6), 346 (6.27), 338 (12.77), 311 (6.82), 282 (32.52), 265 (4.76), 236 (3.67), 184 (5.24), 175 (7.04), 156 (11.69), 136 (33.47), 129 (27.76), 106 (22.56), 90 (35.21), 89 (65.14), 84 (73.14), 78 (90.14), 69 (75.55), 56 (97.69), 43 (98.7), 41 (100). Anal. for C17H11N7O3S (393.38): calcd: C, 51.91; H, 2.82; N, 24.92%; found: C, 51.88; H, 2.8; N, 24.89%.Synthesis of 6-amino-4-(4-chlorophenyl)-1-(5-ethyl-1,3,4-thiadiazol-2-yl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (19c)
Pale yellow crystals; mp 245–247 °C; yield 84%; IR (KBr) ν/cm−1: 3445 (OH), 3338, 3197 (NH2), 2211 (2CN), 1639 (CO). MS m/z (%): 383 (M+ + 1, 98.26), 382 (M+, 100), 381 (10.98), 366 (15.4), 356 (18.74), 337 (34.57), 320 (4.39), 295 (9.77), 294 (2.52), 270 (2.54), 255 (1.86), 199 (3.23), 190 (1.10), 161 (2.38), 138 (3.31), 125 (7.84), 113 (1.87), 86 (1.97), 84 (2.96), 73 (3.12), 56 (1.99). Anal. for C17H11ClN6OS (382.83): calcd: C, 53.34; H, 2.90; N, 21.95%; found: C, 53.32; H, 2.8; N, 21.89%.Synthesis of 6-amino-4-(4-(dimethylamino)phenyl)-1-(5-ethyl-1,3,4-thiadiazol-2-yl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (19d)
Orange crystals; mp 275–277 °C; yield 90%; IR (KBr) ν/cm−1: 3446 (OH), 3430, 3202 (NH2), 2209 (2CN), 1705 (CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.29 (t, 3H, CH3), 3.1 (s, 6H, N(CH3)2), 4.27 (q, 2H, CH2), 6.84 (d, 2H, Ar-H, J = 9.2 Hz), 7.96 (d, 2H, Ar-H, J = 8.8 Hz), 8.12 (s, 1H, NH). MS m/z (%): 392 (M+ + 1, 5.29), 391 (M+, 49.7), 387 (9.7), 368 (22.58), 326 (43.61), 288 (11.82), 272 (34.44), 247 (23.23), 246 (19.36), 228 (58.36), 211 (18.54), 206 (48.02), 198 (100), 196 (44.36), 169 (59.65), 155 (48.75), 140 (39.06), 131 (24.87), 113 (16.55), 105 (49.41), 99 (77.15), 94 (22.16), 77 (34.14), 52 (37.48), 50 (46.43). Anal. for C19H17N7OS (391.45): calcd: C, 58.30; H, 4.38; N, 25.05%; found: C, 58.10; H, 4.35; N, 25.02%.Synthesis of (Z)-2-cyano-N-(5-ethyl-1,3,4-thiadiazol-2-yl)-2-(4-phenyl-1,3-dithiol-2-ylidene)acetamide (21)
To a stirred solution of KOH (0.11 g, 0.002 mol) in DMF (25 mL), compound 1 (0.4 g, 0.002 mol) was added. After stirring for 30 min, carbon disulfide (0.12 mL, 0.002 mol) was added. The reaction mixture was stirred for 12 h, and then phenacyl bromide (0.41 g, 0.002 mol) was added to the resulting mixture. Stirring was continued in the presence of ethanol for an additional 6 h. Then, the reaction mixture was poured into a beaker containing a crushed ice/water mixture. The formed precipitate was filtered off, dried and purified by recrystallization from EtOH to yield 21. Pale yellow powder; mp 275–277 °C; yield 85%; IR (KBr) ν/cm−1: 3421 (NH), 2204 (CN), 1647 (CO). MS m/z (%): 373 (M+ + 1, 4.07), 372 (M+, 8.73), 357 (14.78), 355 (44.95), 333 (21.48), 313 (16.82), 285 (11.9), 270 (88.53), 264 (14.2), 251 (9.09), 199 (7.67), 178 (13.55), 156 (15.94), 126 (11.2), 113 (32.26), 105 (89.81), 96 (57.73), 83 (62.68), 77 (100), 73 (78.59), 69 (40.34), 58 (61.95), 45 (69.49). Anal. for C16H14N4O2S3 (372.48): calcd: C, 51.59; H, 3.25; N, 15.04%; found: C, 51.57; H, 3.21; N, 15.00%.Synthesis of 4-amino-5-benzoyl-N-(5-ethyl-1,3,4-thiadiazol-2-yl)-2-mercaptothiophene-3-carboxamide (22)
To a stirred solution of KOH (0.11 g, 0.002 mol) in DMF (25 mL), compound 1 (0.4 g, 0.002 mol) was added. After stirring for 30 min, carbon disulfide (0.12 mL, 0.002 mol) was added. Stirring was continued for 12 h, and then phenacyl bromide (0.41 g, 0.002 mol) was added to the resulting mixture. The reaction mixture was refluxed in the presence of triethylamine (0.5 mL) for an additional 3 h. Then, the reaction mixture was poured into a beaker containing a crushed ice/water mixture. The formed precipitate was filtered off, dried and purified by recrystallization from EtOH to afford 22. Green powder; mp 285–287 °C; yield 90%; IR (KBr) ν/cm−1: 3413 (NH), 3296 (NH2), 2622 (SH), 1718, 1645 (2CO). 1H NMR (500 MHz, DMSO-d6): δH ppm 1.31 (t, 3H, CH3), 2.95 (q, 2H, CH2), 7.43–7.6 (m, 5H, Ar-H), 8.35 (br s, 2H, NH2). 13C NMR (125 MHz, DMSO-d6): δC ppm 12.37, 23.29, 106.41, 122.18, 126.83, 128.29, 130.71, 140.48, 156.65, 159.56, 162.33, 167.14, 168.66, 185.86. DEPT 13C NMR, H–H COSY, HSQC and HMBC results are in agreement with the proposed structure. MS m/z (%): 390 (M+, 0.53), 357 (15.3), 356 (45.6), 355 (51.44), 339 (5.42), 300 (3.63), 292 (6.64), 286 (6.85), 249 (9.58), 234 (12.13), 201 (5.62), 172 (3.72), 158 (6.35), 146 (10.13), 129 (25.42), 105 (100), 93 (4.04), 86 (10.04), 77 (93.59), 74 (29.31), 70 (12.4), 51 (16.5). Anal. for C16H14N4O2S3 (390.49): calcd: C, 49.21; H, 3.61; N, 14.35%; found: C, 49.16; H, 3.55; N, 14.22%.
Synthesis of (E)-2-cyano-N-(5-ethyl-1,3,4-thiadiazol-2-yl)-2-(4-oxo-1,3-dithiolan-2-ylidene)acetamide (23)
To a stirred solution of KOH (0.11 g, 0.002 mol) in DMF (25 mL), compound 1 (0.4 g, 0.002 mol) was added. After stirring for 30 min, carbon disulfide (0.12 mL, 0.002 mol) was added. The reaction mixture was stirred for 12 h, and then chloroacetyl chloride (0.16 mL, 0.002 mol) was added dropwise to the resulting mixture. Stirring was continued for an additional 6 h. Then, the reaction mixture was poured into a beaker containing a crushed ice/water mixture. The formed precipitate was filtered off, dried and purified by recrystallization from EtOH to give 23. Reddish brown powder; mp 190–192 °C; yield 45%; IR (KBr) ν/cm−1: 3456 (NH), 2209 (CN), 1684, 1630 (2CO). 1H NMR (400 MHz, DMSO-d6): δH ppm 1.29 (t, 3H, CH3), 3.01 (q, 2H, CH2), 4.07 (s, 2H, CH2-dithiolane). MS m/z (%): 312 (M+, 9.15), 289 (6.36), 288 (25.24), 270 (42.28), 270 (42.28), 238 (25.78), 217 (5.68), 207 (6.58), 191 (1.58), 180 (15.27), 160 (25.57), 146 (21.17), 129 (40.19), 110 (22.33), 86 (36.67), 76 (39.55), 64 (100), 60 (76.21), 59 (45.6), 54 (29.34), 45 (54.26). Anal. for C10H8N4O2S3 (312.38): calcd: C, 38.45; H, 2.58; N, 17.94%; found: C, 38.41; H, 2.55; N, 17.90%.Synthesis of 2-cyano-N-(5-ethyl-1,3,4-thiadiazol-2-yl)-3,3-bis(methylthio)acrylamide (24)
To a stirred solution of KOH (0.11 g, 0.002 mol) in DMF (25 mL), compound 1 (0.4 g, 0.002 mol) was added. After stirring for 30 min, carbon disulfide (0.12 mL, 0.002 mol) was added. Stirring was continued for 12 h, and then dimethyl sulfate (0.38 mL, 0.004 mol) was added dropwise to the resulting mixture. Stirring was continued for an additional 6 h. Then, the reaction mixture was poured into a beaker containing a crushed ice/water mixture. The formed precipitate was filtered off, dried and purified by recrystallization from EtOH to furnish 24. Pale yellow powder; mp 210–212 °C; yield 90%; IR (KBr) ν/cm−1: 3445 (NH), 2188 (CN), 1642 (CO). 1H NMR (500 MHz, DMSO-d6): δH ppm 1.39 (t, 3H, CH3), 2.53 (s, 6H, 2SCH3), 3.27 (q, 2H, CH2). 13C NMR (125 MHz, DMSO-d6): δC ppm 11.94, 12.23, 16.71, 21.49, 90.76, 117.89, 157.24, 163.65, 165.91, 171.62. DEPT 13C NMR, H–H COSY, HSQC and HMBC results are in agreement with the proposed structure. MS m/z (%): 300 (M+, 1.67), 253 (3.91), 217 (0.80), 186 (5.96), 170 (39.01), 161 (5.11), 142 (6.59), 110 (23), 91 (34.23), 84 (22.77), 83 (100), 73 (28.82), 56 (21.85), 45 (33.82). Anal. for C10H12N4OS3 (300.42): calcd: C, 39.98; H, 4.03; N, 18.65%; found: C, 39.95; H, 4.00; N, 18.55%.
Synthesis of N-(5-ethyl-1,3,4-thiadiazol-2-yl)-7-imino-5-(methylthio)-1,7-dihydro-1,2,4 triazolo[1,5-a]pyrimidine-6-carboxamide (25)
To a solution of compound 24 (0.4 g, 0.0013 mol) in pyridine (25 mL), 3-amino-1H-1,2,4-triazole (0.112 g, 0.0013 mol) was added. The reaction mixture was refluxed for 3 h, and then left to cool. The formed precipitate was isolated by filtration, then purified by recrystallization from EtOH to afford 25. Deep yellow powder; mp 190–192 °C; yield 52%; IR (KBr) ν/cm−1: 3464, 3433, 3409 (3NH), 1642 (CO). MS m/z (%): 336 (M+, 11.45), 332 (24.08), 325 (32.54), 306 (14.82), 302 (64.48), 300 (37.42), 291 (29.25), 289 (8.13), 257 (12.77), 251 (22.55), 228 (43.51), 225 (21.62), 199 (23.43), 183 (24.76), 175 (54.19), 153 (30.74), 119 (54.27), 95 (49.89), 79 (32.62), 64 (100), 57 (27.07). Anal. for C11H12N8OS2 (336.4): calcd: C, 39.28; H, 3.6; N, 33.31%; found: C, 39.22; H, 3.45; N, 33.28%.Synthesis of 2-cyano-2-(1,3-dihydro-2H-benzo[d]imidazol-2-ylidene)-N-(5-ethyl-1,3,4-thiadiazol-2-yl)acetamide (26)
To a solution of compound 24 (0.4 g, 0.0013 mol) in pyridine (20 mL), o-phenylenediamine (0.15 g, 0.0013 mol) was added. The reaction mixture was refluxed for 3 h, and then left to cool. The formed solid product was isolated by filtration, then purified by recrystallization from EtOH to afford 26. Beige powder; mp 265–267 °C; yield 65%; IR (KBr) ν/cm−1: 3451, 3303, 3130 (3NH), 2219 (CN), 1679 (CO). 1H NMR (500 MHz, DMSO-d6): δH ppm 1.35 (t, 3H, CH3), 3.07 (q, 2H, CH2), 7.47–7.64 (m, 4H, Ar-H), 7.93 (s, 2H, 2NH), 8.35 (s, 1H, NH). 13C NMR (125 MHz, DMSO-d6): δC ppm 13.85, 22.76, 97.26, 116.89, 123.87, 127.70, 132.21, 157.04, 162.61, 165.99, 175.77. DEPT 13C NMR, H–H COSY, HSQC and HMBC results are in agreement with the proposed structure. MS m/z (%): 312 (M+, 9.15), 294 (7.74), 272 (3.49), 257 (6.15), 251 (39.84), 232 (7.57), 205 (29.08), 135 (30.82), 112 (63.99), 107 (100), 93 (36.94), 87 (54.24), 68 (72.37), 44 (88.96). Anal. for C14H12N6OS (312.35): calcd: C, 53.83; H, 3.87; N, 26.91%; found: C, 53.81; H, 3.85; N, 26.88%.
Laboratory bioassay
Laboratory experiments were conducted to study the insecticidal activity of the newly synthesized tested compounds against the 2nd instar larvae of S. littoralis. The experiments were carried out using the leaf dip technique.34 Six concentrations of each compound were formulated as emulsions in solvent, and 0.1% Triton X-100 was used as a surfactant. The emulsions were used immediately after preparation. For larvicidal action, fresh castor bean leaves were dipped in the tested concentrations for 10 seconds. The treated leaves were left in the shade to dry before being offered to the larvae. The larvae were allowed to feed on the treated leaves for 48 hours and then changed to untreated leaves. Three replicates of 10 larvae each were used for each concentration in addition to the control. Control (check) tests were carried out using the same technique. Castor bean leaves were dipped in a solution of 0.1% Triton X-100 and solvent at the same ratio used in the synthesized compound tests. Larval mortality counts were calculated at 1, 2, 3, 4, 5, 6 and 7 days after exposure period. Mortality was corrected according to Abbott's formula,35 and then subjected to probit analysis. The toxicity lines (LC-p lines) were drawn on log concentration–probit paper and statistically analyzed according to Finney's method36 to obtain the LC50 and LC90 values of different tested compounds in order to determine the most effective one. Slope values of the tested compounds were also estimated. In addition, the efficacy of the different compounds was measured by comparing the tested compounds with the most effective compound using the following equation: toxicity index = LC50 of the most effective compound/LC50 of the tested compound × 100, according to Sun.37Conclusions
In the present work, a novel series of different heterocyclic compounds incorporating the thiadiazole moiety has been successfully synthesized and characterized. These compounds were evaluated for their insecticidal activities against the cotton leafworm, S. littoralis. Compounds 10b, 10c and 7 proved to be promising insecticidal agents since they clearly showed higher activities than the other tested compounds.Acknowledgements
Financial support from Chemistry Department, Faculty of Science, Mansoura University and Plant Protection Research Institute, Agricultural Research Centre, Egypt is gratefully acknowledged.Notes and references
- H. Dai, G. Li, J. Chen, Y. Shi, S. Ge, C. Fan and H. He, Bioorg. Med. Chem. Lett., 2016, 26, 3818–3821 CrossRef CAS PubMed.
- Y. Hu, C. Y. Li, X. M. Wang, Y. H. Yang and H. L. Zhu, Chem. Rev., 2014, 114, 5572 CrossRef CAS PubMed.
- Z. S. Li, W. M. Wang, W. Lu, C. W. Niu, Y. H. Li, Z. M. Li and J. G. Wang, Bioorg. Med. Chem. Lett., 2013, 23, 3723 CrossRef CAS PubMed.
- I. Khan, S. Ali, S. Hameed, N. H. Rama, M. T. Hussain, A. Wadood, R. Uddin, Z. UI-Had, A. Khan, S. Ali and M. Z. Choudhary, Eur. J. Med. Chem., 2010, 45, 5200 CrossRef CAS PubMed.
- P. Li, L. Shi, X. Yang, L. Yang, X. W. Chen, F. Wu, Q. C. Shi, W. M. Xu, M. He, D. Y. Hu and B. A. Song, Bioorg. Med. Chem. Lett., 2014, 24, 1677 CrossRef CAS PubMed.
- M. Yusuf, R. A. Khan and B. Ahmed, Bioorg. Med. Chem. Lett., 2008, 16, 8029 CrossRef CAS PubMed.
- S. R. Pattn, B. S. Kittur, B. S. Sastry, S. G. Jadav, D. K. Thakur, S. A. Madamwar and H. V. Shinde, Indian J. Chem., 2011, 50B, 615 Search PubMed.
- W. M. Xu, S. Z. Li, M. He, S. Yang, X. Y. Li and P. Li, Bioorg. Med. Chem. Lett., 2013, 23, 5821 CrossRef CAS PubMed.
- N. Siddiqui, P. Ahuja, S. Malik and S. K. Arya, Arch. Pharm. Chem. Life Sci., 2013, 346, 819 CrossRef CAS PubMed.
- D. V. Dekhane, S. S. Pawar, S. Gupta, M. S. Shingare, C. R. Patil and S. N. Thore, Bioorg. Med. Chem. Lett., 2011, 21, 6527 CrossRef CAS PubMed.
- W. S. Hamama, M. A. Gouda, M. H. Badr and H. H. Zoorob, Med. Chem. Res., 2013, 22, 3556 CrossRef CAS.
- A. Senff-Ribeiro, A. Echevarria, E. F. Silva, C. R. Franco, S. S. Veiga and M. B. Oliveira, Br. J. Cancer, 2004, 91, 297–304 CAS.
- K. A. M. El-Bayouki, M. M. Aly, Y. A. Mohamed, W. M. Basyouni and S. Y. Abbas, Eur. J. Chem., 2011, 2(4), 455–462 CrossRef.
- A. V. Eremeev, I. P. Piskunova and R. S. El’kinson, Khim. Geterotsikl. Soedin., 1985, 9, 1202–1206 (Chem. Abstr., 1987, 107, 7016w) Search PubMed.
- G. H. Elgemeie, A. H. Elghandour, A. M. Elzanate and S. A. Ahmed, J. Chem. Soc. Perkin Trans., 1997, 21, 3285–3290 (Chem. Abstr., 1998, 128, 48171v) RSC.
- M. K. A. Ibrahim, A. M. El-Reedy and M. S. A. El-Gharib, Commun. Fac. Sci. Univ. Ankara, Ser. B: Chem. Chem. Eng., 1994, 36(1–2), 45–52 (Chem. Abstr., 1995, 123, 198686c) CAS.
- Y. A. Ammar, A. M. Sh. El-Sharief, A. G. Al-Sehemi, Y. A. Mohamed, M. A. Senussi and M. S. A. El-Gaby, J. Chin. Chem. Soc., 2005, 52, 553–558 CrossRef CAS.
- K. A. M. El-Bayouki, M. M. Aly, Y. A. Mohamed, W. M. Basyouni and S. Y. Abbas, World J. Chem., 2009, 4, 161–170 CAS.
- G. H. Elgemeie, A. H. Elghandour, A. M. Elzanaty and A. S. Ahmed, Synth. Commun., 2006, 36, 755–764 CrossRef CAS.
- E. F. Dankova, V. A. Bakulev, M. Yu. Kolobov, V. I. Shishkina, Ya. B. Yasman and A. T. Lebedev, Khim. Geterotsikl. Soedin., 1988, 9, 1269–1273 (Chem. Abstr., 1989, 111, 39264z) Search PubMed.
- W. Huang, J. Li, J. Tang, H. Liu, J. Shen and H. Jiang, Synth. Commun., 2005, 35, 1351–1357 CrossRef CAS.
- L. V. Ershov and V. G. Granik, Kim. Geterotsikl. Soedin., 1985, 7, 929–932 (Chem Abstr, 1986, 104, 168389y) Search PubMed.
- J. Stetinova, R. Kada, J. Lesko, M. Dandarova and M. Krublova, Collect. Czech. Chem. Commun., 1996, 61, 921–929 CrossRef CAS.
- E. A. El Rady and A. M. Barsy, J. Heterocycl. Chem., 2006, 43, 243–248 CrossRef CAS.
- S. N. Ibrahim, M. N. Abed and Z. E. Kandeel, Heterocycles, 1984, 22, 1677–1682 (Chem. Abstr., 1985, 102, 6351m) CrossRef.
- M. K. A. Ibrahim, M. M. M. Ramiz and A. H. H. El-Ghandour, J. Chem. Soc. Perkin Trans., 1989, 11(4), 291–296 (Chem. Abstr., 1990, 113, 191303k) CAS.
- A. M. Farag, K. M. Dawood and H. A. El-Menoufy, Heteroat. Chem., 2004, 15, 508–514 CrossRef CAS.
- S. Bondock, A. Tarhoni and A. A. Fadda, Monatsh. Chem., 2008, 139, 153–159 CrossRef CAS.
- S. Bondock, W. Khalifa and A. A. Fadda, Synth. Commun., 2006, 36, 1601–1612 CrossRef CAS.
- A. M. El-Dean, A. A. Geies, T. A. Mohamed and A. A. Atalla, Bull. Fac. Sci., Assiut Univ., 1991, 20, 15–21 (Chem. Abstr., 1992, 116, 106161g) CAS.
- M. H. Refat and A. A. Fadda, J. Heterocycl. Chem., 2015, 53(4), 1129–1137 CrossRef.
- A. A. Fadda, R. Rabie and A. A. Etman, Res. Chem. Intermed., 2015, 41(10), 7883–7897 CrossRef CAS.
- B. A. Bhongade, S. Talath, R. A. Gadad and A. K. Gadad, J. Saudi Chem. Soc., 2016, 20, S463–S475 CrossRef CAS.
- M. M. Sadek, J. Appl. Entomol., 2003, 127(7), 396–404 CrossRef.
- W. S. Abbott, A method for computing the effectiveness of an insecticide, J. Econ. Entomol., 1925, 18, 265–267 CrossRef CAS.
- D. J. Finney, Probit Analysis, Statistical treatment of the sigmoid response curve, Cambridge Univ. Press, London, 7th Edn, 1971 Search PubMed.
- Y. P. Sun, Toxicity index an improved method of comparing the relative toxicity of insecticides, J. Econ. Entomol., 1950, 43, 45–53 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06087d |
| This journal is © The Royal Society of Chemistry 2017 |