
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcid-promoted metal-free protodeboronation of arylboronic acids†
Guoqing Zhangb,
Yang Li*ab and
Jianhui Liu *ab
*ab
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, P. R. China. E-mail: zgqdut8023@mail.dlut.edu.cn; chyangli@dlut.edu.cn
bSchool of Petroleum and Chemical Engineering, Dalian University of Technology, Panjin Campus, Panjin, Liaoning Province 124221, P. R. China
First published on 11th July 2017
Abstract
A facile acid-promoted protodeboronation of arylboronic acids in the absence of metal catalysts or any other additives is described. This protodeboronation is general for a range of arylboronic acids with both electron-donating and electron-withdrawing groups in good to excellent yields under air atmosphere. Density functional theory mechanistic studies showed that the protodeboronation of arylboronic acids followed an intermolecular metathesis via a four-membered ring transition state. The effect of the substituent of arylboronic acids in protodeboronation is also theoretically studied.
Introduction
Arylboronic acids are versatile precursors in transition metal-mediated or metal-free cross-coupling reactions to construct C–C, C–N, C–O, and other C–X bonds.1 They are generally used in the Suzuki–Miyaura cross-coupling reaction2 and transition metal-catalyzed self-coupling reactions3 to form (un)symmetrical biaryl compounds. However, the protodeboronation of arylboronic acids is sometimes a side reaction to compete with Suzuki coupling, due to the strong nucleophilic character of the arylboronic acids. Compared to the dimerization of arylboronic acid, less attention has been paid to the protodeboronation of arylboronic acids. The deliberate removal of the –B(OH)2 that acted as a blocking/directing group in several examples gives the protodeboronation increased value. For example, Aggarwal applied the protodeboronation of allylic boronic esters to afforded trisubstituted alkenes.4 The protodeboronation was also applied by Aggarwal in the asymmetric synthesis to afforded tertiary alkyl stereogenic centers,5 tertiary alcohols and arylethanes.6 Carreño reported the significance of the protodeboronation in the regioselective Friedel-Crafts alkylation in which –B(OH)2 played a temporary regiocontroller.7 Therefore, the protodeboronation has great application potential in the organic synthesis and increasing interest has been given to this research field.Although several methods have been reported, mainly including the use of base and/or metal catalysts (Pd/K2CO3,8 Cu/isopropamide,9 Ag/TEA10), acid-mediated protodeboronation has seldom been described,11 since the acid-mediated protodeboronation would be retarded when the arylboronic acids were substituted with electron-withdrawing groups.12
The study about the protodeboronation kinetics by Lloyd-Jones showed that the protodeboronation rates were pH-dependent.13 Acid-mediated protodeboronation of allylic boronic esters was reported several years ago.4 Recently, a mild reaction condition for the protodeboronation of arylboronic acids, which used AcOH in 1,4-dioxane, was described by Cheon,14 but only electron-donating groups showed better results. He also reported the metal-free protodeboronation of ortho- and para-phenol boronic acids in wet DMSO under thermal conditions.15
Results and discussion
In an effort to prepare thiophene 2 in our recent studies of dye-sensitized solar cells, we found that the standard Knoevenagel condensation of 5-formyl-2-thiopheneboronic acid (1) with cyanoacetic acid did not give the expected thiophene 2, but the protodeboronation product 3 was afforded instead (Scheme 1). This finding inspired us to investigate the protodeboronation of other arylboronic acids.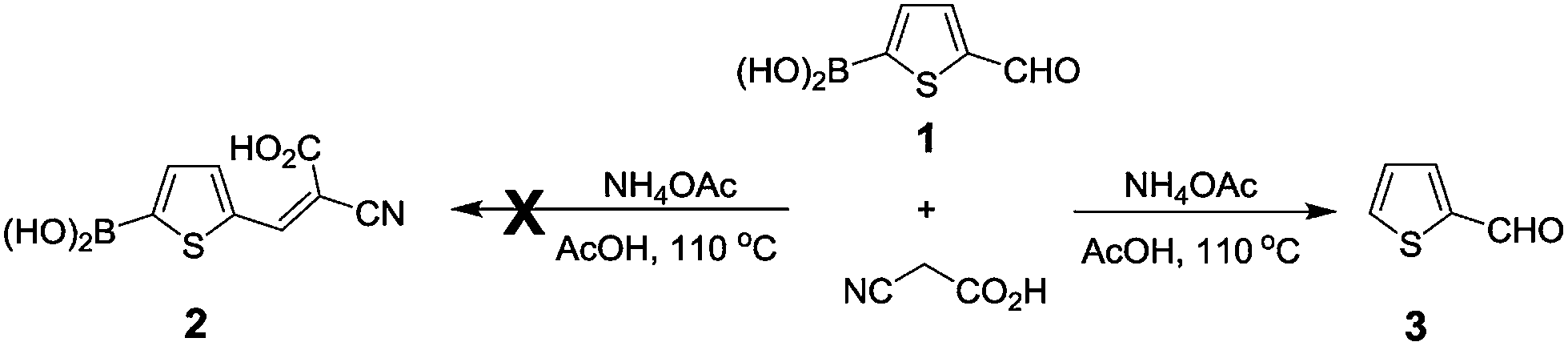 | ||
| Scheme 1 The unexpected protodeboronation rather than condensation. | ||
In our attempted Knoevenagel condensation mentioned above, the protodeboroation product was afforded. Since a good yield (71%) was obtained, we decided to expand this conversion to other substrates. The screen of the reaction conditions was still required before we started our study. In the original Knoevenagel, NH4OAc was used as a weakly basic catalyst to capture the reactive hydrogen. In order to check whether NH4OAc played a role in the protodeboronation, we conducted a control experiment where 2-formylthiophene (3) was still obtained in a yield of 72% without using NH4OAc (Scheme 2). This proved that AcOH alone could promote this reaction and therefore the following studies was performed in AcOH in the absence of NH4OAc. Although the protodeboronation of organoboronic acid by acids was known, but only with formic acid and several inorganic acids.16 No systematic studies were found in the literatures using AcOH. In addition, the early study was focused mainly on the kinetics of the protodeboronation.16 This inspired us to explore a mild and an efficient acid-promoted protodeboronation universale for various arylboronic acid.
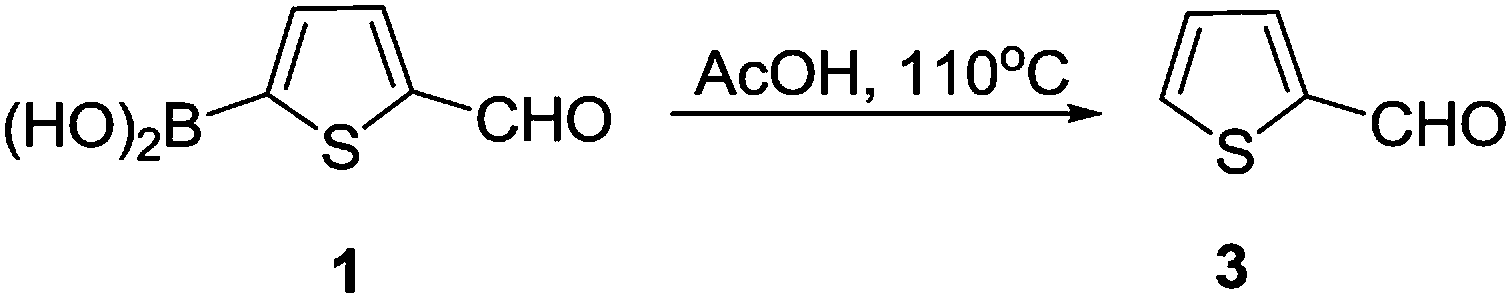 | ||
| Scheme 2 Protodeboronation of 5-formyl-2-thiopheneboronic acid. | ||
The optimization of reaction conditions for the acid-mediated protodeboronation was performed first, and 4-hydroxyphenylboronic acid (4a) was chosen as a substrate. The examination of the solvent indicated that acetic acid is the best acidic medium for the protodeboronation process (yield of 70%), followed by HCOOH with a lower yield of 45%, mixed solvents of AcOH/H2O and HCl/H2O with further decreased yields of no more than 30% (23% and 26%, respectively), and CF3COOH being totally inactive (Table 1, entries 1–5). The influence of the temperature was also investigated. As shown in the same table, the yield of the phenol increased steadily with increasing temperature (Table 1, entries 6–9). Finally, 130 °C was determined as the best temperature for the reaction.

Under these optimized conditions, different arylboronic acids were used as substrates to confirm the usefulness of the acid-promoted protodeboronation, except for several cases where the conversions were conducted at 110 °C due to the lower boiling point of the corresponding protodeboronation products (Table 2, entries 7–11, for 5g, 5h, 5j). The results are summarized in Table 2. Electron-donating groups exhibited a better effect on both rate and yield than electron-withdrawing groups. The protodeboronation of arylboronic acids with electron-donating groups, such as –OH, –OCH3, –Ph, –CH3, were completed in 1–4 h with yields between 78–92% (Table 2, entries 1–7). The position of the groups on the phenyl ring had no obvious effect on the reaction (Table 2, entries 2 vs. 3, 8 vs. 9, 10 vs. 11 and 15 vs. 12). In addition, both the arylboronic acid 4d with the strong steric hindrance of two 2-CH3O and the polycyclic arylboronic acid 4f could afford excellent yields of 88% and 92% (Table 2, entries 4 and 6). Conversely, as expected, electron-withdrawing groups (such as –Br, –Cl, –NO2, –COCH3, –CHO, –COOH, –COOCH3), decreased the yield of the protodeboronation, furnishing yields of 52–71%, except for 4-Cl (85%), 3-COCH3 (80%) and 4-CHO (84%), in a reaction time of 5–20 h (Table 2, entries 8–19). Although no difference was observed in the yields between weaker electron-withdrawing groups such as –Br and –Cl (55–85%) and stronger electron-withdrawing groups such as –NO2, –COCH3, –CHO, –COOH and –COOCH3 (52–84%), the reaction rates clearly differed. Protodeboronation of arylboronic acids with strong electron-withdrawing groups required longer reaction times of 8–20 h (Table 2, entries 12–19). This proved that electron deficiency of the C–B bond did not favor the reaction. Extraordinarily, arylboronic acid substituted with –COOCH3 on the ortho-position did not give the protodeboronated methyl benzoate, but was directly hydrolyzed to benzoic acid 5r in just 2 h (Table 2, entry 20). Heterocyclic boronic acids (4u and 1) could also easily provide the protodeboronation product in excellent yields of 96% and 84% under these conditions (Table 2, entries 21 and 22). In general, the results above demonstrate the universality of this reaction.
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | Compound | Time(h) | Product | Yieldb (%) |
| a Reaction condition: arylboronic acids (0.5 mmol), AcOH (10 mL). The reactions were conducted at 130 °C under air.b Isolated yields.c The reactions were conducted at 110 °C, GC yields.d The product was benzoic acid rather than the expected methyl benzoate. | |||||
| 1 | 4-HO–C6H4 | 4a | 1 | 5a | 81 |
| 2 | 4-CH3O–C6H4 | 4b | 4 | 5b | 84 |
| 3 | 3-CH3O–C6H4 | 4c | 4 | 5b | 85 |
| 4 | 2,6-(CH3O)2–C6H3 | 4d | 3 | 5d | 88 |
| 5 | 4-Ph–C6H4 | 4e | 3 | 5e | 83 |
| 6 | 9-Anthryl | 4f | 3 | 5f | 92 |
| 7 | 4-CH3–C6H4 | 4g | 3 | 5g | 78c |
| 8 | 4-Br–C6H4 | 4h | 5 | 5h | 55c |
| 9 | 3-Br–C6H4 | 4i | 5 | 5h | 70c |
| 10 | 4-Cl–C6H4 | 4j | 5 | 5j | 85c |
| 11 | 3-Cl–C6H4 | 4k | 5 | 5j | 71c |
| 12 | 3-NO2–C6H4 | 4l | 10 | 5l | 62 |
| 13 | 3-CH3CO–C6H4 | 4m | 8 | 5m | 80 |
| 14 | 3-CHO–C6H4 | 4n | 8 | 5n | 52 |
| 15 | 4-NO2–C6H4 | 4o | 10 | 5l | 65 |
| 16 | 4-CH3CO–C6H4 | 4p | 16 | 5m | 69 |
| 17 | 4-CHO–C6H4 | 4q | 8 | 5n | 84 |
| 18 | 4-COOH–C6H4 | 4r | 8 | 5r | 63 |
| 19 | 4-CH3COO–C6H4 | 4s | 20 | 5s | 69 |
| 20 | 2-CH3COO–C6H4 | 4t | 2 | 5r | 66d |
| 21 |  |
4u | 2 | 5u | 96 |
| 22 |  |
1 | 2 | 3 | 84 |
To further explore the scope of the substrates, the protodeboronation of other surrogates such as 4-hydroxyphenylboronic acid pinacol ester (4v) and potassium 4-carboxyphenyltrifluoroborate (4w) were studied. The protodeboronation of 4v and 4w still proceeded although longer reaction time was required (24 h) (Scheme 3). 4v gave the protodeboronation product in a yield of 77%, while 4w affored a lower yield of 36%. The arylboronic acid pinacol ester and potassium aryltrifluoroborate display enhanced chemical stability compared with arylboronic acids.17 The slower protodeboronation rates may be due to the decreased Lewis acidity of the boron atom from arylboronic acid pinacol ester and potassium aryltrifluoroborate, which is consistent with Cheon's studies.14,15
 | ||
| Scheme 3 Protodeboronation of 4-hydroxyphenylboronic acid pinacol ester and potassium 4-carboxyphenyltrifluoroborate. | ||
Aryl iodides, important building blocks in forming C–C and C–X (X = heteroatom) bond, are widely used in organic synthesis.18 As we know, the regioselective iodination of arenes is usually difficult.19 While the 4-iodoanisole (8) was successfully formed in a total yield of 61% through the AcOH-promoted protodeboronation in which the –B(OH)2 acted as a directing group (Scheme 4).
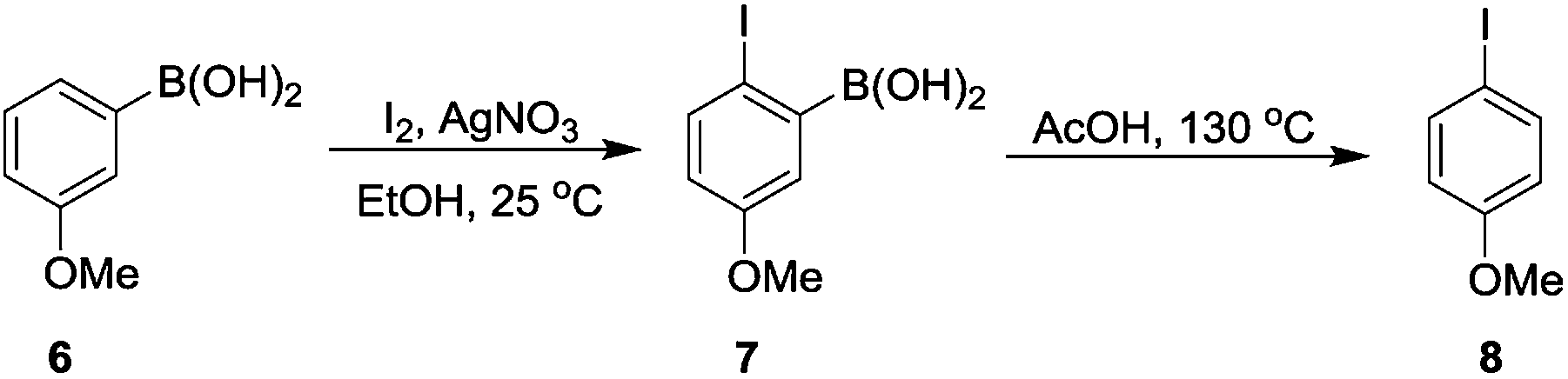 | ||
| Scheme 4 The protodeboronation of arylboronic acid was applied in the synthesis of 4-iodoanisole. | ||
For the protodeboronation of arylboronic acids, as shown in Scheme 5, there are two scenarios to address the aryl–H bond formation: (1) arylboronic acids A react with AcOH to form aryl complex B via a four-membered ring transition state ATS (path 1); (2) arylboronic acids A can be attack by H+ reagent to form complex C, and then OAc− attack the –B(OH)2 motif through B–C bond cleavage and form aryl complex B (path 2). With the help of DFT calculations, the energy profiles of the protodeboronation of arylboronic acids are calculated in Fig. 1. Fig. 1 shows that the arylboronic acids react directly with AcOH to form aryl–H bond via an intermolecular metathesis process. We failed to locate the complex C in path 2, which means the complex C is unlikely to be involved as the stable intermediate in the protodeboronation.
 | ||
| Scheme 5 The proposed mechanism for protodeboronation of arylboronic acids. | ||
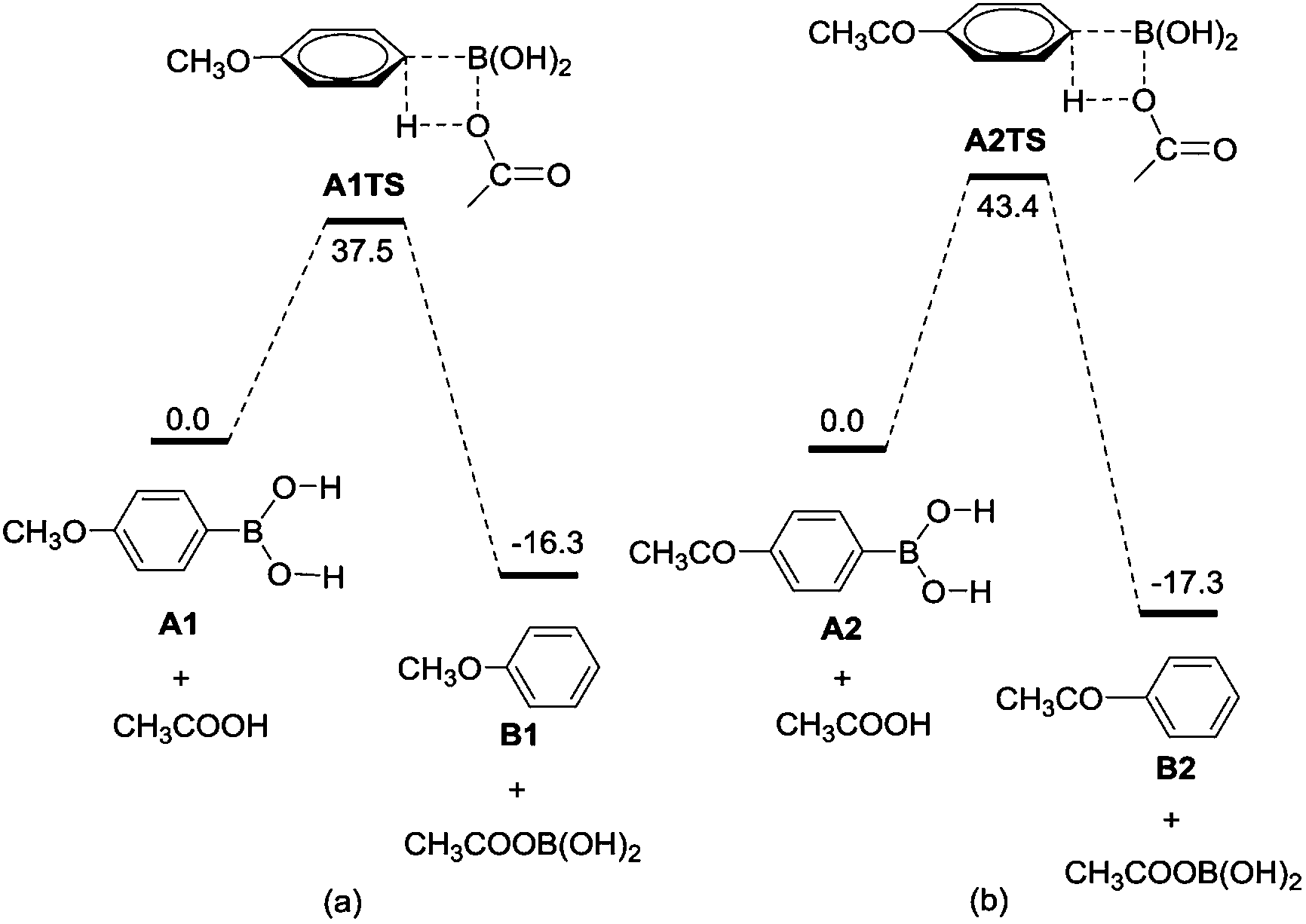 | ||
| Fig. 1 The energy profiles calculated for the protodeboronation of A1 and A2. | ||
In order to understand the effect of the substituent of arylboronic acids in protodeboronation, we compared the reaction profiles starting from the arylboronic acids A1 with electron-donating group (entry 2 in Table 2) and arylboronic acids A2 with electron-withdrawing group (entry 16 in Table 2). From the energy profiles shown in Fig. 1, we can clearly see that protodeboronation of A1 (Fig. 1a, with an energy barrier of 37.5 kcal mol−1) has lower energy barrier than that of A2 (Fig. 1b, with an energy barrier of 43.4 kcal mol−1). Electronically, the boron-bonded carbon in A1 is electron-rich when compared with that in A2 because of the electron property of substituent of arylboronic acids. The natural bond orbital (NBO) analysis shows that the natural atomic charges of the boron-bonded carbon are calculated to be −0.468 in A1 and −0.409 in A2, respectively, suggesting that the AcOH reagent favors attack of the boron-bonded carbon in A1 over that in A2. These results are consistent with the experimental observation that arylboronic acids with electron-donating group have high yield with short reaction time than that with electron-withdrawing group.
Conclusions
In conclusion, we have achieved an acid-mediated protodeboronation from a wide variety of arylboronic acid substrates. Compared to other methods reported before, it is a greener process in that no any metal catalysts and additives are needed. The only use of acetic acid is highly practical. In addition, the electron-withdrawing groups can also be tolerated in the protodeboronation. This method could be an attractive complement to the case in which substituents of the arylboronic acids are susceptible to alkaline medium. An intermolecular metathesis reaction mechanism was developed based on DFT calculations. Additionally, the NBO analysis provided information on the effect of the substituent of arylboronic acids.Acknowledgements
The financial support of this study was from the State Key Laboratory of Fine Chemicals (Panjin) (Grant No. JH2014009) project and the Fundamental Research Funds for the Central Universities. Professor Yang Li thanks the Scientific Research Foundation of Dalian University of Technology (Grant No. DUT16RC(3)037) and Supercomputing Center of Dalian University of Technology for providing access to the supercomputer.Notes and references
- (a) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS PubMed; (b) C. Zhu and J. R. Falck, Adv. Synth. Catal., 2014, 356, 2395 CrossRef CAS PubMed; (c) C. Zhu, G. Li, D. H. Ess, J. R. Falck and L. Kürti, J. Am. Chem. Soc., 2012, 134, 18253 CrossRef CAS PubMed; (d) C. Zhu, R. Wang and J. R. Falck, Org. Lett., 2012, 14, 3494 CrossRef CAS PubMed.
- (a) P. Basnet, S. Thapa, D. A. Dickie and R. Giri, Chem. Commun., 2016, 52, 11072 RSC; (b) G. Xu, W. Fu, G. Liu, C. H. Senanayake and W. Tang, J. Am. Chem. Soc., 2014, 136, 570 CrossRef CAS PubMed; (c) N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513 CrossRef CAS.
- (a) C. A. Contreras-Celedón, J. A. Rincón-Medina, D. Mendoza-Rayo and L. Chacón-García, Appl. Organomet. Chem., 2015, 29, 439 CrossRef; (b) H. Mizuno, H. Sakurai, T. Amaya and T. Hirao, Chem. Commun., 2006, 5042 RSC; (c) C. González-Arellano, A. Corma, M. Iglesias and F. Sánchez, Chem. Commun., 2005, 1990 RSC; (d) A. S. Demir, Ö. Reis and M. Emrullahoglu, J. Org. Chem., 2003, 68, 10130 CrossRef CAS PubMed.
- M. J. Hesse, C. P. Butts, C. L. Willis and V. K. Aggarwal, Angew. Chem., Int. Ed., 2012, 51, 12444 CrossRef CAS PubMed.
- S. Nave, R. P. Sonawane, T. G. Elford and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 17096 CrossRef CAS PubMed.
- C. G. Watson and V. K. Aggarwal, Org. Lett., 2013, 15, 1346 CrossRef CAS PubMed.
- M. Veguillas, M. Ribagorda and M. C. Carreno, Org. Lett., 2011, 13, 656 CrossRef CAS PubMed.
- R. Y. Lai, C. L. Chen and S. T. Liu, J. Chin. Chem. Soc., 2006, 53, 979 CrossRef CAS.
- C. Liu, X. Li, Y. Wu and J. Qiu, RSC Adv., 2014, 4, 54307 RSC.
- C. Liu, X. Li and Y. Wu, RSC Adv., 2015, 5, 15354 RSC.
- (a) H. G. Kuivila and K. V. Nahabedian, J. Am. Chem. Soc., 1961, 83, 2164 CrossRef CAS; (b) B. Floris and G. Illuminati, J. Organomet. Chem., 1978, 150, 101 CrossRef CAS; (c) R. D. Brown, A. S. Buchanan and A. A. Humffray, Aust. J. Chem., 1965, 18, 1521 CrossRef CAS.
- (a) J. Lozada, Z. Liu and D. M. Perrin, J. Org. Chem., 2014, 79, 5365 CrossRef CAS PubMed; (b) K. V. Nahabedian and H. G. Kuivila, J. Am. Chem. Soc., 1961, 83, 2167 CrossRef CAS.
- P. A. Cox, A. G. Leach, A. D. Campbell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2016, 138, 9145 CrossRef CAS PubMed.
- S. J. Ahn, C. Y. Lee, N. K. Kim and C. H. Cheon, J. Org. Chem., 2014, 79, 7277 CrossRef CAS PubMed.
- C. Y. Lee, S. J. Ahn and C. H. Cheon, J. Org. Chem., 2013, 78, 12154 CrossRef CAS PubMed.
- H. G. Kuivila and K. V. Nahabedian, J. Am. Chem. Soc., 1961, 83, 2159 CrossRef CAS.
- (a) M. Schnürch, M. Holzweber, M. D. Mihovilovic and P. Stanetty, Green Chem., 2007, 9, 139 RSC; (b) D. Kuik, J. A. McCubbin and G. K. Tranmer, Synthesis, 2017, 49, 2555 CrossRef CAS; (c) G. A. Molander and L. N. Cavalcanti, J. Org. Chem., 2011, 76, 623 CrossRef CAS PubMed.
- T. S. Mei, D. H. Wang and J. Q. Yu, Org. Lett., 2010, 12, 3140 CrossRef CAS PubMed.
- (a) R. M. Al-Zoubi and D. G. Hall, Org. Lett., 2010, 12, 2480 CrossRef CAS PubMed; (b) C. Y. Zhou, J. Li, S. Peddibhotla and D. Romo, Org. Lett., 2010, 12, 2104 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05979e |
| This journal is © The Royal Society of Chemistry 2017 |