DOI:
10.1039/C7RA05103D
(Paper)
RSC Adv., 2017,
7, 34945-34958
DNA-binding, molecular docking studies and biological activity studies of ruthenium(II) polypyridyl complexes†
Received
6th May 2017
, Accepted 5th July 2017
First published on 12th July 2017
Abstract
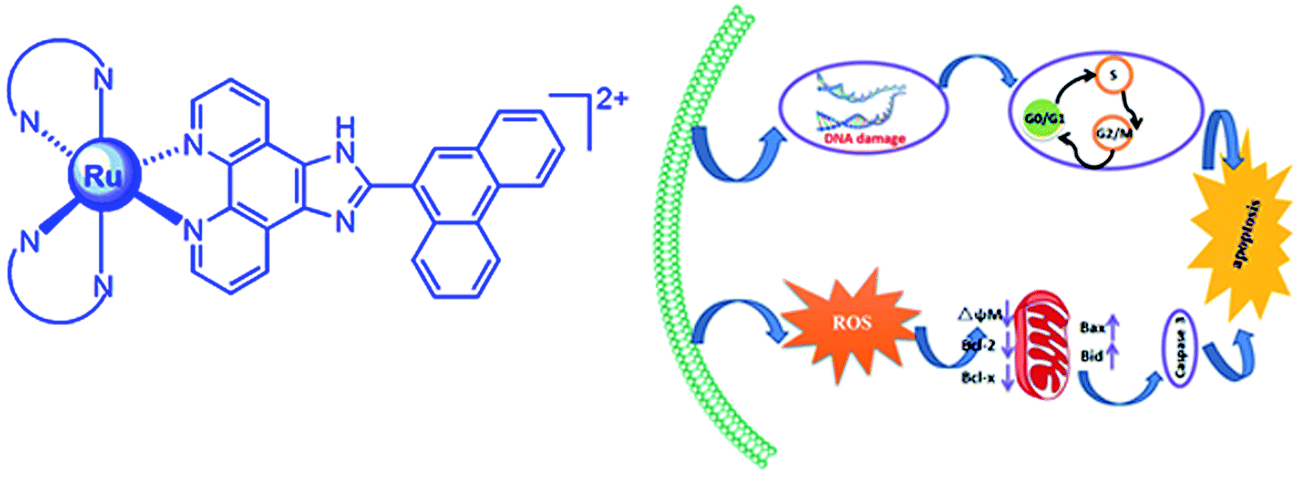
Anticancer properties of chemically synthesized compounds have been evaluated for efficacy and selectivity. A new ligand PTCP (PTCP = 2-phenanthren-9-yl-1H-1,3,7,8-tetraazacyclopenta[l]phenanthrene) and its three new ruthenium(II) polypyridyl complexes [Ru(N–N)2(PTCP)](ClO4)2 (N–N: phen = 1,10-phenanthroline 1; dmp = 2,9-dimethyl-1,10-phenanthroline 2; ttbpy = 4,4′-di-tert-butyl-2,2′-bipyridine 3) were synthesized and characterized by elemental analysis, ESI-MS, IR, 1H NMR and 13C NMR. In this report, we investigated the cytotoxicity in vitro of the complexes against several cancer cell lines SGC-7901, HepG2, HeLa, SiHa and normal cell NIH3T3. The results show that complexes show highly anti-proliferation activity toward SGC-7901 and low cytotoxic activity against normal cell NIH3T3. The relationship between anti-proliferation and molecular interaction mechanism of the complexes was also elucidated. The apoptosis was assayed by flow cytometry. The changes of mitochondrial membrane potential and the ROS levels were measured by flow cytometry. The cell invasion, cell cycle arrest and the expression of Bcl-2 family proteins were studied in detail. N-Acetylcysteine (NAC) was used in several experiments to testify the effect of the complexes on apoptosis. The results demonstrate that the complexes induce apoptosis in SGC-7901 cells through a ROS-mediated mitochondrial dysfunction pathway, which was accompanied by the regulation of Bcl-2 family proteins. In addition, the interaction of the complexes with calf thymus DNA (CT-DNA) shows that the complexes interact with DNA through partial intercalation mode.
1. Introduction
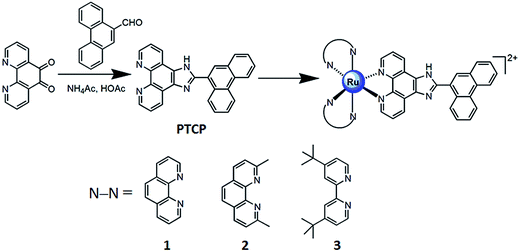
Cisplatin and its derivatives are still used in more than 50% of the treatment drugs for cancer patients.1–4 Platinum-based compounds are the most widely used clinically. However, the clinical drawbacks of cisplatin are also obvious, including the limited applicability, the acquired resistance, and the serious side effects, such as neurotoxicity and nephrotoxicity.5,6 Thus, a number of other metal-based drugs have been sought with the hope of finding an inexpensive metal complex with fewer adverse effects and improved therapeutic value. It is well known that ruthenium compounds with special properties to mimic iron by binding to biological molecules, reduce general toxicity and strong affinity to cancer tissues than normal tissues, which make them be potential candidates as anticancer drugs.7–16 So far, two ruthenium complexes have been proved to display potential anticancer activities; KP1019 and KP1339 have entered clinical trials.17,18 These complexes exhibit some advantages over cisplatin, for instance, their activity against cisplatin-resistant cancer cell lines and higher selectivity for cancer cells over normal cells, which can reduce side effects.19–22 Several mechanisms have been proposed to elucidate the anticancer activities of ruthenium complexes, including the inhibition of metastasis,23,24 interaction with DNA and proteins,25,26 generation of ROS,27 induction of apoptosis.28 Based on our previous work,29–33 we found that ruthenium(II) polypyridyl complexes appear to show high activity against cancer cells. To obtain more information on the anticancer activity of ruthenium complexes, in this study, a new ligand PTCP (PTCP = 2-phenanthren-9-yl-1H-1,3,7,8-tetraazacyclopenta[l]phenanthrene) and its three Ru(II) polypyridyl complexes: [Ru(N–N)2(PTCP)](ClO4)2 (N–N: phen = 1,10-phenanthroline 1; dmp = 2,9-dimethyl-1,10-phenanthroline 2 and ttbpy = 4,4′-di-tert-butyl-2,2′-bipyridine 3, Scheme 1) were synthesized and characterized by elemental analysis, ESI-MS, IR, 1HNMR, 13C NMR. The cytotoxicity in vitro of the ligand and complexes toward SGC-7901, HepG2, HeLa, SiHa and normal NIH3T3 cells was evaluated by MTT method. The apoptosis in SGC-7901 cells induced by the complexes was quantified with Annexin V/PI staining method. The cell cycle arrest, reactive oxygen species and mitochondrial membrane potential (MMP) in SGC-7901 cells were detected by flow cytometry. The matrigel invasion assay was investigated to evaluate the capacity of the complexes to inhibit the invasion of SGC-7901 cells. The expression of Bcl-2 family proteins was studied by western blot. Additionally, the NAC was used in the cell viability, ROS levels and MMP assay to compare with those incubated without NAC, which was used to further explore the apoptotic-inducing mechanism. The DNA-binding of the complexes with CT-DNA was performed by electronic absorption titration, luminescence spectra, viscosity measurements and molecular docking.
 |
| | Scheme 1 The synthetic route of ligand and complexes. | |
2. Results and discussion
2.1. Synthesis and characterization
The ligand PTCP was prepared through condensation of 1,10-phenanthroline-5,6-dione with phenanthrene-9-carbaldehyde using a similar method to that described by Steck and Day.34 The ruthenium(II) complexes were synthesized by the direct reaction with the appropriate precursor complexes in ethylene glycol. In the spectra of ESI-MS for the ruthenium(II) complexes, the expected signal of [M − 2ClO4]2+ was observed. The measured molecular weights were consistent with the expected values. In the 13C NMR spectra for complexes 2 and 3, the signals in the range of 24–36 ppm are attributed to the methyl group. The UV-vis and luminescence spectra of the complexes (5 μM) in PBS solution were detected. The UV-vis spectra of the complexes consist of two well resolved bands in the range 200–600 nm. The high-energy absorption bands below 300 nm are attributed to an intraligand (IL) p–p* transition. The low-energy absorption bands at 450–500 nm are assigned to a metal-to-ligand charge-transfer (MLCT) transition.
2.2. DNA-binding studies
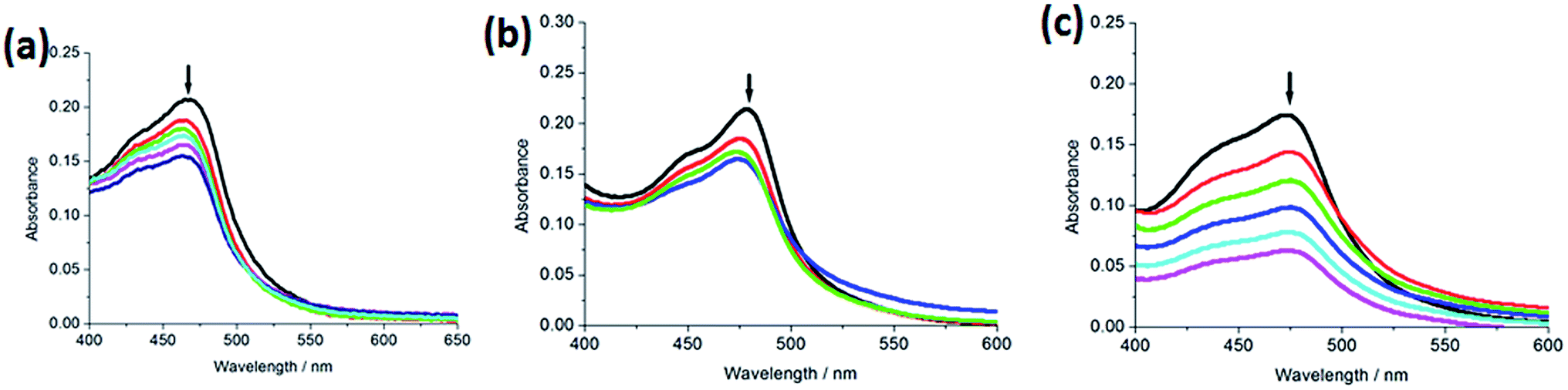
2.2.1. Electronic absorption titration. The absorption spectra of the complexes (5 μM) 1–3 in the absence or presence of increasing amounts of CT-DNA are shown in Fig. 1. With increasing concentration of CT-DNA, a red shift of 2 nm for 1, 5 nm for 2 and 2 nm for 3 was observed at the MLCT bands. The percentage hypochromicity at the MLCT band of complexes 1 (464 nm), 2 (473 nm) and 3 (469 nm) upon binding to DNA was determined to be 17.7%, 33.3% and 28.2%, respectively. In order to quantitatively compare the binding strength of the complexes, the intrinsic binding constant Kb of the complexes with CT-DNA was determined by monitoring the changes of absorbance at the MLCT band with increasing concentration of DNA. The values of Kb are 7.24 (±0.21) × 103 M−1 for 1, 5.82 (±0.15) × 103 M−1 for 2 and 3.63 (±0.11) × 103 M−1 for 3, respectively. The DNA-binding strength of the complexes follows the order of 2 > 1 > 3. Substitution on the 2- and 9-positions of the ancillary phen ligands may cause severe steric constraints near the RuII core when the complex interacts into the DNA base pairs. The methyl groups may come into close proximity of base pairs at the interaction sites. These steric clashes cause a diminution of the intrinsic constant. Thus, complex 2 shows less DNA-binding constant than complex 1. Owing to large steric hindrance of tertiary butyl, complex 3 displays the least DNA-binding constant among the three complexes. The DNA-binding constants are less than those of so-called DNA-intercalative Ru(II) complexes (1.1 × 104 to 4.8 × 104 M−1).35,36 Thus, we consider that the complexes interact with CT-DNA through partial intercalative mode.
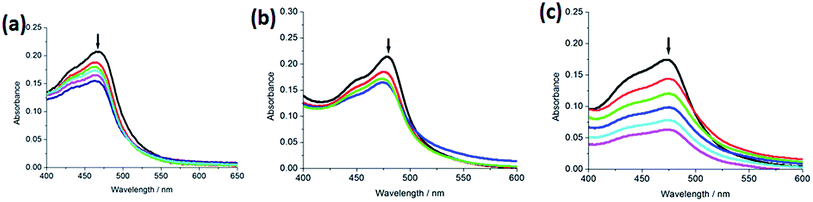 |
| | Fig. 1 Absorption spectra of complex in Tris–HCl buffer upon addition of CT-DNA at room temperature in the presence of complexes 1 (a), 2 (b) and 3 (c), [Ru] = 5 μM, [DNA] = 0–19.8 μM. Arrow shows the absorbance changing upon the increase of DNA concentration. Plots of (εa − εf)/(εb − εf) vs. [DNA] for the titration of DNA with Ru(II) complexes. | |
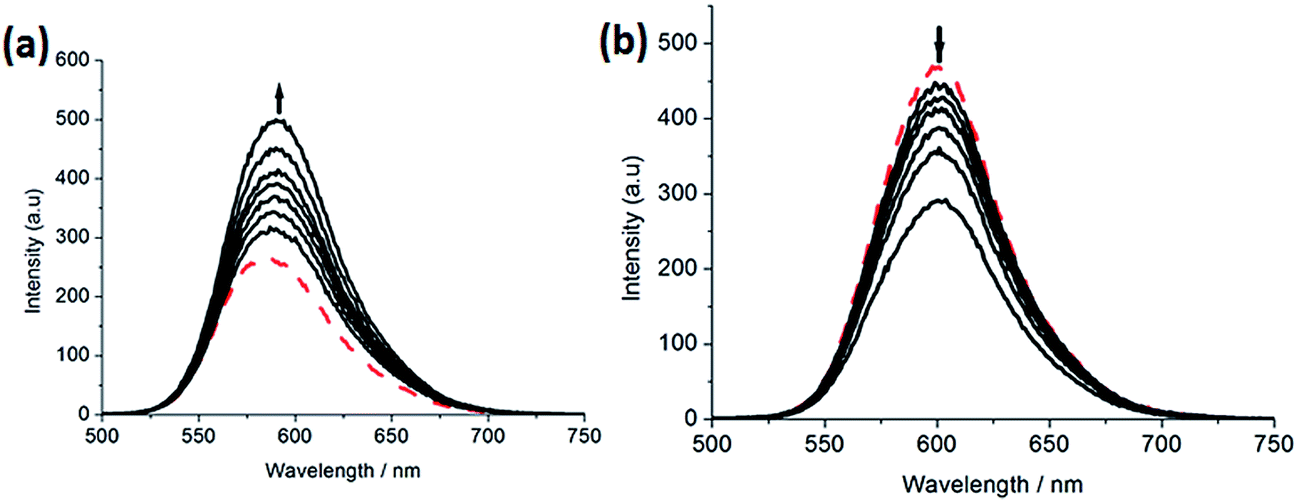
2.2.2. Luminescence spectra. Complex 2 can not emit luminescence. Complexes 1 and 3 can emit luminescence in Tris buffer at ambient temperature, with a maximum appearing at 583 and 601 nm, respectively. As shown in Fig. 2, upon addition of CT-DNA, the emission intensity caused by 1 (a) grows about 1.78 times larger than that in the absence of DNA. This implies that complex 1 can interact with DNA and be protected by DNA efficiently, since the hydrophobic environment inside the DNA helix reduces the accessibility of solvent water molecules to the complex and the complex mobility is restricted at the binding site, leading to a decrease of the vibrational modes of relaxation. However, with the increasing concentration of DNA, the emission intensity induced by complex 3 (b) decreases about 1.73 times than that of the original.
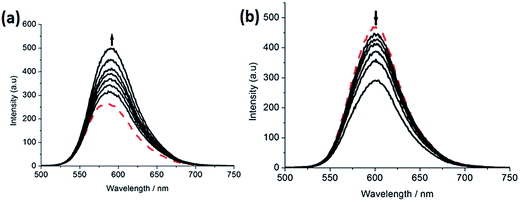 |
| | Fig. 2 Emission spectra of complexes 1 (a) and 3 (b) in Tris–HCl buffer in the absence and presence of CT-DNA. Arrow shows the intensity change upon increasing DNA concentrations. | |
2.3. Viscosity measurements
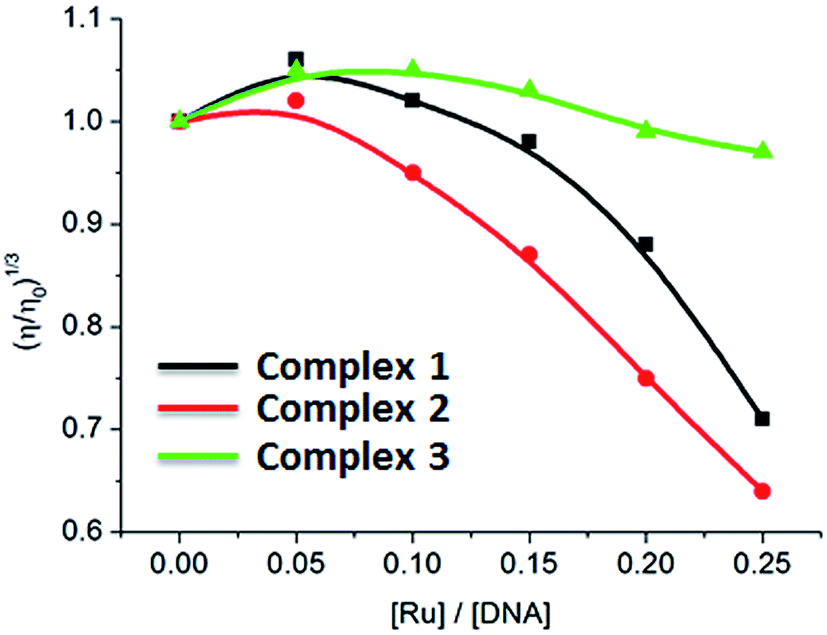
Viscosity measurements that are sensitive to length change of DNA are regarded as the least ambiguous and the most critical tests of binding mode in solution in the absence of crystallographic structural data or NMR spectra.37,38 It is popularly accepted that a classical intercalation mode results in lengthening the DNA helix, as base pairs are separated to accommodate the binding ligand, leading to the increase of DNA viscosity. In contrast, a partial and/or non-classical intercalation of ligand could bend (or kink) the DNA helix, reduce its effective length and, concomitantly, its viscosity.37,38 As shown in Fig. 3, the relative viscosity of DNA increases at a low ratio of [Ru]/[DNA]. However, on increasing concentration of complexes 1–3, the relative viscosity of DNA solution decreases. The viscosity results demonstrate that the complexes interact with DNA through partial intercalation.
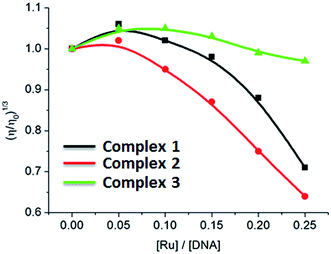 |
| | Fig. 3 Effect of increasing amounts of complexes 1–3 on the relative viscosity of CT-DNA at 25 (±0.1) °C, [DNA] = 3.97 mM. | |
2.4. Molecular docking with DNA
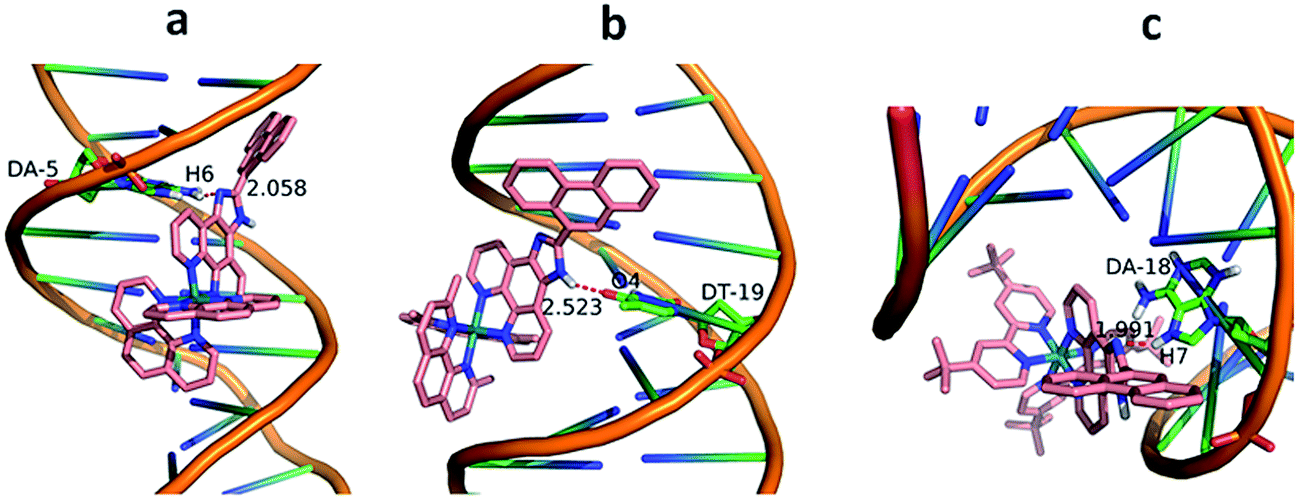
The molecular docking technique can contribute to rational drug design and mechanistic studies by placing a small molecule into the binding site of the DNA target specific region mainly in a non-covalent mode.39 To explore the most feasible binding site, interaction mode and binding affinity docking studies have been performed on complexes 1–3 with B-DNA (PDB ID: 1BNA). As shown in Fig. 4, the complexes interact with DNA through partial intercalation at the major groove. The resulting relative binding energies of docked 1, 2 and 3 with DNA are −37.18, −31.95 and −9.70 kJ mol−1, respectively. The more negative binding energy shows more potent DNA-binding affinity. Thus, the DNA-binding affinities follow the order of 1 > 2 > 3. This is completely consistent with the results obtained from the electronic absorption titration. In addition, the results indicate that there are certain hydrogen-bonding interaction between the complexes and DNA. The length of these hydrogen bonds are 2.058 Å (N (imidazole)⋯H6 (DA-5)) for 1, 2.523 Å (H (imidazole N)⋯O4 (DT-19)) for 2 and 1.991 Å (N (imidazole)⋯H7 (DA-18)) for 3, respectively.
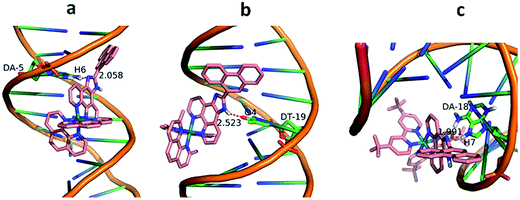 |
| | Fig. 4 Molecular docked models of complexes 1 (a), 2 (b) and 3 (c) with DNA (PDB 1D:1BNA). The hydrogen bonds between the complexes and DNA are labeled using red dashed lines. | |
2.5. Cytotoxic activity in vitro assay
The cytotoxic activity in vitro of the ligand and complexes against cancer cell lines was determined by MTT method, and cisplatin was used as a positive reference. The IC50 values of the ligand and complexes are listed in Table 1. The ligand shows high cytotoxicity toward HepG2, HeLa cancer cell lines and normal NIH3T3 cells. However, when ligand bonded to metal to form complexes, the cytotoxicity activity against normal cell line has been reduced. As expectation, complexes 1, 2 and 3 display high cytotoxicity activity toward SGC-7901 cells and low cytotoxicity toward normal cells (NIH3T3). Unexpectedly, all complexes have no cytotoxicity on HepG2 cells and complexes 3 show high cytotoxic effect against NIH3T3 cells. All the complexes exhibit lower cytotoxic activity than cisplatin toward the selected cell lines, but comparable to that of [Ru(bpy)2(TCPI)]2+ (IC50 = 18.3 ± 2.2 μM, TCPI = 2-(3-1H-1,3,7,8-tetraazacyclopenta[l]phenanthren-2-yl)phenylbenzo[de]isoquinoline-1,3-dione).40 Through comparison of the cytotoxic activity of the same complex against different cancer cell lines or different complexes against the same cancer cell line, we conclude that (1) the same complex shows different cytotoxic effect on the different cell growth, this may be related to the amount which the complex enters into the cell. (2) Different complexes also displays different cytotoxic activity against the same cancer cell, this may be caused by different structures of the different complexes. Comparing the IC50 values of complexes toward the cancer cell lines, we found that SGC-7901 was more sensitive to complexes than others. Thus this cell was selected for the following experiments.
Table 1 The IC50 (μM) values of ligand and complexes toward the selected cell lines
| Complexes |
SGC-7901 |
HepG2 |
HeLa |
SiHa |
NIH3T3 |
| PTCP |
>100 |
0.8 ± 0.3 |
2.7 ± 0.5 |
>100 |
2.7 ± 0.5 |
| 1 |
14.5 ± 1.8 |
>100 |
72.3 ± 4.8 |
61.3 ± 5.7 |
>100 |
| 2 |
12.5 ± 0.6 |
>100 |
>100 |
>100 |
>100 |
| 3 |
18.2 ± 1.7 |
>100 |
>100 |
38.5 ± 3.3 |
9.0 ± 1.3 |
| Cisplatin |
3.4 ± 0.4 |
23.4 ± 2.5 |
7.2 ± 1.2 |
13.4 ± 2.1 |
— |
2.6. Apoptosis assay by flow cytometry
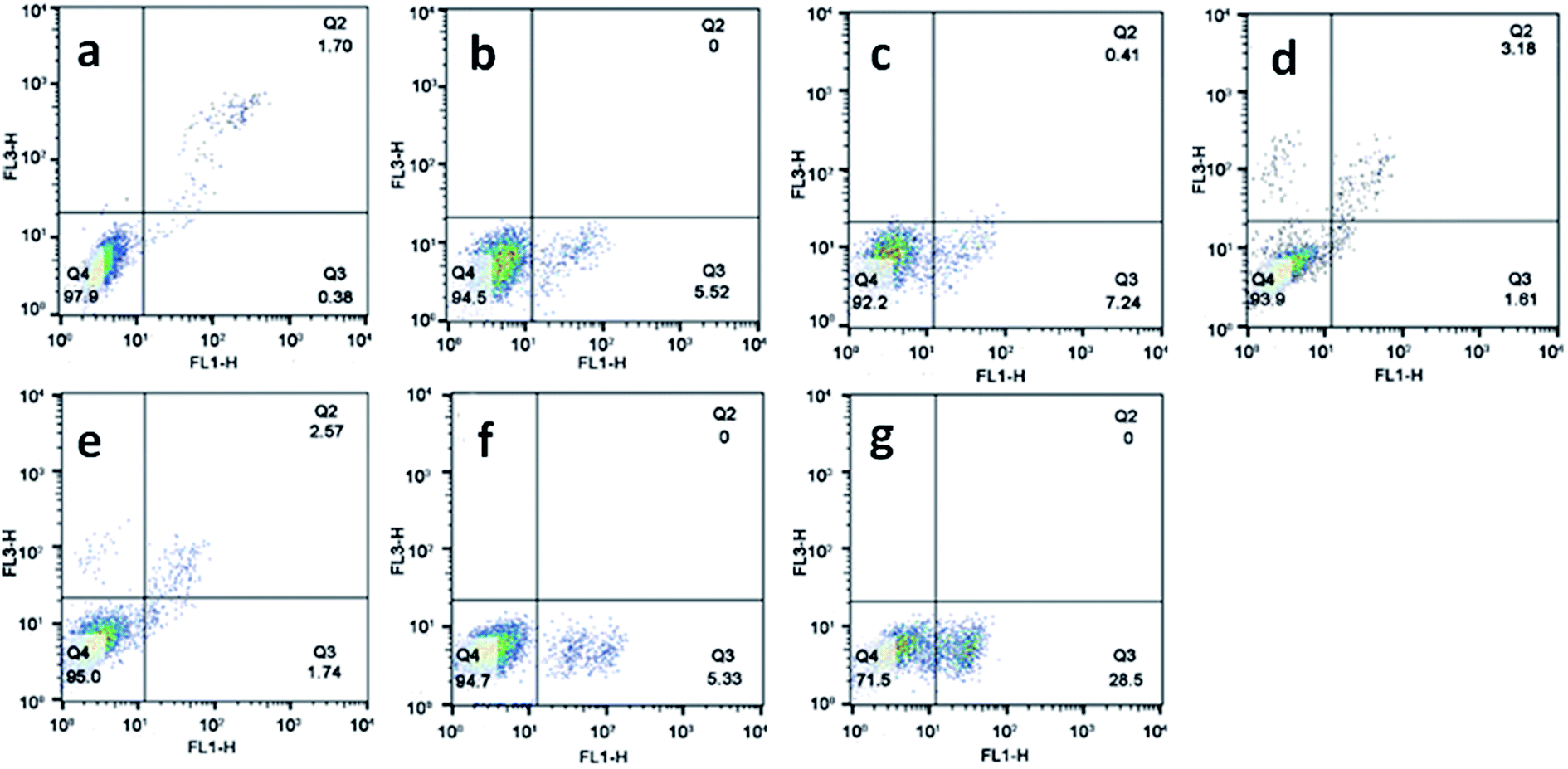
Apoptosis is a normal phenomenon in the process of development and growth in organisms,41 and many diseases including cancer have been connected with the excessive activation or inhibition of apoptotic signaling pathway.42,43 To evaluate the effect of the complexes on apoptosis in SGC-7901 cell, we used flow cytometry analysis to determine the percentage of apoptotic cells. As shown in Fig. 5, in the control (a), the percentage of the apoptotic cells is 0.38%. SGC-7901 cells were treated with 12.5 and 25.0 μM of complexes 1 (b and c), 2 (d and e) and 3 (f and g) for 24 h, the percentages of apoptotic cells are 5.52% and 7.24% for 1, 1.61% and 1.74% for 2, 5.33% and 28.5% for 3, respectively. Complex 3 shows the highest apoptotic effect on SGC-7901 cells among these complexes. Furthermore, the complexes induce apoptosis in a concentration-dependent manner. The apoptotic effect of the complexes against SGC-7901 cell follows the order of 3 > 1 > 2. This is not consistent with the order of cytotoxic activity of 2 > 1 > 3.
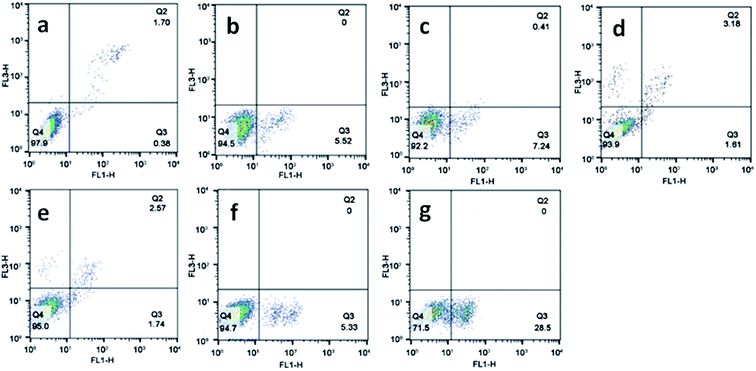 |
| | Fig. 5 The percentage of apoptotic cell was determined by flow cytometry. SGC-7901 cells (a) exposure to 12.5 and 25.0 μM of complexes 1 (b and c), 2 (d and e) and 3 (f and g) for 24 h. | |
2.7. DNA damage studies
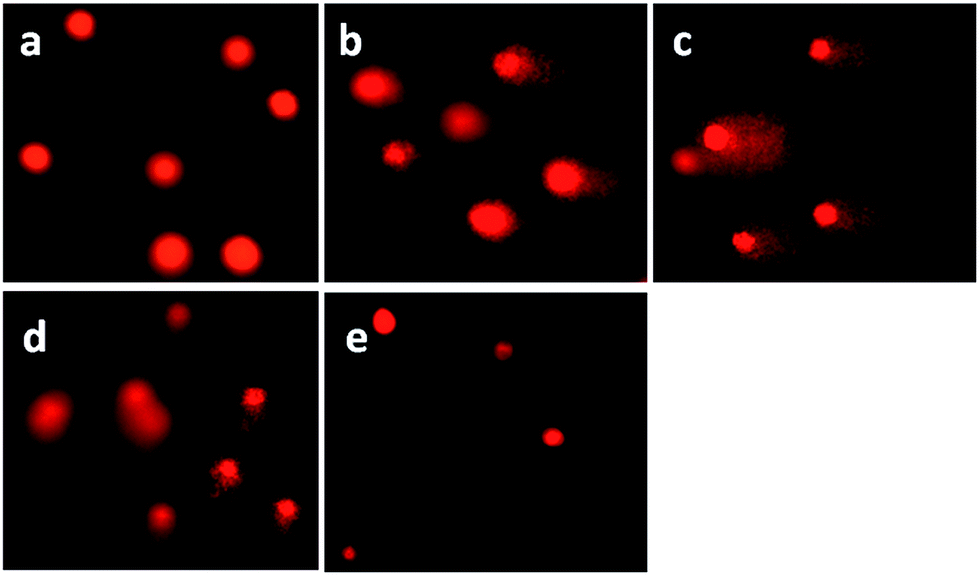
DNA damage was quantified through the single cell gel electrophoresis, using the comet assay.44 DNA fragmentation is a hallmark of apoptosis, mitotic catastrophe or both.45 DNA strand breaks were detected in SGC-7901 cells upon treatment with the complexes in a cell-based alkaline comet assay, cisplatin was used as a positive control. As shown in Fig. 6, in the control (a), SGC-7901 cells fail to show any comet-like appearance. Exposure of SGC-7901 cells to cisplatin (2.5 μM, b) and 12.5 μM of complexes 1 (c), 2 (d) and 3 (e) for 24 h induced considerable strand breakage on chromosomal DNA and led to the appearance of an obscure “halo” around the nucleus of the SGC-7901 cells. The statistically significant and well-formed comets were observed that indicated severe DNA damage. The results of the comet assay reveal that the DNA of a single cell underwent degradation as a consequence of direct DNA damage or rapid apoptosis.46
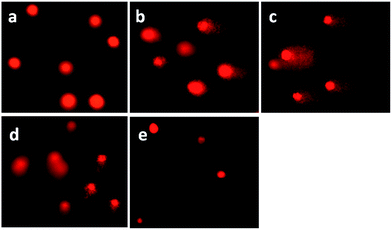 |
| | Fig. 6 Comet assay of SGC-7901 cell (a) exposure to 2.5 μM of cisplatin (b) and 12.5 μM of complexes 1 (c), 2 (d) and 3 (e) for 24 h. Cisplatin was used as a positive control. | |
2.8. Determination of the level of reactive oxygen species (ROS)
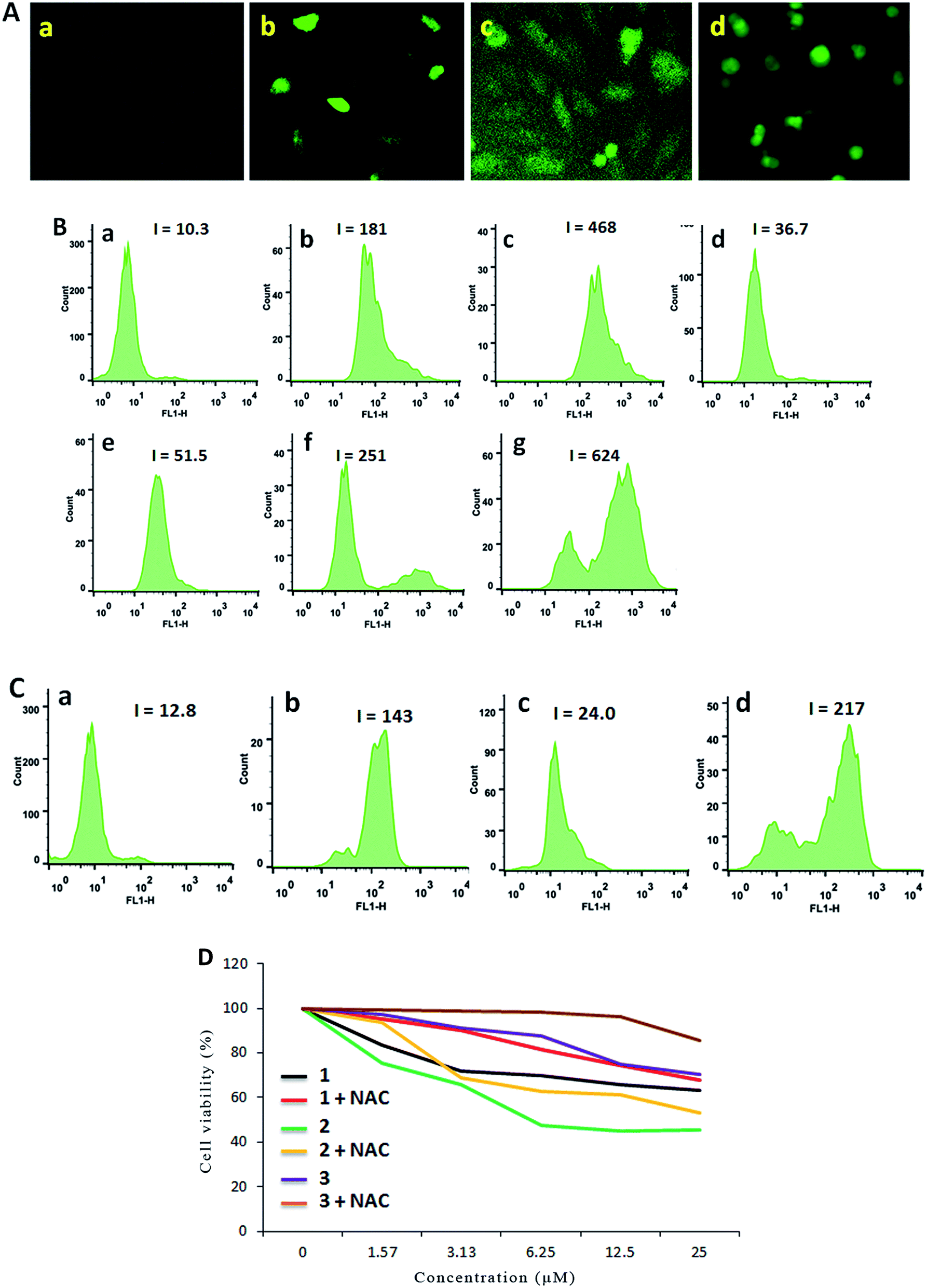
It is well known that pharmacological ROS damage might be a potential element to eliminate cancer cells, which suggests that ROS can induce apoptosis in cancer cells.47 The literature reported that there is an inseparable relationship between the apoptosis or senescence process in cancer cells and the levels of ROS.48 Thus we speculated that the apoptosis of SGC-7901 cells may be mediated by ROS. To verify whether or not our speculation is correct, DCFH-DA was used as a fluorescent probe to determine the change of the level of the ROS induced by the complexes. Inside the cell, DCFH-DA is transferred to a fluorescent product, namely, dichlorofluorescein (DCF).49,50 As shown in Fig. 7A, in the control (a), no obvious green spots were found. After the treatment of SGC-7901 cells with 12.5 μM of complexes 1 (b), 2 (c) and 3 (d) for 24 h, the bright green spots were observed. Comparing the green fluorescent intensity, complex 2 induces weaker green fluorescence than those induced by complexes 1 and 3 under identical conditions. To quantitatively compare the effect of the complexes on ROS levels, the DCF fluorescent intensity is determined by flow cytometry. As shown in Fig. 7B, in the control (a), the DCF fluorescence intensity is 10.3. When SGC-7901 cells were incubated with 25 μM of complexes 1 (c), 2 (e) and 3 (g), the DCF fluorescent intensity increases 45.4, 5.0 and 60.6 times than the original, respectively. The effect of the complexes on ROS levels follows the order of 3 > 1 > 2. This is consistent with the apoptotic effect. Moreover, the ROS levels induced by the complexes show a concentration-dependent manner. N-Acetylcysteine (NAC, 5 mM) is an inhibitor to inhibit the production of ROS. In the presence of NAC, the effect of the complexes on the ROS levels are shown in Fig. 7C, in the control (a), the DCF fluorescence intensity is 12.8. After SGC-7901 cells were exposed to 12.5 μM of complexes 1 (b), 2 (c) and 3 (d), the DCF fluorescence intensity increases 11.2, 1.88 and 16.9 times than the original. The increasing degrees are less than those in the absence of NAC (17.6 times for 1, 3.6 times for 2 and 24.4 times for 3). This demonstrates that the addition of NAC into the complexes lead to a decrease in the ROS levels. Although complex 2 shows higher cytotoxic activity against SGC-7901 cell, this complex displays lower effect on the production of ROS levels. Additionally, we also investigated the relationship between cell viability and ROS levels. As shown in Fig. 7D, in the presence of NAC, the cell viability induced by the complexes increases, which indicates that ROS levels can enhance the ability of the complexes to kill cancer cells.
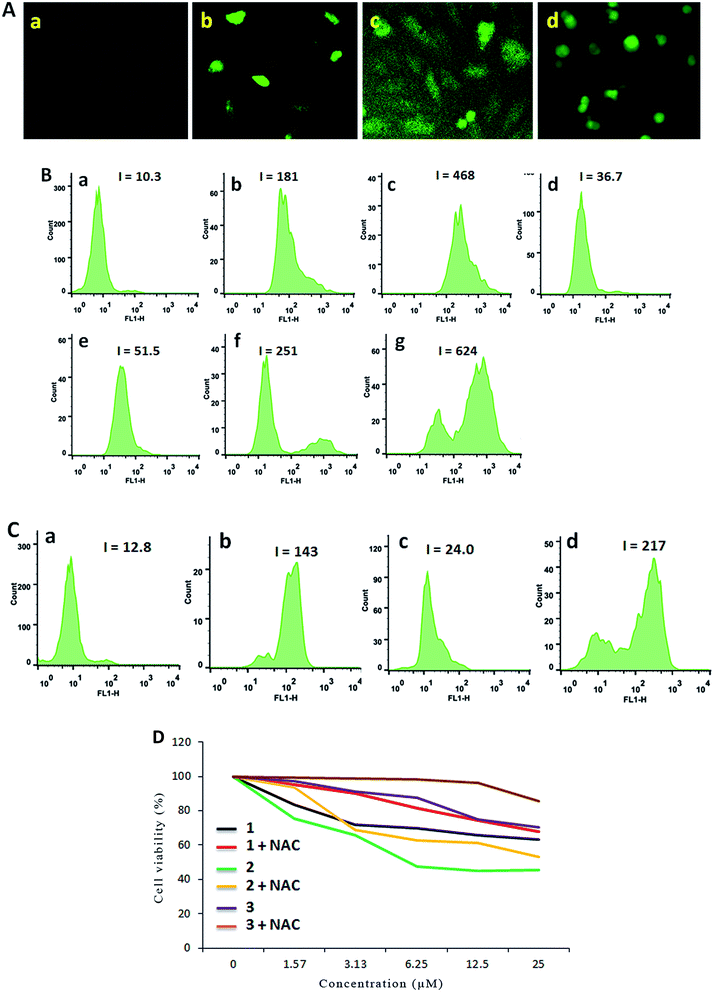 |
| | Fig. 7 (A) Intracellular ROS was detected in SGC-7901 cells (a) exposure to 12.5 μM of complexes 1 (b), 2 (c) and 3 (d) for 24 h. (B) The DCF fluorescence intensity was determined in SGC-7901 cells (a) exposure to 12.5 and 25.0 μM of complexes 1 (b and c), 2 (d and e) and 3 (f and g) for 24 h. (C) In the presence of NAC, the DCF fluorescence intensity was detected in SGC-7901 cells (a) exposed to 12.5 μM of complexes 1 (b), 2 (c) and 3 (d) for 24 h. (D) The cell viability induced by the complexes for 24 h was assayed in the absence or presence of NAC. | |
2.9. Mitochondrial membrane potential detection
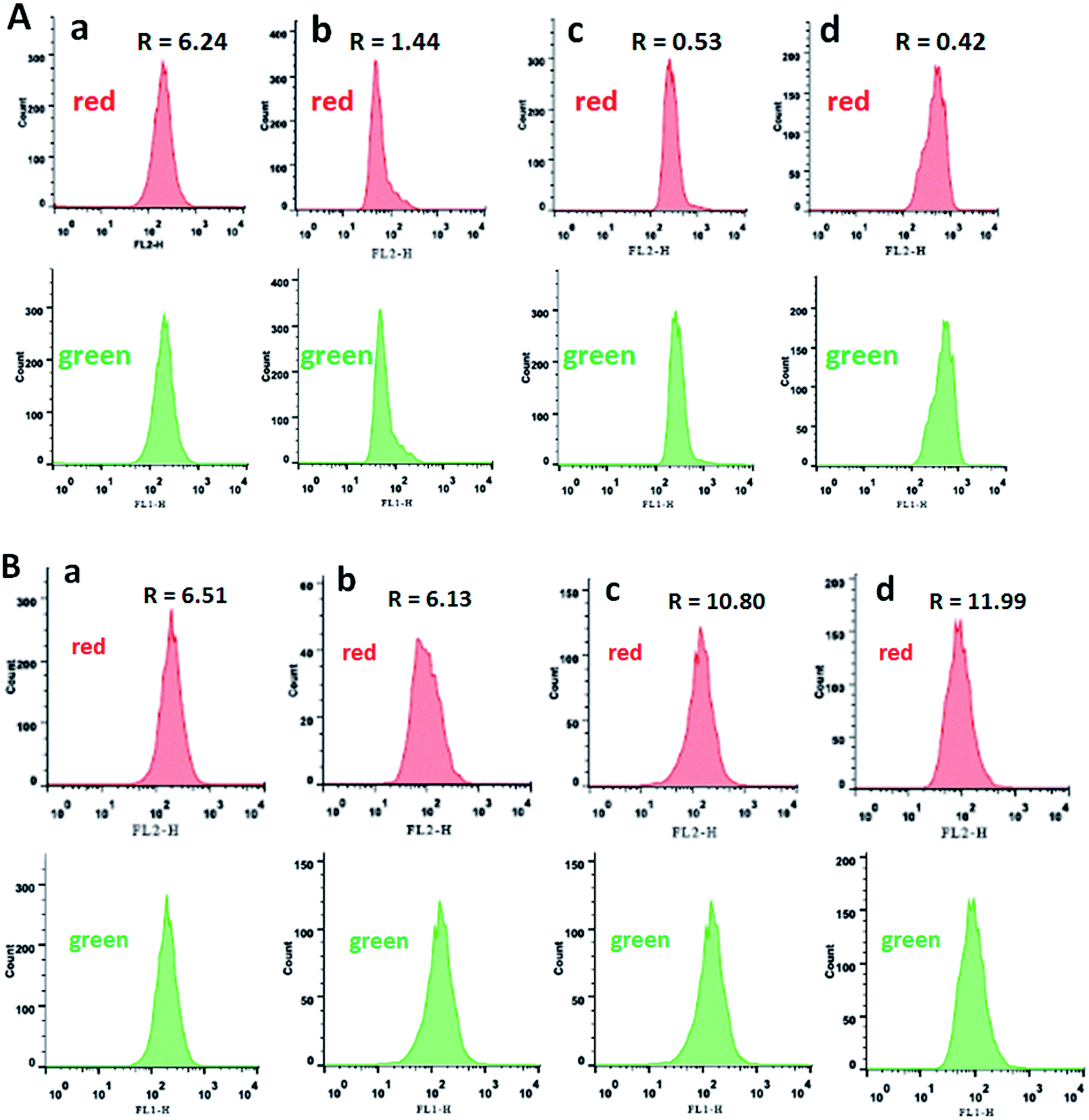
The inner mitochondrial membrane is a main position of production of ROS produced as co-products of the mitochondrial electron transport chain. Mitochondria are a critical organelle involved in maintaining cellular homeostasis and producing cellular energy (adenosine triphosphate, ATP) depending on oxidative phosphorylation,51,52 which induces apoptosis as the intrinsic apoptotic pathway and adjusts calcium homeostasis.53,54 It is confirmed that the mitochondria membrane potential (MMP, ΔΨm) is a major symbol in the early apoptosis, and JC-1 was used as fluorescent probe to measure the MMP.55–57 The above mentioned ROS levels assay shows that apoptosis in SGC-7901 cells treated with complexes 1–3 was induced by excessive release of ROS. According to the mitochondrial membrane potential assay kit, JC-1 accumulates in matrix to form JC-1 aggregates which can emit red fluorescence corresponding to high mitochondrial membrane potential. On the contrast, JC-1 forms monomer to emit green fluorescence corresponding to low mitochondrial membrane potential. The ratio of red/green fluorescence intensity was determined by flow cytometry. As shown in Fig. 8A, in the control (a, up and down), the ratio of the red/green fluorescence is 6.24. The complexes (12.5 μM) treated SGC-7901 cells for 24 h, the ratios of the red/green are 1.44, 0.53 and 0.42 for 1–3, respectively. The reduction in the ratio indicates that the red fluorescence decreases and the green fluorescence increases, which suggests that the complexes can induce a reduction in the mitochondrial membrane potential. To explore the influence of NAC on the changes in mitochondrial membrane potential, in the presence of NAC, the ratios of the red/green fluorescence are shown in Fig. 8B. Comparing the ratio of the red/green fluorescence in the presence of NAC with those in the absence of NAC, the ratios in the presence of NAC increase, which indicates that high ROS level induces more changes in the mitochondrial membrane potential.
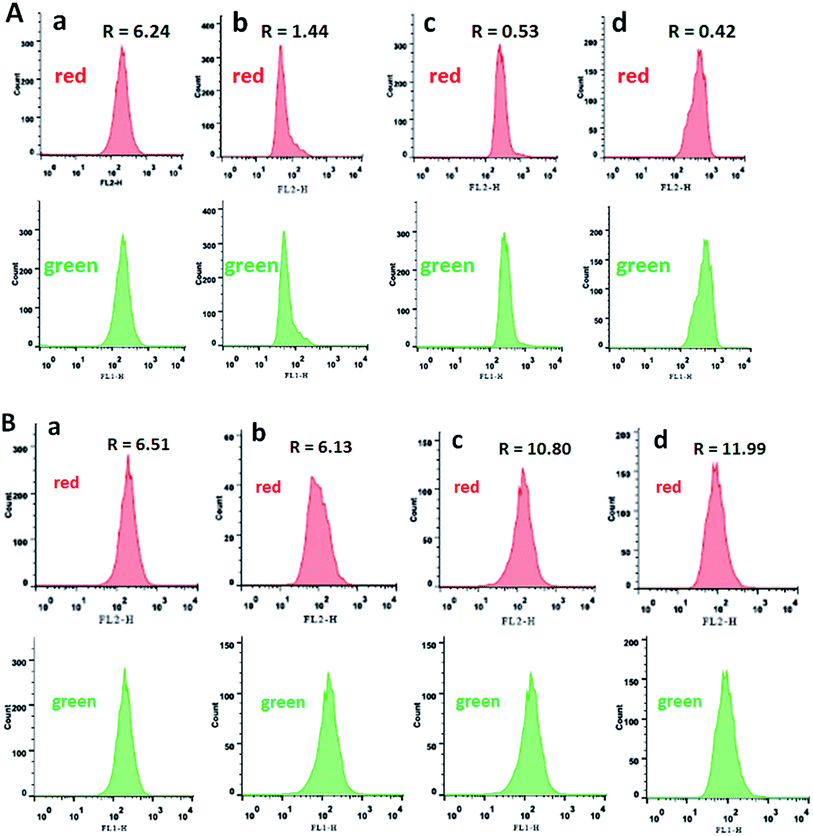 |
| | Fig. 8 The ratio of red/green fluorescence was determined in the absence (A) or presence (B) of NAC while SGC-7901 cells (a) were treated with 12.5 μM of complexes 1 (b), 2 (c) and 3 (d) for 24 h. | |
2.10. Cell cycle arrest studies
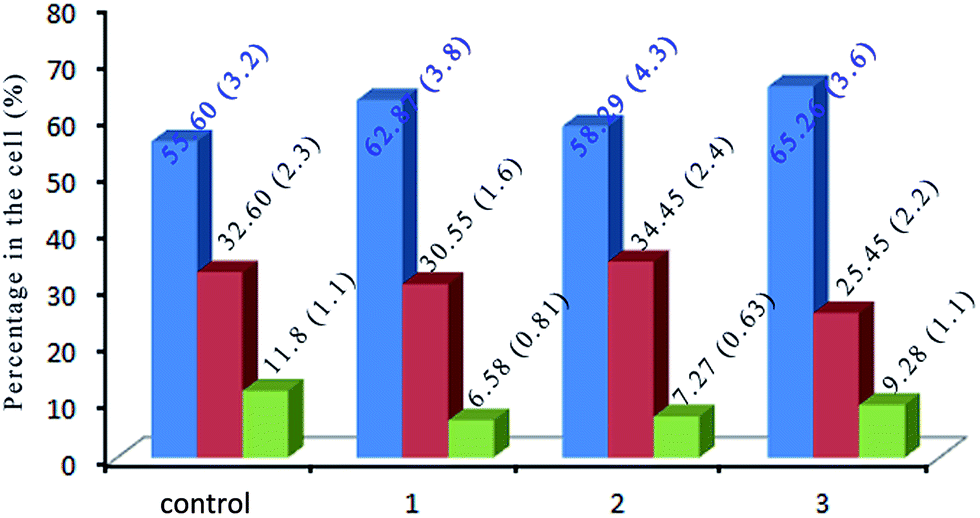
DNA is the most highlight of the target for anticancer drugs to exert toxic effects.58 Most anti-tumor drugs eliminate cancer cells by inducing apoptosis,59,60 while others cause cell death via disturbing the mitotic process.61 In order to evaluate the effect of the complexes on cell cycle distribution of SGC-7901, the cell cycle arrest of SGC-7901 cells were investigated by flow cytometry. As shown in Fig. 9, in the control, the percentage in the cells at G0/G1 is 55.60 (±3.2)%. After the treatment of SGC-7901 cells with 12.5 μM of the complexes, an increase of 7.27% for 1, 2.69% for 2 and 9.66% for 3 in the percentage at G0/G1 phase was observed, which was accompanied by the corresponding reduction of 5.22% for 1, 4.53% for 2 and 2.52% for 3 in the percentage of cells in the G2/M phase. The data demonstrates that the complexes inhibit the cell growth in the G0/G1 phase.
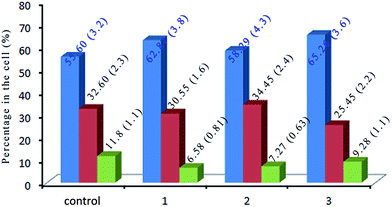 |
| | Fig. 9 The cell cycle distribution of SGC-7901 cells (control) exposure to 12.5 μM of complexes 1, 2 and 3 for 24 h. | |
2.11. Cell invasion studies
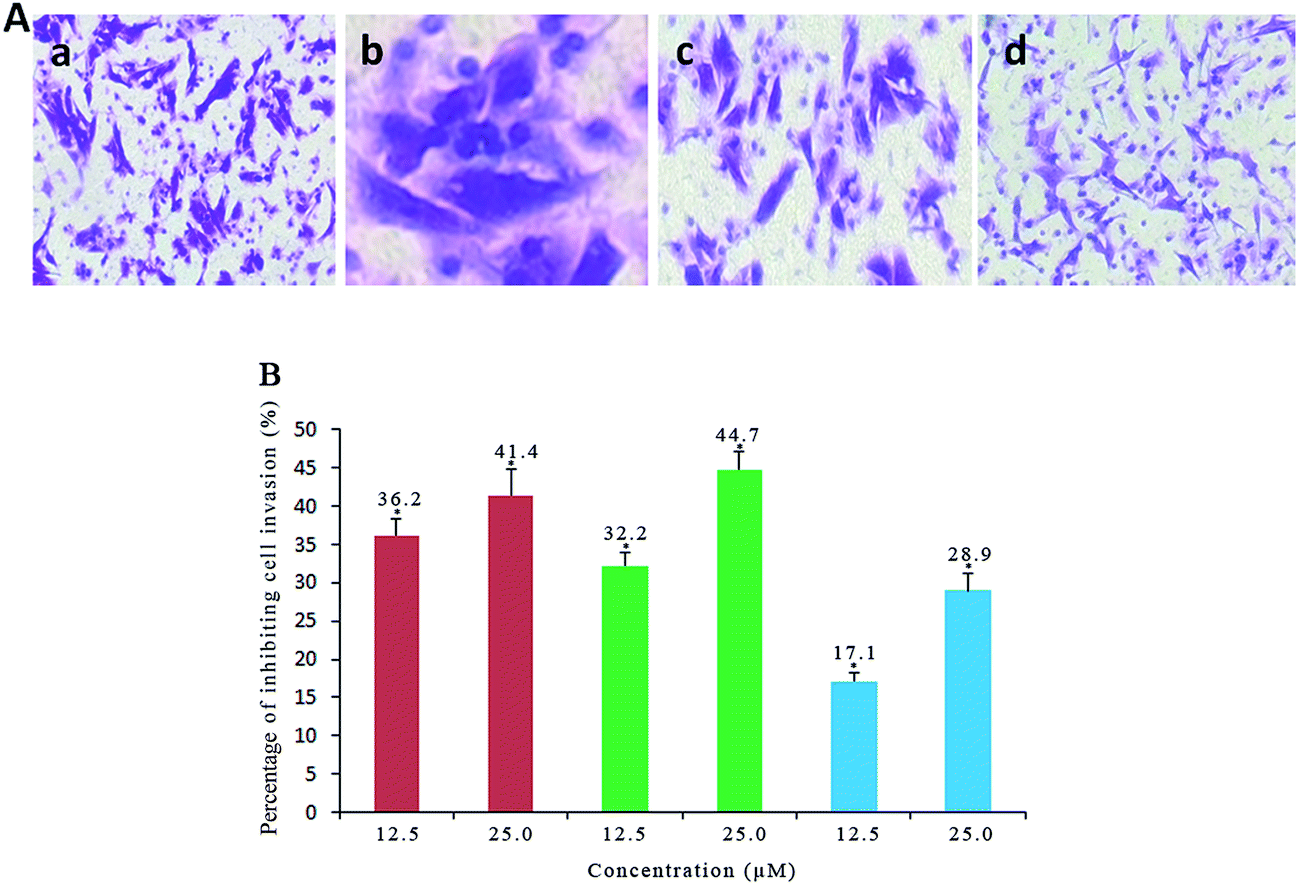
Cell invasion is a process associated with cancer metastasis, which refers to the cell migration through an extracellular matrix.62 To investigate the efficiency of complexes 1, 2 and 3 on inhibiting the cell invasion, the Matrigel invasion assay was performed. As shown in Fig. 10A and B, the percentage of inhibiting the cell invasion is 41.4% for 1, 44.7% for 2 and 28.9% for 3 after SGC-7901 cells were exposed to 25.0 μM of the complexes, respectively. It is clear that the inhibiting effects of the complexes display a concentration-dependent manner. The effect of the complexes at 25.0 μM on the cell invasion follows the order of 2 > 1 > 3. This order is consistent with the cytotoxic activity of the complexes toward SGC-7901 cells.
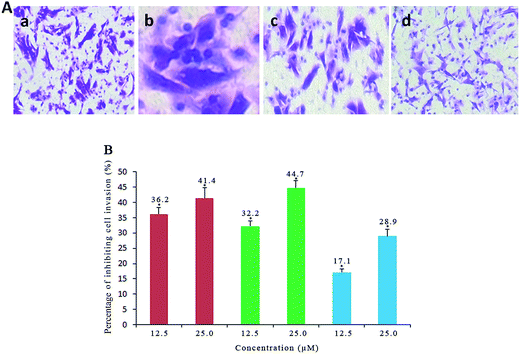 |
| | Fig. 10 (A) Microscope images of invading SGC-7901 cells (a) that have migrated through the Matrigel induced by 12.5 μM of the complexes 1 (b), 2 (c) and 3 (d) for 24 h. (B) The percentage of invading SGC-7901 cells induced by different concentration of the complexes 1 (red), 2 (green) and 3 (blue) for 24 h. *p < 0.05 represents significant differences compared with control. | |
2.12. Autophagy induced by the complexes
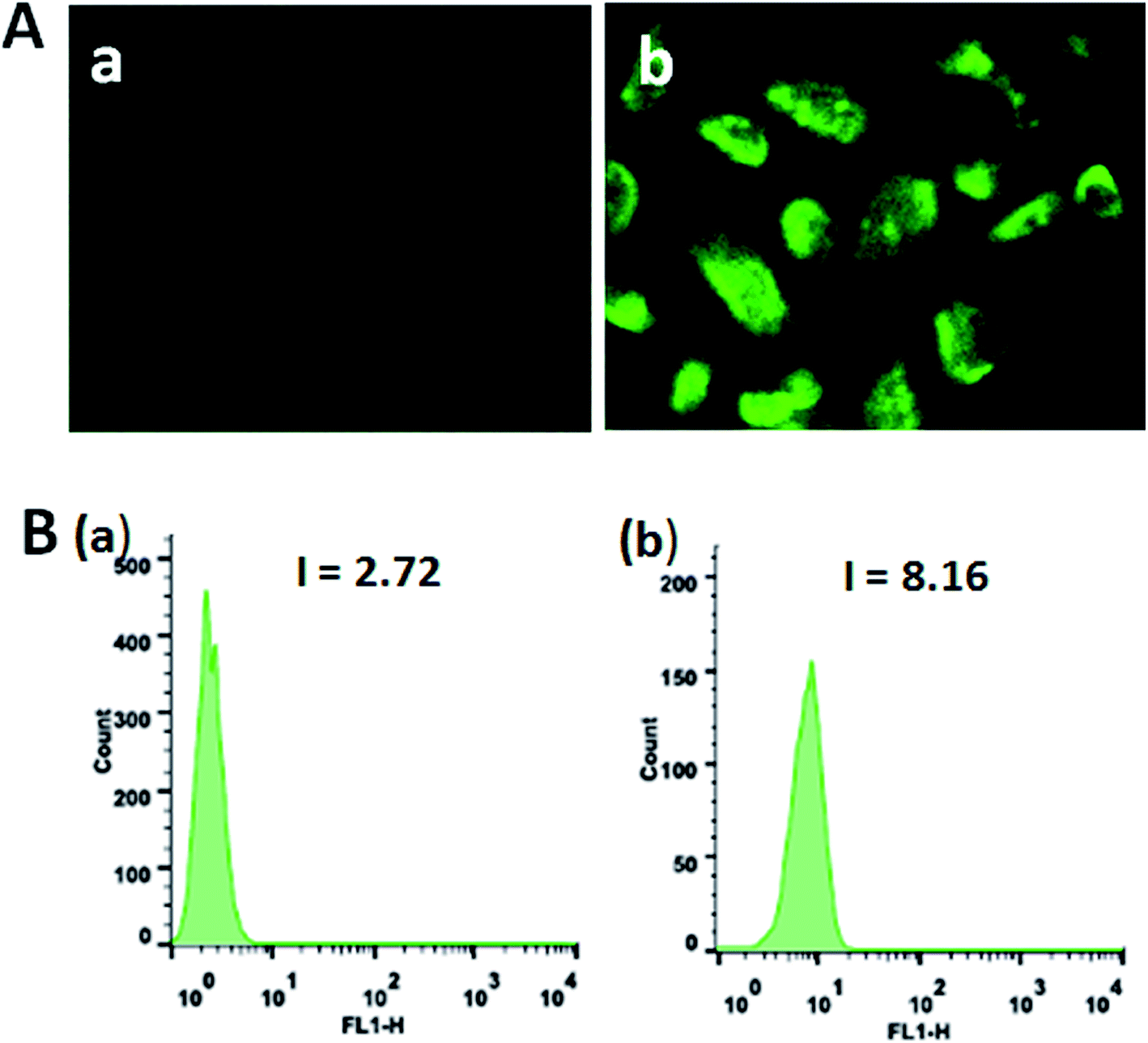
Autophagy is an evolutionarily conserved process degrading cellular proteins and cytoplasmic organelles, in which the phagophores surround and pack organelles to form autophagosomes.63 The effect of the complexes on autophagy was studied using monodansylcadaverine (MDC) as fluorescent probe. MDC is a specific, in vivo marker for autophagic vacuoles, and MDC incorporation is an indicator of autophagic activity.64 As shown in Fig. 11A, in the control (a), no obvious spots are found. The treatment of SGC-7901 cells with 12.5 μM of complexes 1 and 2 for 24 h, no fluorescence spots are observed (data not present), which indicates that complexes 1 and 2 can not induce autophagy. However, SGC-7901 cells were treated with complex 3 (b), the MDC labeling green fluorescent points in the cytoplasm were discovered, which suggests that the autophagic vacuoles were formed. The MDC fluorescence intensity was determined by flow cytometry. As shown in Fig. 11B, in the control (a), the fluorescence intensity is 2.72. After SGC-7901 cells were exposed to 12.5 μM of complex 3 for 24 h, the fluorescence intensity is 8.16. The MDC fluorescence intensity increases 3 times than the original, which also indicates that complex 3 can induce autophagy.
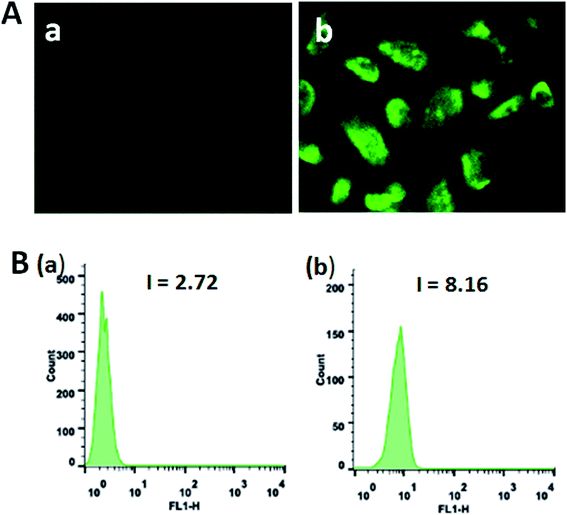 |
| | Fig. 11 (A) Autophagy was assayed with MDC staining in SGC-7901 cells (a) induced by 12.5 μM of complex 3 (b) for 24 h. (B) The MDC green fluorescence intensity in SGC-7901 cells (a) exposed to 12.5 μM of complex 3 (b) for 24 h. | |
2.13. Western blot analysis
The transduction of apoptotic signals requires the activation of a cascade of cysteine proteases and caspases. Caspase 3, in particular, plays a central role in the execution of apoptosis.65 Bcl-2 family proteins maintain the balance of cell death and survival, which plays a key role in the regulation of apoptosis via the control of mitochondrial membrane.66 NAC was used to investigate whether ROS influence the expression of those proteins or not. After treatment of SGC-7901 cells with 12.5 μM of complexes 1–3, the results are shown in Fig. 12. Complexes 1–3 result in an increase in the expression of caspase-3, Bax and Bid compared to the control. In the presence of NAC, the expression of caspase 3, Bcl-2 and Bcl-x down-regulates, whereas the expression of Bax and Bid up-regulates. This shows that NAC can regulate the expression of Bcl-2 family proteins.
 |
| | Fig. 12 Western blot analysis of caspase 3, Bcl-2, Bcl-x, Bax and Bid in SGC-7901 cells treated with 12.5 μM of complexes 1–3 in the absence and presence of NAC for 24 h. SGC-7901 (a), SGC-7901 + NAC (b), 1 (c), 1 + NAC (d), 2 (e), 2 + NAC (f), 3 (g) and 3 + NAC (h). β-actin was used as internal control. | |
3. Conclusions
In summary, we have synthesized three ruthenium(II) complexes and investigate DNA-binding and the probable anticancer mechanism in vitro of the complexes against SGC-7901. These complexes interact with CT-DNA through partial intercalative mode at DNA major groove. The complexes show high cytotoxic activity toward SGC-7901 cells. They can increase the ROS levels and induce a decrease in the mitochondrial membrane potential. The complexes inhibit the cell growth at G0/G1 phase, and down-regulate the expression of Bcl-2 and Bcl-x, and up-regulate the expression of Bax and Bid. Take above together, we consider that the complexes induce apoptosis in SGC-7901 cell through the following two pathways (Fig. 13): (1) firstly, the complexes enhance ROS levels, and then the increase of ROS levels induces a decrease in the mitochondrial membrane potential. Secondly, the changes of mitochondrial membrane potential activate the expression of caspase 3. Thirdly, the activation of caspase stimulates the cell apoptosis. (2) The complexes can cause DNA damage, and then the complexes inhibit the cell growth at G0/G1 phase. Finally, the complexes induce cell apoptosis. This work will be helpful for design and synthesis of new ruthenium(II) complexes as potent anticancer drugs.
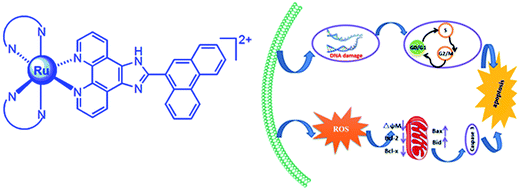 |
| | Fig. 13 The molecular mechanism of the complexes inducing apoptosis in SGC-7901 cell. | |
4. Experimental
4.1. Materials and methods
The reagents and solvents used in the experiments were purchased commercially and used without further purification unless special statement. Ultrapure water was used in all experiments Calf Thymus DNA (CT-DNA) was obtained from the Sino-American Biotechnology Company. pBR 322 DNA was obtained from Shanghai Sangon Biological Engineering & Services Co., Ltd. Doubly distilled water was used to prepare buffers (5 mM Tris(hydroxymethylaminomethane)–HCl, 50 mM NaCl, pH = 7.2). A solution of calf thymus DNA in the buffer gave a ratio of UV absorbance at 260 and 280 nm of ca. 1.8–1.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating that the DNA was sufficiently free of protein.67 The DNA concentration per nucleotide was determined by absorption spectroscopy using the molar absorption coefficient (6600 M−1 cm−1) at 260 nm.68
:1, indicating that the DNA was sufficiently free of protein.67 The DNA concentration per nucleotide was determined by absorption spectroscopy using the molar absorption coefficient (6600 M−1 cm−1) at 260 nm.68
DMSO and RPMI 1640 (Roswell Park Memorial Institute) were purchased from GIBCO. Cell lines of SGC-7901, HepG2, HeLa, SiHa and NIH3T3 were purchased from the American Type Culture Collection. RuCl3·3H2O was purchased from the Kunming Institution of Precious Metals. 1,10-Phenanthroline was obtained from the Guangzhou Chemical Reagent factory. Microanalyses (C, H, and N) were investigated with a Perkin-Elmer 240Q elemental analyzer. Electrospray ionization mass spectra (ESI-MS) were recorded on a LCQ system (Finnigan MAT, USA) using acetonitrile as mobile phase. The spray voltage, tube lens offset, capillary voltage, and capillary temperature were set at 4.50 kV, 30.00 V, 23.00 V, and 200 °C, respectively, and the quoted m/z values are for the major peaks in the isotope distribution. 1H NMR and 13C NMR spectra were recorded on a Varian-500 spectrometer with DMSO-d6 as solvent and tetramethylsilane as an internal standard at 500 MHz at room temperature.
4.2. Synthesis and characterization of ligand and complexes
4.2.1. Synthesis of ligand PTCP. A mixture of 1,10-phenanthroline-5,6-dione (0.315 g, 1.5 mmol),69 phenanthrene-9-carbaldehyde (0.309 g, 1.5 mmol) and ammonium acetate (2.31 g, 30 mmol) in glacial acetic acid (30 mL) was refluxed with stirring for 3 h. After completion of the reaction, the pink precipitate was collected and washed with water and dried in vacuo. Yield: 86%. Anal. calcd for C27H16N4: C, 81.80; H, 4.07; N, 14.13%. Found: C, 81.66; H, 4.02; N, 14.22%. ESI-MS: m/z = 397 [M + H]. IR (KBr, cm−1): 3057m, 1943w, 1689s, 1607m, 1545m, 1494s, 1427m, 1397m, 1246w, 1196w, 1072s, 1032m, 1012m, 894s, 804s, 738s, 681m, 624w.
4.2.2. Synthesis of [Ru(phen)2(PTCP)](ClO4)2 (1). A mixture of [Ru(phen)2Cl2]·2H2O70 (0.284 g, 0.5 mmol) and PTCP (0.198 g, 0.5 mmol) in ethylene glycol (30 mL) was refluxed under argon for 8 h to give a clear red solution. After cooling to room temperature, a red precipitate was obtained by the addition of an excess of saturated aqueous NaClO4 solution. The crude product was purified by column chromatography on neutral alumina with a mixture of CH3CN–toluene (3:1, v/v) as eluent. The red band was collected. The solvent was removed under reduced pressure and a red powder was obtained. Yield: 70%. Anal. calcd for C51H32N8Cl2O8Ru: C, 57.96; H, 3.05; N, 10.60%. Found: C, 58.03; H, 3.21; N, 10.68%. IR (KBr, cm−1): 3056m, 1970m, 1681w, 1601m, 1577w, 1542m, 1492s, 1445w, 1408s, 1366s, 1315w, 1245w, 1199s, 1144m, 1087s, 899w, 843s, 807m, 768m, 721s, 623s. 1H NMR (DMSO-d6): δ 9.14 (d, 2H, J = 8.0 Hz), 9.04 (d, 1H, J = 8.0 Hz), 8.98 (d, 2H, J = 7.0 Hz), 8.80 (dd, 2H, J = 4.5, J = 4.5 Hz), 8.78 (dd, 2H, J = 4.5, J = 6.5 Hz), 8.48 (s, 1H), 8.41 (s, 4H), 8.22 (d, 1H, J = 7.0 Hz), 8.18 (dd, 2H, J = 5.0, J = 5.5 Hz), 8.12 (dd, 2H, J = 5.5, J = 5.0 Hz), 8.07 (dd, 2H, J = 5.5, J = 5.0 Hz), 7.86 (d, 4H, J = 6.0 Hz), 7.81–7.77 (m, 6H). 13C NMR (DMSO-d6, 125 MHz): 152.85, 152.68, 152.39, 150.45, 147.27, 147.18, 145.59, 136.82, 130.48, 130.42, 130.38, 129.86, 129.80, 129.27, 128.98, 128.53, 128.37, 128.08, 127.71, 127.66, 127.53, 126.86, 126.74, 126.67, 126.51, 126.36, 126.31, 126.04, 124.29, 124.25, 123.52, 123.23, 123.15, 122.70. ESI-MS (CH3CN): m/z 429.2 ([M − 2ClO4]2+).
4.2.3. Synthesis of [Ru(dmp)2(PTCP)](ClO4)2 (2). This complex was synthesized in a manner identical to that described for 1, with [Ru(dmp)2Cl2]·2H2O71 in place of [Ru(phen)2Cl2]·2H2O. Yield: 72%. Anal. calcd for C55H40N8Cl2O8Ru: C, 59.36; H, 3.62; N, 10.07%. Found: C, 59.25; H, 3.81; N, 10.15%. IR (KBr, cm−1): 3058w, 2966w, 2926w, 1964m, 1625w, 1604w, 1589w, 1542m, 1508s, 1443s, 1403m, 1367s, 1349w, 1305m, 1246w, 1199m, 1088s, 903w, 856s, 810s, 771w, 727s, 623s. 1H NMR (DMSO-d6): δ 9.00 (d, 3H, J = 8.5 Hz), 8.94 (d, 4H, J = 6.0 Hz), 8.48 (s, 1H), 8.45 (dd, 4H, J = 8.5, J = 9.0 Hz), 8.26 (d, 2H, J = 8.5 Hz), 8.16 (d, 1H, J = 7.0 Hz), 8.01 (d, 2H, J = 8.5 Hz), 7.84–7.73 (m, 4H), 7.54 (dd, 2H, J = 5.5, J = 5.5 Hz), 7.42 (d, 4H, J = 8.0 Hz), 1.96 (s, 6H), 1.75 (s, 6H). 13C NMR (DMSO-d6, 125 MHz): 167.95, 166.40, 152.63, 150.74, 148.89, 147.81, 145.78, 138.13, 136.68, 130.82, 130.37, 130.32, 130.27, 129.76, 129.54, 129.23, 128.88, 128.18, 127.62, 127.55, 127.50, 127.43, 127.12, 126.87, 126.75, 126.59, 126.52, 126.07, 125.29, 123.45, 123.11, 25.61, 24.53. ESI-MS (CH3CN): m/z 457.0 ([M − 2ClO4]2+).
4.2.4. Synthesis of [Ru(ttbpy)2(pttcp)](ClO4)2 (3). This complex was synthesized in a manner identical to that described for 1, with [Ru(ttbpy)2Cl2]·2H2O70 in place of [Ru(phen)2Cl2]·2H2O. Yield: 70%. Anal. calcd for C63H64N8Cl2O8Ru: C, 61.36; H, 5.23; N, 9.09%. Found: C, 60.51; H, 5.62; N, 9.01%. IR (KBr, cm−1): 2965s, 2871w, 1964m, 1614s, 1541m, 1481s, 1445m, 1413s, 1368s, 1316w, 1251m, 1201m, 1090s, 898m, 839m, 809m, 769m, 726s, 624s. 1H NMR (DMSO-d6): δ 9.13 (dd, 2H, J = 8.5, J = 8.5 Hz), 9.02 (dd, 2H, J = 8.5, J = 8.5 Hz), 8.95 (d, 1H, J = 8.5 Hz), 8.88 (dd, 4H, J = 7.0, J = 6.5 Hz), 8.49 (s, 1H), 8.20 (d, 1H, J = 8.0 Hz), 8.03 (d, 2H, J = 5.0 Hz), 7.97 (dd, 2H, J = 5.5, J = 5.5 Hz), 7.85–7.81 (m, 2H), 7.77 (d, 2H, J = 6.0 Hz), 7.71 (d, 2H, J = 6.0 Hz), 7.64 (dd, 2H, J = 6.0, J = 6.0 Hz), 7.52 (d, 2H, J = 6.5 Hz), 7.36 (dd, 2H, J = 6.0, J = 7.0 Hz), 1.44 (s, 18H), 1.36 (s, 18H). 13C NMR (DMSO-d6, 125 MHz): 161.89, 161.71, 156.57, 156.41, 153.27, 150.85, 150.76, 149.46, 145.19, 130.39, 130.36, 130.23, 129.65, 129.25, 129.14, 128.89, 128.38, 128.19, 127.60, 127.44, 126.87, 126.61, 126.32, 124.86, 124.51, 123.46, 123.11, 121.85, 121.79, 35.55, 35.42, 30.11, 30.01. ESI-MS (CH3CN): m/z 517.0 ([M − 2ClO4]2+).
4.3. DNA-binding studies
The absorption titrations of ruthenium(II) complex in buffer were performed by using a fixed ruthenium complex concentration (20 μM) to which the DNA stock solution were added. Ruthenium–DNA solutions were allowed to incubate for 5 min before the absorption spectra were recorded. In order to further illustrate the binding strength of the complex, the intrinsic binding constant K with CT-DNA was obtained by monitored the change in the absorbance at metal-to-ligand transfer (MLCT), with increasing concentration of DNA, the following equation was applied.72| | |
[DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/[Kb(εb − εf)]
| (1) |
where [DNA] is the concentration of DNA in base pairs, εa, εf and εb correspond to the apparent absorption coefficient Aobsd/[Ru], the extinction coefficient for the free ruthenium complex and the extinction coefficient for the ruthenium complex in the fully bound form, respectively. In plots of [DNA]/(εa − εf) versus [DNA], Kb is given by the ratio of slope to the intercept.
Viscosity measurements were carried out using an Ubbelodhe viscometer maintained at a constant temperature at 25.0 (±0.1) °C in a thermostatic bath. DNA samples approximately 200 base pairs in average length were prepared by sonicating in order to minimize complexities arising from DNA flexibility.73 Flow time was measured with a digital stopwatch, and each sample was measured three times, and an average flow time was calculated. Relative viscosities for DNA in the presence and absence of complexes were calculated from the relation η = (t − t0)/t0, where t is the observed flow time of the DNA-containing solution and t0 is the flow time of buffer alone.74,75 Data were presented as (η/η0)1/3 versus binding ratio,76 where η is the viscosity of DNA in the presence of complex and η0 is the viscosity of DNA alone.
4.4. Molecular docking
The optimized structures of the complexes were performed using density function theory (DFT) B3LYP. The crystal data of the B-DNA dodecamer d(CGCGAATTCGCG)2 (PDB 1D:1BNA) were downloaded from the Protein Data Bank.77 The water molecules and the ligands were removed from the 1BNA, and Gasteiger charges were added to the complexes by Autodock 4.2 Tools (ADT) before performing docking calculations. The binding site was centered on the DNA molecule and a grid box was created with 60 × 60 × 60 points in which almost involved the entire DNA molecule. The rigid docking protocol and 100 runs of the Lamarckian genetic algorithm for searching ligand conformations were performed.
4.5. Cytotoxicity in vitro assay
Cell viability was determined by MTT assay.78 Cancer cells (8 × 103 cells per well) were seeded in 96-well for 24 h. Cells was incubated with the tested compounds to achieve final concentrations ranging from 10−6 to 10−4 M. Control wells were prepared by addition of culture medium (100 μL) and cisplatin was used as a positive control. After 48 h incubation, 10 μL of MTT dye solution (5 mg mL−1) was added to each well. After incubation at 37 °C for 4 h, buffer (100 μL) containing dimethylformamide (50%) and sodium dodecyl sulfate (20%) was added to transform MTT to a purple formazan dye. The optical density of each well was then measured for three times to obtain the mean values. The IC50 values were analyzed by software of SPSS.
4.6. DNA damage assay
DNA damage was investigated by means of comet assay. SGC-7901 cells in culture medium were incubated with 12.5 μM of the complex for 24 h at 37 °C. The cells were harvested by a trypsinization process at 24 h. A total of 100 μL of 0.5% normal agarose in PBS was dropped gently onto a fully frosted microslide, covered immediately with a coverslip, and then placed at 4 °C for 10 min. The coverslip was removed after the gel had been set. A mixture of 50 μL of the cell suspension (200 cells per μL) mixed with 50 μL of 1% low melting agarose was preserved at 37 °C. A total of 100 μL of this mixture was applied quickly on top of the gel, coated over the microslide, covered immediately with a coverslip, and then placed at 4 °C for 10 min. The coverslip was again removed after the gel had been set. A third coating of 50 μL of 0.5% low melting agarose was placed on the gel and allowed to place at 4 °C for 15 min. After solidification of the agarose, the coverslips were removed, and the slides were immersed in an ice-cold lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, 90 mM sodium sarcosinate, NaOH, pH 10, 1% Triton X-100 and 10% DMSO) and placed in a refrigerator at 4 °C for 2 h. All of the above operations were performed under low lighting conditions to avoid additional DNA damage. The slides, after removal from the lysis solution, were placed horizontally in an electrophoresis chamber. The reservoirs were filled with an electrophoresis buffer (300 mM NaOH, 1.2 mM EDTA) until the slides were just immersed in it, and the DNA was allowed to unwind for 30 min in electrophoresis solution. Then the electrophoresis was carried out at 25 V and 300 mA for 20 min. After electrophoresis, the slides were removed, washed thrice in a neutralization buffer (400 mM Tris, HCl, pH 7.5). Cells were stained with 20 μL of EB (20 μg mL−1) in the dark for 20 min. The slides were washed in chilled distilled water for 10 min to neutralize the excess alkali, air-dried and scored for comets by fluorescent microscopy.
4.7. Annexin V-FITC apoptosis detection
SGC-7901 cells were seeded into 6-well plates at a density of 1 × 106 cells per well and incubated for 24 h. The different concentration of compounds were added into the above well for 24 h, cells were collected and washed with PBS twice, and then stained with fluorescein isothiocyanate (FITC)-conjugated Annexin V and then PI. Cells were quantified by a FACS Calibur flow cytometry (Beckman Dickinson & Co., Franklin Lakes, NJ).
4.8. Measurement of mitochondrial membrane potential (MMP, ΔΨm)
SGC-7901 cells (2 × 105 per well) were treated using compounds for 24 h. JC-1 (1 mg mL−1) as fluorescence probe for determination of MMP was added to stain cells at 37 °C for 30 min. Then the cells were washed twice with PBS and measured by a flow cytometer. Data was analyzed by FlowJo V10.2 software.
4.9. Reactive oxygen species (ROS) detection
Intracellular ROS levels were measured with a fluorescent dye 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). SGC-7901 cells were seeded into 6-well plates at a density of 1 × 106 cells per well. After incubation for 24 h, the medium was replaced with medium containing different concentrations of compounds for 24 h. Then the cells were stained with 20 μM DCFH-DA in PBS for 30 min in the dark. Finally, the cells were harvested and washed twice with PBS, then the data were determined by flow cytometry and analyzed with FlowJo V10.2 software.
4.10. Cell cycle arrest studies
SGC-7901 cells were seeded into six-well plates (Costar, Corning Corp., New York) at a density of 1 × 106 cells per well and incubated for 24 h. The cells were cultured in RPMI 1640 supplemented with 10% of FBS, and incubated at 37 °C and 5% CO2. The medium was removed and replaced with medium (final DMSO concentration 0.05% v/v) containing 12.5 μM complexes 1, 2 and 3. After incubation for 24 h, the cell layer was trypsinized and washed with cold PBS and fixed with 70% ethanol. Twenty μL of RNAse (0.2 mg mL−1) and 20 μL of propidium iodide (0.02 mg mL−1) were added to the cell suspensions and the mixtures were incubated at 37 °C for 30 min. The samples were then analyzed with a FACS Calibur flow cytometry. The number of cells analyzed for each sample was 10000.
4.11. Anti-metastasis effect
The BD BioCoat™ Matrigel™ invasion chamber (BD Biosciences) was used according to the manufacturer's instructions. Compounds were dissolved in cell media at the desired concentration and added into Matrigel. Twenty-five thousands of SGC-7901 cells in serum free media were then seeded in the top chamber of the two chamber Matrigel system. To the low compartment, RPMI and 5% FBS were added as chemo-attractant. Cells were allowed to invade for 24 h. After incubation, non-invading cells were removed from the upper surface and cells on the lower surface were fixed with 4% paraformaldehyde and stained with 0.1% of crystal violet. Membranes were photographed and the invading cells were counted under a light microscope. Mean values from three independent assays were calculated.
4.12. Autophagy studies
SGC-7901 cells were seeded onto chamber slides in 12-well plates and incubated for 24 h. The cells were cultured in RPMI 1640 supplemented with 10% of FBS and incubated at 37 °C in 5% CO2. The medium was removed and replaced with medium (final DMSO concentration, 0.05% v/v) containing the complex for 24 h. The medium was removed again, and the cells were washed with ice-cold PBS twice. Then the cells were stained with MDC (monodansylcadaverine) solution (50 μM) for 10 min and washed with PBS twice. The cells were observed and imaged under fluorescence microscope.
4.13. Western blot analysis
SGC-7901 cells were seeded in 3.5 cm dishes for 24 h and incubated with 12.5 μM of complexes 1, 2 and 3 in the presence of 10% FBS. Then cells were harvested in lysis buffer. After sonication, the samples were centrifuged for 20 min at 13000g. The protein concentration of the supernatant was determined by BCA assay. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis was done loading equal amount of proteins per lane. Gels were then transferred to poly(vinylidene difluoride) membranes (Millipore) and blocked with 5% non-fat milk in TBST (20 mM Tris–HCl, 150 mM NaCl, 0.05% Tween 20, pH 8.0) buffer for 1 h. The membranes were incubated with primary antibodies at 1:5000 dilution in 5% non-fat milk overnight at 4 °C, and after washed four times with TBST for a total of 30 min, then the secondary antibodies conjugated with horseradish peroxidase at 1:5000 dilution for 1 h at room temperature and washed four times with TBST. The blots were visualized with the Amersham ECL Plus western blotting detection reagents according to the manufacturer's instructions. To assess the presence of comparable amount of proteins in each lane, the membranes were stripped finally to detect the β-actin.
5. Data analysis
All data was expressed as means ± SD. Statistical significance was evaluated by a t-test. Differences were considered to be significant when a *p value was less than 0.05.
Acknowledgements
This work was supported by the National Nature Science Foundation of China (No. 81403111), the Natural Science Foundation of Guangdong Province (No. 2016A030313728) and Project of Innovation for Enhancing Guangdong Pharmaceutical University, Provincial Experimental Teaching Demonstration Center of Chemistry & Chemical Engineering.
References
- B. Rosenberg, L. Vancamp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 2, 183–194 RSC.
- P. J. Dyson and G. Sava, Dalton Trans., 2006, 16, 1929–1933 RSC.
- M. Markman, Expert Opin. Drug Saf., 2003, 2, 597–607 CrossRef CAS PubMed.
- P. C. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS PubMed.
- Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 38, 4764–4776 RSC.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- A. K. Singha, D. S. Pandeyb, Q. Xua and P. Braunstein, Coord. Chem. Rev., 2014, 270, 31–56 CrossRef.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. J. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- T. Khamrang, R. Kartikeyan, M. Velusamy, V. Rajendiran, R. Dhivya, B. Perumalsamy, M. A. Akbarsha and M. Palaniandavar, RSC Adv., 2016, 6, 114143–114258 RSC.
- P. C. Hu, Y. Wang, Y. Zhang, H. Song, F. F. Gao, H. Y. Lin, Z. H. Wang, L. Wei and F. Yang, RSC Adv., 2016, 6, 29963–29976 RSC.
- T. S. Kamatchi, P. Kalaivani, F. R. Fronczek, K. Natarajan and R. Prabhakaran, RSC Adv., 2017, 7, 46531–46547 RSC.
- L. M. Chen, F. Peng, G. D. Li, X. M. Jie, K. R. Cai, C. Cai, Y. Zhong, H. Zeng, W. Li, Z. Zhang and J. C. Chen, J. Inorg. Biochem., 2016, 156, 64–74 CrossRef CAS PubMed.
- W. X. Chen, X. D. Song, S. F. He, J. Sun, J. X. Chen, T. Wu and Z. W. Mao, J. Inorg. Biochem., 2016, 164, 91–98 CrossRef CAS PubMed.
- C. Zhang, B. J. Han, C. C. Zeng, S. H. Lai, W. Li, B. Tang, D. Wan, G. B. Jiang and Y. J. Liu, J. Inorg. Biochem., 2016, 157, 62–72 CrossRef CAS PubMed.
- I. Bratsos, S. Jedner, T. Gianferrara and E. Alessio, Chimia, 2007, 61, 692–697 CrossRef CAS.
- X. Meng, M. L. Leyva, M. Jenny, I. Gross, S. Benosman, B. Fricker, S. Harlepp, P. H. Ebraud, A. Boos, P. Wlosik, P. Bischoff, C. Sirlin, M. Pfeffer, J. P. Loeffler and C. Gaiddon, Cancer Res., 2009, 69, 5458–5466 CrossRef CAS PubMed.
- G. Gupta, P. Nowak-Sliwinska, N. Herrero, P. J. Dyson and B. Therrien, Inorg. Chim. Acta, 2014, 423, 524–529 CrossRef CAS.
- G. Sava, A. Bergamo and P. J. Dyson, Dalton Trans., 2011, 40, 9069–9075 RSC.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- K. J. Kilpin, S. Crot, T. Riedel, J. A. Kitchen and P. J. Dyson, Dalton Trans., 2014, 43, 1443–1448 RSC.
- G. Sava, S. Zorzet, C. Turrin, F. Vita, M. Soranzo, G. Zabucchi, M. Cocchietto, A. Bergamo, S. Giovine, G. Pezzoni, L. Sartor and S. Garbisa, Cancer Res., 2003, 91, 1898–1905 Search PubMed.
- A. Bergamo, A. Masi, P. J. Dyson and G. Sava, Internet J. Oncol., 2008, 33, 1281–1289 CAS.
- V. Moreno, M. Font-Bardia, T. Calvet, J. Lorenzo, F. X. Aviles, M. H. Garcia, T. S. Morais, A. Valente and M. P. J. Robalo, J. Inorg. Biochem., 2011, 105, 241–249 CrossRef CAS PubMed.
- A. Martinez, J. Suarez, T. Shand, R. S. Magliozzo and R. A. J. Sanchez-Delgado, J. Inorg. Biochem., 2011, 105, 39–45 CrossRef CAS PubMed.
- S. Kapitza, M. A. Jakupec, M. Uhl, B. K. Keppler and B. Marian, Cancer Lett., 2005, 226, 115–121 CrossRef CAS PubMed.
- L. Li, Y. Wong, T. Chen and W. Zheng, Dalton Trans., 2012, 41, 1138–1141 RSC.
- S. H. Lai, W. Li, X. Z. Wang, C. Zhang, C. C. Zeng and Y. J. Liu, RSC Adv., 2016, 6, 63143–63155 RSC.
- W. Li, B. J. Han, J. H. Yao, G. B. Jiang, G. J. Lin, Y. Y. Xie, H. L. Huang and Y. J. Liu, Spectrochim. Acta, Part A, 2015, 150, 127–134 CrossRef CAS PubMed.
- C. C. Zeng, S. H. Lai, J. H. Yao, C. Zhang, H. Yin, W. Li and B. J. Han, Eur. J. Med. Chem., 2016, 122, 118–126 CrossRef CAS PubMed.
- B. J. Han, G. B. Jiang, J. H. Yao, W. Li, J. Wang, H. L. Huang and Y. J. Liu, Spectrochim. Acta, Part A, 2015, 135, 840–849 CrossRef CAS PubMed.
- W. Li, B. J. Han, J. H. Yao, G. B. Jiang and Y. J. Liu, RSC Adv., 2015, 5, 24534–24543 RSC.
- E. A. Steck and A. R. Day, J. Am. Chem. Soc., 1943, 65, 452–456 CrossRef CAS.
- Q. X. Zhen, B. H. Ye, Q. L. Zhang, J. G. Liu, H. Li, L. N. Ji and L. Wang, J. Inorg. Biochem., 1999, 76, 47–53 CrossRef CAS PubMed.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3051–3058 CrossRef CAS.
- C. V. Kumar and E. H. Asuncion, J. Am. Chem. Soc., 1993, 115, 8547–8553 CrossRef CAS.
- S. Satyanarayana, J. C. Dabroniak and J. B. Chaires, Biochemistry, 1992, 31, 9319–9324 CrossRef CAS PubMed.
- R. Rohs, I. Bloch, H. Sklenar and Z. Shakked, Nucleic Acids Res., 2005, 33, 7048–7057 CrossRef CAS PubMed.
- C. C. Zeng, C. Zhang, S. H. Lai, H. Yin, B. Tang, D. Wan and Y. J. Liu, Inorg. Chem. Commun., 2016, 70, 210–218 CrossRef CAS.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- C. Tse, A. R. Shoemaker, J. Adickes, M. G. Anderson, J. Chen, S. Jin, E. F. Johnson, K. C. Marsh, M. J. Mitten, P. Nimmer, L. Roberts, S. K. Tahir, Y. Xiao, X. Yang, H. Zhang, S. Fesik, S. H. Rosenberg and S. W. Elmore, Cancer Res., 2008, 68, 3421–3428 CrossRef CAS PubMed.
- A. J. Souers, J. D. Leverson, E. R. Boghaert, S. L. Ackler, N. D. Catron, J. Chen, B. D. Dayton, H. Ding, S. H. Enschede, W. J. Fairbrother, D. C. Huang, S. G. Hymowitz, S. Jin, S. L. Khaw, P. J. Kovar, L. T. Lam, J. Lee, H. L. Maecker, K. C. Marsh, K. D. Mason, M. J. Mitten, P. M. Nimmer, A. Oleksijew, C. H. Park, C. M. Park, D. C. Phillips, A. W. Roberts, D. Sampath, J. F. Seymour, M. L. Smith, G. M. Sullivan, S. K. Tahir, C. Tse, M. D. Wendt, Y. Xiao, J. C. Xue, H. Zhang, R. A. Humerickhouse, S. H. Rosenberg and S. W. Elmore, Nat. Med., 2013, 19, 202–208 CrossRef CAS PubMed.
- R. R. Tice, E. Agurell, D. Anderson, B. Burlinson, A. Hartmann, H. Kobayashi, Y. Miyamae, E. Rojas, J. C. Ryu and Y. F. Sasaki, Environ. Mol. Mutagen., 2000, 35, 206–221 CrossRef CAS PubMed.
- C. Alapetite, T. Wachter, E. Sage and E. Moustacchi, Int. J. Radiat. Biol., 1996, 69, 359–369 CrossRef CAS PubMed.
- P. Y. Zhang, J. Q. Wang, H. Y. Huang, L. P. Qiao, L. N. Ji and H. Chao, Dalton Trans., 2013, 42, 8907–8917 RSC.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- T. Ozben, J. Pharm. Sci., 2007, 96, 2181–2196 CrossRef CAS PubMed.
- A. Kawiak, J. Zawacka-Pankau, A. Wasilewska, G. Stasilojc, J. Bigda and E. Lojkowska, J. Nat. Prod., 2012, 75, 9–14 CrossRef CAS PubMed.
- L. R. Silveira, L. Pereira-Da-Silva, C. Juel and Y. Hellsten, Free Radical Biol. Med., 2003, 35, 455–464 CrossRef CAS PubMed.
- A. Heller, G. Brockhoff and A. Goepferich, Eur. J. Pharm. Biopharm., 2012, 82, 1–18 CrossRef CAS PubMed.
- S. E. Abu-Gosh, N. Kolvazon, B. Tirosh, I. Ringel and E. Yavin, Mol. Pharmaceutics, 2009, 6, 1138–1144 CrossRef CAS PubMed.
- S. Fulda, L. Galluzzi and G. Kroemer, Nat. Rev. Drug Discovery, 2010, 9, 447–464 CrossRef CAS PubMed.
- J. S. Armstrong, Br. J. Pharmacol., 2007, 151, 1154–1165 CrossRef CAS PubMed.
- L. Wang, K. Y. Lu, H. H. Hao, X. Y. Li, J. Wang, K. Wang, J. Wang, Z. Yan, S. L. Zhang, Y. H. Du and H. R. Liu, PLoS One, 2013, 8, e81296 Search PubMed.
- E. Cavalieri, A. Rigo, M. Bonifacio, A. C. D. Prati, E. Guardalben, C. Bergamini, R. Fato, G. Pizzolo, H. Suzuki and F. Vinante, J. Transl. Med., 2011, 9, 1–13 CrossRef PubMed.
- Y. S. Shin, H. A. Shin, S. U. Kang, J. H. Kim, Y. T. Oh, K. H. Park and C. H. Kim, PLoS One, 2013, 8, e6915 Search PubMed.
- F. Kopper, C. Bierwirth, M. Schon, M. Kunze, I. Elvers and D. Kranz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16856–16861 CrossRef PubMed.
- L. Gamet-Payrastre, P. Li, S. Lumeau, G. Cassar, M. A. Dupont, S. Chevolleau, N. Gasc, J. Tulliez and F. Terce, Cancer Res., 2000, 60, 1426–1433 CAS.
- J. Klucar and M. Al-Rubeai, FEBS Lett., 1997, 400, 127–130 CrossRef CAS PubMed.
- V. M. Dirsch, D. S. Antlsperger, H. Hentze and A. M. Vollmar, Leukemia, 2002, 16, 74–83 CrossRef CAS PubMed.
- R. Horwitz and D. Webb, Curr. Biol., 2003, 13, R756–R759 CrossRef CAS PubMed.
- F. Zhao, Y. Zhao, Y. Liu, X. L. Chang, C. Y. Chen and Y. L. Zhao, Small, 2011, 7, 1322–1377 CrossRef CAS PubMed.
- A. Biederbick, H. F. Kern and H. P. Elsasser, Eur. J. Cell Biol., 1995, 66, 3–14 CAS.
- A. Kawiak, J. Zawacka-Pankau, A. Wasilewska, G. Stasilojc, J. Bigda and E. Lojkowska, J. Nat. Prod., 2012, 75, 9–14 CrossRef CAS PubMed.
- M. Degli Espost and C. Dive, Biochem. Biophys. Res. Commun., 2003, 304, 455–461 CrossRef.
- J. Marmur, J. Mol. Biol., 1961, 3, 208–218 CrossRef CAS.
- M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- W. Paw and R. Eisenberg, Inorg. Chem., 1997, 36, 2287–2293 CrossRef CAS PubMed.
- B. P. Sullivan, D. J. Salmon and T. J. Meyer, Inorg. Chem., 1978, 17, 3334–3341 CrossRef CAS.
- J. P. Collin and J. P. Sauvage, Inorg. Chem., 1986, 25, 135–141 CrossRef CAS.
- A. Wolfe, G. H. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- J. B. Chaires, N. Dattagupta and D. M. Crothers, Biochemistry, 1982, 21, 3933–3940 CrossRef CAS PubMed.
- S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Biochemistry, 1993, 32, 2573–2584 CrossRef CAS PubMed.
- S. Satyanaryana, J. C. Dabrowial and J. B. Chaires, Biochemistry, 1982, 31, 9319–9324 CrossRef.
- G. Cohen and H. Eisenberg, Biopolymers, 1969, 8, 45–55 CrossRef CAS.
- S. Tabassum, W. M. Al-Asbahy, M. Afzal, F. Arjmanda and V. Bagchib, Dalton Trans., 2012, 41, 4955–4964 RSC.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05103d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ac
*ac