DOI:
10.1039/C7RA05947G
(Paper)
RSC Adv., 2017,
7, 33113-33119
Norsampsone E, an unprecedented decarbonyl polycyclic polyprenylated acylphloroglucinol with a homoadamantyl core from Hypericum sampsonii†
Received
27th May 2017
, Accepted 23rd June 2017
First published on 29th June 2017
Abstract
Norsampsone E (1), an unprecedented decarbonyl polycyclic polyprenylated acylphloroglucinol, together with one new and two known analogues (2–4), was isolated from the aerial parts of Hypericum sampsonii. Compound 1 featured an unusual homoadamantyl skeleton with the loss of C-4 carbonyl and followed by the formation of a new C–C bond between C-3 and C-5. Their structures and absolute configurations were elucidated by extensive NMR spectroscopy, single-crystal X-ray experiments, ECD calculations and CD comparison methods, and further confirmed using the single-crystal X-ray structures of biogenetically related congeners 3 and 4. Besides, compounds 1 and 2 showed direct binding to the RXRα-LBD protein in the SPR assay with a KD of 10.28 μM and 31.70 μM, respectively. Furthermore, compound 1 (5–20 μM) showed potent RXRα transcriptional inhibitory activity in a dose dependent manner, while compound 2 (5–20 μM) slightly enhanced RXRα transactivation.
Introduction
Polycyclic polyprenylated acylphloroglucinols (PPAPs) have been of great interest to the scientific community due to their intricate “diamond-like” architectures and large portfolio of biological activities.1–3 As a vast family of natural products from the Hypericum genus, PPAPs possess stunningly complex structures containing bicyclo[3.3.1]nonane, homoadamantyl and adamantyl cores.1–3 Recently, a bewildering array of PPAPs derivatives with intriguing novel skeletons were characterized by natural product researchers, which were biosynthesized from conventional PPAPs through carbon migration, rearrangement, ring breakage, carbon reduction and so on.4–14 Besides, driven by their fascinating and challenging structures, PPAPs have been a vibrant area in synthesis research.15–19
Retinoid X receptor alpha (RXRα), a ligand-dependent transcription factor, regulates a broad spectrum of physiological functions including cell growth, differentiation, and apoptosis.20 RXRα has been considered as an intriguing drug target and ligands for RXRα show promise as therapeutic agents for many diseases, especially in the treatment of cancer.21 Targretin, a small synthetic molecule targeting RXRα, was approved by the FDA for the treatment of cutaneous T-cell lymphoma.22 Though many ligands of RXRα have been reported, their druggability have been hampered by severe side effects.23 Thus, discovery of novel small-molecule modulators of RXRα with high efficacy and low toxicity from natural products is an effective strategy for the development of RXRα based drugs.
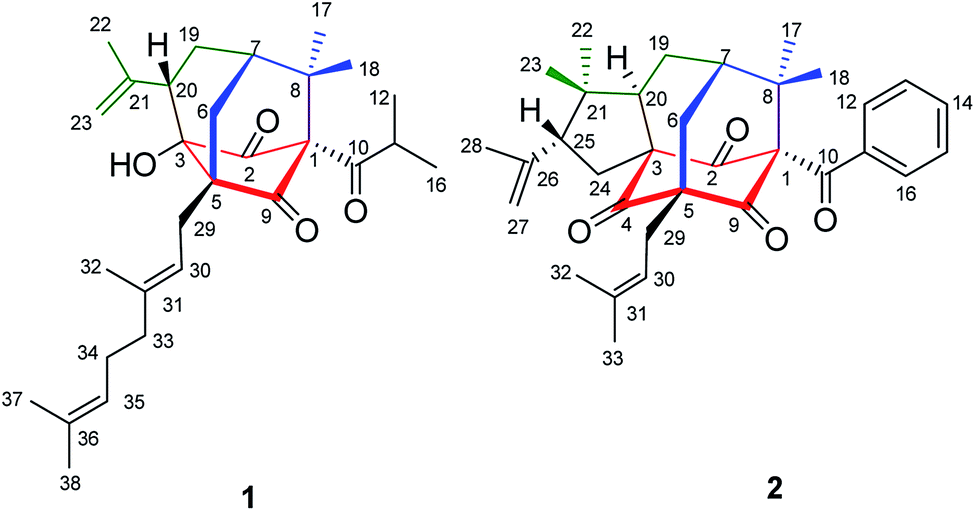
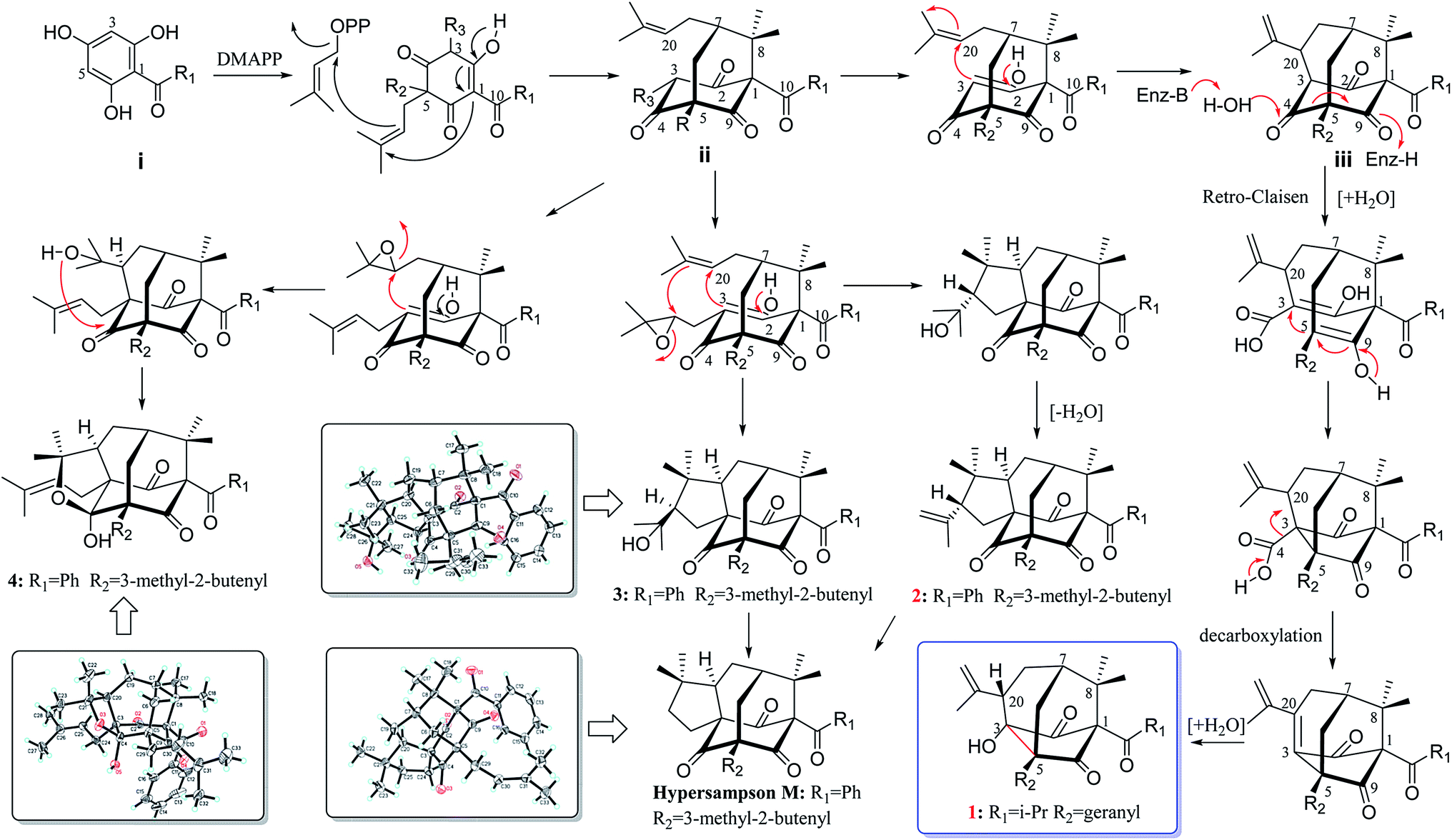
Hypericum sampsonii, a rich source of complex caged PPAPs, shows powerful aid in treating many ailments such as hematemesis, enteritis, traumatic hemorrhage, swellings and cancer.24 In our previous investigations on the aerial parts of H. sampsonii, several novel PPAPs were discovered including four new decarbonyl PPAPs norsampsons A–D with the loss of C-2 carbonyl in phloroglucinol ring of bicyclo[3.3.1]nonane skeleton.11,12,25–27 Besides, some of them show potent RXRα transcriptional inhibitory activities, suggesting that PPAPs derivatives may be the small-molecule modulators of RXRα. In this paper, we reported the isolation and structural elucidation of another decarbonyl PPAPs norsampsone E (1) which featured an unprecedented homoadamantyl skeleton with the loss of C-4 carbonyl and followed by the formation of a new C–C bond between C-3 and C-5, along with one new analogue hypersampsone X (2) (Fig. 1). Besides, single crystal X-ray structures of hypersampsone Q (3) (CCDC 1551384†) and sampsonione B (4) (CCDC 1551380†) was reported for the first time and further confirmed the absolute configurations of the new compounds (Scheme 1).
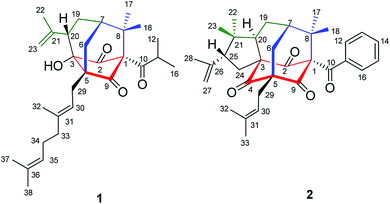 |
| | Fig. 1 Structures of compounds 1–2. | |
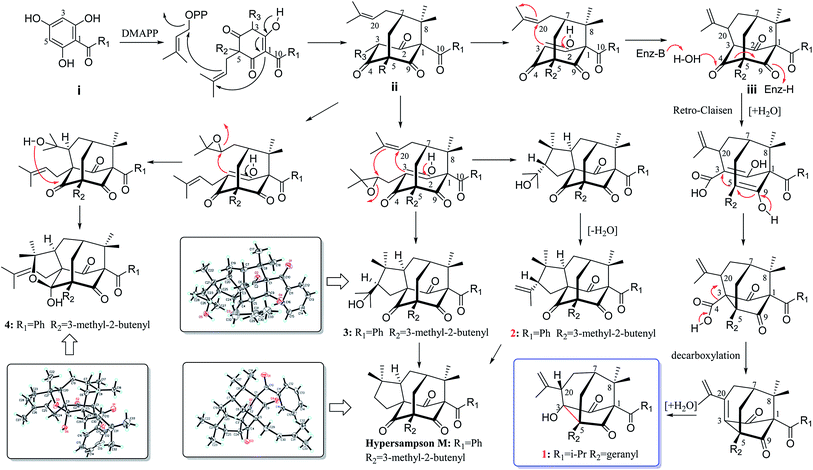 |
| | Scheme 1 Plausible Biogenetic Pathway for of 1–4. | |
Results and discussion
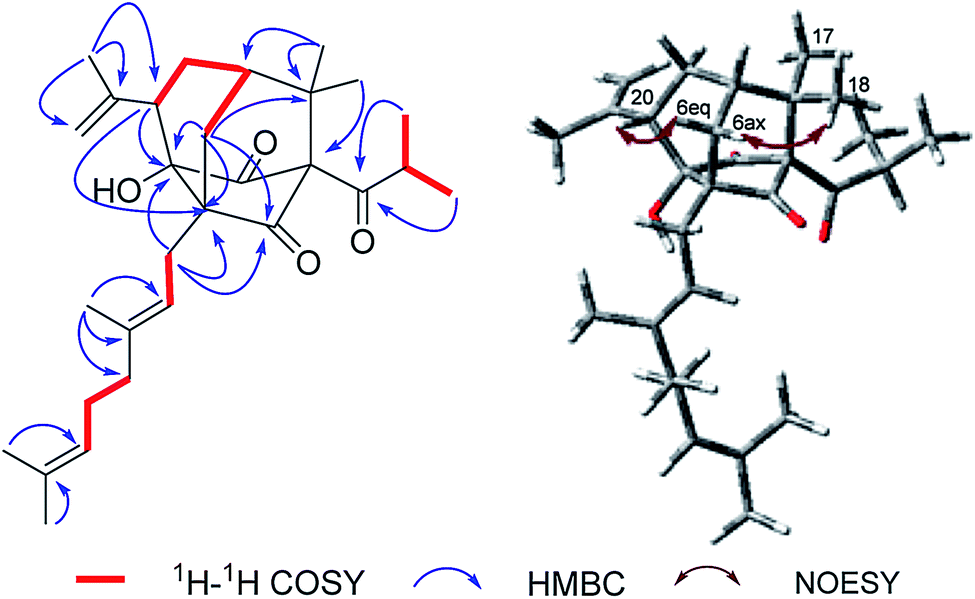
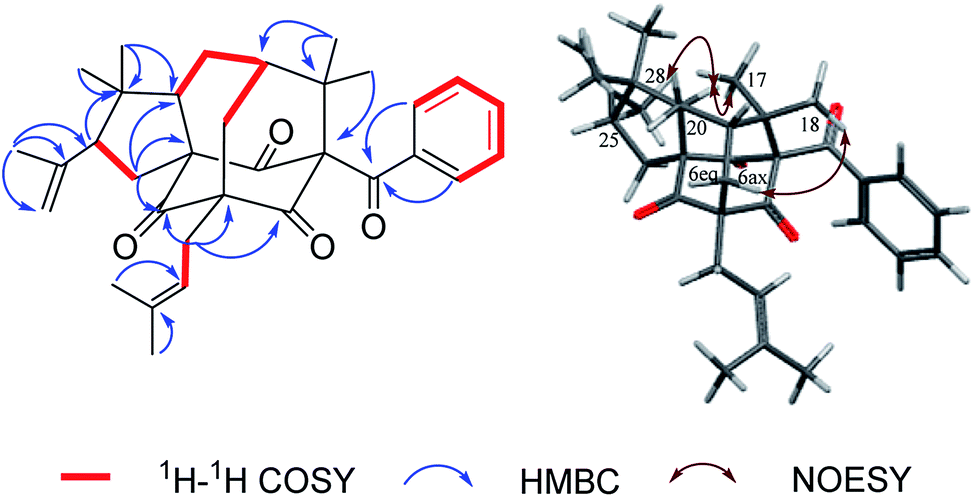
Norsampsone E (1), was isolated as a colorless oil. The HR-ESI-MS spectrum of 1 showed the molecular ion peak at m/z 455.3161 [M + H]+, corresponding to the molecular formula C29H42O4, with 9 degrees of unsaturation. The 1H NMR and 13C NMR spectra exhibited an isopropyl group [(δC 42.6; δH 2.27, sept, J = 6.4 Hz), (δC 19.8; δH 1.06, d, J = 6.4 Hz), (δC 19.6; δH, 1.02, d, J = 6.4 Hz)], a terminal double bond [(δC 118.4; δH 5.23, br.s, δH 4.95, br.s), (δC 142.6)], three carbonyl groups [δC 211.7, 211.0, 208.4], a geranyl group [δC 25.9, 118.7, 139.0, 16.3, 40.3, 26.6, 124.2, 131.8, 17.8, 25.9] and additional three singlet methyls [δC 21.9–24.5; δH 1.20–1.84], which accounted for six degrees of unsaturation. The remaining three degrees of unsaturation required a tricyclic core structure in 1. The above analysis indicated that compound 1 was a derivative of PPAPs. The NMR data of 1 were similar to those of sampsonione H,28 except for an isobutyryl group at C-1 instead of a benzoyl group, the significant downshift of C-3 (δC 82.7) as well as obvious upfield shift of C-5 (δC 58.4) and C-20 (δC 42.0), together with the absence of C-4 carbonyl and C-24-25. Additionally, the HMBC correlations from H-6eq to C-5/C-8/C-9/C-19, H-6ax to C-3, H-20 to C-2/C-3/C-5 and those from H3-23 to C-20/C-21/C-22 further suggested the existence of an decarbonyl homoadamantyl core structure with the loss of C-4 carbonyl and followed by the formation of a new C–C bond between C-3 and C-5. HMBC correlations from H-29 to C-3/C-5/C-6/C-9 allowed the linkage of the geranyl group to the C-5 of the core structure. By now, the gross structure was poor in one hydroxyl groups compared with the molecular formula of 1. Taking the chemical shift of C-3 (δC 82.7) into account, the hydroxyl group should be attached to the C-3 of the core structure (Fig. 2). Thus, the planar structure of 1 was established.
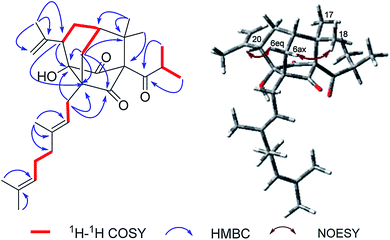 |
| | Fig. 2 Key 1H–1H COSY, HMBC and NOESY correlations of 1. | |
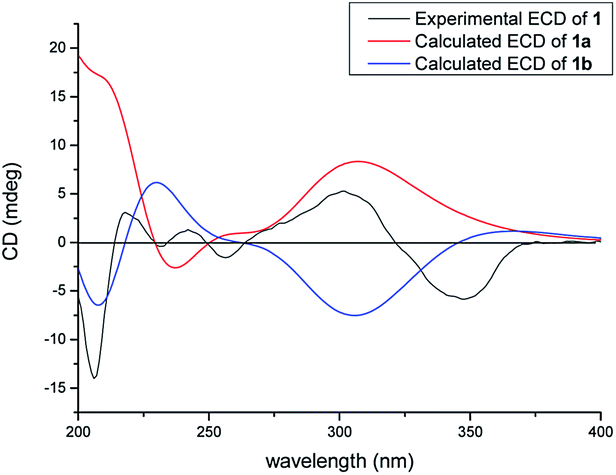
In the NOESY spectrum, the correlations of H3-18/H-6ax and H-6eq/H-20 indicated the β-orientation of H-20. Therefore, the relative configuration of 1 was elucidated as shown in Fig. 2. Since compound 1 was obtained as colorless oil with low polarity, crystals suitable for single X-ray experiment were failed to be obtained. Recently, the absolute configuration of an oily PPAPs derivative hypercohin A with a hydroxyl group was successfully determined by the single X-ray crystal of its p-bromobenzoate ester which was much easier to crystallize.14 Thus, the p-bromobenzoate ester of compound 1 was try to prepared in order to obtain its single crystal X-ray structure. However, it was failed as well, which may be due to the existence of a tertiary alcohol hydroxyl instead of a secondary alcohol hydroxyl as well as the large space steric hindrance. Fortunately, the calculated electronic circular dichroism (ECD) method has demonstrated great success in determining the absolute configurations of the PPAPs type compounds.4–14 Thus, the calculated ECD method was used to determine the absolute configuration of 1. On the basis of the above deduction of the relative configuration of 1, a pair of enantiomers (1R,3S,5S,7S,20S)-1a and (1S,3R,5R,7R,20R)-1b were calculated for their ECD spectrum by the TDDFT [B3LYP/6-31þþG(2d,3p)] method using the Gaussian 09 software package.29 As a consequence, the overall pattern of the calculated ECD curve of 1a was in agreement with the measured CD spectrum of 1 (Fig. 3). Thus, the absolute configurations of chiral carbons in 1 were assigned as 1R,3S,5S,7S,20S, which was further confirmed by its consistence with the absolute configurations of previously reported homoadamantyl PPAPs from the same plant.11,12,25
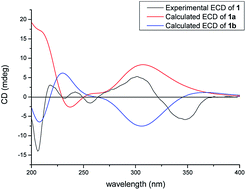 |
| | Fig. 3 Calculated and experimental ECD spectra of 1. | |
Hypersampsone X (2) was obtained as colorless oil. Its molecular formula was assigned as C33H40O4 on the basis of HRESIMS with a pseudo-molecular ion at 501.3016 [M + H]+. The 13C and DEPT NMR spectra displayed 33 carbons including a benzoyl group [δC 192.7, 135.0, 132.1, 129.1 × 2, 128.2 × 2], three carbonyl groups [δC 205.0, 206.7, 204.2], an isopentenyl group [δC 29.7, 119.2, 134.9, 18.2, 26.2], a terminal double bond [δC 118.4, 142.6] and five methyls [δC 23.0–27.3], suggesting that 2 was a PPAP derivative possessing a homoadamantyl skeleton. Extensively comparison of the NMR data of 2 with those of hypersampsone M11 revealed that they were structurally similar except for an additional 1-methylethenyl group substituted at C-25, which can be further confirmed by the obvious downshift of C-25 (δC 55.2) and the HMBC correlations from H3-28 to C-25/C-26/C-27 and H-25 to C-21 (Fig. 4).
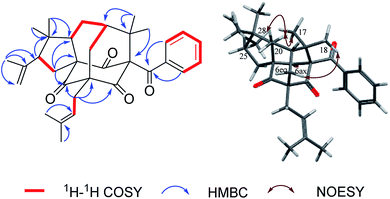 |
| | Fig. 4 Key 1H–1H COSY, HMBC and NOESY correlations of 2. | |
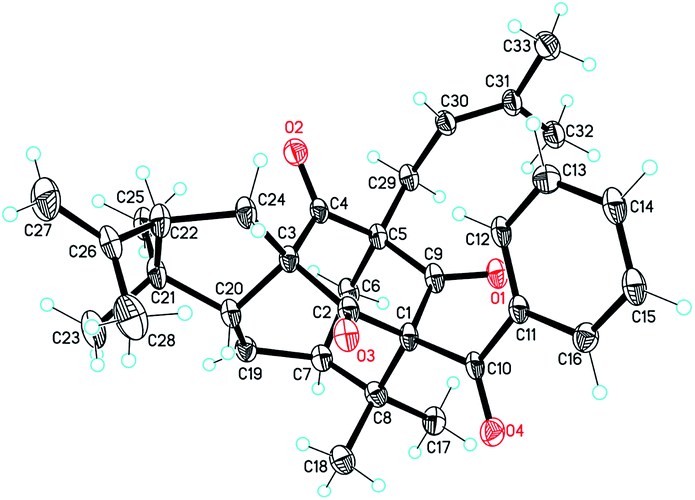
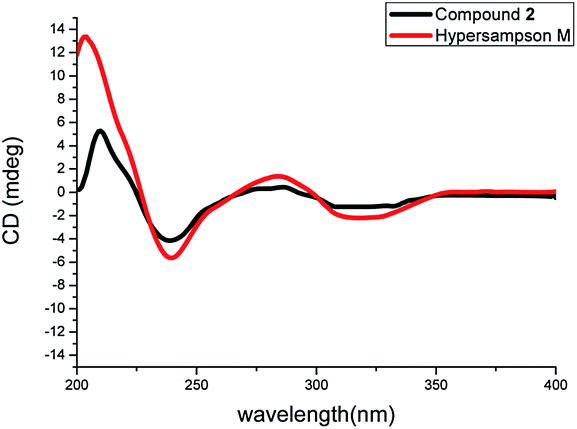
The NOESY correlations of H3-18/H-6ax, H3-17/H-20 and H-20/H3-28 indicated the α-orientation of H-20 and the β-orientation of H-25 in 2 (Fig. 4). Moreover, crystals suitable for a single-crystal X-ray diffraction experiment were fortunately obtained, which further confirmed the structure and relative configuration of 2 (Fig. 5). The CD spectrum of compound 2 has a highly agreement with biogenetic related analogue hypersampsone M (Fig. 6), whose absolute configuration has been unambiguously determined as 1R,3R,5S,7S,20R by the single crystal X-ray structure in our previously reported work.11 Thus, the absolute configuration of 2 was determined as 1R,3R,5S,7S,20R,25R, which was consistent with the absolute configuration of hypersampsone M.
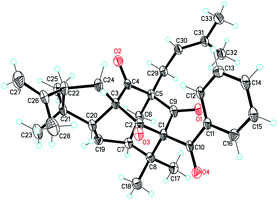 |
| | Fig. 5 The single X-ray crystal structure of 2. (CCDC 1551387†). | |
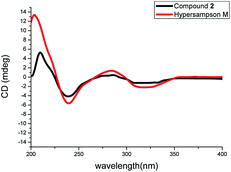 |
| | Fig. 6 The comparison of the CD spectra of 2 and hypersampson M. | |
Biosynthetically, PPAPs are probably derived from the acylphloroglucinol core structure (i). Prenylation of i would afford monocyclic polyprenylated acylphloroglucinols (MPAPs), which may be further cyclized to bicyclic polyprenylated acylphloroglucinols (BPAPs) type metabolites. The BPAPs type intermediate ii undergoes the cyclization of C-3 and C-20 to yield the homoadamantyl (iii) core structure.30 Norsampsone E (1) could be considered as the homoadamantyl type PPAPs with the loss of C-4 carbonyl in the phloroglucinol ring and followed by the formation of a new C–C bond between C-3 and C-5. The plausible biogenetic pathway of 1 was proposed to be generated from iii through the Retro-Claisen and decarboxylation reactions (Scheme 1). Compounds 2–4 was probably biosynthesized from the same precursor ii by epoxidation, intramolecular cyclization, and dehydration reactions (Scheme 1).25,30
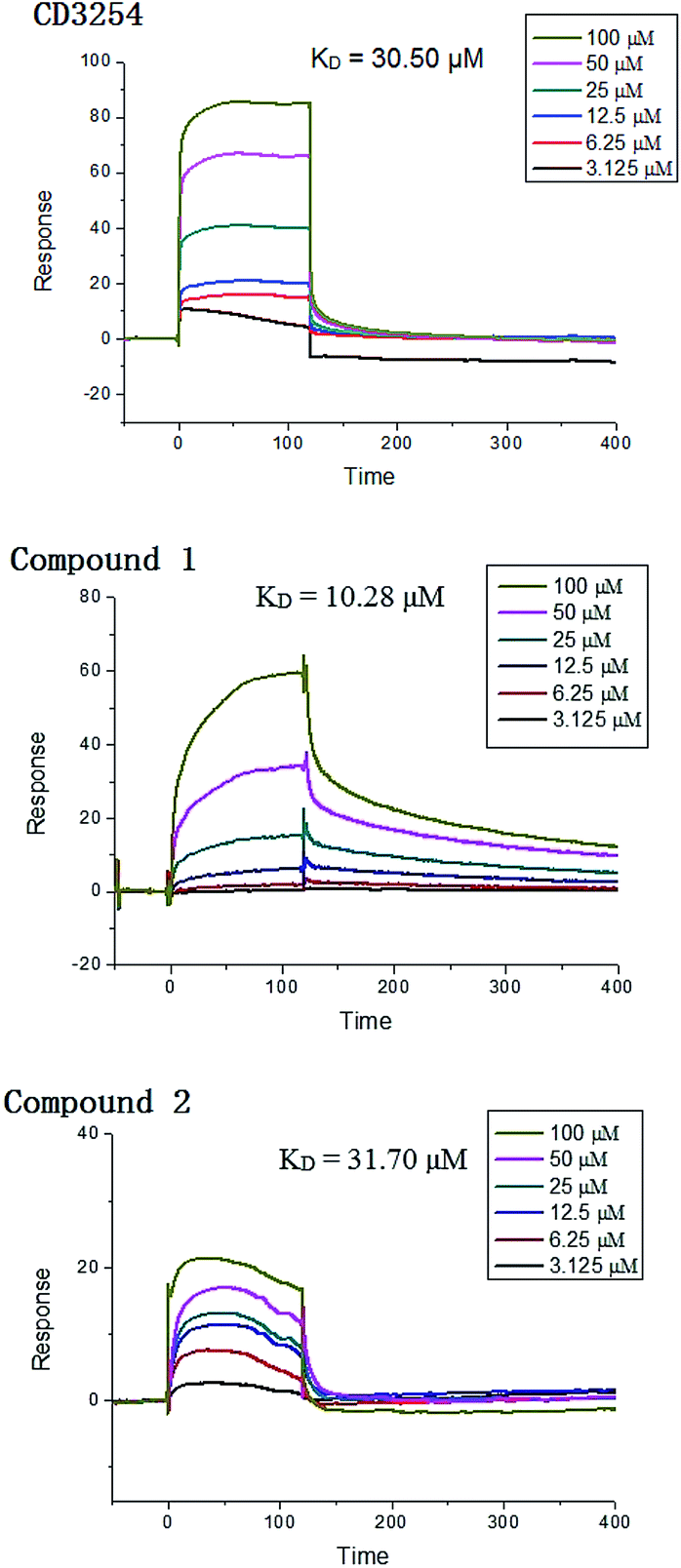
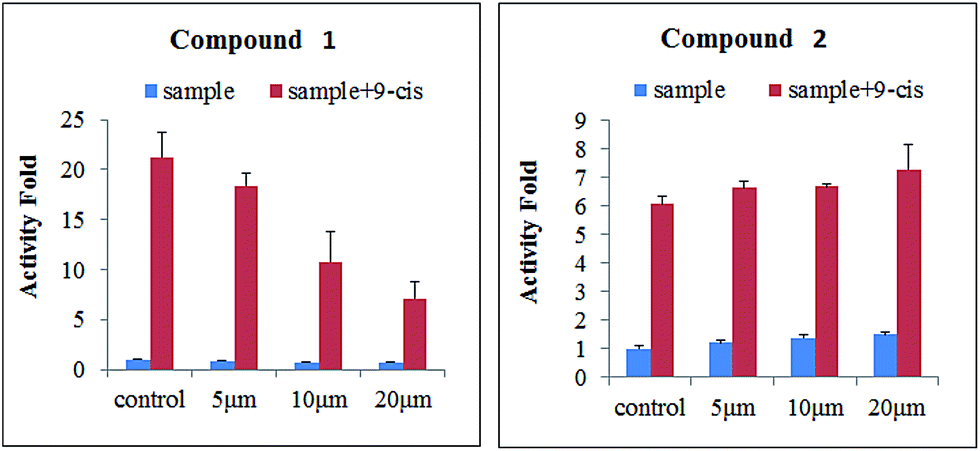
Surface plasmon resonance (SPR) technology has been widely used to monitor the kinetics of protein/protein and ligand/protein interactions, which has been successfully used to study the interactions between ligands and nuclear receptor-RXRα.23 Compounds 1–2 were evaluated for their binding affinity toward RXRα-LBD (ligand-binding domain) by SPR assay, which indicated that these two compounds had direct interaction towards RXRα-LBD with concentration-dependent SPR signals. As a positive control, the interaction between the known ligand CD3254 and RXRα was also investigated, which show directed interaction in a fast association and fast dissociation process (KD = 30.50 μM) (Fig. 7). However, compound 1 could bind to RXRα-LBD in a particularly slow association and slow dissociation mode with a KD of 10.28 μM, calculated by the kinetic analysis, while compound 2 was similar to CD3254 in a faster association and dissociation mode with a KD of 31.70 μM by steady-state binding analysis (Fig. 7). Besides, dual luciferase reporter gene assay was used to investigate the effects of compounds on RXRα transcriptional activity. As a result, compound 1 (5–20 μM) showed potent RXRα transcriptional-inhibitory activity in a dose dependent manner, while compound 2 (5–20 μM) slightly enhanced RXRα transactivation (Fig. 8). The above analysis indicated that compounds 1–2 might bind to RXRα with different mode of action, which needs further investigation.
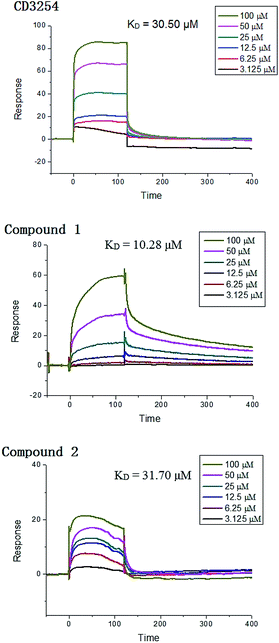 |
| | Fig. 7 SPR results of CD3254, compound 1 and 2 binding to RXRα-LBD. | |
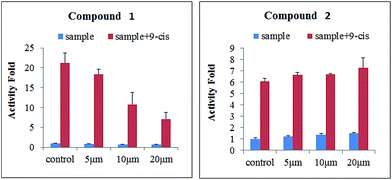 |
| | Fig. 8 Effects of compounds 1–2 (5, 10, and 20 μM) on the transcriptional activities of RXRα. | |
Conclusions
Up to now, several new skeleton PPAPs with the ring breakage or carbon reduction of phloroglucinol ring have been reported, including norsampsons A–D with the loss of C-2 carbonyl of the normal BPAPs, hyphenrones A–D formed by cleavage of the C-1/C-9 bond of the normal BPAPs, and hypersubone A represents the first seco-adamantyl PPAP by the cleavage of the C-1/C-9 bond of normal adamantine. However, there were no reports about the ring breakage or carbon reduction of normal homoadamantyl PPAPs. To the best of our knowledge, norsampsone E (1) stands for the first example of decarbonyl homoadamantyl PPAPs, which not only enriched the structural diversity of PPAPs, but also shed new light on the synthesis of this type compounds. Furthermore, the proposed plausible biogenetic pathways for 1–2 enriched the awareness of biosynthesis of homoadamantyl PPAPs. Besides, the SPR assay and RXRα transcriptional activity results indicated that compounds 1–2 might be the small-molecule modulators of RXRα and could be considered as potential lead compounds for RXRα based drugs with different mode of action, which were worthy of further investigation.
Experimental
General experimental procedures
Optical rotations were measured on a Jasco P-1020 polarimeter with a 1 cm cell at room temperature. UV spectra were recorded on a JASCO V-550 UV/Vis spectrometer. IR spectra were obtained using a JASCO FT/IR-480 plus spectrometer. CD spectra were measured on a Jasco J-810 spectropolarimeter at room temperature. HR-ESI-MS spectra were acquired using a Waters Synapt G2 mass spectrometer. The NMR spectra were measured with a Bruker AV-300/400/600 spectrometer at room temperature. Silica gel (200–300 mesh, Qingdao Marine Chemical Ltd., China), octadecylsilanized (ODS) silica gel (YMC Ltd., Japan) were used for open column chromatography (CC). TLC was performed on precoated silica gel plates (SGF254, 0.2 mm, Yantai Chemical Industry Research Institute, China). The semi-preparative HPLC was carried out on a Shimadzu LC-6AD Liquid Chromatography system with a SPD-20A Detector (Shimadzu, Japan) using a reversed-phase C18 column (5 μm, 10 × 250 mm; YMC, Japan).
Plant material
The aerial parts of Hypericum sampsonii were collected from Wuming, Guangxi Province, China, in June, 2014, and authenticated by Professor Songji Wei (Guangxi University of Traditional Chinese Medicine, Guangxi, China).
Extraction and isolation
The air-dried aerial parts of H. sampsonii (20 kg) were extracted by supercritical fluid of carbon dioxide. The crude extract (443.7 g) was partitioned with petroleum ether against MeOH–H2O (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10). The petroleum ether soluble extract (200.4 g) was separated by chromatography on a silica gel column (3.5 × 13.2 cm; cyclohexane to ethyl acetate, 100:0 → 0:100) to afford eleven fractions (Fr. 1–Fr. 11). Fr. 2 was further purified by column chromatography (silica gel CC, 3.5 × 13.2 cm; petroleum ether to acetone, 100:0 → 0:100) to yield six fractions (Fr. 2.1–Fr. 2.6). Fr. 2.4 (6.7 g) was further subjected to ODS column chromatography eluted with MeOH–H2O (70:30 → 100:0) and purified by semi-preparative HPLC on ODS column with 80% ACN-H2O to afford compound 1 (26.3 mg) and compound 2 (6.9 mg). Fr. 2 (8.3 g) was subjected to ODS column chromatography eluted with MeOH–H2O (60:40 → 100:0) to obtained nine fractions (Fr. 2.1–Fr. 2.9). Fr. 2.5 (138.5 mg) was purified by semi-preparative HPLC on ODS column with 75% ACN-H2O to afford compound 4 (9.6 mg). Fr. 8 (23.7 g) was subjected to ODS column chromatography eluted with MeOH–H2O (60:40 → 100:0) to obtained ten fractions (Fr. 8.1–Fr. 8.10). Fr. 8.9 (6.9 g) was further subjected to a silica gel column using petroleum ether to acetone gradient elution to yield 11 fractions (Fr. 8.9.1–8.9.11). Fr. 8.9.6 (297.4 mg) was purified by preparative HPLC on ODS column with 75% MeOH–H2O to yield compounds 3 (7.8 mg).
:10). The petroleum ether soluble extract (200.4 g) was separated by chromatography on a silica gel column (3.5 × 13.2 cm; cyclohexane to ethyl acetate, 100:0 → 0:100) to afford eleven fractions (Fr. 1–Fr. 11). Fr. 2 was further purified by column chromatography (silica gel CC, 3.5 × 13.2 cm; petroleum ether to acetone, 100:0 → 0:100) to yield six fractions (Fr. 2.1–Fr. 2.6). Fr. 2.4 (6.7 g) was further subjected to ODS column chromatography eluted with MeOH–H2O (70:30 → 100:0) and purified by semi-preparative HPLC on ODS column with 80% ACN-H2O to afford compound 1 (26.3 mg) and compound 2 (6.9 mg). Fr. 2 (8.3 g) was subjected to ODS column chromatography eluted with MeOH–H2O (60:40 → 100:0) to obtained nine fractions (Fr. 2.1–Fr. 2.9). Fr. 2.5 (138.5 mg) was purified by semi-preparative HPLC on ODS column with 75% ACN-H2O to afford compound 4 (9.6 mg). Fr. 8 (23.7 g) was subjected to ODS column chromatography eluted with MeOH–H2O (60:40 → 100:0) to obtained ten fractions (Fr. 8.1–Fr. 8.10). Fr. 8.9 (6.9 g) was further subjected to a silica gel column using petroleum ether to acetone gradient elution to yield 11 fractions (Fr. 8.9.1–8.9.11). Fr. 8.9.6 (297.4 mg) was purified by preparative HPLC on ODS column with 75% MeOH–H2O to yield compounds 3 (7.8 mg).
Norsampsone E (1). Colorless oil; [α]23D −63.0 (c 0.30, CHCl3); UV (CH3OH) λmax (logε) 201 (4.32) nm; IR (KBr) νmax 3415, 2922, 2866, 1727, 1635, 1456, 1372, 1247, 1224, 1067 cm−1; CD (CH3OH) λmax (Δε) 327 (−1.14), 254 (0.10) nm; HR-ESI-MS m/z 455.3161 [M + H]+ (calcd for C29H43O4, 455.3161). 1H (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) data, see Table 1.
Table 1 1H NMR and 13C NMR data for 1–2 (CDCl3)
| No. |
1a |
2b |
| δC |
δH (J in Hz) |
δC |
δH (J in Hz) |
| Recorded at 100 M for 13C and 400 M for 1H. Recorded at 150 M for 13C and 600 M for 1H. |
| 1 |
85.6 |
|
81.0 |
|
| 2 |
211.0 |
|
205.0 |
|
| 3 |
82.7 |
|
74.1 |
|
| 4 |
|
|
206.7 |
|
| 5 |
58.4 |
|
69.1 |
|
| 6ax |
35.8 |
1.97, dd (13.6, 4.0) |
43.2 |
2.54, m |
| 6eq |
|
2.20, dd (13.6, 2.0) |
|
1.89, m |
| 7 |
41.7 |
1.69, m |
44.1 |
2.12, m |
| 8 |
50.0 |
|
51.0 |
|
| 9 |
211.7 |
|
204.2 |
|
| 10 |
208.4 |
|
192.7 |
|
| 11 |
42.6 |
2.27, sept (6.4) |
135.0 |
|
| 12 |
19.8 |
1.06, d (6.4) |
129.1 |
7.12, br.d (7.4) |
| 13 |
|
|
128.2 |
7.39, br.t (7.4) |
| 14 |
|
|
132.2 |
7.24, br.t (7.4) |
| 15 |
|
|
128.2 |
7.39, br.t (7.4) |
| 16 |
19.6 |
1.02, d (6.4) |
129.1 |
7.12, br.d (7.4) |
| 17 |
21.9 |
1.20, s |
23.0 |
1.41, s |
| 18 |
24.5 |
1.26, s |
25.4 |
1.45, s |
| 19 |
26.3 |
1.88, br.s |
29.2 |
2.28, m |
| 20 |
42.0 |
2.70, br.t (10.0) |
57.4 |
2.03, br. d (8.4) |
| 21 |
142.6 |
|
44.5 |
|
| 22 |
22.1 |
1.84, s |
27.1 |
0.87, s |
| 23 |
118.4 |
5.23, br.s 4.96, br.s |
27.3 |
0.94, s |
| 24 |
|
|
34.6 |
2.65, m |
| 25 |
|
|
55.2 |
3.16, m |
| 26 |
|
|
145.4 |
|
| 27 |
|
|
112.0 |
4.91, br.s 4.84, br.s |
| 28 |
|
|
24.1 |
1.80, s |
| 29 |
25.9 |
2.41, m |
29.7 |
2.55, m |
| 30 |
118.7 |
5.30, br.t (7.6) |
119.2 |
5.06, br.t (10.8) |
| 31 |
139.0 |
|
134.9 |
|
| 32 |
16.3 |
1.63, s |
18.2 |
1.66, s |
| 33 |
40.3 |
2.01, m |
26.2 |
1.66, s |
| 34 |
26.6 |
2.08, m |
|
|
| 35 |
124.2 |
5.06, br.t (6.8) |
|
|
| 36 |
131.8 |
|
|
|
| 37 |
17.8 |
1.58, s |
|
|
| 38 |
25.9 |
1.64, s |
|
|
Hypersampsone X (2). Colorless oil; [α]23D +27.1 (c 0.50, CHCl3); UV (CH3OH) λmax (logε) 206 (3.91), 246 (3.56) nm; IR (KBr) νmax 2922, 2975, 2361, 1736, 1698, 1677, 1457, 1377, 1224, 683 cm−1; CD (CH3OH) λmax (Δε) 327 (−12.3), 280 (0.14), 244 (−16.7) nm; HR-ESI-MS m/z 501.3016 [M + H]+ (calcd for C33H41O4, 501.2981). 1H (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, see Table 1.
Quantum chemical ECD calculation method
In theoretical calculations, the geometry of the molecules was optimized with Gaussian 09 package29 at B3LYP/6-31G(d) computational level. The minimum nature of the structure was confirmed by frequency calculations at the same computational level. Then ECD calculations were carried out in the methanol solvent medium using time-dependent density functional theory (TDDFT) with B3LYP functional and DGDZVP basis set.
X-ray crystallographic analysis of 2, 3, and 4
Crystal data for hypersampsone X (2). C33H40O4 (M = 500.65): orthorhombic, space group P212121, a = 7.7835(6) Å, b = 12.1558(8) Å, c = 28.405(3) Å, α = 90°, β = 90°, γ = 90°, V = 2687.6(4) Å3, Z = 4, T = 100.00(10) K, μ(Cu Kα) = 0.626 mm−1, Dcalc = 1.237 g cm−3, 19267 measured reflections, 5321 independent reflections [Rint = 0.1215]. The final R1 was 0.0977 and wR2 was 0.2371 [I > 2σ(I)]. The goodness of fit on F2 was 1.069 (CCDC 1551387†).
Crystal data for hypersampsone Q (3). C33H42O5 (M = 518.66): orthorhombic, space group P212121, a = 14.15822(19) Å, b = 17.0208(3) Å, c = 24.2524(4) Å, β = 99°, V = 5844.45(16) Å3, Z = 8, T = 150.05(10) K, μ(Cu Kα) = 0.618 mm−1, Dcalc = 1.179 g mm−3, 20563 measured reflections, 8816 independent reflections [Rint = 0.0304]. The final R1 was 0.0398 and wR2 was 0.1032 [I > 2σ(I)]. The goodness of fit on F2 was 1.043. Flack parameter = −0.04(8) (CCDC 1551384†).
Crystal data for sampsonione B (4). C33H42O5 (M = 518.67): orthorhombic, space group P212121, a = 9.8352(5) Å, b = 11.5100(7) Å, c = 49.946(3) Å, α = 90°, β = 90°, γ = 90°, V = 5654.1(6) Å3, Z = 8, T = 150.05(10) K, μ(Cu Kα) = 0.639 mm−1, Dcalc = 1.219 mg mm−3, 35128 measured reflections, 9014 independent reflections [Rint = 0.0710]. The final R1 was 0.042 and wR2 was 0.0833 [I > 2σ(I)]. The goodness of fit on F2 was 1.016. Flack parameter = 0.18(16) (CCDC 1551380†).
Acknowledgements
This research was financially supported by the China Postdoctoral Science Foundation funded project (No. 2016M592094), the Fundamental Research Funds for the Central Universities (20720160117), and grants from Natural Science Foundation of China (NSFC) (No. 81602988).
Notes and references
- S. B. Wu, C. L. Long and E. J. Kennelly, Nat. Prod. Rep., 2014, 31, 1158–1174 RSC
 .
. - D. Marianna and A. T. Emmanuel, Biomimetic Org. Synth., 2011, 433–467 Search PubMed .
- R. Ciochina and R. B. Grossman, Chem. Rev., 2006, 106, 3963–3968 CrossRef CAS PubMed .
- W. J. Xu, J. Luo, R. J. Li, M. H. Yang and L. Y. Kong, Org. Chem. Front., 2017, 4, 313–317 RSC .
- X. W. Yang, Y. P. Li, J. Su, W. G. Ma and G. Xu, Org. Lett., 2016, 18, 1876–1879 CrossRef CAS PubMed .
- D. S. Tian, P. Yi, L. Xia, X. Xiao, Y. M. Fan, W. Gu, L. J. Huang, Y. B. David, Y. T. Di, C. M. Yuan and X. J. Hao, Org. Lett., 2016, 18, 5904–5907 CrossRef CAS PubMed .
- Y. M. Fan, P. Yi, C. Yan, T. Huang, W. Gu, Y. Ma, L. J. Huang, J. X. Zhang, C. M. Yuan and X. J. Hao, Org. Lett., 2015, 17, 2066–2069 CrossRef CAS PubMed .
- Y. Liao, X. Liu, J. Yang, Y. Z. Lao, X. W. Yang, X. N. Li, J. J. Zhang, Z. J. Ding, H. X. Xu and G. Xu, Org. Lett., 2015, 17, 1172–1175 CrossRef CAS PubMed .
- X. W. Yang, Y. Q. Ding, J. J. Zhang, X. Liu, L. X. Yang, X. N. Li, D. Ferreira, L. A. Walker and G. Xu, Org. Lett., 2014, 16, 2434–2437 CrossRef CAS PubMed .
- J. J. Zhang, J. Yang, Y. Liao, X. W. Yang, J. Z. Ma, Q. L. Xiao, L. X. Yang and G. Xu, Org. Lett., 2014, 16, 4912–4915 CrossRef CAS PubMed .
- W. J. Tian, Y. Yu, X. J. Yao, H. F. Chen, Y. Dai, X. K. Zhang and X. S. Yao, Org. Lett., 2014, 16, 3448–3451 CrossRef CAS PubMed .
- W. J. Tian, Y. Q. Qiu, X. J. Yao, H. F. Chen, Y. Dai, X. K. Zhang and X. S. Yao, Org. Lett., 2014, 16, 6346–6349 CrossRef CAS PubMed .
- H. C. Zhu, C. M. Chen, J. Yang, X. N. Li, J. J. Liu, B. Sun, S. X. Huang, D. G. Li, G. M. Yao, Z. W. Luo, Y. Li, J. W. Zhang, Y. B. Xue and Y. H. Zhang, Org. Lett., 2014, 16, 6322–6325 CrossRef CAS PubMed .
- X. W. Yang, X. Deng, X. Liu, C. Y. Wu, X. N. Li, B. Wu, H. R. Luo, Y. Li, H. X. Xu, Q. S. Zhao and G. Xu, Chem. Commun., 2012, 48, 5998–6000 RSC .
- C. P. Ting and T. J. Maimone, J. Am. Chem. Soc., 2015, 48, 10516–10519 CrossRef PubMed .
- H. Fiene, B. Nicole and P. Bernd, J. Am. Chem. Soc., 2014, 136, 4026–4030 CrossRef PubMed .
- J. G. Alexander, H. B. Jonathan and A. P. John Jr, J. Am. Chem. Soc., 2014, 136, 11799–11804 CrossRef PubMed .
- H. B. Jonathan and A. P. John Jr, Angew. Chem., 2014, 126, 7966–7971 CrossRef .
- N. S. Simpkins, Chem. Commun., 2013, 49, 1042–1051 RSC .
- L. Q. Chen, Z. G. Wang, A. E. Aleshin, F. Chen, J. B. Chen, F. Q. Jiang, G. Alitongbieke, Z. P. Zeng, Y. Ma, M. F. Huang, H. Zhou, G. Cadwell, J. F. Zheng, P. Q. Huang, R. C. Liddington, X. K. Zhang and Y. Su, Chem. Biol., 2014, 5, 596–607 CrossRef PubMed .
- N. Bushue and Y. J. Y. Wan, Adv. Drug Delivery Rev., 2010, 62, 1285–1298 CrossRef CAS PubMed .
- M. I. Dawson and X. K. Zhang, Curr. Med. Chem., 2002, 9, 623–637 CrossRef CAS PubMed .
- D. Xu, L. J. Cai, S. J. Guo, L. Xie, M. M. Yin, Z. W. Chen, H. Zhou, Y. Su, Z. P. Zeng and X. K. Zhang, Bioorg. Med. Chem. Lett., 2017, 27, 1055–1061 CrossRef CAS PubMed .
- Jiangsu New Medical College, Dictionary of Chinese Crude Drugs, Shanghai Scientific Technological Publishers, Shanghai, 1977, pp. 345–346 Search PubMed .
- W. J. Tian, Y. Q. Qiu, X. J. Jin, H. F. Chen, X. J. Yao, Y. Dai and X. S. Yao, Tetrahedron, 2014, 70, 7912–7916 CrossRef CAS .
- W. J. Tian, Y. Q. Qiu, X. J. Jin, H. F. Chen, X. J. Yao, Y. Dai and X. S. Yao, RSC Adv., 2016, 6, 50887–50894 RSC .
- W. J. Tian, Y. Q. Qiu, H. F. Chen, X. J. Jin, X. J. Yao, Y. Dai and X. S. Yao, Fitoterapia, 2017, 116, 39–44 CrossRef CAS PubMed .
- Z. Y. Xiao, Q. Mu, W. K. P. Shiu, Y. H. Zeng and S. Gibbons, J. Nat. Prod., 2007, 70, 1779–1782 CrossRef CAS PubMed .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009 .
- L. H. Hu and K. Y. Sim, Tetrahedron, 2000, 56, 1379–1386 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Biological assays details and NMR spectra of compounds 1–2. CCDC 1551380, 1551384 and 1551387. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra05947g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Guang-Hui Wanga,
Yi Dai*b,
Hai-Feng Chen*a and
Xin-Sheng Yao
c,
Guang-Hui Wanga,
Yi Dai*b,
Hai-Feng Chen*a and
Xin-Sheng Yao