DOI:
10.1039/C7RA05892F
(Paper)
RSC Adv., 2017,
7, 35181-35190
High catalytic activity over novel Mg–Fe/Ti layered double hydroxides (LDHs) for polycarbonate diols (PCDLs): synthesis, mechanism and application
Received
26th May 2017
, Accepted 1st July 2017
First published on 13th July 2017
Abstract
A series of novel activated Mg–Fe/Ti layered double hydroxides (LDHs) with high crystallinity, which were used for the transesterification between dimethyl carbonate (DMC) and aliphatic diols as acid–base bifunctional catalysts were successfully synthesized by co-precipitation. The structures of the LDHs were characterized by both X-ray diffraction (XRD) and Fourier transform infrared spectroscopy (FTIR), indicating that the formation of LDHs is strongly affected by the initial pH of the suspension. A general precipitation–recombination–crystallization mechanism was proposed to explain the LDHs formation and growth. The results of the CO2 and NH3 temperature-programmed desorption (CO2/NH3-TPD) tests showed that with increasing the amount of Ti4+ cations, the basicity of LDHs decreased, while the acidity of LDHs increased. An acid–base cooperative manner to catalyze the transesterification reaction was found to improve the catalytic activity of LDHs. The polycarbonate diols (PCDLs) with a high number-average molecular weight (Mn) and low hydroxyl value were obtained via a two-step transesterification method, which can avoid the DMC/methanol azeotrope during the reaction. With LDH-10 catalyst, the yield of methanol is up to 89.12%, and the PCDL shows the highest Mn (3030) and lowest hydroxyl value (43.5).
1. Introduction
Polyurethanes produced from polycarbonate diols (PCDLs), which were employed as soft segments show better mechanical performance, biocompatibility, hydrolytic resistance, oxidizing resistance and weather resistance than those of the conventional ones based on polyether or polyester diols.1–6
Over recent years, PCDLs have been synthesized by many routes, such as the conventional phosgene method,7 ring-opening polymerization of cyclic carbonates,8 the copolymerization of oxiranes with CO2,9 and the transesterification between organic carbonate (dimethyl carbonate (DMC), diethyl carbonate (DEC) and/or diphenyl carbonate (DPC)) and diols.10–12 Among them, the transesterification between DMC and aliphatic diols is regarded as a promising green route to prepare PCDLs because DMC is a green compound and has been commercially produced via the oxidative carbonylation of methanol instead of conventional phosgene route. Unfortunately, due to the formation of azeotrope between methanol and DMC during the reaction, it is difficult to control the molecular weight of the target product.13,14
To improve the controllability of the transesterification between dimethyl carbonate (DMC) and aliphatic diols, various kinds of homogeneous catalysts and heterogeneous catalysts have been developed. Homogeneous catalysts show high catalytic activity, but it is usually difficult to be removed from the product. Moreover, the strong alkaline of alkali metal may cause some side reactions, and titanates are unstable and easy to hydrolysis in the air. Heterogeneous catalysts including metal–organic frameworks (MOFs)15–17 and layered double hydroxides (LDHs)13,18–24 are new environmentally friendly catalysts, which can be easily removed from the reaction mixture and recycled. Wang et al.15 synthesized polycarbonal diol catalyzed by metal–organic framework Zn4O[CO2–C6H4–CO2]3. The results show high catalytic activity in the preparation of PCDL via the transesterification between DPC and 1,6-hexandiol (1,6-HD). However, the metal–organic framework is made of metal ions and organic ligands by coordination links, which leading to the poor of hydrolytic stability and thermal stability.25,26 LDHs is a group of anionic clays, which are represented by a general formula [M1−x2+Mx3+(OH)2]x+(An−)x/n·mH2O, where M2+ and M3+ are divalent and trivalent cations respectively, An− is the exchangeable interlayer anion with n negative charge. Due to the ion-exchange properties of the An− ions with other anions or anionic complexes, and acid–basic sites, the LDHs have the potential application as catalysts.18,19 Mg–Al LDHs were synthesized by mixing divalent metal oxide with the relevant trivalent metal chloride solution.20,21 The results indicated that the MgO content has a great impact on the catalyst structure and acid–basic properties, which can be adjusted to further improve the product selectivity. Zazoua et al.22 synthesized Mg–M (M: Al, Fe, Cr) LDHs by a co-precipitation method, which indicated that the order of activity was attributed to both metal and acid–base properties of the catalysts. LDHs with varying Mg/Fe rations were investigated by Mónika et al.23 It was found that the quadrupole splitting decreased with increasing Mg(II)/Fe(III) ratio reflecting the effect of substitution of Fe(III) with the Mg(II) site in the layered structure. Hosni et al.24 synthesized Mg–Al–Ti LDHs containing divalent, trivalent and tetravalent cations, which was demonstrated as efficient visible-light photocatalysts. In the literature,27 it was found that the activity and stability of Ce1−xTixO2−δ catalysts were promoted by doping Ti content, which attributed to the enhanced redox properties together with the modification of surface acid–base sites, providing by the proper formation of Ce–O–Ti linkage bonds.
Although LDHs have previously been studied much, the report about the LDHs used in the transesterification between DMC and aliphatic diols is rare. Feng et al.13 reported the environmentally benign route for synthesis of PCDLs catalyzed by calcined Mg–Al hydrotalcites as heterogeneous catalysts. The results suggested that the use of the heterogeneous catalysts was advantageous because they can overcome the shortcomings associated with conventional homogeneous ones, which were commonly adopted for the PCDL preparation. But the problem of the formation of azeotrope between DMC and methanol during the reaction was not solved. And the formation and catalytic mechanisms of the LDHs were not been studied deeply.
In this work, a series of novel Mg–Fe/Ti LDHs with different Fe3+/Ti4+ molar ratio were prepared and applied for the synthesis of PCDLs. The formation and catalyzing mechanisms of the Mg–Fe/Ti LDHs were systematically studied. X-ray diffraction (XRD), Fourier transform infrared spectrometry (FTIR), transmission electro microscopy (TEM), thermogravimetric (TG), and CO2/NH3 temperature-programmed desorption (CO2/NH3-TPD) were used to elucidate the relationship between the catalyst structure, acid–base property and catalytic activity. The PCDLs with high number-average molecular weight (Mn) and low hydroxyl value was obtained via two-step polycondensation, which can effectively solved the problem of the formation of azeotrope during the transesterification.
2. Experimental
2.1 Materials
All reagents were commercially available and analytical grade without any further purification. The magnesium nitrate hexahydrate (Mg(NO3)2·6H2O, 99.0%, Guangzhou chemical reagents co., LTD.), iron nitrate nonahydrate (Fe2(NO3)3·9H2O, 98.5.0%, Tianjin Fu Chen chemical reagents co., LTD.), titanium sulfate (Ti(SO4)2, 96.0%, Shanghai Runjie Chemical Reagents co., LTD.), sodium hydroxide (NaOH, 96%, Tianjin Fu Chen chemical reagents co., LTD.), and sodium carbonate (Na2CO3, Tianjin Fu Chen chemical reagents co., LTD.) were used to prepare the catalyst Mg–Fe/Ti LDHs.
The dimethyl carbonate (DMC, 99%, Shanghai Runjie Chemical Reagents co., LTD.), 1,5-pentanediol (1,5-PD, 98%, Shanghai Runjie Chemical Reagents co., LTD.), 1,6-hexanediol (1,6-HD, 99.5%, Shanghai Runjie Chemical Reagents co., LTD.) were used to synthesize PCDLs.
2.2 Synthesis of Mg–Fe/Ti LDHs
Co-precipitation method was adopted for the preparation of Mg–Fe/Ti LDHs with various Fe3+/Ti4+ molar ratios. All of the samples were labeled as LDH-x (x = 1, 2, …13), as shown in Table 1. The detailed synthetic procedure for Mg–Fe/Ti LDHs is described as follows: a aqueous solution containing of NaOH (2 M) and Na2CO3 (1.5 M), which was used as precipitant, was dropwise added into a salt solution containing of Mg(NO3)2·6H2O, Fe(NO3)3·9H2O, and Ti(SO4)2 (the molar ratio of Mg2+/(Fe3+ + Ti4+) is 3.0, and the molar ratio of Fe3+/Ti4+ varies from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 40:60.) under constant stirring at a pH between 9 and 13. After that, the slurry was aged at 80 °C for 24 h. The resulting precipitates were filtered and repeatedly washed with deionized water to eliminate excess Na+, NO3−, SO42−, and CO32− ions until the pH value of the washing water was 7, followed by drying in a vacuum oven at 60 °C for 12 h.
:0 to 40:60.) under constant stirring at a pH between 9 and 13. After that, the slurry was aged at 80 °C for 24 h. The resulting precipitates were filtered and repeatedly washed with deionized water to eliminate excess Na+, NO3−, SO42−, and CO32− ions until the pH value of the washing water was 7, followed by drying in a vacuum oven at 60 °C for 12 h.
Table 1 Chemical compositions and lattice parameter of Mg–Fe/Ti LDHs
| Samples |
Denoted |
pH |
Mg:Fe:Ti |
Lattice parameters |
d(003) (Å) |
| In solution |
a (Å) |
c (Å) |
| Mg–Fe |
LDH-1 |
9 |
3:1:0 |
3.121 |
23.049 |
7.683 |
| Mg–Fe |
LDH-2 |
10 |
3:1:0 |
3.115 |
23.127 |
7.709 |
| Mg–Fe |
LDH-3 |
11 |
3:1:0 |
3.097 |
23.439 |
7.813 |
| Mg–Fe |
LDH-4 |
12 |
3:1:0 |
3.109 |
23.433 |
7.811 |
| Mg–Fe |
LDH-5 |
12.5 |
3:1:0 |
3.110 |
23.436 |
7.812 |
| Mg–Fe |
LDH-6 |
13 |
3:1:0 |
3.113 |
23.430 |
7.81 |
| Mg–Fe/Ti |
LDH-7 |
12.5 |
3:0.95:0.05 |
3.114 |
23.373 |
7.791 |
| Mg–Fe/Ti |
LDH-8 |
12.5 |
3:0.90:0.10 |
3.116 |
23.367 |
7.789 |
| Mg–Fe/Ti |
LDH-9 |
12.5 |
3:0.85:0.15 |
3.117 |
23.352 |
7.784 |
| Mg–Fe/Ti |
LDH-10 |
12.5 |
3:0.80:0.20 |
3.119 |
23.325 |
7.775 |
| Mg–Fe/Ti |
LDH-11 |
12.5 |
3:0.75:0.25 |
3.120 |
23.313 |
7.771 |
| Mg–Fe/Ti |
LDH-12 |
12.5 |
3:0.60:0.40 |
3.123 |
23.286 |
7.762 |
| Mg–Fe/Ti |
LDH-13 |
12.5 |
3:0.40:0.60 |
3.128 |
23.271 |
7.757 |
2.3 Synthesis of PCDLs
PCDLs were synthesized by two-step polycondensation of DMC, 1,5-PD, and 1,6-HD via transesterification as shown in Scheme 1. Due to the azeotropy of DMC and methanol during the reaction, excessive DMC (270 g, 3 mol) was first reacted with 1,5-PD (31.2 g, 0.3 mol) by LDHs (1.0 wt% based on the total mass of raw material, used as the catalyst) in a 500 mL four-necked round-bottom flask equipped with a N2 inlet, mechanical stirrer, reflux condenser, thermometer under atmospheric-pressure nitrogen atmosphere in an oil bath at a temperature between 90 and 100 °C for 5 h. The bis(methylcarbonate)α,ω-alkylene (BMCA) was produced, which mainly consisted of dimmers of the dimethylene carbonate units. The pressure in the flask was then reduced to about 0.05 MPa at the same temperature to facilitate the polymerization by removing the unreacted DMC and ethanol produced by the condensation reactions until there were no volatile byproducts distilled off. Subsequently, the LDHs catalyst was removed by filtration.
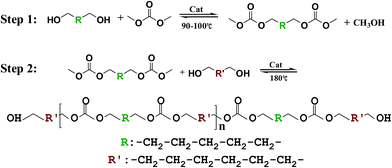 |
| | Scheme 1 The synthesis of PCDL. | |
After the reaction, different stoichiometric ratio of bis(methylcarbonate)α,ω-alkylene, 1,6-HD, and LDHs reacted under atmospheric-pressure nitrogen atmosphere at 100 °C over 1 h using the same equipment as the one in the first step. The reactor temperature was gradually increased to 180 °C for 4 h. Then, the reaction temperature remained 180 °C, while the pressure was reduced to ∼0.05 MPa for 2 h. Once more, the pressure was reduced to ∼0.005 MPa until there was no ethanol distilled off. Eventually, the LDHs were filtered out when the products turned into liquid.
2.4 Characterization
The X-ray diffraction (XRD) measurements were conducted on an X-ray diffractometer using Cu Ka radiation (Bruker, Germany). The Fourier transform infrared (FTIR) spectra were recorded between 4000 and 400 cm−1 with a VERTEX 33 FTIR spectrophotometer (Bruker, Germany). Transmission electron microscope (TEM) of JEM 2100F (Jeol, Japan) was applied to analyze the microstructure of the LDHs materials. Thermogravimetric (TG) experiments were carried out on a TA instruments (Netzsch STA 449 F3 Jupiter, Selb, Germany) with the scanning range from 30 to 800 °C. The molecular weight (number-average molecular weights 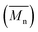 and weight-average molecular weights
and weight-average molecular weights 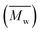 ), and the polydispersity index
), and the polydispersity index 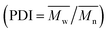 (PDI) of PDCLs were determined by gel permeation chromatography (GPC) performed on a Viscotek GPC Max (USA) equipped with Triple Detector Array TDA 305. The 1H NMR spectra were performed on an AVANCE III HD 600 (Bruker, Germany) using deuterated chloroform (CDCl3) as the solvent.
(PDI) of PDCLs were determined by gel permeation chromatography (GPC) performed on a Viscotek GPC Max (USA) equipped with Triple Detector Array TDA 305. The 1H NMR spectra were performed on an AVANCE III HD 600 (Bruker, Germany) using deuterated chloroform (CDCl3) as the solvent.
The temperature-programmed desorption of CO2 and NH3 (CO2/NH3-TPD) were used to determine the basic and acid properties of Mg–Fe/Ti LDHs. The desorption of CO2 and NH3 were monitored on line by a thermal conductivity detector (TCD) during heating the sample from 60 to 700 °C at a ramp rate of 10 K min−1 (AutoChem II 2920, Micromeritics, America).
3. Results and discussion
3.1 Characterization of LDHs
3.1.1 X-ray diffraction (XRD). Fig. 1 showed the powder XRD patterns of the Mg–Fe LDHs, which were obtained for pH value varying from 9 to 13. The basal peaks attributed to (0 0 3), (0 0 6) and (0 0 9) crystal planes, and non-basal peaks corresponding to (0 1 2), (0 1 5), (0 1 8) and (1 1 0) diffraction peaks can be indexed to a hexagonal symmetry (JCPDS: 70-2150), according to the typical structure of LDH materials. With increasing the pH value, the diffraction peaks of LDHs gradually become sharper, indicating an increase in the crystallinity of Mg–Fe LDHs. The similar phenomenon was also found in the previously reports,28–30 which maybe due to the Mg2+ cation precipitate at the higher pH value. Especially, the peaks at (0 0 3) and (0 0 6) planes shift to lower scattering angles for the pH value varying from 9 to 11, and remain unchanged subsequently, as shown in Fig. 1(b). The basal interlayer spacings (d003), which were calculated from the (003) diffraction peaks by Bragg formula were listed in Table 1. It is clearly seen that d003 increase from 7.683 Å to 7.813 Å with the pH value increasing from 9 to 11, which maybe attribute to (i) the second phase noted as FeO(OH) (JCPDS: 22-0353) and Mg5(CO3)4(OH)2(H2O)4 (JCPDS: 70-1177) or (ii) the lower amount of compensating anion (CO32−) in the interlayer for pH < 11 than that for pH ≥ 11. As previous reported,31 the initial pH of the solution is important to avoid the presence of unreacted precursors and secondary crystalline phases. When pH = 12.5, a good crystal shape and high crystallinity of LDH was obtained and remained almost same with further increasing the pH value, which indicated that 12.5 is the suitable pH value for obtaining the high crystallinity of LDHs.
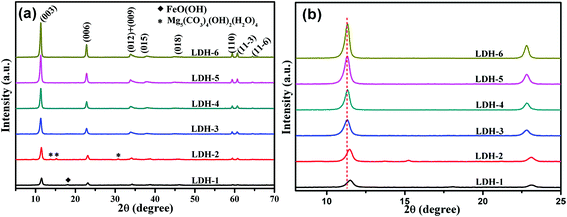 |
| | Fig. 1 XRD patterns of Mg–Fe LDHs with different pH values. | |
As is known to all, LDH is an acid–base bifunctional catalyst containing both of acidic and basic active sites. The catalytic activity is influenced by the strength matching of the acidic and basic active sites on the acid–base cooperative catalysis, which can be changed by using different types of materials. In order to obtain acid–base bifunctional catalysts with high catalytic activity, which were used for the transesterification between DMC and aliphatic diols, the tetravalent metal ion (Ti4+) were introduced into the Mg–Fe LDH to adjust the strength matching of the acidic and basic active sites of Mg–Fe/Ti LDHs. XRD patterns for Mg–Fe/Ti LDHs with different Fe3+/Ti4+ molar ratio at pH = 12.5 was shown in Fig. 2. All of the peaks can be indexed to typical LDH structure without any secondary phases. It is clearly seen that the intensity of the reflection peaks decreases with increasing the Ti4+/(Fe3+ + Ti4+) molar ratio, corresponding to a decrease of the crystallinity of the LDH. From Fig. 2(b), the basal reflection (2θ ≈ 11.4°) of LDHs displayed a little shift to higher scattering angle with the incorporation of Ti4+ cation, indicating that the basal interlayer spacing (d003) decrease with the incorporation of tetravalent metal cation (Ti4+). It is similar with the previous reports on Ti-containing LDHs.24,32 The decrease in the basal interlayer spacing (d003) of Mg–Fe/Ti LDHs maybe attribute to the decrease of gravitational potential energy between hydroxide of the layers and carbonates. The basal interlayer spacings is proportional to the gravitational potential energy, which can be calculated by the following equation:33
| |
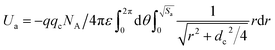 | (1) |
where the
qc and
q are the quantity of positive charges of the layer and negative charges. The
ε,
NA, and
dc represent permittivity of vacuum, Avogadro's constant, and interlayer spacings, respectively. The
Sa is the area of the layer board, and
r is the anionic spacings. With the increase of the Ti
4+/(Fe
3+ + Ti
4+) molar ratio, the charge density on the layer become higher, and the anionic space decrease, which caused the decrease of the gravitational potential energy, leading to the decrease of basal interlayer spacing.
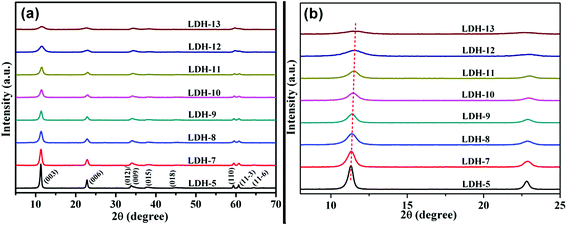 |
| | Fig. 2 XRD patterns for Mg–Fe/Ti LDHs with different Fe3+/Ti4+ molar ratio at pH = 12.5. | |
As a hexagonal crystal system, the lattice parameters “a” and “c” were calculated from (110) and (003) reflections, respectively. From Table 1, the “a” parameter corresponding to the distance of cations in the layer become larger with increase the Ti4+/(Fe3+ + Ti4+) molar ratio, which could be attribute to the larger ion radii of Ti4+ (0.068 nm) than that of Fe3+ (0.0645 nm). The decrease of the lattice parameter “c”, which is the triple of the sum of the thickness of a brucite-like layer and an interlayer maybe due to the decrease of the basal interlayer spacings.
3.1.2 Fourier transform infrared spectrometry (FTIR). The FT-IR spectra of Mg–Fe/Ti LDHs with different Ti4+/(Fe3+ + Ti4+) molar ratio in the region between 400 and 4000 cm−1, illustrated in Fig. 3, which show that there appears a strong band centered around 3500 cm−1 attributed to a superposition of the OH stretching vibration of hydrogen-bonded hydroxyl groups from both the hydroxyl group of the layers and interlayer water molecules. The extreme broadness of the OH band is ascribed to the presence of hydrogen bonding between hydroxides of layers, interlayer water and anions in the interlayer gap.24,32,34
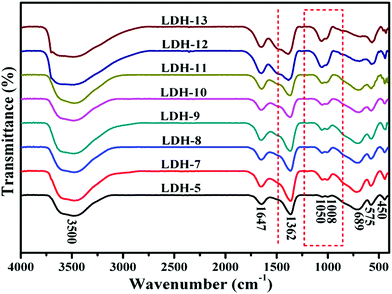 |
| | Fig. 3 FT-IR spectra of the Mg–Fe/Ti LDHs at pH = 12.5. | |
A low-resolution band around 1647 cm−1 attributed to the bending vibration of water molecules, which indicates that a small amount of interlayer water were in Mg–Fe/Ti LDHs due to abundant anions compensated positive charge of the layer. For carbonate LDHs, which consist of Mg and Fe (sample LDH-5), interlayer carbonate anions are symmetrically hydrogen-bonded to water molecules.32 Only a single band is observed at 1362 cm−1, which means that the symmetric stretching mode of interlayer carbonate anions is close to free anions.35 With the increase of Ti4+/(Fe3+ + Ti4+) molar ratio, the band is split into two located at 1490 cm−1 and 1385 cm−1, which maybe attribute to the restricted symmetry in the interlayer space. Two valence positive charges were yielded via the incorporating of Ti4+ cations into Mg–Fe LDHs layer, which caused the stronger electrostatic attraction between the layer and the interlayer carbonate anions, influencing the symmetries of the interlayer anions.35
Lowering the carbonate symmetry (site group splitting) would result in the activation of the vibrational mode at 1050 cm−1 and 1008 cm−1 corresponding to the stretching vibration of carbonate anions. And the band become stronger with increasing the Ti4+/(Fe3+ + Ti4+) molar ratio, indicate that the amount of CO32− as compensating anion in the interlayer increased. The band at 689 cm−1 is attributed to the overlap of the asymmetric bending vibration of C–O. Band at lower wave numbers might be due to lattice vibrations implying M–O, M–O–M, and O–M–O bonds in the layers. The band between 575–450 cm−1 is due to the metal–oxygen vibrations in the layers and is typical of this kind of layered solids.35
3.1.3 Transmission electron microscope (TEM) micrographs and the selected area electron diffraction (SAED) patterns. The transmission electron microscope (TEM) micrographs and selected area electron diffraction (SAED) patterns showed that the prepared Mg–Fe/Ti LDHs were nearly hexagonal, plate-like particle. Different morphologies were developed depending on the chemical composition of the LDHs.36,37 It is clearly seen that the crystal size of LDH-10 is much lower than that of LDH-5, corresponding to the decrease of crystallinity from Fig. 2(a). Moreover, several particles of LDH-10 that stay roughly perpendicularly to the plane are seen as dark lines (Fig. 4(b)). The periodic diffraction concentric rings verify the polycrystalline nature of the LDHs by the SAED patterns, as shown in Fig. 4(c) and (d). Three rings can be attributed to the (110), (018) and (012) crystal planes of Mg–Fe/Ti LDHs, according to JCPDS PDF no. 70-2150. All of these results, which are similar with the previous report,38 coincide with those of the XRD patterns.
 |
| | Fig. 4 (a), (b) TEM images of the LDH-5 and LDH-10; (c), (d) SAED patterns of the LDH-5 and LDH-10. | |
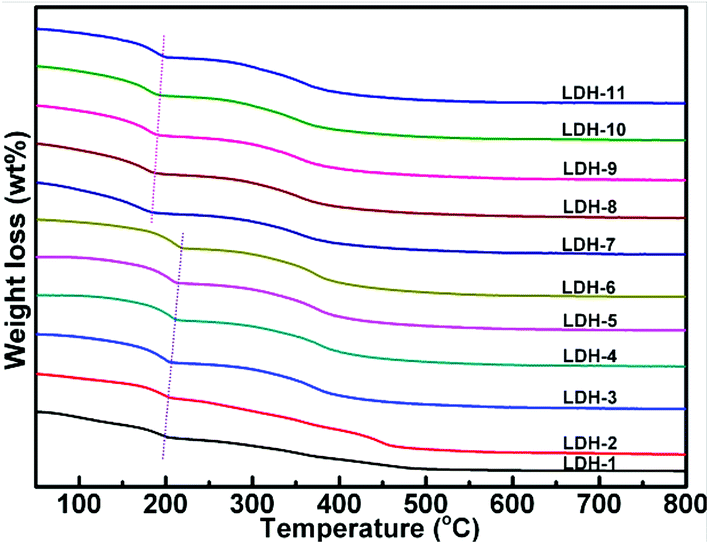
3.1.4 Thermogravimetric (TG) analysis. Fig. 5 shows the thermal behavior of Mg–Fe/Ti LDHs with CO32− as the compensating anion. It can be observed that the TG curves are very similar, showing two weight loss steps. The total weight loss percentages oscillated between 31.72% and 40.90%. The first mass loss should be due to the interlayer water in Mg–Fe/Ti LDHs and the surface weakly held water without collapse of the hydrotalcite structure. The second mass loss is likely corresponded to the removal of CO2 from the CO32− physically bound in the Mg–Fe/Ti LDHs and H2O from dehydroxylation of OH groups in the hydrotalcite. As a result, the Mg–Fe–Ti mixed oxides were formed, which means the layered double hydroxide structure is collapsed. It is clear that the thermal stability of Mg–Fe LDHs is higher than that of Mg–Fe/Ti LDHs, which maybe due to the strong electronegativity of Ti(IV).32
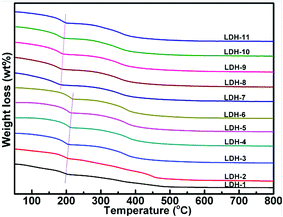 |
| | Fig. 5 TG curves of Mg–Fe/Ti LDHs. | |
3.2 Formation mechanism of Mg–Fe/Ti LDHs
So far most papers addressing the question of the formation mechanism of LDHs were concerned with Al3+ based LDHs, such as the topotactical mechanism for reconstruction from calcined Mg–Al LDH system,39–42 dissolution–crystallization for Mg–Al salt system,42 dissolution–precipitation–recrystallization mechanism for Mg–Al LDH system43,44 and dissolution–deposition–diffusion mechanism for Al2O3–MgO system,45 while no reports have been concerned with the formation mechanism of Mg–Fe/Ti LDHs. Based on the above results and the previous findings of other groups, a proposed formation mechanism of LDH synthesis is schematically illustrated in Fig. 6.
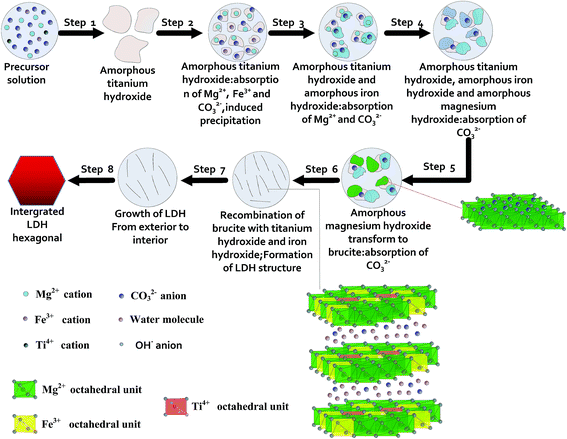 |
| | Fig. 6 Schematic illustration of proposed crystal evolution process of Mg–Fe/Ti LDHs. | |
Noticeably, these reactions are all related with hydroxide anion (OH−) and thus affected by the solution pH, especially the initial pH, playing an important role in the formation process. In our experiments, the initial pH of the solution increased gradually from 2 to 13, and then kept constant. The color of the solution changed from canary-yellow to reddish-brown, then to milk-white. According to the very different solubility of the three cations,46,47 the precipitation pH of metal cations is in the order of Ti4+ < Fe3+ < Mg2+, sequential hydrolysis of Ti4+ favors the precipitation of amorphous titanium hydroxide (step 1). Meanwhile, CO32−, OH−, and a small quantity of Fe3+ and Mg2+ ions were absorbed on the surface of titanium hydroxide. When the pH was not enough high to support the independent precipitation of Fe3+ and Mg2+, induced precipitation generated (step 2). The reaction can be expressed as this interpretation:
| Ti(OH)4 + Fe3+ + Mg2+ + 5OH− = Ti(OH)4·Fe(OH)3·Mg(OH)2↓ |
With increasing the pH value of the solution, the precipitation of amorphous iron hydroxide and amorphous magnesium hydroxide generated successively, and CO32− ions were absorbed on the surface of hydroxide (step 3, 4).
According to the literature,48 the crystallization of LDHs occurred by means of the diffusing of aluminum atoms into the magnesium hydroxide structure. However, from the deposition mechanism, the crystallization of LDHs is more likely to form by the recombination of different hydroxides. Then, an in situ phase transformation from amorphous magnesium to lamellan brucite occurs, forming numerous nanoflakes (step 5).49 In the next step, the compound products about Mg–Fe–Ti pairs bridged by oxygen atoms were obtained via the recombination of lamellar brucite with amorphous titanium hydroxide and amorphous iron hydroxide, as follow:
| Ti(OH)4·Fe(OH)3·Mg(OH)2 → (OH)3Ti–O–Mg–O–Fe(OH)2 + 2H2O |
| (OH)3Ti–O–Mg–O–Fe(OH)2 + 5Mg(OH)2 → [(OH)Mg–O–]3Ti–O–Mg–O–Fe[–O–Mg(OH)]2 + 5H2O |
| [(OH)Mg–O–]3Ti–O–Mg–O–Fe[–O–Mg(OH)]2 + 7H2O → {[(OH)Mg–OH–]3Ti–OH–Mg–OH–Fe[–OH–Mg(OH)]2}7+ + 7OH− |
Subsequently, the dehydration–condensation reaction of the adjacent hydroxy in the compound product continues until all the octahedral coordination groups of metal cations staking into sheets. Meanwhile, Ti(OH)62−, Fe(OH)63−, and Mg(OH)64−, which were formed because of the dissociation of the corresponding hydroxides, are continuously to migrate on the surface of the compound product, then, adsorption, association and orientation adjustment. The stacking of sheets begins to form layered structure, which leads to the unbalance of the sheet charge and destroys the interlamellar hydrogen bonds; therefore, CO32− ions are intercalated into the interlayer to balance the charge. (step 6). The crystallization and growth of LDH crystallites occur from the exterior to the interior of the aggregates (step 7), and finally, an integrated and perfect hexagonal LDHs is formed (step 8).
The entire evolution process is the results of precipitation of titanium hydroxide, iron hydroxide and magnesium hydroxide, phase transformation of amorphous magnesium hydroxide, recombination of brucite with amorphous titanium hydroxide and amorphous iron hydroxide, intercalation of carbonate ions, stacking of the sheets, crystallization and the growth of LDHs.
3.2.1 Temperature-programmed desorption (TPD). Taking into account the basicity and acidity dependence of the PCDL synthesis via transesterification, the basicity and acidity of the Mg–Fe/Ti LDHs catalysts were characterized using CO2 and NH3-TPD under the temperature ranging from 60 to 700 °C, as shown in Fig. 7. It is clearly seen that the desorption peaks, which represent Lewis basic sites and Brønsted acid sites were observed, indicating the coexistence of acidic and basic sites on the catalysts.27,50 Brønsted acid sites mainly represented the surface hydroxyl groups and Lewis basic sites are corresponded to oxygen groups. From Fig. 7(a), the desorption peaks with maxima shift to low temperature, indicating the strength of base decrease. Instead, the strength of acid increase with increase the content of the Ti4+ cations, as shown in Fig. 7(b). The total of basicity and acidity of Mg–Fe/Ti LDHs is shown in Fig. 8. The reason for the variation is maybe due to the substitute of Fe3+ by Ti4+, which increased the amount of charge on the layers, leading to the variation of distribution of carbonate anions in the interlayer. The relationship between the acid–base property and catalytic activity was illustrated in the following text.
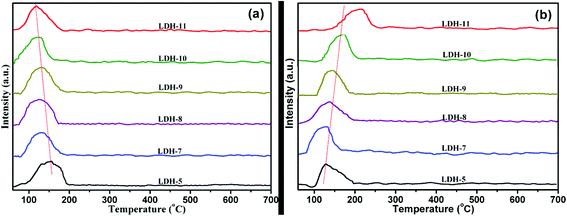 |
| | Fig. 7 CO2-TPD profiles (a) and NH3-TPD profiles (b) of Mg–Fe/Ti LDHs. | |
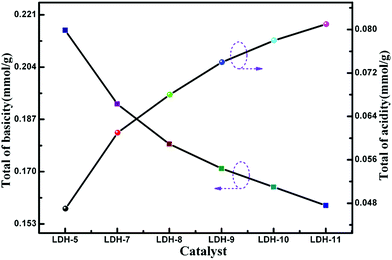 |
| | Fig. 8 The total of basicity and acidity of Mg–Fe/Ti LDHs. | |
3.2.2 Catalytic activity of Mg–Fe/Ti LDHs. Fig. 9 shows the catalytic activity of Mg–Fe/Ti LDHs with different nTi4+/(nTi4+ + nFe3+) molar ratio in the transesterification process, which was determined by the yield of methanol (Z) defined as the follow formula:where m is the amount of methanol produced and the value of 19.2 is the amount of methanol produced when 0.3 mol BMCA is completely reacted with 1,6-HD. The catalytic activity was enhanced obviously with the increasing of Ti4+ content when the nTi4+/(nF3+ + nTi4+)% was below 20, beyond this point, the catalytic activity decreased, which indicated that the excess doped Ti catalysts (nTi4+/(nF3+ + nTi4+)% >20) are unfavorable for the transesterification reaction. As shown in Fig. 8, the change trend of basicity and acidity of the LDH-10 catalyst is opposite. It is implied that an acid–base cooperative manner to catalyze the transesterification reaction played an important part in improving the catalytic activity of LDHs. Climent et al.51 hold that the acid–base bifunctional catalysts with suitable weak acid–base pairs exhibited larger catalytic activity than other acid or basic solid catalyst. The catalytic activity is influenced by the strength matching of the acidic and basic active sites on the acid–base cooperative catalysis. The catalytic activity of LDH-10 in the polycondensation process is evaluated by the number-average molecular weight (Mn) and the hydroxyl value, as listed in Table 2. Obviously, when LDH-10 was used as the catalyst, the product PCDL is viscous liquid and shows highest Mn and lowest hydroxyl value, which illustrates that LDH-10 is an efficient catalyst for the synthesis of PCDL via the transesterification between DMC and aliphatic diol.
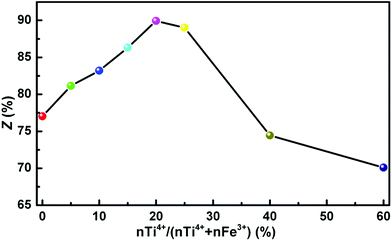 |
| | Fig. 9 Effect of Mg–Fe/Ti LDHs with different nTi4+/(nTi4+ + nFe3+) molar ratio on the transesterification process. | |
Table 2 Effect of different catalysts on the property of PCDLsa
| Catalyst |
Mn (g mol−1) |
Hydroxyl value (mg KOH per g) |
Polydispersity |
Appearance |
| Reaction conditions: n(BMCA) = 0.3 mol, n(1,6-HD)/n(DMC) = 1.2, w(catalyst) = 1.0 wt%. |
| LDH-5 |
1105 |
106.4 |
1.45 |
Viscous liquid |
| LDH-7 |
1320 |
96.8 |
1.49 |
Viscous liquid |
| LDH-8 |
1864 |
74.1 |
1.57 |
Viscous liquid |
| LDH-9 |
2382 |
54.7 |
1.64 |
Viscous liquid |
| LDH-10 |
3030 |
43.5 |
1.78 |
Viscous liquid |
| LDH-11 |
3005 |
44.1 |
1.79 |
Viscous liquid |
| LDH-12 |
753 |
202.2 |
1.31 |
Viscous liquid |
| LDH-13 |
615 |
208.3 |
1.27 |
Viscous liquid |
3.3 Reaction mechanism
The CO2 and NH3-TPD results indicated that the LDH-10 is bifunctional catalyst containing both of Brønsted acidic and Lewis basic sites. An acid–base cooperative manner to catalyze the transesterification reaction was also observed. The possible mechanism for PCDL synthesis reaction is proposed in Fig. 10.
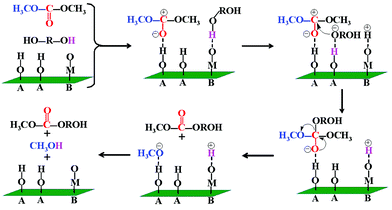 |
| | Fig. 10 Proposed mechanism for PCDL synthesis reaction of LDH-10 as catalysts ((A) Brønsted acid site; (B) Lewis base site). | |
The hydroxyl in 1,6-HD/1,5-PD was polarized and broken by an oxygen atom as the Lewis basic site of the catalyst. The electron of the hydrogen atom shifted toward to the oxygen atom, generating nucleophilic species HD−/PD−. Meanwhile, the carbonyl oxygen in DMC was activated by a hydrogen atom as the Brønsted acid site of the catalyst, increasing the electropositive of carbon atoms in the carbonyl. Subsequently, the nucleophilic species HD−/PD− attacked the carbon atom in the carbonyl group of DMC, forming a intermediate compound. Through the charge transfer, the methoxy group dropped from the intermediate compound. Then the methoxy group was combined with hydrogen on the LDH-10 catalyst, yielding methanol. In this way, the transesterification process continuously proceeded on the catalyst and PCDLs were finally obtained.
3.4 Characterization of PCDL
The composition of PCDL, which was synthesis using the catalyst of LDH-10, was verified by the FTIR spectrum and 1H NMR spectrum, as shown in Fig. 11. From Fig. 11(a), the peaks observed at 3542 and 3464 cm−1 are attributed to the stretching vibration of hydroxyl group. The peaks at 2938 and 2864 cm−1 attributed to the symmetric and asymmetric stretching vibration of methylene group. The peaks at 1467 and 1405 cm−1 are correspond to the bending vibration of methylene group. The peak at 1740 cm−1 reflects the stretching vibration of carbonate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. The peak at 1255 cm−1 represents the stretching vibration of aliphatic carbonate O–C–O.
O. The peak at 1255 cm−1 represents the stretching vibration of aliphatic carbonate O–C–O.
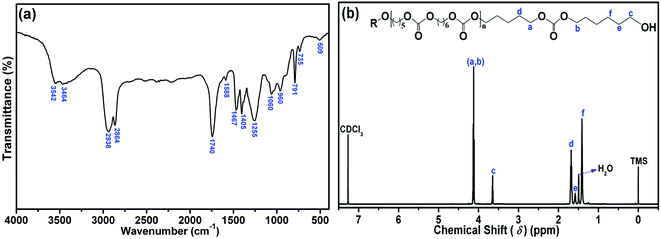 |
| | Fig. 11 The FTIR spectrum (a) and 1H NMR spectrum (b) of PCDL catalyzed by LDH-10. | |
The peaks in the 1H NMR spectrum, as shown in Fig. 11(b), correspond to δ = 4.12 (a/b, OC(O)OCH2), 3.65 (c, HOCH2), 1.68 (d, OC(O)OCH2CH2), 1.58 (e, HOCH2CH2), 1.41 (f, HOCH2CH2CH2), respectively. All those data from Fig. 10 verify that the product is PCDL.
4. Conclusions
In this work, a series of novel Mg–Fe/Ti LDHs as acid–base bifunctional catalysts, which were used for the transesterification between dimethyl carbonate (DMC) and aliphatic diols were prepared by co-precipitation. The formation of LDHs strongly depended on the initial pH of the suspension, and 12.5 is the suitable pH value for obtaining LDHs with high crystallinity. With increase the amount of Ti4+ cations, the basicity of LDHs decreased, while the acidity of LDHs increased. A general precipitation–recombination–crystallization mechanism was proposed according to the whole experimental results associated with the previous findings of other groups. The evaluation of these catalysts in the synthesis of PCDL was carried out using dimethyl carbonate (DMC) and aliphatic diols as comonomers. An acid–base cooperative manner to catalyze the transesterification reaction was found to improve the catalytic activity of LDHs. Two-step transesterification method, which can avoid the azeotrope between DMC and methanol during the reaction was adopted to synthesized the PCDLs. By using LDH-10 catalyst, the yield of methanol is up to 89.12%, and the PDCL shows the highest Mn (3030) and lowest hydroxyl value (43.5).
Acknowledgements
This work is financially supported by the University-Industry Cooperation Project of Dongguan (Grant No. 2013509117202).
References
- H. G. Fang, H. L. Wang, J. Sun, H. B. Wei and Y. S. Ding, RSC Adv., 2016, 16, 13589–13599 RSC.
- J. Yang, Y. L. Gao, J. H. Li, M. M. Ding, F. Chen, H. Tan and Q. Fu, RSC Adv., 2013, 3, 8291–8297 RSC.
- W. Kuran, M. Sobczak, T. Listos, C. Debek and Z. Florjanczyk, Polymer, 2000, 41, 8531–8541 CrossRef CAS.
- V. Costa, A. Nohales, P. Félix, C. Guillem, D. Gutiérrez and C. M. Gómez, J. Appl. Polym. Sci., 2015, 132, 41704–41714 CrossRef.
- V. Garcıá-Pacios, V. Costa, M. Colera and J. M. Martín-Martínez, Int. J. Adhes. Adhes., 2010, 30, 456–465 CrossRef.
- A. Eceiza, M. D. Martin, K. D. L. Caba, G. Kortaberria, N. Gabilondo, M. A. Corcuera and I. Mondragon, Polym. Eng. Sci., 2008, 28, 297–306 Search PubMed.
- R. Nehring and W. Seeliger, US Pat. No. 3631200, U.S. Patent and Trademark Office, Washington, DC, 1971.
- H. Hocker and H. Heul, Polymeric Materials Encyclopedia, 1996, pp. 16–47 Search PubMed.
- H. Sugimoto and A. Ogawa, React. Funct. Polym., 2007, 67, 1277–1283 CrossRef CAS.
- S. Matsumura, S. Harai and K. Toshima, Macromol. Chem. Phys., 2000, 201(14), 1632–1639 CrossRef CAS.
- Z. Z. Jiang, C. Liu, W. C. Xie and R. A. Gross, Macromolecules, 2007, 40(22), 7934–7943 CrossRef CAS.
- P. Pawlowski and G. Rokicki, Polymer, 2004, 45(10), 3125–3137 CrossRef CAS.
- Y. X. Feng, N. Yin, Q. F. Li, J. W. Wang, M. Q. Kang and X. K. Wang, Ind. Eng. Chem. Res., 2008, 47, 2140–2145 CrossRef CAS.
- L. P. Wang, F. Wang and L. X. Xu, Indian J. Chem., 2015, 5, 607–612 Search PubMed.
- L. P. Wang, B. Xiao, G. Y. Wang and J. Q. Wu, Sci. China: Chem., 2011, 54, 1468–1473 CrossRef CAS.
- A. M. Rasero-Almansa, M. Iglesias and F. Sánchez, RSC Adv., 2016, 6, 106790–106797 RSC.
- A. Taher, D. W. Kim and I. M. Lee, RSC Adv., 2017, 7, 17806–17812 RSC.
- W. T. Reichle, J. Catal., 1985, 94(2), 547–557 CrossRef CAS.
- A. Corma, S. Iborra, S. Miquel and J. Primo, J. Catal., 1998, 173, 315–321 CrossRef CAS.
- R. Chitrakar, S. Tezuka, A. Sonoda, K. Sakane and T. Hirotsu, Ind. Eng. Chem. Res., 2008, 47, 4905–4908 CrossRef CAS.
- M. X. Gao, M. H. Zhang and Y. H. Li, RSC Adv., 2017, 7, 11929–11937 RSC.
- H. Zazoua, A. Saadi, K. Bachari, D. Halliche and C. Rabia, Res. Chem. Intermed., 2014, 40, 931–946 CrossRef CAS.
- M. Sipiczki, E. Kuzmann, Z. Homonnay, J. Megyeri, K. Kovács, I. Pálinkó and P. Sipos, Hyperfine Interact., 2013, 214, 145–149 CrossRef.
- K. Hosni, O. Abdelkarim, N. Frini-Srasra and E. Srasra, Korean J. Chem. Eng., 2015, 32, 104–112 CrossRef CAS.
- D. Saha and S. G. Deng, J. Phys. Chem. Lett., 2010, 1, 73–78 CrossRef CAS.
- W. Kleist, M. Maciejeuski and A. Baiker, Thermochim. Acta, 2010, 499, 71–78 CrossRef CAS.
- F. P. Lu, B. B. Jiang, J. D. Wang, Z. L. Huang, Z. W. Liao, Y. R. Yang and J. Zheng, RSC Adv., 2017, 7, 22017–22026 RSC.
- M. R. Weir, J. Moore and R. A. Kydd, Chem. Mater., 1997, 9, 1686–1690 CrossRef CAS.
- A. Seron and F. Delorme, J. Phys. Chem. Solids, 2008, 69, 1088–1090 CrossRef CAS.
- Q. Wang, H. H. Tay, Z. H. Guo, L. W. Chen, Y. Liu, J. Chang, Z. Y. Zhong, J. Z. Luo and A. Borgna, Appl. Clay Sci., 2012, 55, 18–26 CrossRef CAS.
- J. S. Valente, M. Sanchez-Cantú, E. Lima and F. Figueras, Chem. Mater., 2009, 21, 5809–5818 CrossRef CAS.
- W. H. Zhang, X. D. Guo, J. He and Z. Y. Qian, J. Eur. Ceram. Soc., 2008, 28, 1623–1629 CrossRef CAS.
- Z. M. Ni, G. X. Pan, L. G. Wang, C. P. Fang and D. Li, Chin. J. Inorg. Chem., 2006, 22, 91–95 CAS.
- K. T. Ehlsissen, A. Delahaye-Vidal, P. Genin, M. Figlarz and P. Willmann, J. Mater. Chem., 1993, 3, 883–888 RSC.
- M. J. Hernandez-Moreno, M. A. Ulibarri, J. L. Rendon and C. J. Serna, Phys. Chem. Miner., 1985, 12, 34–38 CAS.
- J. Pérez-Ramirez, S. Abelló and N. M. van der Pers, J. Phys. Chem. C, 2007, 111, 3642–3650 Search PubMed.
- C. P. Kelkar and A. A. Schutz, Microporous Mater., 1997, 10, 163–172 CrossRef CAS.
- C. Ruby, R. Aïssa, A. Géhin, J. Cortot, M. Abdelmoula and J. M. Génin, C. R. Geosci., 2006, 338, 420–432 CrossRef CAS.
- T. Sato, H. Fujita, T. Endo and M. Shimada, React. Solids, 1988, 5, 219–228 CrossRef CAS.
- T. Stanimirova and G. Kirov, Appl. Clay Sci., 2003, 22, 295–301 CrossRef CAS.
- T. S. Stanimirova, G. Kirov and E. Dinolova, J. Mater. Sci. Lett., 2001, 20, 453–455 CrossRef CAS.
- J. W. Boclair and P. S. Braterman, Chem. Mater., 1999, 11, 298–302 CrossRef CAS PubMed.
- J. S. Valente, M. Sánchez-Cantú, E. Lima and F. Figueras, Chem. Mater., 2009, 21, 5809–5818 CrossRef CAS.
- Y. M. Yang, X. F. Zhao, Y. Zhu and F. Z. Zhang, Chem. Mater., 2012, 24, 81–87 CrossRef CAS.
- Z. P. Xu and G. Q. Lu, Chem. Mater., 2005, 17, 1055–1062 CrossRef CAS.
- A. L. McKenzie, C. T. Fishel and R. J. Davis, J. Catal., 1992, 138, 547–561 CrossRef CAS.
- B. Grégoire, C. Ruby and C. Carteret, Dalton Trans., 2013, 15687–15698 RSC.
- W. T. Reichle, CHEMTECH, 1986, 16(l), 58–63 CAS.
- A. A. Eliseev, A. V. Lukashin, A. A. Vertegel, V. P. Tarasov and A. Y. D. Tret'yakov, Dokl. Chem., 2002, 387, 339–343 CrossRef CAS.
- J. I. D. Cosimo, C. R. Apestegui, M. J. L. Ginés and E. Iglesia, J. Catal., 2000, 190, 261–275 CrossRef.
- M. J. Climent, A. Corma, S. Iborra and A. Velty, J. Mol. Catal. A: Chem., 2002, 182, 327–342 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Lijuan Yang,
Xiaohong Peng* and
Zhijun Jin
,
Lijuan Yang,
Xiaohong Peng* and
Zhijun Jin

 and weight-average molecular weights
and weight-average molecular weights  ), and the polydispersity index
), and the polydispersity index 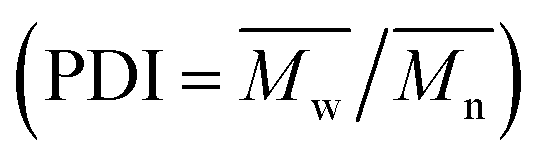 (PDI) of PDCLs were determined by gel permeation chromatography (GPC) performed on a Viscotek GPC Max (USA) equipped with Triple Detector Array TDA 305. The 1H NMR spectra were performed on an AVANCE III HD 600 (Bruker, Germany) using deuterated chloroform (CDCl3) as the solvent.
(PDI) of PDCLs were determined by gel permeation chromatography (GPC) performed on a Viscotek GPC Max (USA) equipped with Triple Detector Array TDA 305. The 1H NMR spectra were performed on an AVANCE III HD 600 (Bruker, Germany) using deuterated chloroform (CDCl3) as the solvent.