
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly selective production of heavy hydrocarbons over cobalt–graphene–silica nanocomposite catalysts†
Jian Huang ,
Weixin Qian,
Hongfang Ma,
Haitao Zhang and
Weiyong Ying*
,
Weixin Qian,
Hongfang Ma,
Haitao Zhang and
Weiyong Ying*
Engineering Research Center of Large Scale Reactor Engineering and Technology, Ministry of Education, State Key Laboratory of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: wying@ecust.edu.cn; Fax: +86 21 64252192; Tel: +86 21 64252151
First published on 3rd July 2017
Abstract
Herein, cobalt–graphene–silica nanocomposites were prepared by a sol–gel method to produce heavy hydrocarbons for Fischer–Tropsch synthesis. The catalysts were characterized by N2 physisorption, XRD, TEM, TPR, TPD, XPS, and DRIFTS techniques. The activity of catalysts and the selectivity of products were examined in a tubular fixed-bed reactor. It can be concluded that the introduction of graphene into cobalt–silica nanocomposites significantly enhanced the amount and stability of adsorbed CO at low temperatures, resulting in higher concentrations of CO species on the catalyst surface. Moreover, graphene can weaken the cobalt–silica interaction, leading to higher degree of reduction of cobalt oxides and higher adsorption amounts of H2. In addition, the introduction of graphene led to the formation of cobalt with smaller particle sizes, which contributed to great enhancement of CO conversion. The selectivity to methane distinctly decreased to 4.2% from 8.1%, whereas the selectivity to C5+ products increased from 84.5% to 92.4%. The α value increased from 0.89 for the Co–Si catalyst to 0.94 for the Co–0.1GSi catalyst. In addition, with the increase in the graphene content, the fraction of heavy hydrocarbons (C19–29) for catalysts evidently increased to 36.0% from 28.3%, but the fraction of naphtha (C5–12) clearly reduced to 17.7% from 27.7%.
1. Introduction
Due to the anticipated depletion of fossil energy sources, efforts have been made to replace conventional fuels with renewable energy sources via Fischer–Tropsch synthesis (FTS).1 The FTS process involves the formation of linear hydrocarbon molecules from the synthesis gas derived from coal, natural gas, or biomass through gasification, water steam reforming, or partial thermal oxidation.2,3 FTS has been considered as a promising technology to produce ultrapure transportation fuels and high value-added chemicals due to the global shortage of transportation fuels and environmental pollution.4–6 The synthesis gas is converted in the presence of iron, cobalt, or ruthenium catalysts with highest activity, and the product distribution is extremely reliant on the FTS operating conditions and the type of the catalyst used.7–9 Therefore, the selective synthesis of specific products is still one of the most critical issues in the FTS industry.10Moreover, cobalt is a favored active component of the FTS catalysts for the production of heavy hydrocarbons due to its high activity for carbon monoxide hydrogenation, low selectivity towards carbon dioxide, and long life span as compared to iron.11,12 With high cobalt loadings and low metal–support interactions, agglomeration and large cluster formation of cobalt nanoparticles during the process of calcination are difficult to avoid.13 Therefore, controlling the size of metal nanoparticles on supports is an effective means to obtain highly thermostable catalysts.14
Catalytic properties and the microstructure of the FTS catalysts are highly correlated with the carriers such as silica, alumina, titania, and zeolites.14–17 The influence of the supports and their porosity has been extensively investigated on conventional oxide carriers.18–20 Supports with wide pores could accelerate the formation of larger Co3O4 particles, but large cobalt particle sizes would lead to less Co dispersion.21 It is believed that traditional oxides supports are highly stable and comparatively inert. However, their metal–support interaction is still relatively strong, and they have a major impact on the textural and chemical properties of the deposited surface layer as compared to carbon material supports such as carbon nanotubes22 or graphene.23 Therefore, the metal–support interactions can be adjusted via improvement of the supports to some extent.24
Recently, graphene has exhibited unique characteristics and outstanding adjustability in supporting various metallic-supported catalysts.25,26 It can be regarded as a significantly potential support material for metal particle catalysts because of its unique electronic, thermal, textural, and chemical properties.27 Furthermore, with a large surface area and high adsorption capacity, graphene can serve as a valuable substrate to interact with various species.26,28 Cobalt–graphene-supported catalysts were prepared by an impregnation method; these catalysts showed higher cobalt dispersion and decreased the average size of the clusters of cobalt oxides, exhibiting higher electronic interaction.29 Moreover, it was found that graphene, as a carrier, could lower the reduction temperature of metal oxides.30,31 However, it is difficult to utilize graphene-supported catalysts in industry due to the fact that graphene has low mechanical strength and is difficult to shape. Therefore, these were anticipated to combine the unique electronic and structural characteristics of graphene with high stability and comparative inertness of conventional carriers.
In this study, we focused on the effects of graphene on cobalt–graphene–silica nanocomposites, which were prepared by the sol–gel method and tested under FTS conditions in a fixed tube reactor to study the effects on the selectivity and distribution of products. Characterization methods such as N2 adsorption, X-ray diffraction (XRD), transmission electron microscopy (TEM), temperature-programmed reduction (TPR), X-ray photoelectron spectroscopy (XPS), temperature-programmed desorption (TPD), and in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) were applied to characterize the catalysts. The effects of graphene content on the texture, reduction properties, chemisorption, and FTS performance of the catalysts was investigated.
2. Experimental
2.1. Catalyst preparation
Co–graphene–silica nanocomposites were prepared in situ by a sol–gel method with commercial silica sol (34 wt% SiO2, pH = 3.0), graphene (99.5 wt%, Chengdu Organic Chemicals Co., LTD., Chinese Academy of Sciences), and cobalt(II) nitrate hexahydrate (>99 wt% Sinopharm Chemical Reagent Co., Ltd) as precursor solutions. In a typical preparation process, 12.59 g of cobalt nitrate hexahydrate was completely dissolved in 15 mL of deionized water and mixed with 30 g silica sol and x (0, 10.2, 51, and 102) mg of graphene under vigorous stirring at 25 °C for 2 h. The mixtures were placed in a drying oven and heated to 110 °C in an air atmosphere at a heating rate of 10 °C min−1 for 24 h to form a viscous gel. Subsequently, the obtained dried viscous gel was calcined at 400 °C for 6 h at the given heating rate of 2 °C min−1 under a N2 stream. The prepared samples were denoted as Co–xGSi (x = 0.1, 0.5, and 1.0), where x wt% represents the content of graphene and G represents graphene. The sample without graphene was denoted as Co–Si. The cobalt loadings of the catalysts were measured via ICP-AES technology (Agilent 725ES, USA), detailed in Table 1. To compare the differences in the texture properties of the catalysts and supports, silica and graphene–silica nanocomposites (0.5 wt% graphene) were prepared by a similar method and signified as SiO2 and 0.5GSi.| Sample | Co loadinga (%) | SBET (m2 g−1) | Vpb (cm3 g−1) | Dpc (nm) | dCo3O4d (nm) | dCoe (nm) | dCof (nm) |
|---|---|---|---|---|---|---|---|
| a Measured by ICP-AES technology.b BJH desorption pore volume.c BJH adsorption average pore sizes.d Estimated from the Co3O4 (311) plane by Scherrer's equation.e Calculated by dCo = 0.75dCo3O4.f Estimated by H2-TPD and d = 96/D (D% is dispersion of cobalt). | |||||||
| Co–Si | 21.8 | 130 | 0.35 | 10.2 | 22.7 | 17.0 | 9.7 |
| Co–0.1GSi | 21.6 | 132 | 0.38 | 11.4 | 21.4 | 16.1 | 8.6 |
| Co–0.5GSi | 21.4 | 135 | 0.39 | 11.3 | 18.0 | 13.5 | 8.2 |
| Co–1.0GSi | 21.3 | 153 | 0.44 | 11.0 | 17.0 | 12.8 | 7.9 |
| 0.5GSi | — | 185 | 0.29 | 6.2 | — | — | — |
| SiO2 | — | 164 | 0.29 | 6.7 | — | — | — |
2.2. Catalyst characterization
N2 physisorption was measured using a Micrometrics ASAP 2020 physisorption device at liquid N2 temperature. Specific surface areas were measured by the Brunauer Emmett Teller (BET) method. Total pore volume and pore sizes were measured by the Barrett–Joyner–Halenda (BJH) procedure.XRD profiles of the catalysts were obtained using a Rigaku D/Max2550VB/PC with Cu Kα radiation (λ = 0.154 nm) at 40 kV and 100 mA. The average Co3O4 crystallite sizes were estimated by the Scherrer equation using the Co3O4 peak at 36.9° and a K factor of 0.89. These particle sizes could be switched to the corresponding particle sizes of Co0 according to dCo = 0.75dCo3O4.32
TEM images and elemental mapping of the catalysts were obtained using a JEOL JEM 2100FX microscope equipped with an energy dispersive X-ray detector (EDX, Oxford-INCA) at 200 kV. The samples were dispersed in absolute ethanol, dropped on a copper grid, and dried under infra-red lamp for 15 min. Carbon species of the samples were determined by an energy dispersive X-ray spectroscope (EDS, Falcon) equipped with a CDU X-ray detector. The samples for EDS measurements were coated on a copper stage by a conducting resin.
H2-TPR tests of the catalysts were performed using a Micrometrics AutoChem II 2920 equipment. Before the tests, about 50 mg of the catalyst sample was swept in an argon flow at 350 °C for 2.0 h to remove the residual water in the catalysts. After being cooled down to 50 °C, each sample was heated up to 800 °C at a heating rate of 10 °C min−1 in a 10% H2/90% Ar (v/v) flow with a flux rate of 30 mL min−1. A thermal conductivity detector (TCD) was applied to measure H2 consumption.
XPS spectra of the samples were obtained by a VG ESCALAB 250Xi electron spectrometer with an Al-Kα X-ray source operating at 10 mA and 12 kV. The binding energies of the cobalt species were referenced to C 1s at 284.8 eV. The curves of the samples were fitted to the Lorentzian/Gaussian lines after subtracting a Shirley-shaped background.
Both H2-TPD and CO-TPD-GC-MSD measurements were performed using a Micrometrics AutoChem II 2920 device, and about 200 mg of the catalyst was reduced in a H2 flow (50 mL min−1) at 400 °C for 4 h and purged under a He flow for 30 min. After cooling to 50 °C, H2 or CO was introduced until saturation, and then, the catalyst was swept by a He flow until the baseline was flat. Subsequently, the sample was heated under a He flow at a rate of 10 °C, and a TCD detector was used to detect the desorbed CO or H2. The dispersion of cobalt can be estimated by the desorption amount of H2 on the Co0 surface. The tail gas of the CO-TPD measurements was detected via GC-MSD (Agilent 7890A-5975C).
In situ DRIFTS measurements were performed using a Nicolet 6700 spectrometer equipped with an MCT detector (Thermo, USA). The sample was reduced in H2 for 12 h at 400 °C and then swept by pure N2 for 30 min. The backgrounds were obtained when the vacuum degree of the in situ cell was below 10−4 Pa. Then, 5% CO/He was introduced at 205 °C for 30 min. Subsequently, 10% H2/Ar was introduced at 30 mL min−1, and the outlet of the in situ cell was closed. The spectra of the catalysts were obtained using a resolution of 4 cm−1 and an accumulation of 64 scans at 0, 5, 15, 30, and 60 min.
2.3. Catalyst evaluation
The performance of the cobalt catalysts was tested in a fixed-bed reactor (ID 10 mm). The catalyst loading was 1.00 g, and its particle size was 48–75 μm. In addition, the catalyst was mixed with 2.6 g of quartz grains with the same particle size. All the catalysts were reduced at atmospheric pressure, 400 °C, and a space velocity of 3000 cm3 STP per g per h under a flow of pure H2 for 24 h. After cooling to 150 °C, the syngas was introduced, and the experimental reaction conditions were maintained at a temperature of 205 °C, a pressure of 2.0 MPa, and space velocity of 2800 cm3 STP per g per h in the reaction system. The reaction products were introduced into a hot trap (180 °C) and a cold trap (0 °C) to separate water, oil, and wax.Moreover, two Agilent 7890A GCs equipped with different columns and detectors were applied to analyze the FTS products; then, two TCDs were applied to detect CO, H2, N2, and CO2, which were separated using 5 A molecular sieves and Propack Q packed columns. A flame ionization detector (FID) was applied to detect C1–6 hydrocarbons separated using a HP-AL/S capillary column. The components of the gas products were quantified by an external standard method with the standard gas containing CO, H2, N2, CO2, and C1–6 hydrocarbons. The oil product was analyzed using a HP-5 capillary column, and the wax product was dissolved in CS2 and separated using a DB-5ht capillary column, and both oil and wax were detected via FID. The quantification of wax and oil was calculated by an area percentage.
All the reported data about catalyst performance were obtained after 50 h on stream to ensure steady-state behavior and representative product samples. The conversion of CO denoted as XCO could be calculated by
 | (1) |
The selectivity of Cn hydrocarbons denoted as SCn could be calculated by
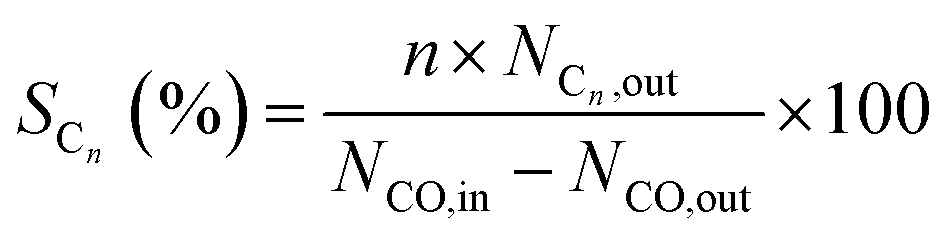 | (2) |
3. Results and discussion
3.1. Catalyst characterization
The pore size distributions of Co–xGSi and SiO2 are shown in Fig. S1 (details in the ESI†). It is clear that a pore size distribution of around 10–20 nm was observed for each catalyst sample, whereas a pore size distribution of around 5–8 nm was observed for SiO2.
The difference between both types of samples was that the precursors of Co–xGSi contained nitrate species, which could be considered as a pore-forming agent due to the fact that cobalt nitrate could be thermally decomposed as nitric oxides during heating, leading to larger pore sizes. In consideration of the introduction of graphene, the pore size distributions of catalysts slightly narrowed, and the average pore size slightly enlarged.
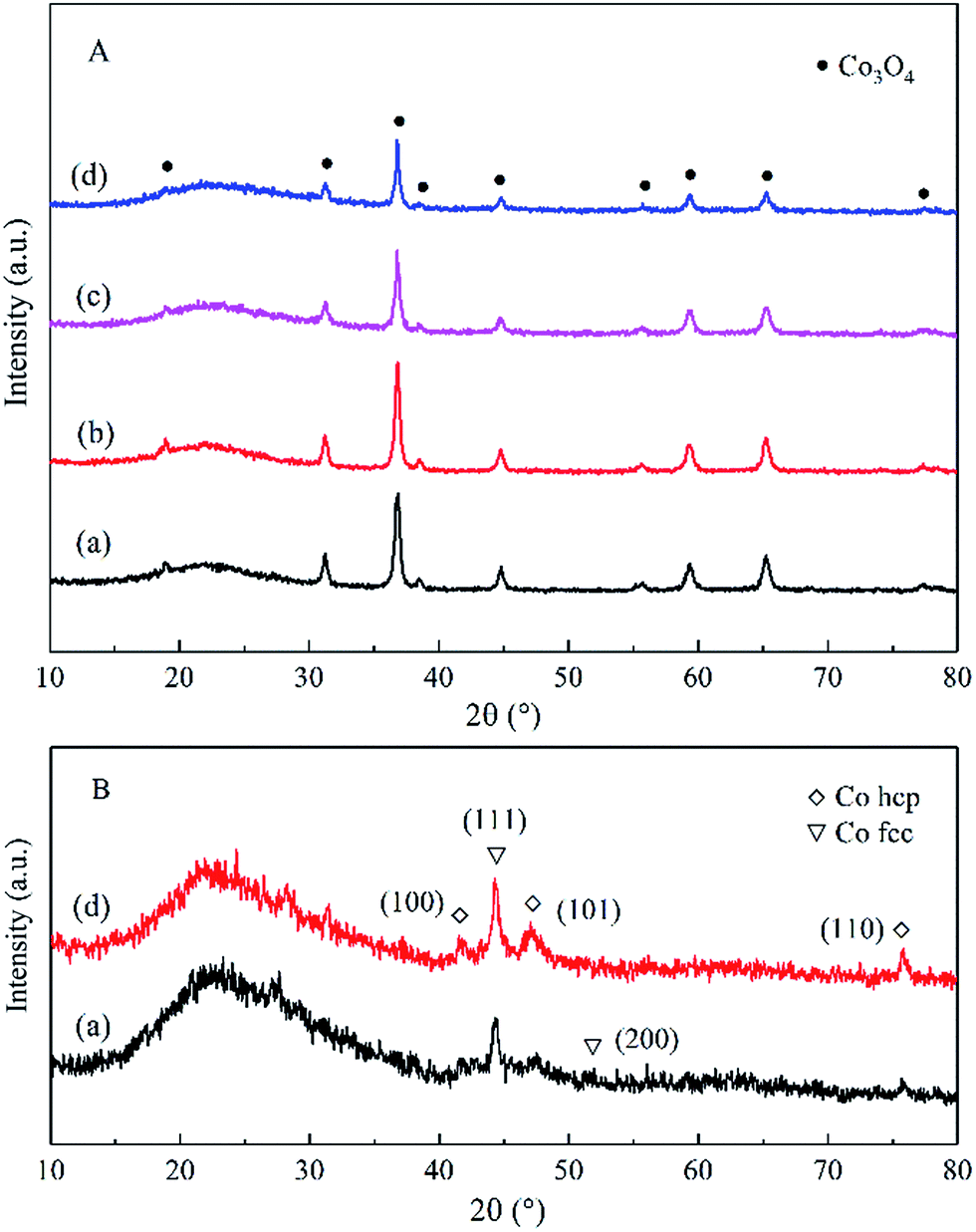 | ||
| Fig. 1 XRD patterns of the calcined catalysts (A) and reduced catalysts (B): (a) Co–Si, (b) Co–0.1GSi, (c) Co–0.5GSi, and (d) Co–1.0GSi. | ||
The only difference between the XRD results of the calcined catalysts was the half-peak width and intensity of the Co3O4 peaks. The Co3O4 crystallite sizes of the samples diminished with the addition of graphene, as shown in Table 1. The results showed that introduction of graphene in cobalt–silica nanocomposites resulted in significant decrease in the sizes of the Co3O4 nanoparticles as compared to the sample without carbon species.9 According to Fig. 1(B), the cobalt species of reduced catalysts were mainly hexagonal close packed (hcp) and face-centered cubic (fcc) crystal phases.34
 | ||
| Fig. 2 TEM images of the calcined Co–Si (a and b) and Co–1.0GSi (c and d). TEM images of the reduced Co–1.0GSi (e). | ||
The grains sizes of the catalysts were approximately 20 nm, and the dispersion of cobalt oxides of both catalysts was relatively high, as observed in Fig. 2(a)–(d). Thus, the sizes of the catalysts grains were in the nanoscale, and the synthesized samples were nanocomposites. The TEM image of the reduced Co–1.0GSi catalyst is shown in Fig. 2(e). From this TEM image, it was observed that the crystal texture of the metallic cobalt phase was mainly hcp cobalt (101) and less fcc cobalt (200) was observed. According to the XRD results, it was demonstrated that the hcp phase of the reduced cobalt catalysts was much more easily exposed than the fcc cobalt (200) phase on the catalyst surface.34
The influence of carbon species on resisting the formation of a Co2SiO4 spinel may be due to cobalt that was scattered by graphene species, which obstructed the immediate strong interaction between cobalt and silica particles. This hypothesis was studied in more detail via the EDS-mapping technology. Elemental mapping of the calcined Co–1.0GSi sample is presented in Fig. 3. As can be clearly deduced from the mapping, the surface of the sample has more exposed cobalt particles. Carbon species of the Co–1.0GSi sample was determined by EDS, as shown in Fig. S2.† The carbon content was 1.24 wt%, and the cobalt content was almost in agreement with the results of ICP-AES.
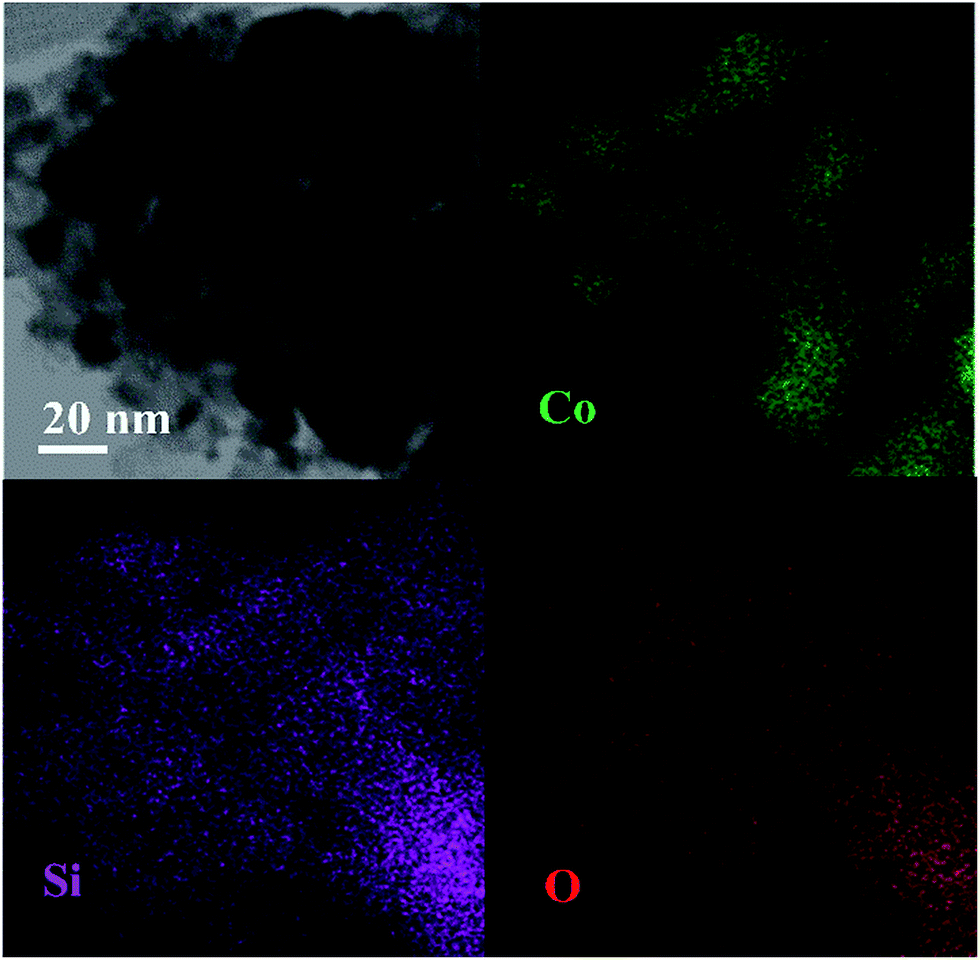 | ||
| Fig. 3 EDS-mapping of the reduced Co–1.0GSi catalyst: elemental mapping of cobalt (Co), silicon (Si), and oxygen (O). | ||
| Co3O4 + H2 → 3CoO + H2O | (3) |
| CoO + 3H2 → 3Co0 + 3H2O | (4) |
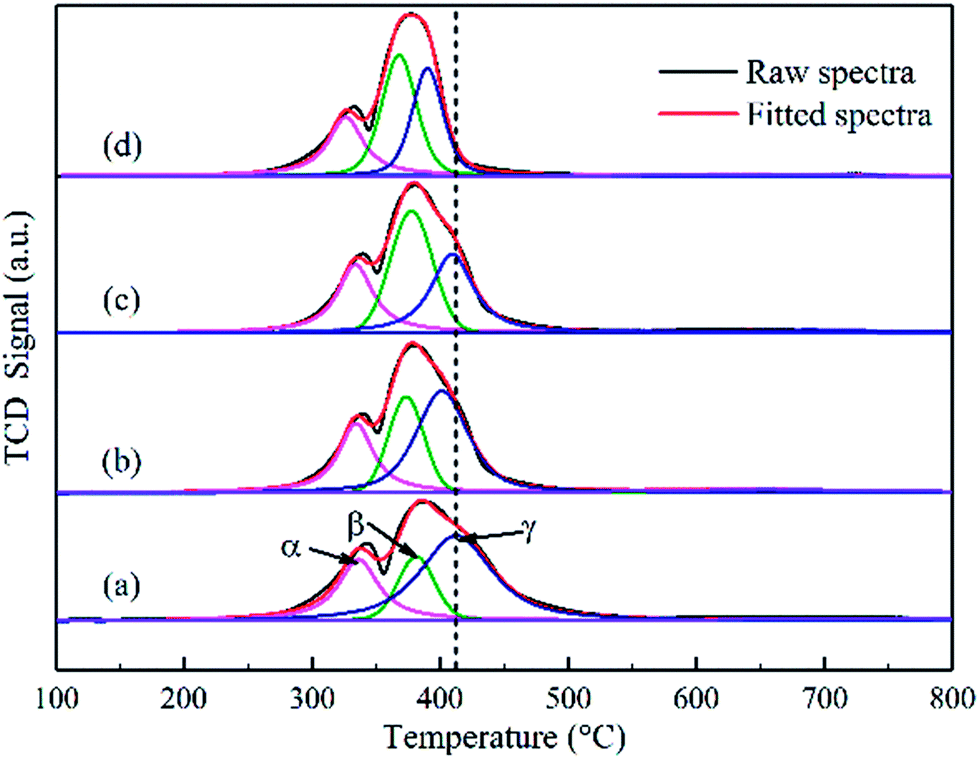 | ||
| Fig. 4 Hydrogen temperature-programmed reduction of the calcined catalysts: (a) Co–Si, (b) Co–0.1GSi, (c) Co–0.5GSi, and (d) Co–1.0GSi. | ||
The first reduction peaks below 350 °C can be ascribed to the reduction of Co3O4 to CoO, and the second peak is attributed to the reduction of CoO to metallic cobalt. For Co3O4 species, the hydrogen consumption ratio between the reduction of Co3+ to Co2+ with that of Co2+ to Co0 is one to three, according to eqn (3) and (4). Thus, the reduction curves of the catalysts can be decomposed as three peaks (noted as α, β, and γ), with the hypothesis that a proportion of one part by the first peak areas at low temperatures to three parts by another peak area can be used, as presented in Table S1 (details in the ESI†). Based on this assumption, the results of the TPR profiles showed that the reduction temperature of Co–xGSi catalysts decreased from 335 °C to 326 °C (α: Co3+ to Co2+), 376 °C to 368 °C (β: partial Co2+ to Co0), and 410 °C to 390 °C (γ: remaining Co2+ to Co0).
Moreover, the hydrogen consumption increased from 19% to 42% at the β peak, but decreased from 56% to 33% at the γ peak. From Table S1,† it was observed that the degree of reduction increased from 86.9% to 97.2% with an increase in the graphene content. Accordingly, most cobalt oxide was converted into metallic cobalt during the reduction process, which could provide more metal cobalt clusters and more active sites. Therefore, it also indicates that the addition of graphene can decrease the interaction between cobalt oxide and the silica support and significantly facilitate the reduction of Co2+ to Co0. This could be due to the fact that graphene located at the interface between cobalt and silica decreased the formation probability of cobalt species, which was difficult to reduce.35,36
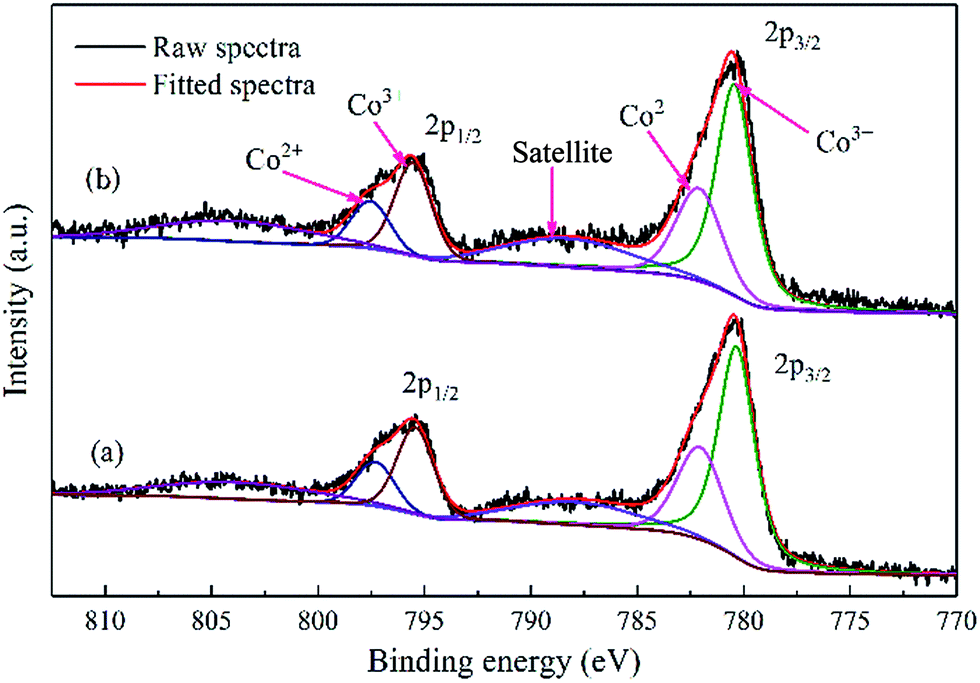 | ||
| Fig. 5 XPS spectra of the calcined cobalt catalysts: (a) Co–Si and (b) Co–1.0GSi. | ||
The binding energy of Co 2p3/2 for Co–1.0GSi was 779.9 eV, slightly larger than that of Co–Si (779.8 eV), as presented in Table S2,† since the charge transfer became relatively facile in the presence of graphene.20 Therefore, the charge density of cobalt atoms on the catalyst surface decreased with the addition of graphene.
From Table S2 (details in the ESI†), it is apparent that the atomic ratio of Co/Si for the Co–1.0GSi sample is 8.3% larger than that (6.7%) of the Co–Si sample. This may be due to the fact that the exposure probability of cobalt species on the catalyst surface from silica becomes much larger in the preparation process with the addition of graphene. Hence, the catalysts with graphene had higher reducibility and higher atomic ratio of Co/Si, which was consistent with the results of H2-TPR and EDS-mapping.
| Sample | Temperature (°C) | DoR (%) | CO-des uptakea (μmol gcat−1) | H2-des uptakeb (μmol gcat−1) | ||
|---|---|---|---|---|---|---|
| Peak I | Peak II | Peak I | Peak II | |||
| a CO-des uptake was corrected by measuring the component of tail gas.b H2-des uptake was measured by H2-TPD. | ||||||
| Co–Si | 138 | 638 | 86.9 | 7.33 | 22.05 | 85.3 |
| Co–0.1GSi | 199 | 668 | 89.5 | 13.21 | 24.57 | 105.0 |
| Co–0.5GSi | 195 | 640 | 91.9 | 10.70 | 23.85 | 114.7 |
| Co–1.0GSi | 149 | 648 | 97.2 | 8.10 | 22.41 | 122.8 |
In addition, the CO adsorption on cobalt particles was measured by CO-TPD, and two CO desorption peaks were observed, as shown in Fig. 6. Based on the carbide mechanism,39 CO was directly dissociated after adsorption on the catalysts, resulting in C and O species. Consequently, with the addition of graphene, the low temperature peaks (peak I) of the catalysts were attributed to the molecular adsorption of CO, dramatically increasing from 138 to 199 °C, and the high temperature peaks (peak II) were ascribed to the dissociation of adsorbed CO. Moreover, the desorption amount of CO increased from 7.33 μmol gcat−1 to 13.21 μmol gcat−1 with 0.1 wt% content of graphene. However, with the increasing graphene content from 0.1 wt% to 1.0 wt%, the desorption amount of CO decreased from 13.21 μmol gcat−1 to 8.10 μmol gcat−1.
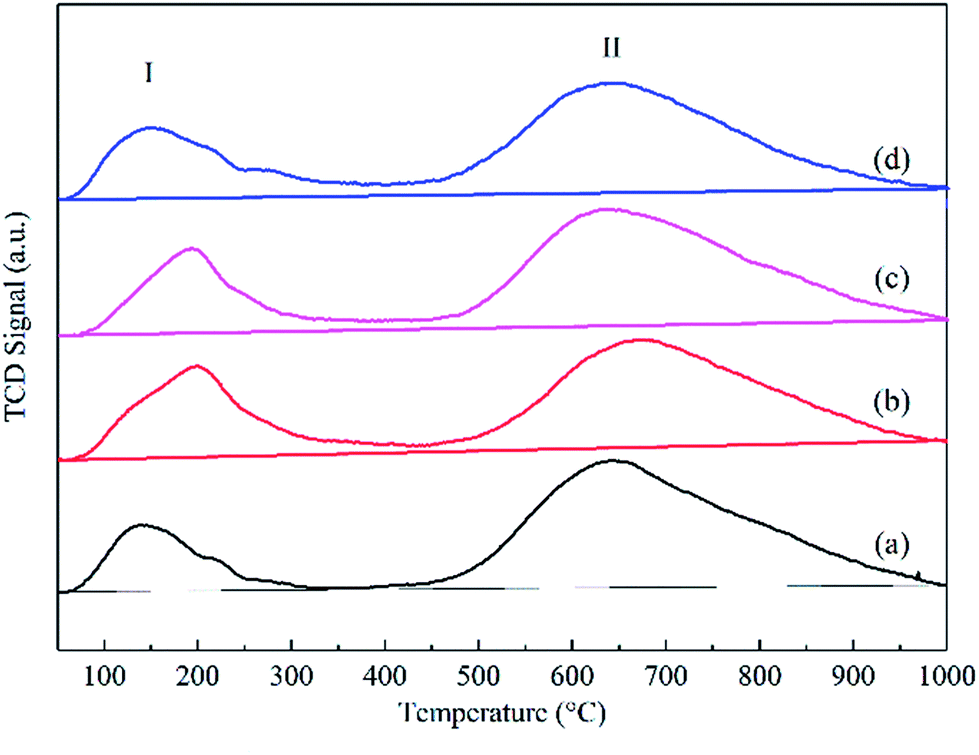 | ||
| Fig. 6 Carbon monoxide temperature-programmed desorption curves of the reduced catalysts: (a) Co–Si, (b) Co–0.1GSi, (c) Co–0.5GSi, and (d) Co–1.0GSi. | ||
The results of CO-TPD show that via a proper content (0.1–0.5 wt%) of graphene, the molecular adsorption intensity of CO was significantly increased, and the adsorbed CO molecules were more thermally stable, whereas the dissociative adsorption of CO was evidently increased. Moreover, higher reduction and smaller particle sizes of the Co–xGSi catalysts can offer more adsorption sites with more cobalt clusters, verified by the results of XRD and H2-TPR.
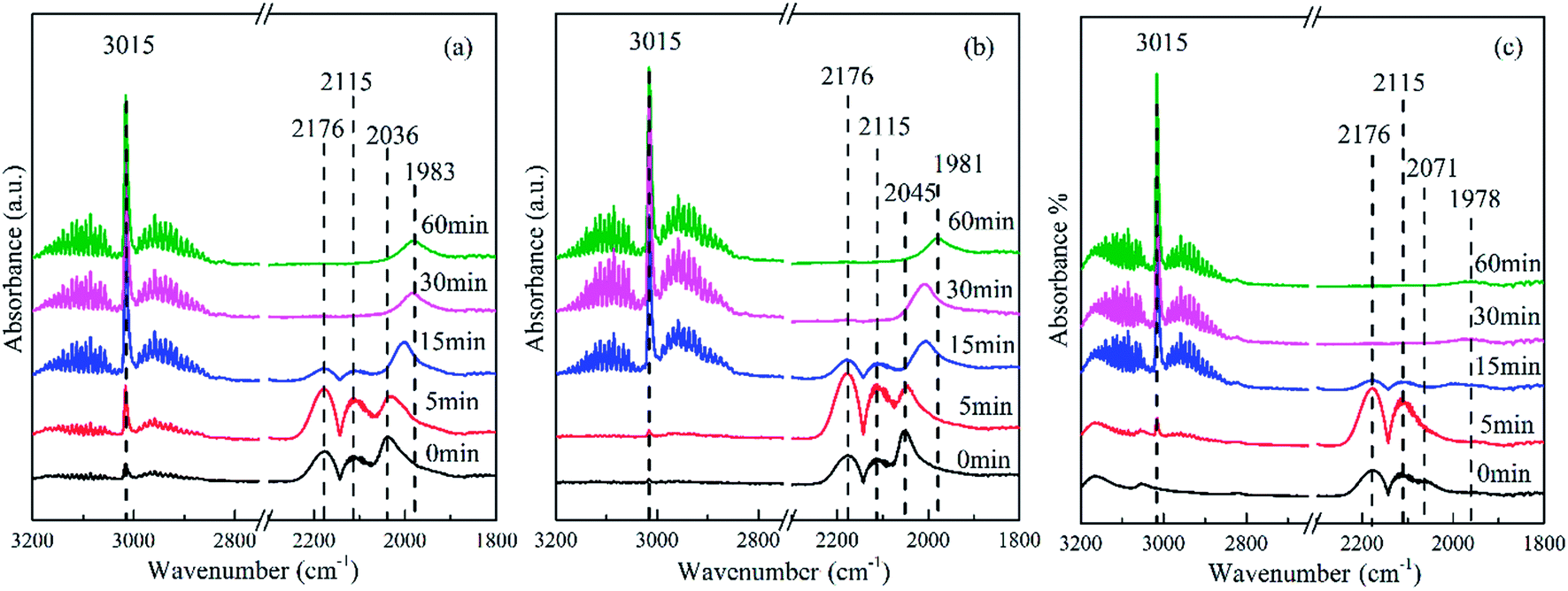 | ||
| Fig. 7 In situ FTIR spectra of the hydrogenation reaction over the reduced catalysts with adsorbed CO at 205 °C in a closed system: (a) Co–Si, (b) Co–0.1GSi, and (c) Co–1.0GSi. The spectra were obtained at 0, 5, 15, 30, and 60 min. | ||
The band at 2176 and 2115 cm−1 was assigned to gaseous CO and was observed in all the spectra of the catalysts.40 The band at 3015 cm−1 was attributed to CH4, and the band at 2850–3000 cm−1 was ascribed to the stretching of CHx species. The most intense band at 2036 cm−1 observed in Fig. 7(a) was attributed to the adsorption of CO on Coδ+ species,41 where the electron density of cobalt became lower than that of the catalysts with graphene. However, the band at 2072 cm−1 observed in Fig. 7(c) was assigned to linear CO adsorption on top of metal Co particles.40 With an increasing content of graphene, this band was significantly shifted from 2036 to 2072 cm−1, indicating an increase of reduced cobalt species,42 which was consistent with the results of H2-TPR. Moreover, the intensity of this band for the Co–0.1GSi catalyst was distinctly larger as compared to that for other catalysts. It was proven that the amount of CO molecular adsorption was significantly enhanced by the addition of 0.1 wt% graphene, confirmed by the results of CO-TPD shown in Table 2.
Moreover, with the hydrogenation reaction proceeding in situ in the cell, the gaseous CO was gradually consumed and the bands of CO adsorption clearly shifted towards lower wavenumber because of low coverage at low CO concentrations, as shown in Fig. 7. This also demonstrated that the presence of H2 accelerated the exclusion of adsorbed CO on weak sites.43
3.2. Catalyst performance
The performance of the graphene-modified catalysts is shown in Fig. 8, where the activity and selectivity were obtained after 50 h. In addition, the FTS activity of the Co–xGSi catalysts measured as the CO consumption rate per unit weight of the catalysts (weight-time yield, WTY) is presented in Table 3. The introduction of graphene in cobalt–silica nanocomposites obviously enhanced the FTS activity and selectivity to C5+ products, leading to higher CO conversion and C5+ product selectivity. An increase in CO conversion from 41.0% to 56.7% was observed for the Co–1.0GSi catalyst as compared to that for the Co–Si catalyst. This was due to the smaller cobalt particle sizes and higher reducibility of the catalysts, as reported in Tables 1 and 2; more hcp cobalt exposed on the catalyst surface is shown in Fig. 2(e), and more adsorption amount of H2 and CO is shown in Table 2. | ||
| Fig. 8 CO conversion and product selectivity over Co–xGSi at 205 °C, 2.0 MPa, H2/CO = 2, and 2800 cm3 STP per g per h space velocity. | ||
| Samplea | XCO (%) | Product selectivity (% C) | WTYb | α | Product distribution (wt%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CO2 | CH4 | C2–4 | C5+ | C5–12 | C13–18 | C19–29 | C30+ | ||||
| a Catalyst weight was 1.00 g.b Activity per unit weight catalyst (weight–time yield, WTY), (μmol CO per gcat per s). | |||||||||||
| Co–Si | 41.0 | 0.7 | 8.1 | 6.7 | 84.5 | 5.1 | 0.88 | 27.7 | 23.0 | 28.3 | 21.0 |
| Co–0.1GSi | 49.6 | 0.0 | 4.2 | 3.4 | 92.4 | 5.7 | 0.91 | 25.4 | 22.9 | 29.7 | 22.0 |
| Co–0.5GSi | 50.8 | 0.2 | 4.8 | 4.1 | 90.9 | 6.3 | 0.92 | 23.6 | 21.7 | 31.5 | 23.2 |
| Co–1.0GSi | 56.7 | 0.5 | 7.9 | 6.3 | 85.3 | 6.6 | 0.94 | 17.7 | 20.2 | 36.0 | 26.1 |
| Co/50C–SiO2–air9 | 71 | — | 6.2 | 5.0 | 88.8 | — | — | — | — | — | — |
| Co/graphene29 | ∼60 | 1.42 | 8.2 | 1.6 | 88.7 | — | 0.85 | — | — | — | — |
With the addition of 0.1 wt% and 0.5 wt% graphene, the selectivity to C5+ products was 92.4% and 90.9%, respectively, probably due to the fact that the concentration and stability of adsorbed CO was increased under the reaction conditions, which was in agreement with the results of CO desorption presented in Fig. 6 and Table 2. While a decline in the selectivity to methane from 8.1% to 4.2% was seen for the Co–0.1GSi catalyst in comparison with that in the Co–Si catalyst, probably due to the stronger intensity of adsorbed CO and higher concentrations of carbon species. Fig. 8 also shows that the CO2 formation rate slightly decreased, but increased at higher graphene contents, which can be ascribed to the increased partial pressure of water due to the higher CO conversion in the FTS reaction.44
The performance of the Co–0.1GSi catalyst at the temperatures of 205–235 °C was investigated, as shown in Fig. 9. With the increasing temperatures, the conversion of CO and the selectivity to CH4 and CO2 notably increased, and the selectivity to C5+ products slightly decreased. The CO conversion of the Co–0.1GSi catalyst at 225 °C was 78.9% larger than that of Co/graphene and Co/50C–SiO2–air under similar reaction conditions, as listed in Table 3. Moreover, the selectivity to CH4 of the Co–0.1GSi catalyst was 6.0% lower than that of the graphene-supported cobalt catalyst. Compared to the Co/graphene catalyst, the Co–0.1GSi catalyst had a lower water gas-shift reaction. The samples with an addition of small amount of graphene possess high activity and high selectivity and avoid high cost and low mechanical strength of graphene. Therefore, this study provides a possible method for the industrial application of graphene in FTS.
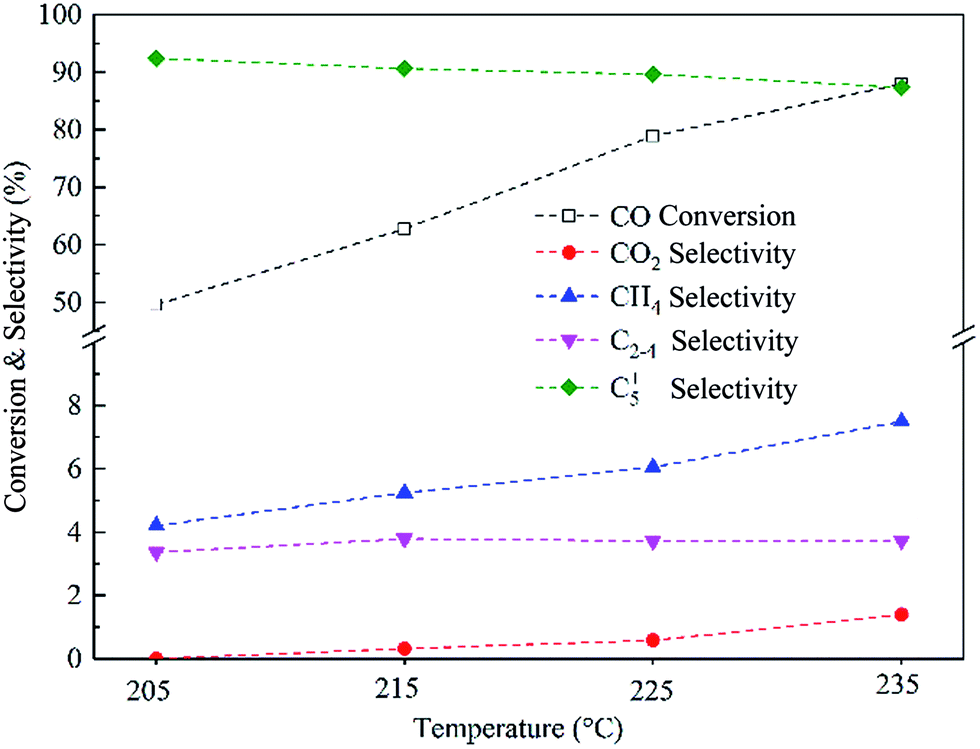 | ||
| Fig. 9 The CO conversion and product selectivity over the Co–0.1GSi catalyst at different reaction temperatures, 2.0 MPa, H2/CO = 2, and 2800 cm3 STP per g per h space velocity. | ||
The heightened FTS performance of cobalt–graphene–silica nanocomposites was attributed to their larger surface area, providing more active surface, higher reducing properties, and smaller cobalt particle sizes; this resulted in a larger density of active sites, more stable CO adsorbed on the catalyst surface, and higher concentrations of hydrogen species, promoting hydrogenation of CO.
The hydrocarbon chain growth probability factor (α) described by the Anderson–Schulz–Flory (ASF) distribution model was determined for the catalysts based on the weight fraction of hydrocarbon species, as shown in Fig. 10. The α value was estimated via the ASF plot from the C10–35 hydrocarbon range due to the fact that the corresponding correlation coefficients were comparatively high (greater than 0.99). However, the α value of the C36+ hydrocarbon for each sample was almost the same. The α value increased from 0.89 for the Co–Si catalyst to 0.94 for the Co–0.1GSi catalyst, as shown in Table 3.
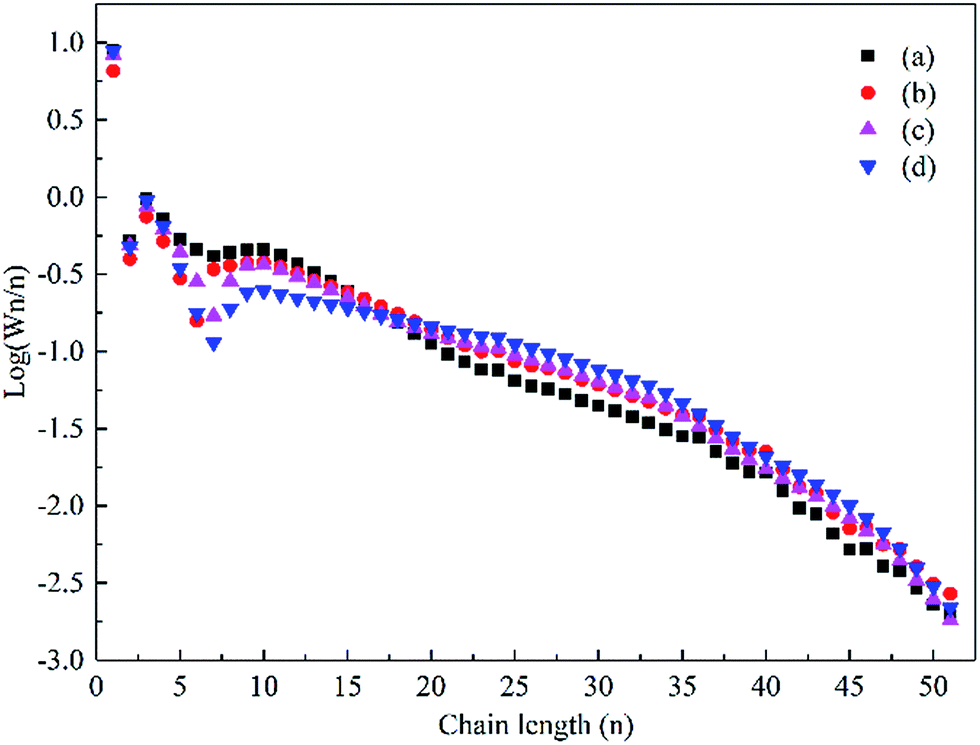 | ||
| Fig. 10 The ASF distribution of the hydrocarbon products over the Co–xGSi catalysts: (a) Co–Si, (b) Co–0.1GSi, (c) Co–0.5GSi, and (d) Co–1.0GSi. | ||
The product distribution presents a variation for heavy hydrocarbons, as detailed in Table 3. Compared to that for the Co–Si catalyst, the fraction of C19–29 for the Co–1.0GSi catalyst significantly increased to 36.0%, but the fraction of naphtha (C5–12) was clearly reduced to 17.7%. As for the Co–xGSi catalysts, higher amounts of adsorbed CO could reduce the H2/CO ratio on the catalyst surface, resulting in lower concentrations of adsorbed hydrogen species, which in turn led to the production of heavier hydrocarbons.45 Therefore, it was demonstrated that graphene not only influenced the selectivity, but also affected the distribution of products in FTS.
4. Conclusions
Herein, catalysts, cobalt–graphene–silica nanocomposites, were prepared by a sol–gel method and investigated for FTS. Surface areas and pore volumes slightly enhanced due to the textural properties of the catalysts with the addition of graphene. Moreover, smaller cobalt particle sizes were obtained. The introduction of graphene could weaken the interaction between cobalt oxide and the silica support and visibly facilitate the reduction of the Co–xGSi catalyst from Co2+ to Co0. This reduction was due to the fact that graphene located at the interface between cobalt and silica decreased the probability of the formation of cobalt species that were difficult to reduce. The concentration of hydrogen species and adsorbed CO species was higher than those of the catalyst without graphene. Moreover, the amount and stability of adsorbed CO were significantly enhanced by 0.1–0.5 wt% graphene. In addition, graphene could distinctly increase the FTS reaction activity, C5+ product selectivity, and the fraction of heavier hydrocarbons (C19+).Acknowledgements
We gratefully acknowledge the financial support of the National High Technology Research and Development Plan of China (863 plan, 2011AA05A204) and the Fundamental Research Funds for the Central Universities (No. 222201717013).References
- Y. Wang, Y. Jiang, J. Huang, J. Liang, H. Wang, Z. Li, J. Wu, M. Li, Y. Zhao and J. Niu, Fuel, 2016, 174, 17 CrossRef CAS.
- J. Eilers, S. A. Posthuma and S. T. Sie, Catal. Lett., 1990, 7, 253 CrossRef CAS.
- M. E. Dry, Catal. Today, 2002, 71, 227 CrossRef CAS.
- J. C. Park, J. I. Kwon, S. W. Kang, D. H. Chun, H.-T. Lee, H. Jung and J.-I. Yang, RSC Adv., 2017, 7, 8852 RSC.
- J. Kang, X. Wang, X. Peng, Y. Yang, K. Cheng, Q. Zhang and Y. Wang, Ind. Eng. Chem. Res., 2016, 55, 13008 CrossRef CAS.
- X. Peng, K. Cheng, J. Kang, B. Gu, X. Yu, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 4553 CrossRef CAS PubMed.
- H. Zhang, H. Ma, H. Zhang, W. Ying and D. Fang, Catal. Lett., 2012, 142, 131 CrossRef CAS.
- J. Xie, H. M. Torres Galvis, A. C. Koeken, A. Kirilin, A. I. Dugulan, M. Ruitenbeek and K. P. de Jong, ACS Catal., 2016, 6, 4017 CrossRef CAS PubMed.
- K. Cheng, V. Subramanian, A. Carvalho, V. V. Ordomsky, Y. Wang and A. Y. Khodakov, J. Catal., 2016, 337, 260 CrossRef CAS.
- S. Chen, C. Wang, J. Li, Y. Zhang, J. Hong, X. Wen and C. Liu, Catal. Sci. Technol., 2015, 5, 4985 CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692 CrossRef CAS PubMed.
- S. Krishnamoorthy, M. Tu, M. P. Ojeda, D. Pinna and E. Iglesia, J. Catal., 2002, 211, 422 CrossRef CAS.
- E. Marceau, M. Che, J. Čejka and A. Zukal, ChemCatChem, 2010, 2, 413 CrossRef CAS.
- T. M. Eggenhuisen, P. Munnik, H. Talsma, P. E. de Jongh and K. P. de Jong, J. Catal., 2013, 297, 306 CrossRef CAS.
- J. Du, J. Yan, J. Hong, Y. Zhang, S. Chen and J. Li, RSC Adv., 2015, 5, 60534 RSC.
- J. Li, T. Wang, L. Wu and X. Li, RSC Adv., 2016, 6, 89770 RSC.
- K. Cheng, L. Zhang, J. Kang, X. Peng, Q. Zhang and Y. Wang, Chem.–Eur. J., 2015, 21, 1928 CrossRef CAS PubMed.
- J. W. Bae, S. J. Park, M. H. Woo, J. Y. Cheon, K. S. Ha, K. W. Jun, D. H. Lee and H. M. Jung, ChemCatChem, 2011, 3, 1342 CrossRef CAS.
- J. Zhang, J. Chen, J. Ren, Y. Li and Y. Sun, Fuel, 2003, 82, 581 CrossRef CAS.
- A. Y. Khodakov, Catal. Today, 2009, 144, 251 CrossRef CAS.
- J. Yang, E. Z. Tveten, D. Chen and A. Holmen, Langmuir, 2010, 26, 16558 CrossRef CAS PubMed.
- T. O. Eschemann, W. S. Lamme, R. L. Manchester, T. E. Parmentier, A. Cognigni, M. Rønning and K. P. de Jong, J. Catal., 2015, 328, 130 CrossRef CAS.
- W. Luo, W. H. Doh, Y. T. Law, F. Aweke, A. Ksiazek-Sobieszek, A. Sobieszek, L. Salamacha, K. Skrzypiec, F. Le Normand, A. Machocki and S. Zafeiratos, J. Phys. Chem. Lett., 2014, 5, 1837 CrossRef CAS PubMed.
- S. L. Soled, E. Iglesia, R. A. Fiato, J. E. Baumgartner, H. Vroman and S. Miseo, Top. Catal., 2003, 26, 101 CrossRef CAS.
- B. F. Machado and P. Serp, Catal. Sci. Technol., 2011, 2, 54 Search PubMed.
- J. Guo, F. Li, Y. Sun, X. Zhang and L. Tang, Electrochim. Acta, 2015, 167, 32 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- S. Karimi, A. Tavasoli, Y. Mortazavi and A. Karimi, Appl. Catal., A, 2015, 499, 188 CrossRef CAS.
- N. M. Julkapli and S. Bagheri, Int. J. Hydrogen Energy, 2015, 40, 948 CrossRef CAS.
- S. O. Moussa, L. S. Panchakarla, M. Q. Ho and M. S. Elshall, ACS Catal., 2014, 4, 535 CrossRef CAS.
- A. n. Martínez, C. López, F. Márquez and I. Díaz, J. Catal., 2003, 220, 486 CrossRef.
- M. Nurunnabi and S. Q. Turn, Fuel Process. Technol., 2015, 130, 155 CrossRef CAS.
- G. Jacobs, J. A. Chaney, P. M. Patterson, T. K. Das and B. H. Davis, Appl. Catal., A, 2004, 264, 203 CrossRef CAS.
- J. Gong and X. Bao, Chem. Soc. Rev., 2017, 46, 1770 RSC.
- B. Jongsomjit, J. Panpranot and J. G. Goodwin Jr, J. Catal., 2003, 215, 66 CrossRef CAS.
- J. S. Girardon, A. Constant-Griboval, L. Gengembre, P. A. Chernavskii and A. Y. Khodakov, Catal. Today, 2005, 106, 161 CrossRef CAS.
- C. V. Schenck, J. G. Dillard and J. W. Murray, J. Colloid Interface Sci., 1983, 95, 398 CrossRef CAS.
- J. Girardon, E. Quinet, A. Gribovalconstant, P. Chernavskii, L. Gengembre and A. Khodakov, J. Catal., 2007, 248, 143 CrossRef CAS.
- G. Kadinov, C. Bonev, S. Todorova and A. Palazov, J. Chem. Soc., Faraday Trans., 1998, 94, 3027 RSC.
- T. Li, H. Wang, Y. Yang, H. Xiang and Y. Li, Fuel Process. Technol., 2014, 118, 117 CrossRef CAS.
- F. M. Mendes, C. A. Perez, F. B. Noronha, C. D. Souza, D. V. Cesar, H. J. Freund and M. Schmal, J. Phys. Chem. B, 2006, 110, 9155 CrossRef CAS PubMed.
- C. Qiu, B. Wu, S. Meng and Y. Li, Acta Chim. Sin., 2015, 73, 690 CrossRef CAS.
- A. Tavasoli, R. M. M. Abbaslou, M. Trepanier and A. K. Dalai, Appl. Catal., A, 2008, 345, 134 CrossRef CAS.
- J. Yang, W. Ma, D. Chen, A. Holmen and B. H. Davis, Appl. Catal., A, 2014, 470, 250 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05887j |
| This journal is © The Royal Society of Chemistry 2017 |