DOI:
10.1039/C7RA05873J
(Paper)
RSC Adv., 2017,
7, 43319-43326
Screening of inhibitors of Taenia solium glycogen synthase Kinase-3β
Received
25th May 2017
, Accepted 7th August 2017
First published on 7th September 2017
Abstract
Glycogen synthase kinase-3β (GSK-3β) has been found to be a potential target for the effective treatment of diseases such as type 2 diabetes, Alzheimer's disease, and schizophrenia. GSK-3β has also been suggested as a target for the treatment of protozoan parasitic infections such as those caused by Leishmania spp., Plasmodium falciparum, and Trypanosoma brucei. In this study, we determined the sequence of GSK-3β from T. solium (TsGSK-3β), which encodes 434 amino acid residues, as it could be a potential drug target. We modeled the TsGSK-3β tertiary structure by applying the crystal structure of the HsGSK-3β protein (PDB ID: 1UV5) as the template. Fourteen inhibitors of TsGSK-3β were screened by structure-based molecule docking. Recombinant TsGSK-3β protein was expressed and purified. We obtained 14 pharmaceutical compounds and performed surface plasmon resonance imaging to determine their binding affinities to TsGSK-3β and HsGSK-3β. Three compounds were identified to be potential novel inhibitors, as they showed significant interaction with TsGSK-3β but no binding to HsGSK-3β. Our study provides useful information for finding innovative drugs for treatment of T. solium infections.
Introduction
Taenia solium is a neglected parasitic zoonotic agent causing taeniasis and cysticercosis/neuro cysticercosis (NCC) in humans and is prevalent in many developing countries. Humans are the intermediate and definitive hosts. An adult tapeworm in the small intestine causes taeniasis, whereas the cysticercus (larval stage) usually develops in different organs, causing cysticercosis, and lodges in the central nervous system, causing NCC. Pigs are the obligate intermediate hosts. Taeniasis and cysticercosis were designated among 17 neglected tropical diseases by the Word Health Organization (WHO) in 2010.1 Approximately 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths have been reported to annually result from NCC, and approximately 2.5 million people are infected with T. solium in the world.2 Epilepsy cases caused by NCC reached 30% in many endemic areas in low and lower-middle income countries, with an estimated disease burden of 2–5 million lost in disability-adjusted life years.3
000 deaths have been reported to annually result from NCC, and approximately 2.5 million people are infected with T. solium in the world.2 Epilepsy cases caused by NCC reached 30% in many endemic areas in low and lower-middle income countries, with an estimated disease burden of 2–5 million lost in disability-adjusted life years.3
Praziquantel and niclosamide are the main drugs that are considered effective for the treatment of taeniasis.4–6 Their efficacy is high in the endemic areas. Raziquantel is more frequently used than niclosamide currently, and niclosamide is not widely used in some endemic areas.7 As a traditional Chinese remedy, pumpkin seeds combined with areca nut extract are effective for treatment of taeniasis.8 Praziquantel and albendazole are two potent cysticidal drugs used in the treatment of NCC.9 Praziquantel may cause severe side effects in individuals with NCC.10 Albendazole is preferred over praziquantel in some cases, particularly in cases of subarachnoid and ventricular cysts.11–13 Thus far, the major limitation in taeniasis/cysticercosis therapy is the limited number of suitable drugs available. In recent years, praziquantel has become ineffective against tapeworm and cysticercus, and NCC treatment failure using praziquantel has been reported.14 Further increase in praziquantel resistance could compromise the treatment and prevention of taeniasis/cysticercosis. To date, no effective marketed drug substitute for praziquantel and albendazole has been found.
Therefore, there is an urgent need to develop strategies for efficient screening of additional anti-tapeworm drugs, which should ideally represent novel chemical types and/or act on novel biological targets. Glycogen synthase kinase-3 (GSK-3) is a widely conserved family of protein kinases that was identified to regulate glycogen metabolism in the 1970s.15 In higher eukaryotes, GSK-3 consists of two isoforms, GSK-3α and GSK-3β, which are highly homologous in their catalytic domains but show remarkable differences in their terminal regions.16 GSK-3β is a key regulator in many cellular processes, including embryo development, metabolic homeostasis, cell survival, and neuronal growth and differentiation.17,18 Inhibition of GSK-3β is being considered a potential intervention to treat diseases such as type 2 diabetes, cancer, Alzheimer's disease, atherosclerosis, cardiac hypertrophy, osteoporosis, and schizophrenia.19–24 Some GSK-3β inhibitors are in the clinical phase of drug discovery and development, such as LY-317615 (enzastaurin) and NP-12 (NP-031112), which are under phase I clinical trials.25 AZD1080, an ATP competitive inhibitor, was tested in the treatment of Alzheimer's disease (AD) and related tauopathies; however, development was stopped because of a poor therapeutic window.26,27 Tideglusib, a non-ATP-competitive inhibitor, has been administered to patients with progressive supranuclear palsy (PSP) and mild-to moderate AD in phase II clinical trials.28,29 In recent years, some novel potent antiparasitic compounds have been discovered by researchers. Xingi et al. reported that leishmanial GSK-3β is the major target of 6-Br-5-methylindirubin-3′oxime, which affects cell-cycle progression and induces apoptosis-like death.30 Fugel et al. discovered a new class of Plasmodium falciparum GSK-3 (PfGSK-3) inhibitors, 3,6-diamino-4-(2-halophenyl)-2-benzoylthieno[2,3-b]pyridine-5-carbonitriles, which exhibit high selectivity for GSK-3 orthologs of other species, including human protein kinases, and antiplasmodial activity against erythrocyte stages of P. falciparum at micromolar concentrations. Droucheau et al. also reported that two compounds (indirubin-3′-monoxime and hymenialdisine) were potent inhibitors of PfGSK-3 and these compounds could inhibit the proliferation of P. falciparum in vitro.31 Ojo et al. showed that GSK-3β was a drug target for Trypanosoma brucei.32 Oduor et al. identified two compounds (PF-04903528 and 0181276) with approximately 7-fold selectivity for T. brucei GSK-3β over HsGSK-3β.33
In this study, we aimed to determine the RNA and amino acid sequences of T. solium glycogen synthase kinase-3β (TsGSK-3β) and construct a 3D model structure. Thereafter, we investigated novel and effective small molecule inhibitors of TsGSK-3β through a structure-based virtual screening approach. We identified novel small molecules that only bind to TsGSK-3β, by small molecule microarrays (SMMs) and surface plasmon resonance imaging (SPRi). We anticipate that it will be possible to identify small molecules that inhibit the activity of TsGSK-3β and not affect HsGSK-3β activity, thereby ensuring effective treatment of T. solium infections in humans.
Results and discussion
Sequence analysis
The core sequence of TsGSK-3β is composed of 673 bp, as shown by PCR. Based on the core sequence, two sequences from 5′- and 3′-RACE nested PCR were obtained. The full-length nucleotide sequence of TsGSK-3β is 1305 bp, and has been deposited to GenBank with an accession number of KY550711. The predicted TsGSK-3β protein consists of 434 amino acid residues with a predicted molecular weight of 48688.81 Da and a theoretical isoelectric point of 8.02. The deduced TsGSK-3β amino acid sequence analysis showed that the protein has no signal peptide or transmembrane region.
Homology modeling of TsGSK-3β
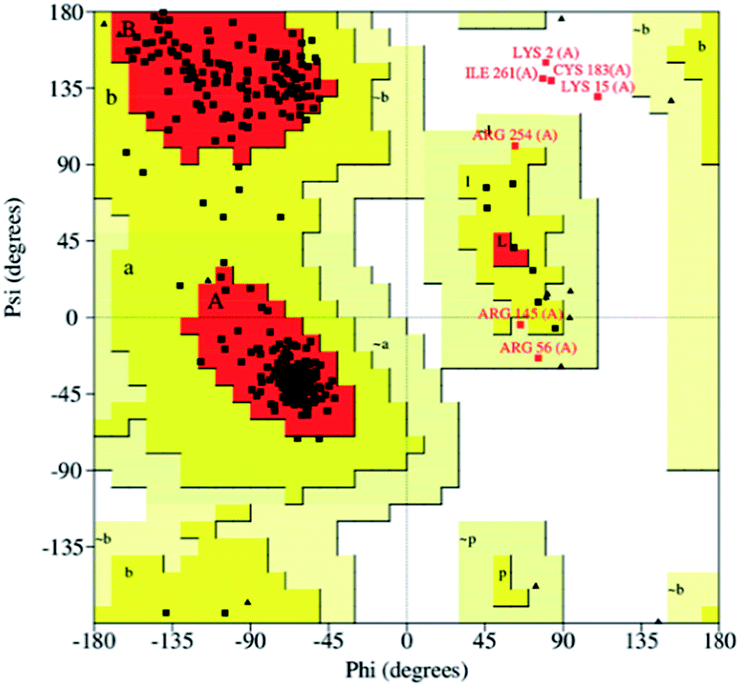
To obtain the TsGSK-3β tertiary structure, homology modeling was employed. The multiple sequence alignment analysis showed that sequence identity and sequence similarity were 72.6% and 84.5% between the protein sequences, respectively. Homology modeling tends to be reliable when the identity is higher than 50%.34 Therefore, the three-dimensional structure of TsGSK-3β can be modeled by computer homology modeling techniques. Five tertiary structure models were generated using the Build Homology Models program. The tertiary structure model with the lowest PDF total energy was subjected to the next structural assessment. The Ramachandran plot is used to check the detailed residue-by-residue stereochemical quality of a protein structure.35 Profile 3D evaluates a 3D model compared with its one-dimensional amino acid sequence.36 In our model, the Ramachandran plot revealed that 97.7% of all amino acids resided in the reasonable region (Fig. 1), and the Profile 3D calculated value reached 140.91, which was close to the Verify Expected High Score (156.705). These results show that the simulated three-dimensional structure of TsGSK-3β was rational and can be used for virtual drug screening.
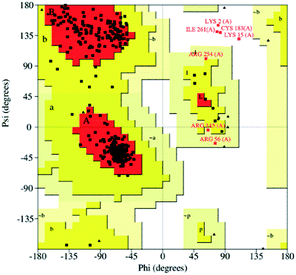 |
| | Fig. 1 Ramachandran plot of the TsGSK-3β homology model. Red color indicates the most favored regions, yellow indicates allowed regions, pale yellow indicates the generously allowed regions, and white indicates disallowed regions. | |
Identification of the binding region
Previous studies indicated that the ATP-binding pocket of HsGSK-3β includes a hinge region (Asp133, Tyr134, Val135), a hydrophobic pocket (Ile62, Gly63, Phe67, Val70), a polar region (Lys85, Glu97, Arg141, Glu185, Cys199, Asp200), and a gatekeeper (Leu132).37,38 These amino acid residues are conserved, and correspond to residues Glu98, Phe99, Val100, Ile27, Gly28, Phe32, Val35, Lys50, Glu62, Arg106, Glu150, Cys164, Asp165, and Leu97 in TsGSK-3β. The amino acids at positions 98 (Asp–Glu) and 99 (Tyr–Phe) in the TsGSK-3β active pocket were different from those of HsGSK-3β. The center of the pocket containing these conserved residues was chosen for further molecular docking studies.
Virtual screening of novel compounds
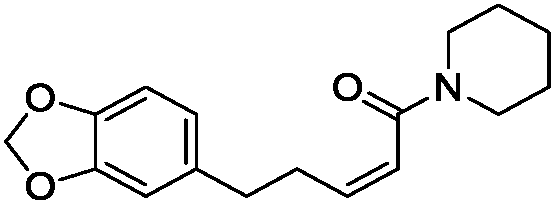
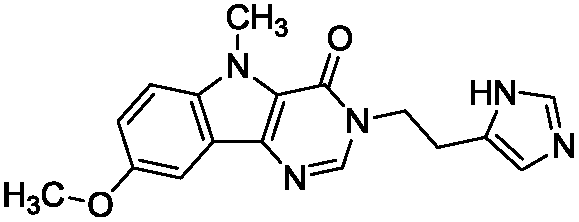
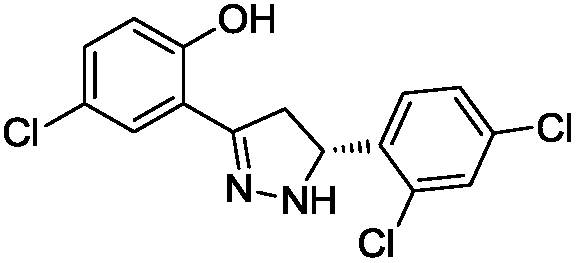
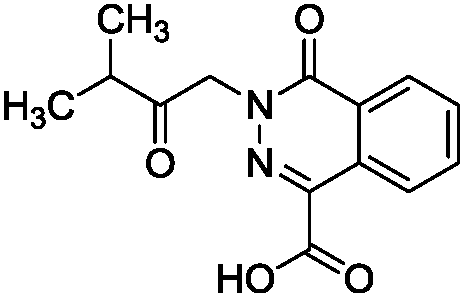
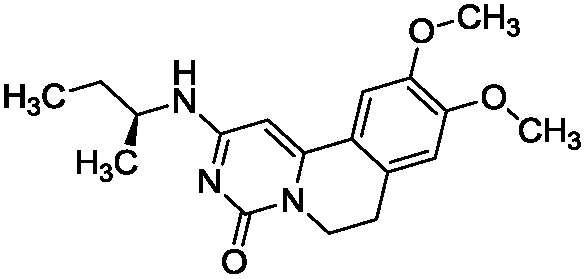
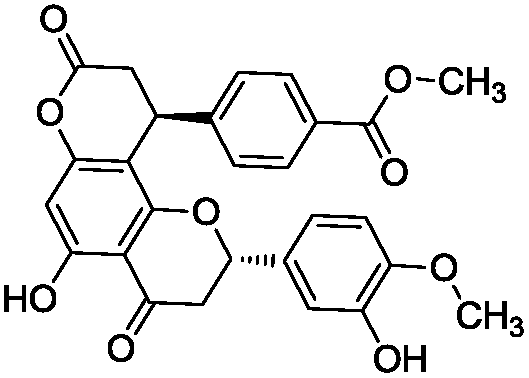
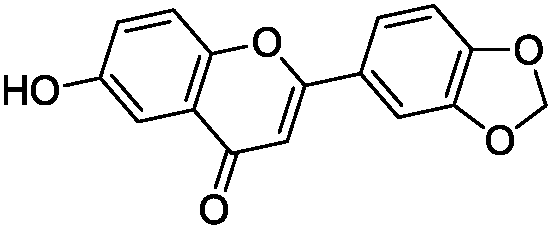
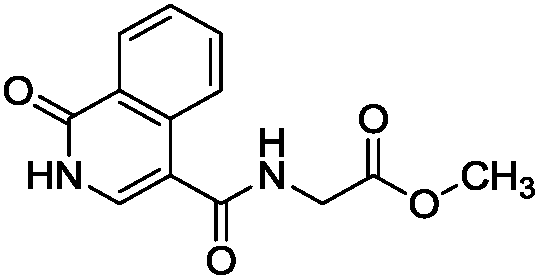
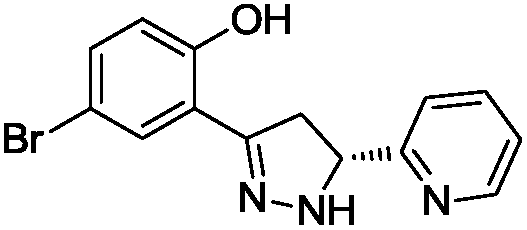
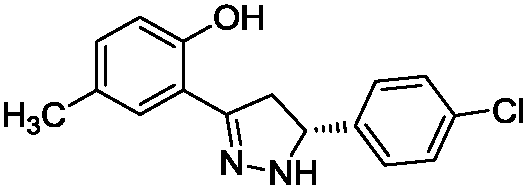
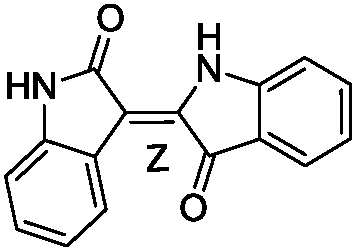
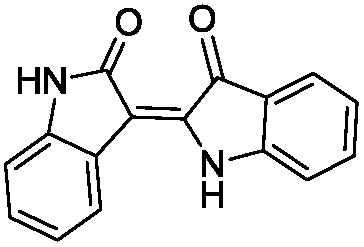
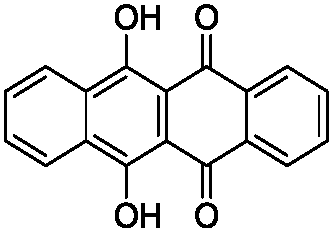
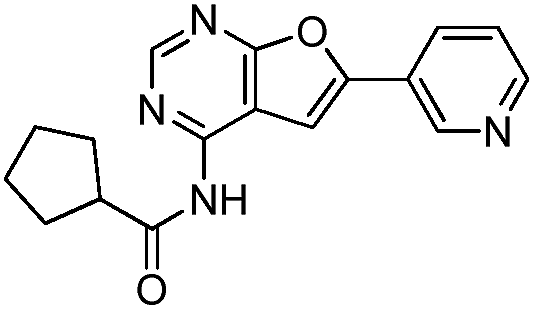
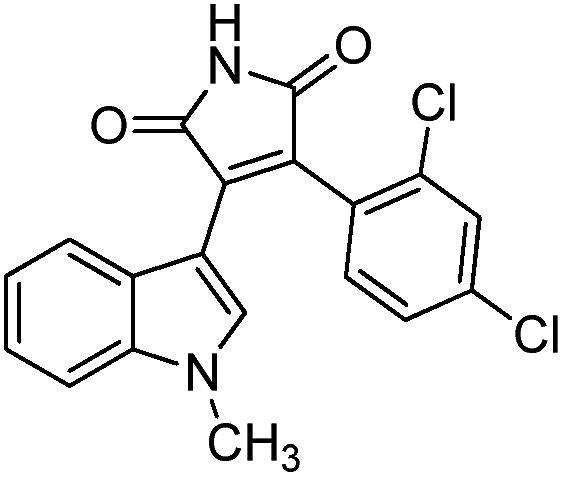
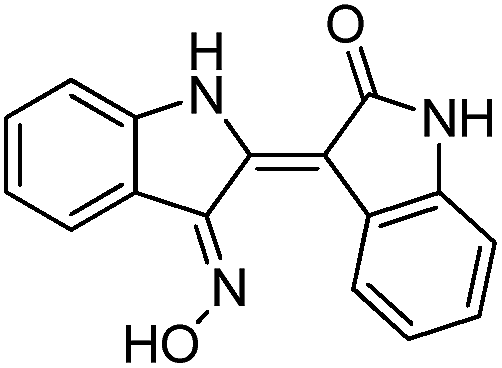
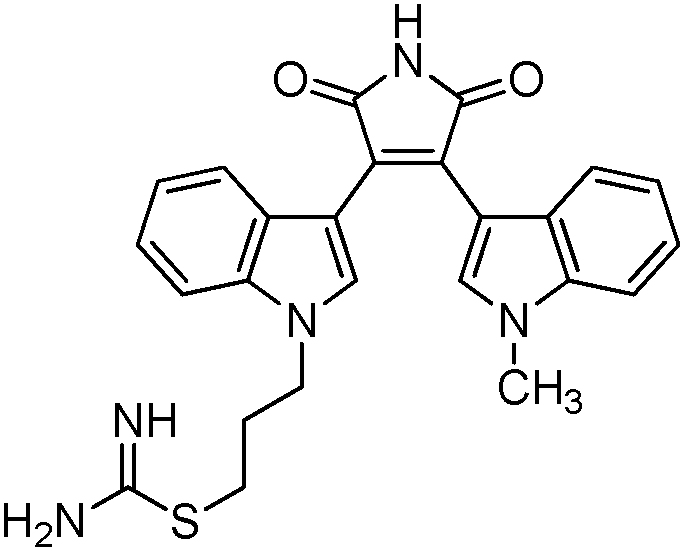
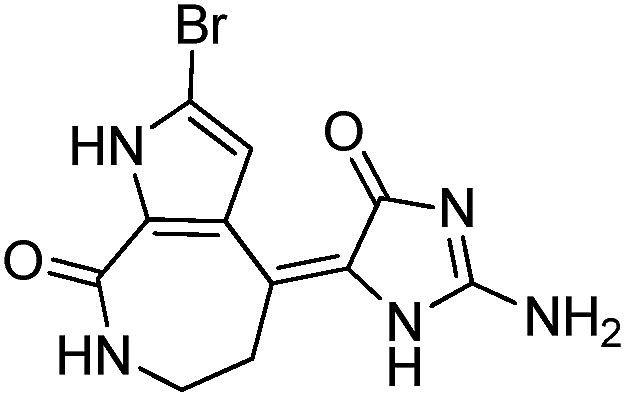
To identify novel and potent inhibitors, we performed docking-based virtual screening of the ATP-binding pocket of TsGSK-3β. We selected 100 compounds as candidate molecules according to the docking scores. These candidate molecules were first screened through Lipinski's rule-of five; the residues were further subjected to prime molecular mechanics/generalized-born/surface area (MM-GBSA) calculations and vision analysis. MM-GBSA-based relative binding free energy quantification has emerged as an effective tool to characterize the binding effect in large biomolecular systems.39 Its calculated value is an important reference parameter for our screening of molecules. After analysis of all of the candidate molecules, 14 potential pharmaceutical molecules (Table 1) were selected by molecular docking and purchased from Topscience company (Shanghai, China) for further research.
Table 1 Characteristics of selected small molecules based on virtual screening
| Compound no. |
Molecular structure |
Docking score |
MM-GBSA (kcal mol−1) |
| 1 |
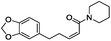 |
−7.579 |
−74.109 |
| 2 |
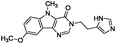 |
−8.406 |
−65.381 |
| 3 |
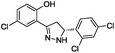 |
−7.276 |
−72.597 |
| 4 |
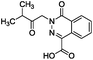 |
−8.003 |
−55.950 |
| 5 |
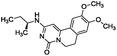 |
−9.248 |
−90.243 |
| 6 |
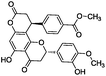 |
−7.921 |
−74.803 |
| 7 |
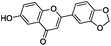 |
−8.556 |
−65.742 |
| 8 |
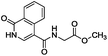 |
−9.132 |
−69.681 |
| 9 |
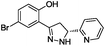 |
−7.089 |
−63.555 |
| 10 |
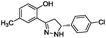 |
−6.849 |
−62.203 |
| 11 |
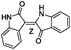 |
−10.106 |
−75.123 |
| 12 |
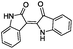 |
−9.699 |
−64.577 |
| 13 |
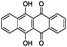 |
−8.336 |
−70.510 |
| 14 |
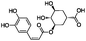 |
−8.716 |
−69.720 |
Protein expression and western blot analysis
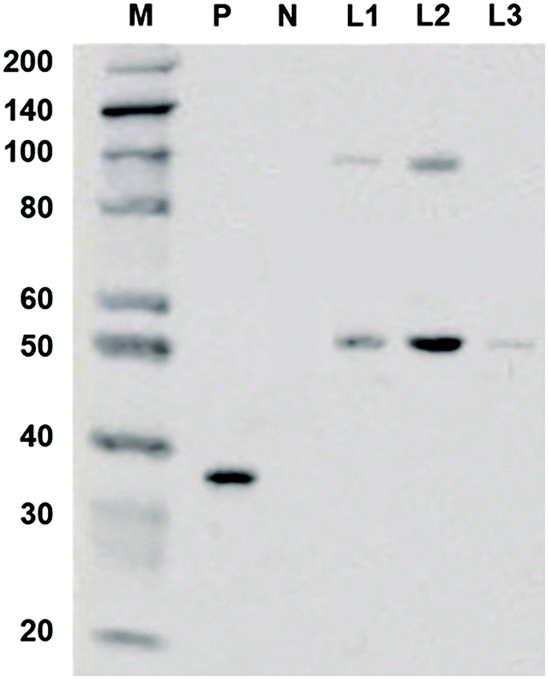
The coding sequence of TsGSK-3β was cloned and inserted into the pcDNA3.1+ vector. The recombinant expression vector pcDNA3.1+–TsGSK-3β was constructed and transformed into HEK293 cells. The protein was identified by detecting the expression of 6 × His genes after transient transfection into eukaryotic cells. Western blotting (Fig. 2) showed an approximately 50 kDa band for the concentrated cell culture supernatant (lane L1), indicating that the recombinant TsGSK-3β protein was successfully expressed in the transfected HEK293 cells. The samples in lane L2 were eluted with 50 mM imidazole elution buffer, and showed two distinct bands; the samples in lane L3 were eluted with 250 mM imidazole elution buffer, and showed only one band. The above results indicate that fusion proteins with high purity (lane L3) were obtained by separation and purification of the supernatants, which facilitated the inhibitory activity test in vivo.
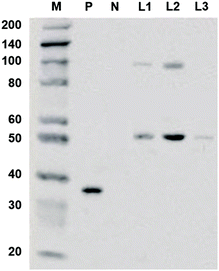 |
| | Fig. 2 Western blotting of expressed recombinant protein. M: marker, the marker has the following sizes: 200, 140, 100, 80, 60, 50, 40, 30 (weak), 20; P: positive control, recombinant GAPDH-His protein (36KD); N: negative control, recombinant GAPDH protein (36KD); L1: cell culture supernatant concentrated 5:1; L2: Ni-IDA sample eluted with 50 mM imidazole elution buffer; L3: Ni-IDA through sample during binding. | |
Detection of microarray chip based on SPRi
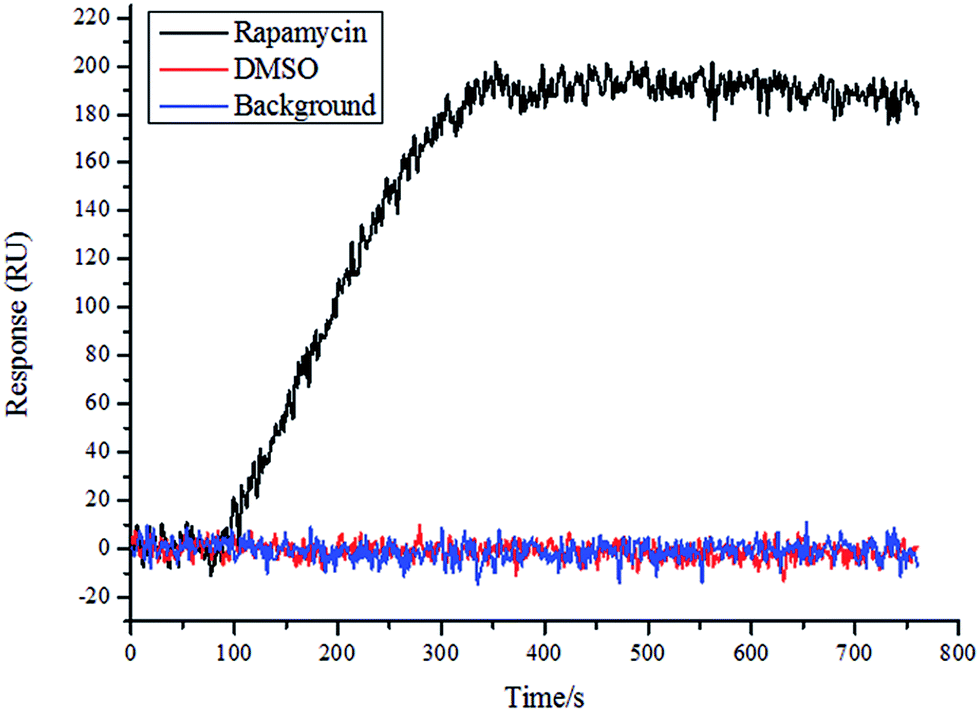
To determine the quality of the chip, rapamycin and DMSO were used as positive and negative controls, respectively, and a solution of FKPB12 (100 nM; Abcam, UK) was allowed to flow in the SPRi instrument. As shown in Fig. 3, the rapamycin spot bound the FKPB12 protein specifically. Conversely, DMSO showed no specific binding to the FKPB12 protein. These results showed that the chip quality was good and the chip could be used for follow-up tests.
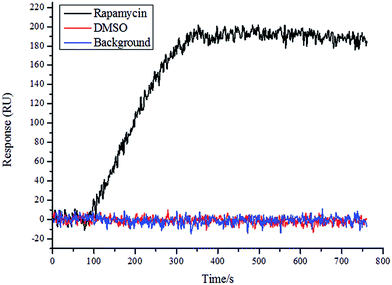 |
| | Fig. 3 Detection of microarray chip based on SPRi. SPRi graph showing interaction of rapamycin (positive control) and DMSO (negative control) with FKBP12 protein on the microarray chip. | |
Binding affinity assay by SPRi
The amino acids in the ATP-binding pocket play an important role in the selectivity and inhibitory activity. Changing any residue of the active pocket may affect the inhibitor's selectivity. As mentioned above, two amino acids in the TsGSK-3β active pocket were different from those in the active pocket of HsGSK-3β. We were interested in determining whether these amino acid differences could lead to different interactions with the same inhibitor molecules. SPR technology has proven to be an effective method for analyzing molecular interactions over the last 20 years.40–42 SPRi has been developed from conventional SPR technology and is a technique with label-free detection, high throughput, and parallel and image-based analysis; it plays an important role in drug discovery research.43,44 Furthermore, SPRi enables rapid identification of biomolecular interactions together with their kinetic parameters in real time.45,46 These advantages of SPRi decrease the assay time and increase the analytical precision from several available replicate data points in one measurement.47–49
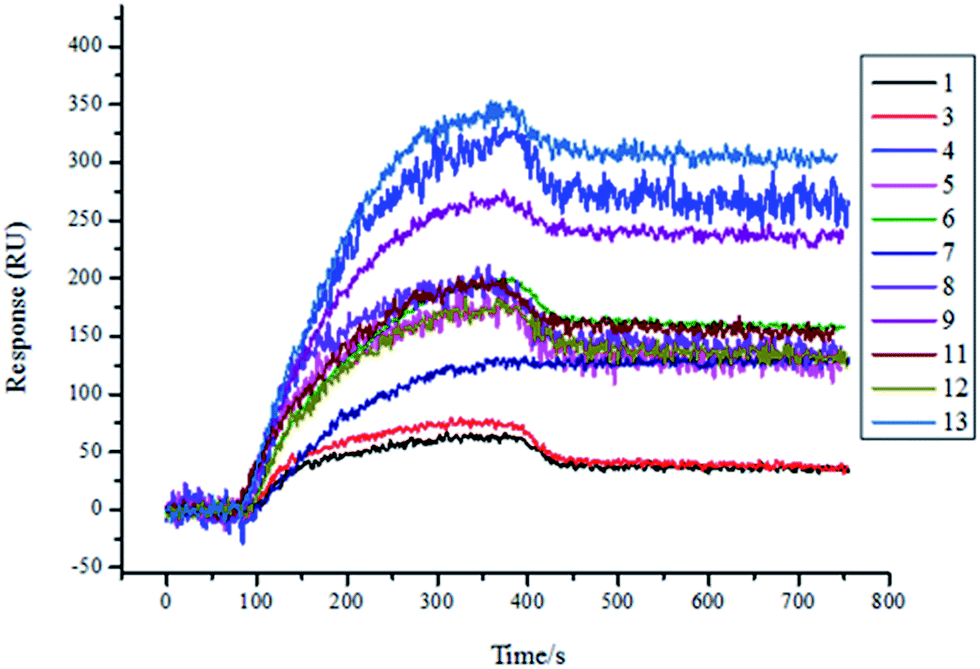
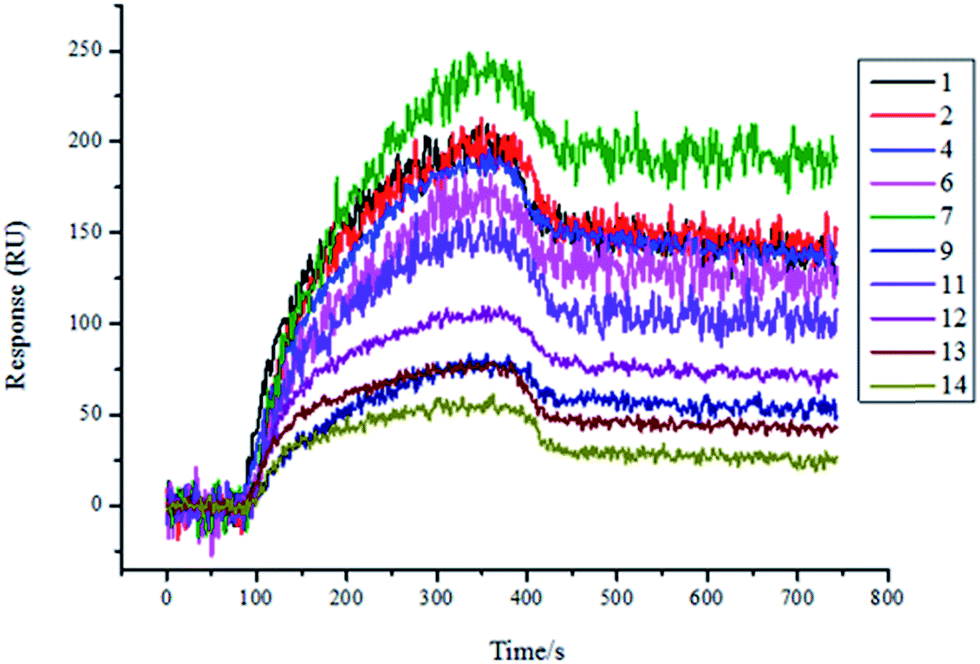
Therefore, we expressed and purified the recombinant protein TsGSK-3β and purchased HsGSK-3β (SignalChem, BC, Canada). The two proteins were used as analytes and were flowed through the flow cell at different concentrations (125 nM, 250 nM, 500 nM, 1000 nM) on the same SPRi chip. The affinity between molecules and proteins was increased along with increasing protein concentration (data not shown). The resultant arrays were regenerated with glycine-HCl (pH = 2.0) solution and reused several times, with great reproducibility. As shown in Fig. 4, 11 compounds (numbers 1, 3, 4, 5, 6, 7, 8, 9, 11, 12, and 13) showed significant binding to TsGSK-3β, and the kinetic parameters for these compounds that bind TsGSK-3β were measured (Table 2). As shown in Fig. 5, HsGSK-3β bound specifically to 10 compounds (numbers 1, 2, 4, 6, 7, 9, 11, 12, 13, and 14) whose kinetic parameters of interaction were obtained (Table 3). The binding affinity assay suggested that three compounds (numbers 3, 5, and 8) bound to TsGSK-3β but not to HsGSK-3β; we subsequently docked three compounds to TsGSK-3β and analyzed their interactions with key amino acid residues. Docking results showed that compound 3 formed one hydrogen bond with Val100, compound 5 formed two hydrogen bonds with Lys50 and Val100, and compound 8 formed two hydrogen bonds with Lys50 and Glu98 at the TsGSK-3β active sites. All data indicated that these hits could be used to further develop highly selective novel drugs against T. solium.
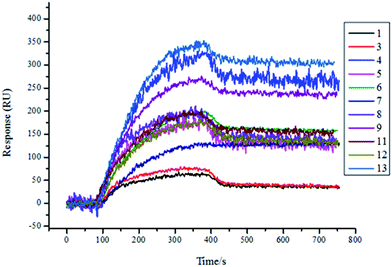 |
| | Fig. 4 Screening results of TsGSK-3β (1000 nM) protein binding molecules by SPRi. 11 molecules (1, 3, 4, 5, 6, 7, 8, 9, 11, 12, 13) bound to TsGSK-3β. | |
Table 2 Kinetic parameters of TsGSK-3β and molecules from SPRi
| Compound no. |
Avg Kon (1/Ms) |
Avg Koff (1/s) |
Avg KD (M) |
SD of KD |
| 1 |
1.31 × 104 |
2.37 × 10−4 |
1.81 × 10−8 |
3.16 × 10−9 |
| 3 |
2.12 × 104 |
7.23 × 10−4 |
3.41 × 10−8 |
1.07 × 10−10 |
| 4 |
8.27 × 103 |
3.72 × 10−4 |
4.50 × 10−8 |
2.14 × 10−9 |
| 5 |
2.01 × 104 |
2.35 × 10−4 |
1.17 × 10−8 |
6.63 × 10−9 |
| 6 |
1.27 × 103 |
5.21 × 10−4 |
4.10 × 10−7 |
3.26 × 10−12 |
| 7 |
6.31 × 103 |
7.44 × 10−6 |
1.18 × 10−9 |
4.25 × 10−12 |
| 8 |
4.69 × 103 |
4.73 × 10−4 |
1.01 × 10−7 |
1.41 × 10−10 |
| 9 |
4.72 × 103 |
4.14 × 10−5 |
8.77 × 10−9 |
1.59 × 10−9 |
| 11 |
3.58 × 103 |
7.21 × 10−4 |
2.01 × 10−7 |
2.92 × 10−9 |
| 12 |
3.71 × 103 |
2.82 × 10−4 |
7.61 × 10−8 |
1.38 × 10−8 |
| 13 |
3.38 × 103 |
3.94 × 10−4 |
1.17 × 10−7 |
1.48 × 10−8 |
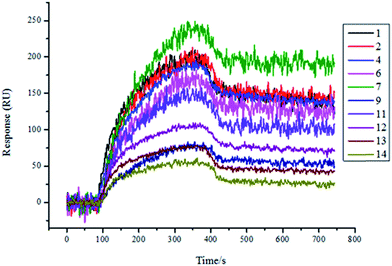 |
| | Fig. 5 Screening results for HsGSK-3β (1000 nM) protein-binding molecules by SPRi. 10 molecules (1, 2, 4, 6, 7, 9, 11, 12, 13, and 14) bound to HsGSK-3β. | |
Table 3 Kinetic parameters of HsGSK-3β and molecules from SPRi
| Compound no. |
Avg Kon (1/Ms) |
Avg Koff (1/s) |
Avg KD (M) |
SD of KD |
| 1 |
7.51 × 103 |
5.54 × 10−4 |
7.38 × 10−8 |
1.76 × 10−9 |
| 2 |
5.96 × 103 |
3.86 × 10−4 |
6.48 × 10−8 |
1.68 × 10−9 |
| 4 |
1.53 × 104 |
3.82 × 10−4 |
2.50 × 10−8 |
1.06 × 10−9 |
| 6 |
2.88 × 103 |
3.59 × 10−4 |
1.25 × 10−7 |
3.02 × 10−9 |
| 7 |
4.57 × 103 |
2.63 × 10−4 |
5.76 × 10−8 |
8.60 ×10−10 |
| 9 |
9.81 × 103 |
6.53 × 10−4 |
6.66 × 10−8 |
3.87 × 10−9 |
| 11 |
5.35 × 103 |
4.36 × 10−4 |
8.15 × 10−8 |
9.12 ×10−10 |
| 12 |
4.79 × 103 |
6.69 × 10−4 |
1.40 × 10−7 |
7.38 × 10−9 |
| 13 |
2.63 × 103 |
8.92 × 10−4 |
3.39 × 10−7 |
1.55 × 10−8 |
| 14 |
8.12 × 103 |
1.35 × 10−3 |
1.67 × 10−7 |
1.16 × 10−8 |
Experimental
Total RNA extraction and cDNA synthesis of TsGSK-3β
Total RNA was isolated from the proglottid of T. solium, using TRIzol reagent (Thermo Fisher, USA) following the product instructions. cDNA was synthesized according to the user manual of SMARTer® RACE 5′/3′ Kit (Clontech, USA).
Rapid amplification of cDNA ends (RACE) for TsGSK-3β
A pair of primers (forward primer: 5′-CATCAAAGTTATTGGAAATGGCTCA-3′ and reverse primer: 5′-GCGAATCTGCTCGTGGGAAGG-3′) was designed based on the sequence (systematic name: TsM_000742900) of GeneDB (http://www.genedb.org/). PCR was performed at 95 °C for 5 min; then at 94 °C for 1 min, 50 °C for 30 s, and 72 °C for 1 min for 30 cycles; and then at 72 °C for a final extension for 10 min. The PCR products were sequenced. Subsequently, 5′-RACE gene-specific primers (GSP1: 5′-GCGAATCTGCTCGTGGGAAGGTGTGCC-3′; nested GSP1: 5′-CAGAAGCAACTCCGCAAATACGCAACC-3′) and 3′-RACE gene-specific primers (GSP2: 5′-CGAGTTTGTTCCCGAGACAGTCTACAG-3′; nested GSP2: 5′-TGCCATCGGGACATCAAACCCCAAAAC-3′) were designed based on the acquired PCR sequence. The 5′- and 3′-RACE PCR experiments were performed according to the manual for the SMARTer RACE 5′/3′ Kit (Clontech, USA) with no modifications.
Sequence assembly
The PCR products were separated by 1% agarose gel electrophoresis stained with ethidium bromide and purified using an Agarose Gel DNA Extraction Kit (Takara, Dalian, China). The purified target DNA products were ligated into the pMD18-T Vector (Takara, Dalian, China) for transformation of E. coli JM109 (Takara, Dalian, China) and positive clones were selected for sequencing. The full-length cDNA sequence of TsGSK-3β was assembled using the SeqMan module of DNAStar 7.0 software. The properties of the putative protein were predicted using the Expert Protein Analysis System (ExPASy) (http://www.expasy.org/).
Homology modeling of TsGSK-3β
The TsGSK-3β gene-coding amino acid sequence was subjected to template searching using the BLAST program of the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/). The crystal structure of HsGSK-3β (PDB ID: 1UV5) was chosen as the template protein for computer-aided molecular modeling. The full-length amino acid sequence of TsGSK-3β was used as the query sequence and the crystal structure of HsGSK-3β protein was used as the template. The Sequence Analysis and Protein Modeling modules of Discovery Studio 2.5 software (BIOVIA, San Diego, CA, USA) were employed for sequence alignment and to build the 3D homology model, respectively. The ligand was copied while modeling. Furthermore, the Ramachandran plot and Profile 3D were used for model quality assessment to obtain an accurate model.
Protein model preparation
The TsGSK-3β homology model was processed with default settings, using the Protein Preparation Wizard module of Schrödinger 9.3 software (Schrödinger, Cambridge, MA, USA).
Ligand library preparation
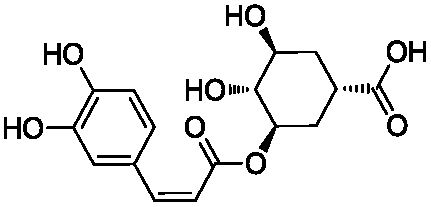
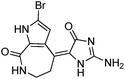
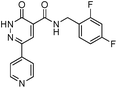
We selected 6 compounds with different molecular frameworks and moderate molecule weights from the literature.50 Biological activity experiments have shown that these compounds have a good inhibitory effect on HsGSK-3β in vitro. Their molecular formulae and IC50 values are shown in Table 4. All 6 compounds were built in ChemDraw 7.0 and converted to 3D structure, and the energy state was minimized using the LigPrep module of Schrödinger software. The 6 compounds as query molecular structure were submitted to http://lilab.ecust.edu.cn/chemmapper/#userconsent#. New compounds were screened based on three-dimensional molecular similarity methods, using the Hit Explorer module. A small library of compounds was obtained from the ZINC and NCI databases. All compounds were prepared using the LigPrep module of Schrödinger software for further docking analysis.
Table 4 Chemical structures of query molecules with their IC50 values
| Compound no. |
Molecular structure |
IC50 value |
| 1 |
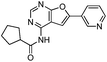 |
5 nM |
| 2 |
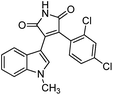 |
34 nM |
| 3 |
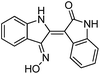 |
22 nM |
| 4 |
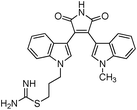 |
2.8 nM |
| 5 |
 |
10 nM |
| 6 |
 |
65 nM |
Molecular docking
The grid box was generated and the co-crystal ligand was set as the centroid using the Glide module in Schrödinger software. Molecular docking was implemented using the Ligand Docking tap of the Glide module. Docking parameters: precision was set as extra precision (XP), ligand sampling was set as flexible, the number of poses per ligand to include option was set as 5, and other parameters were used with the default settings. After docking, the top 50 small molecules were selected from each database according to docking scores. Additionally, some of the molecules were filtered from 100 compounds that violated Lipinski's rule of five. The residue compounds were further subjected to Prime MM-GBSA calculations and vision analysis.
Cloning and expression of recombinant TsGSK-3β
The recombinant TsGSK-3β gene was cloned from T. solium cDNA using the following primer pair: 5′-CGGGATCCACATGAGCGCTAAAGACGGTAAAG-3′ (forward primer) and 5′-GGAATTCATGATGATGATGATGATGCGATTGCTGCTGCGTTTCAG-3′ (reverse primer). The coding sequence of the 6 × His tag was included in the reverse primer and incorporated by PCR into the TsGSK-3β gene at the C-terminal. PCR cycling conditions were 95 °C for 5 min; then 94 °C for 1 min, 60 °C for 40 s, 72 °C for 1 min for 30 cycles; and then a final extension at 72 °C for 10 min. PCR products with the correct sequence were inserted into the eukaryotic expression vector pcDNA3.1+ (Invitrogen, USA) between the BamHI and EcoRI restriction sites. The recombinant expression vector was transiently transfected into the HEK293 cells (CRL-1573, ATCC, USA), using FuGENE HD transfection reagent (Promega, US). The pcDNA3.1+–TsGSK-3β-His vector was expressed in HEK293 cells and the resulting TsGSK-3β was concentrated and purified on a His affinity column (GE Healthcare, US) per the instructions of the manufacturer. The resultant protein was eluted with different concentrations of imidazole buffers (10 mM, 50 mM, 250 mM).
Western blot analysis of TsGSK-3β protein expression
The protein samples were separated by 12% SDS-PAGE and then transferred onto a nitrocellulose membrane. The membrane was blocked in 5% w/v skim milk for 1 h at room temperature. Subsequently, the membrane was incubated with His-tag monoclonal antibodies (Proteintech Group, Rosemont, USA) overnight at 4 °C. After three washes with PBS containing 0.1% v/v Tween-20, the membrane was incubated with horseradish peroxidase-labeled anti-rabbit secondary antibody (Proteintech Group, Rosemont, IL, USA) for 1 h at room temperature. DAB (3,3′-diaminobenzidine) detection reagent (Solarbio, Beijing, China) was used for protein visualization on the nitrocellulose membrane. Recombinant GAPDH-His protein (36 kDa) and recombinant GAPDH protein were used as the loading control. The purified protein concentrations were determined using the BCA protein assay kit (Beyotime, Shanghai, China). The purified TsGSK-3β protein was added with 10% (v/v) glycerin and aliquoted to 100 μl per tube and then maintained at −80 °C until use.
SMM preparation
A photo-cross-linking technique was used to immobilize the drug molecule onto the standard Plexera Nanocapture sensor chip. The chip was self-assembled with SH-PEG-COOH:SH-PEG-OH in a 1:10 ratio, and functionalized with a diazarine photo-cross-linker after activation with EDC/NHS (Aladdin, Shanghai, China) (0.4/0.2 M). The printed area was subsequently blocked by 1 M ethanolamine (pH 8.4) to prevent nonspecific adsorption of proteins on the surface. Fourteen drug molecule solutions (10 mM in 100% DMSO) were spotted in multiplex (3 spots) on the sensor chip using a commercial Genetix spotter and left for complete evaporation of DMSO in a dark room at room temperature for 30 min. Then, the slides were exposed to UV irradiation of 2.4 J cm−2 (365 nm) in a UV chamber (Amersham Life Science). The slides were subsequently washed with dimethylformamide, ethanol, and distilled water for 15 min to remove non-specifically adsorbed compounds. Air-dried slides were assembled in a flow cell and then mounted on a SPRi instrument (PLEXERA PlexArray HT) for measurement.
SPRI method
The 2 protein samples were injected at a rate of 2 μl s−1 at 25 °C. Oval regions of interests (ROIs) were automatically set with the data collection software in the imaging area. ROIs of rapamycin (Sigma, USA) and DMSO (Sigma, USA) were used as the positive control and negative control for measurement of specific signals, respectively. The 2 proteins were diluted in PBST containing Tween 20 (0.05%), pH 7.4, and used as analytes with an association and dissociation flow rate of 2 μl s−1 at different concentrations by serial dilution. A solution of glycine-HCl (pH 2.0) was used to regenerate the surface and remove bound proteins from the small molecules, enabling the sensor chip to be reused for additional analyte injections.
Binding experiments and data analysis
All small molecules were stored as stock solutions in 100% dimethyl sulfoxide at −20 °C. Protein samples were stored in PBST at −80 °C. PBST was used as both an analyte and running buffer. A typical sample injection cycle consists of a 300 s association phase with the analyte solution and a 300 s dissociation phase with the running buffer at a flow rate of 2 μl s−1. Proteins were flowed as analytes at four different concentrations (125 nM, 250 nM, 500 nM, 1000 nM). The average results from 10 different spots were used to plot standard deviations. The average kinetics of the complexes was obtained from a 1:1 Langmuir model of kinetics fitting from four different concentrations. All of the experiments were repeated three times to ensure data repeatability. All of the data analysis was performed using the PLEXEA data analysis module and the ORIGINLab software.
Conclusions
In this study, we determined the amino acid sequence of Ts-GSK-3β to identify novel molecular inhibitors of Ts-GSK-3β as a strategy for treatment of tapeworm infections and NCC. We selected 3 compounds that bound to TsGSK-3β but not to HsGSK-3β, using virtual screening combined with SPRi technology. However, the effects of TsGSK-3β inhibitors on T. solium, especially on the cysticercus, have not been tested. Further studies are needed to determine the activity of the 3 inhibitors against cysticercus in vitro. Next, we plan to perform biological experiments to validate these inhibitors. We believe that the findings of our study may be useful for the design of more potent and selective compounds for the treatment of T. solium that do not damage the definitive host.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Special Basic Funds for Central Research Institutes of Public Interests (Grant No. 1610312016101). The authors are thankful to Shanghai Key Laboratory of Chemical Biology (East China University of Science and Technology) for providing facilities for this research and GeneDB database for providing the nucleotide sequence data.
References
- P. J. Hotez, D. H. Molyneux, A. Fenwick, E. Ottesen, S. Ehrlich Sachs and J. D. Sachs, PLoS Med., 2006, 3, e102 Search PubMed.
- Z. Pawlowski, J. Allan and E. Sarti, Int. J. Parasitol., 2005, 35, 1221–1232 CrossRef PubMed.
- P. R. Torgerson and C. N. Macpherson, Vet. Parasitol., 2011, 182, 79–95 CrossRef PubMed.
- J. C. Allan, M. Velasqueztohom, C. Fletes, R. Torresalvarez, G. Lopezvirula, P. Yurrita, D. A. H. Soto, A. Rivera and J. Garcianoval, Trans. R. Soc. Trop. Med. Hyg., 1997, 91, 595–598 CrossRef CAS PubMed.
- Z. S. Pawłowski, Acta Trop., 1990, 48, 83 CrossRef.
- Z. S. Pawlowski, Parasitol. Int., 2006, 55, S105–S109 CrossRef CAS PubMed.
- W. H. Organization, Drug Intell. Clin. Pharm., 2015, 12, 169–171 Search PubMed.
- T. Li, A. Ito, X. Chen, C. Long, M. Okamoto, F. Raoul, P. Giraudoux, T. Yanagida, M. Nakao and Y. Sako, Acta Trop., 2012, 124, 152–157 CrossRef CAS PubMed.
- O. H. Del Brutto, Sci. World J., 2012, 2012, 159821 Search PubMed.
- M. V. Johansen, C. Trevisan, S. Gabriël, P. Magnussen and U. C. Braae, Parasitology, 2016, 144, 1 Search PubMed.
- J. Sotelo, O. H. D. Brutto, P. Penagos, F. Escobedo, B. Torres, J. Rodríguezcarbajal and F. Rubiodonnadieu, J. Neurol., 1990, 237, 69–72 CrossRef CAS PubMed.
- O. M. Takayanagui and E. Jardim, JAMA Neurol., 1992, 49, 290–294 CAS.
- O. H. Del Brutto, J. Neurol., Neurosurg. Psychiatry, 1997, 62, 659 CrossRef CAS.
- A. W. Parvaiz, A. Koul, M. Hayat and B. A. Sofi, Infect. Dis., 1999, 31, 603–604 Search PubMed.
- N. Embi, D. B. Rylatt and P. Cohen, Eur. J. Biochem., 1980, 107, 519–527 CrossRef CAS PubMed.
- I. N. Gaisina, J. Med. Chem., 2009, 52, 1853–1863 CrossRef CAS PubMed.
- J. H. Youn, T. W. Kim, E. J. Kim, S. Bu, S. K. Kim, Z. Y. Wang and T. W. Kim, Mol. Cells, 2013, 36, 564–570 CrossRef CAS PubMed.
- M. A. Mines, Eur. J. Pharmacol., 2013, 708, 56–59 CrossRef CAS PubMed.
- R. J. Gum, L. L. Gaede, S. L. Koterski, M. Heindel, J. E. Clampit, B. A. Zinker, J. M. Trevillyan, R. G. Ulrich, M. R. Jirousek and C. M. Rondinone, Diabetes, 2003, 52, 21–28 CrossRef CAS PubMed.
- S. C. Hsiung, M. Adlersberg, V. Arango, J. J. Mann, H. Tamir and K. P. Liu, J. Neurochem., 2003, 87, 182–194 CrossRef CAS PubMed.
- K. Inoki, H. Ouyang, T. Zhu, C. Lindvall, Y. Wang, X. Zhang, Q. Yang, C. Bennett, Y. Harada, K. Stankunas, C. Y. Wang, X. He, O. A. MacDougald, M. You, B. O. Williams and K. L. Guan, Cell, 2006, 126, 955–968 CrossRef CAS PubMed.
- C. Morisco, K. Seta, S. E. Hardt, Y. Lee, S. F. Vatner and J. Sadoshima, J. Biol. Chem., 2001, 276, 28586–28597 CrossRef CAS PubMed.
- E. Smith and B. Frenkel, J. Biol. Chem., 2005, 280, 2388–2394 CrossRef CAS PubMed.
- L. A. Robertson, A. J. Kim and G. H. Werstuck, Can. J. Physiol. Pharmacol., 2006, 84, 39–48 CrossRef CAS PubMed.
- S. Phukan, V. S. Babu, A. Kannoji, R. Hariharan and V. N. Balaji, Br. J. Pharmacol., 2010, 160, 1–19 CrossRef CAS PubMed.
- B. Georgievska, J. Sandin, J. Doherty, A. Mörtberg, J. Neelissen, A. Andersson, S. Gruber, Y. Nilsson, P. Schött and P. I. Arvidsson, J. Neurochem., 2013, 125, 446–456 CrossRef CAS PubMed.
- V. Palomo and A. Martinez, Expert Opin. Ther. Pat., 2017, 27, 657 CrossRef CAS PubMed.
- G. U. Höglinger, H. J. Huppertz, S. Wagenpfeil, M. V. Andrés, V. Belloch, T. León and S. T. Del, Mov. Disord., 2014, 29, 479–487 CrossRef PubMed.
- S. T. Del, K. C. Steinwachs, H. J. Gertz, M. V. Andrés, B. Gómezcarrillo, M. Medina, J. A. Vericat, P. Redondo, D. Fleet and T. León, J. Alzheimers Dis., 2013, 33, 205 Search PubMed.
- E. Xingi, D. Smirlis, V. Myrianthopoulos, P. Magiatis, K. M. Grant, L. Meijer, E. Mikros, A. L. Skaltsounis and K. Soteriadou, Int. J. Parasitol., 2009, 39, 1289–1303 CrossRef CAS PubMed.
- E. Droucheau, A. Primot, V. Thomas, D. Mattei, M. Knockaert, C. Richardson, P. Sallicandro, P. Alano, A. Jafarshad and B. Baratte, Biochim. Biophys. Acta, Proteins Proteomics, 2004, 1697, 181–196 CrossRef CAS PubMed.
- K. K. Ojo, J. R. Gillespie, A. J. Riechers, C. L. M. J. Verlinde, F. S. Buckner, M. H. Gelb, M. M. Domostoj, S. J. Wells, A. Scheer, T. N. C. Wells and W. C. V. Voorhis, Antimicrob. Agents Chemother., 2008, 52, 3710–3717 CrossRef CAS PubMed.
- R. O. Oduor, K. K. Ojo, G. P. Williams, F. Bertelli, J. Mills, L. Maes, D. C. Pryde, T. Parkinson, W. C. Van Voorhis and T. P. Holler, PLoS Neglected Trop. Dis., 2011, 5, e1017 CAS.
- D. Baker and A. Sali, Science, 2001, 294, 93 CrossRef CAS PubMed.
- V. B. Chen, W. B. Arendall III, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 66, 12 CrossRef PubMed.
- R. Luthy, J. U. Bowie and D. Eisenberg, Nature, 1992, 356, 83–85 CrossRef CAS PubMed.
- B. S. Darshit, B. Balaji, P. Rani and M. Ramanathan, J. Mol. Graphics Modell., 2014, 53, 31–47 CrossRef CAS PubMed.
- D. I. Osolodkin, N. V. Zakharevich, V. A. Palyulin, V. N. Danilenko and N. S. Zefirov, Parasitology, 2011, 138, 725 CrossRef CAS PubMed.
- I. Massova and P. A. Kollman, Perspect. Drug Discovery Des., 2000, 18, 113–135 CrossRef CAS.
- A. M. Hutchinson, Mol. Biotechnol., 1995, 3, 47 CrossRef CAS PubMed.
- L. S. Jung, K. E. Nelson, P. S. Stayton and C. T. Campbell, Langmuir, 2003, 16, 9421–9432 CrossRef.
- L. S. Jung, K. E. Nelson, C. T. Campbell, P. S. Stayton, S. S. Yee, V. Pérez-Luna and G. P. López, Sens. Actuators, B, 1999, 54, 137–144 CrossRef CAS.
- F. Pillet, C. Thibault, S. Bellon, E. Maillart, E. Trévisiol, C. Vieu, J. M. François and V. A. Leberre, Sens. Actuators, B, 2010, 147, 87–92 CrossRef CAS.
- J. M. Mcdonnell, Curr. Opin. Chem. Biol., 2001, 5, 572 CrossRef CAS PubMed.
- P. J. Hotez, D. H. Molyneux, A. Fenwick, E. Ottesen, S. S. Ehrlich and J. D. Sachs, PLoS Med., 2007, 4, e102 CrossRef.
- S. O. Jung, H. S. Ro, B. H. Kho, Y. B. Shin, M. G. Kim and B. H. Chung, Proteomics, 2005, 5, 4427–4431 CrossRef CAS PubMed.
- G. Spoto and M. Minunni, J. Phys. Chem. Lett., 2012, 3, 2682 CrossRef CAS PubMed.
- J. M. Brockman, B. P. Nelson and R. M. Corn, Annu. Rev. Phys. Chem., 2000, 51, 41–63 CrossRef CAS PubMed.
- J. Fasoli and R. M. Corn, Langmuir, 2015, 31, 9527–9536 CrossRef CAS PubMed.
- S. Vadivelan, B. N. Sinha, S. Tajne and S. A. Jagarlapudi, Eur. J. Med. Chem., 2009, 44, 2361–2371 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Jiagao Chengc,
Shuai Wangb,
Xichen Zhang*a and
Xuepeng Cai,*ab
ab,
Jiagao Chengc,
Shuai Wangb,
Xichen Zhang*a and
Xuepeng Cai,*ab