DOI:
10.1039/C7RA05808J
(Paper)
RSC Adv., 2017,
7, 42884-42890
Fabrication of highly stable and sensitive electrochemical sensor from hemoglobin–Au nanocomposites and its analytical applications†
Received
23rd May 2017
, Accepted 26th July 2017
First published on 5th September 2017
Abstract
To improve electron transfer, a combination of electroactive hemoglobin (Hb) and Au nanoparticles (Au NPs) was designed to obtain nanocomposites (NCs). Cyclic voltammetry and redox probe characterization showed that Hb–Au NCs incorporating the layer-by-layer technique to improve the ration of electroactive Hb, showed a better conjugated structure, lower standard entropy, and faster electron transfer. Au entrapped in Hb on the glassy carbon electrode (GCE) also exhibited a wider linear range, and excellent reproducibility and recovery for analytical applications of hydrogen peroxide, oxygen, and trichloroacetic acid. Therefore, our research could potentially become a promising alternative by combining bioactive molecules with nanoparticles, and broaden the applications of mediator-free biosensors.
1. Introduction
Hemoglobin (Hb) is a biomolecule with high, long-term stability.1,2 Fast electron transfer between Hb and electrode surfaces has been reported as not possible owing to deep embedding of the electroactive heme group.3,4 In recent decades, much effort has been made to increase electron transfer kinetics, with the most commonly used methods including specific surface modifications of electrodes with polymer films,5 surfactants,6–8 nanoparticles (NPs),9,10 and biomembranes.11 Au nanomaterials have excellent properties with respect to volume, surface, quantum size, macro quantum tunneling effect, and catalytic properties.12 Furthermore, Au NPs exhibit better catalytic activity toward hydrogen peroxide (H2O2) than metallic gold due to their high surface-area-to-volume ratio and their interface-dominated properties.13 The immobilization of biomolecules, such as horseradish peroxidase, laccase, and cytochrome c, on Au NP surfaces to construct biosensors has been widely investigate.14–16 However, Hb and Au NP nanocomposites have rarely been reported.
Recently, the analysis of hydrogen peroxide (H2O2), oxygen (O2), and trichloroacetic acid (TCA) using various methods has attracted considerable attention. Traditional methods17–19 usually detect these species using expensive and sophisticated equipment, involving time-consuming and multistep procedures that have low sensitivity, poor reproducibility, and poor repeatability. Therefore, electrochemical biosensors are better choices for meeting these various demands. Furthermore, the application of Hb-loaded biosensors to electrochemical detection of multiple targets has seldom been reported.
Herein, we present a novel approach to constructing a biosensor for H2O2, O2, and TCA based on Hb electrochemistry. Au NPs and Hb–Au NCs were alternately immobilized on a GCE using layer-by-layer technology. The resultant {Au NP/Hb–Au NC}n films showed direct and reversible electrochemistry of heme Fe(III)/Fe(II) redox couples. The n (value of layer-by-layer) was optimized to 3 using a variety of electrochemical techniques. Owing to the outstanding bioelectrical catalytic abilities of the {Au NPs/Hb–Au NCs}3 films, the modified biosensor exhibited sensitive electrochemical responses to H2O2, O2, and TCA. This type of bionanocomposite maintains the natural biological activity of Hb and offers a more accurate and rapid alternative approach to electrochemical measurements for further research.
2. Experimental
2.1. Materials and reagents
Hb from bovine blood (lyophilized powder protein, Mw 64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500) was purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Potassium ferricyanide and HAuCl4·4H2O (99.999%; Au%, >48%) were purchased from Sigma-Aldrich, USA. Sodium citrate (Na3C6H5O7) was purchased from Shanghai No. 1 Reagent Factory. TCA (C2HCl3O2) was purchased from Tokyo Chemistry Industry Corporation. Chitosan (95% deacetylation, Mw 5.09105), H2O2 (30%) and other common chemicals were purchased from Aladdin Industrial Corporation. Other reagents were of analytical reagent grade. Phosphate buffers (0.10 M) with different pH values were prepared by mixing standard solutions of Na2HPO4 and NaH2PO4, and adjusting the pH by adding 0.10 M H3PO4 or 0.10 M NaOH solutions. Double-distilled deionized (DI) water was used in all experiments. Deoxygenated water was obtained from DI water by bubbling through highly pure nitrogen for at least 30 min.
500) was purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Potassium ferricyanide and HAuCl4·4H2O (99.999%; Au%, >48%) were purchased from Sigma-Aldrich, USA. Sodium citrate (Na3C6H5O7) was purchased from Shanghai No. 1 Reagent Factory. TCA (C2HCl3O2) was purchased from Tokyo Chemistry Industry Corporation. Chitosan (95% deacetylation, Mw 5.09105), H2O2 (30%) and other common chemicals were purchased from Aladdin Industrial Corporation. Other reagents were of analytical reagent grade. Phosphate buffers (0.10 M) with different pH values were prepared by mixing standard solutions of Na2HPO4 and NaH2PO4, and adjusting the pH by adding 0.10 M H3PO4 or 0.10 M NaOH solutions. Double-distilled deionized (DI) water was used in all experiments. Deoxygenated water was obtained from DI water by bubbling through highly pure nitrogen for at least 30 min.
2.2. Apparatus
Scanning electron microscopy (SEM) images were obtained with a JSM-6510 microscope (JEOL, Japan). Transmission electron microscopy (TEM) images were obtained with a JEM-2100 microscope (JEOL, Japan). Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were performed using a CHI660D electrochemical workstation (Shanghai Chenhua Instrument Company, China), and were operated in the electrochemical cell consisting of a modified GCE (Φ = 3 mm) as the working electrode, a platinum wire as the counter electrode, and a saturated calomel electrode (SCE) as the reference electrode. Phosphate buffer containing different concentrations of the detected materials was prepared by adding different amounts of the detected materials to deoxygenated phosphate buffer (8 mL, 0.1 M, pH 7.0) using a microsyringe. CV signals were measured at room temperature (scan potential, −0.9 to 0.3 V; scan rate, varied from 10 to 500 mV s−1; sensitivity (A/V), 1 × 10−6) for different electrochemical characterizations of GCE-CS–{Au NPs/Hb–Au NCs}3. DPV signals were measured at room temperature (scan potential, −0.9 to 0.3 V; pulse amplitude, 0.05 V; pulse width, 0.1 s; sensitivity (A/V), 1 × 10−6) for quantitatively detection of the target analytes. Electrochemical impedance spectroscopy (EIS) was performed in 0.10 M KCl solution, containing 5 mM Fe(CN)63−/Fe(CN)64− (1:1) as a supporting electrolyte, at its open circuit potential using the AUTOLAB PGSTAT 302N electrochemical working station (Metrohm Co. Ltd., Switzerland) (start frequency, 1.0 × 105 Hz; end frequency, 5.0 × 10−2 Hz; frequency number, 60; amplitude of potential perturbation, 10 mV). A CHI 440B electrochemical analyzer (CHInstruments, Co., USA) was used to perform electrochemical quartz crystal microbalance (EQCM) measurements. Gold-coated quartz crystal resonator electrodes were soaked in freshly prepared “piranha” solution (30% H2O2 and 98% H2SO4, 3:7 v/v) for 15 min, and then washed with pure water. The zeta potential of the Hb–Au NCs was measured using a Zetasizer Nano ZS-90 (Malvern, England). All measurements were performed at room temperature (25 ± 0.2 °C) in a Faraday cage, unless otherwise specified. All solutions were deoxygenated by bubbling through highly pure nitrogen for at least 30 min under a nitrogen atmosphere, and were maintained throughout the measurement period.
2.3. Synthesis of Hb–Au NCs and biosensor construction
Au NPs were prepared according to our previous work.20 The prepared Au NPs were stored in brown glass bottles at 4 °C. Hb–Au NCs were synthesized from Hb solution (3.0 mg mL−1) and Au NPs (0.1 mg mL−1) at the optimized volume ratio of 3:1, shown in Fig. S1.† The mixed solution was sonicated for 30 min, centrifuged at 5000 rpm for 10 min, and washed twice with DI water. Finally, the precipitates were dispersed in pH 7.0 PBS to form 3.0 mg mL−1 dispersions, which were stored in a refrigerator at 4 °C. The zeta potential of the Hb–Au NCs was 2.4 ± 0.1 mV, as measured by a Zetasizer Nano ZS-90, which indicated that the Hb–Au NCs were positively charged.
The bare GCE was polished with abrasive paper and alumina (0.25 and 0.05 μm) slurry on microcloth pads, ultrasonicated in DI water for 30 s, and then dried under nitrogen. The detailed assembly process is illustrated in Scheme 1. The chitosan (CS)-modified glassy carbon electrode (GCE) was prepared according to our previous work.21 The CS modified glassy carbon electrode (CS-GCE) was then dipped into Au NPs at 4 °C for 30 min, washed with DI water and dried at room temperature to form Au NP–CS modified GCE, denoted as Au–CS-GCE. Afterwards, the modified GCE was dropcast with 20 μL of 3.0 mg mL−1 Au–Hb NCs, dried at room temperature, and washed with DI water. The procedure was repeated for the optimal number of {Au NPs/Hb–Au NCs}3 bilayers, using layer-by-layer technology to form GCE-CS–{Au NPs/Hb–Au NCs}3.
 |
| | Scheme 1 Assembly process of {Au NPs/Hb–Au NCs}n immobilized on the GCEs. | |
In the application of real samples, 0.1 M zinc acetate and potassium ferrocyanide were added to a solution of milk powder (from a local market; 100 mL, 0.1 g mL−1) to remove proteins by centrifuging at 4000 rpm for 5 min. A 10 mL aliquot of the above supernatant was used for H2O2 recovery experiments. The tape was used for TCA and O2 recovery research without specific treatment. Recovery tests were carried out by adding different concentrations of standard H2O2, TCA, or O2 into the samples and comparing with the results of the national standard method. The standard methods for detecting H2O2, TCA, or O2 were iodine titrimetry, colorimetry, and gas chromatography, respectively.
3. Results and discussion
3.1. Morphology of Hb–Au NCs
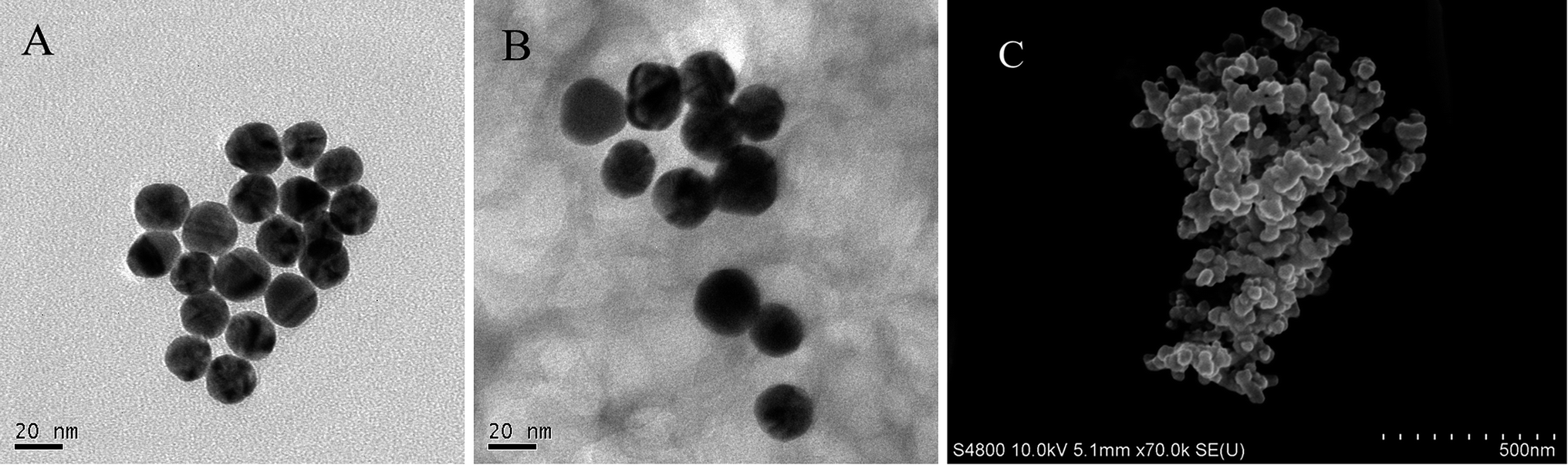
TEM was used to determine the morphology and structure of the Au NPs and Hb–Au NCs. As shown in Fig. 1(A), the Au NPs had average diameters of about 25 nm and an almost uniform and clear surface. The morphology of the Hb–Au NCs is shown in Fig. 1(B), in which an Au cluster was surrounded by polymer materials, and the surfaces of the Au NPs were hazy and aggregated, in contrast to the pure Au NPs. The Hb–Au NCs were also characterized by SEM. Hb–Au NCs were formed as a film on the electrode surface, into which the Au NPs were then enmeshed, as shown in Fig. 1(C). The porous structure of the Hb–Au NCs might allow the small ions in the buffers to pass through the films easily, which would be beneficial to Hb electron transfer in the films with underlying electrodes.
 |
| | Fig. 1 TEM images of (A) Au NPs and (B) Hb–Au NCs at 100000× magnification. SEM images of (C) Hb–Au NCs at 70000× magnification on a GCE. | |
3.2. Electrochemical characterization of Hb–Au NCs
No cyclic voltammetry (CV) peaks were observed for the bare GCE (curve a), CS-GCE (curve b), and Au–CS-GCE (curve c) in the selected potential range, as shown in Fig. S2.† A pair of well-defined, nearly reversible redox peaks at −0.312 V and −0.376 V were observed when Hb–Au NCs were modified on the Au–CS-GCE (curve d), indicating the successful assembly of Hb–Au NCs. According to the results of EIS in Fig. S3,† with the assembly of {Au NPs/Hb–Au NCs}n, the diameter of impedance increased with the layer number (curve e–h), indicating successful assembly of the Hb–Au NPs.22,23
The value of the redox current of {Au NPs/Hb–Au NCs}n reached a clear maximum at n = 3 (Fig. 2). The assembly process was mainly based on electrostatic interactions and the “A–N” chemical bond between the Hb–Au NCs and Au NPs.24,25 Hb is positively charged in pH 6.0 PBS due to its isoelectric point (pI) of 7.4, and can be adsorbed on oppositely-charged Au NPs by electrostatic attraction to form Hb–Au NCs. The zeta potential of the Hb–Au NCs was measured as 2.4 ± 0.1 mV, indicating that the Hb–Au NCs held positive charges. Therefore, we used negatively charged Au NPs to realize layer-by-layer Hb–Au NCs on the GCE surface. The EQCM technique has also been shown to be valuable for studying surface-related processes in liquids, including protein adsorption,22,23 using the Sauerbrey equation,24
| |
 | (1) |
where
A is the geometric area of the QCM electrode (0.196 cm
2). According to the EQCM results (Table S1
†), the Δ
f values of the {Au NPs/Hb–Au NCs}
n films absorption steps (
n = 8) were 44.1 ± 3.2 ({Au NPs/Hb–Au NCs}
1), 89.1 ± 5.2 ({Au NPs/Hb–Au NCs}
2), 135.1 ± 6.9 ({Au NPs/Hb–Au NCs}
3), and 180.1 ± 4.0 Hz ({Au NPs/Hb–Au NCs}
4). The corresponding total film masses were 59.13, 119.88, 181.98, and 244.08 μg, respectively. As the geometric area of the EQCM electrode was 0.196 cm
2 and the molecular mass of Hb was 64
500, the surface concentrations of total Hb in each film were 4.67 × 10
−9, 9.48 × 10
−9, 14.3 × 10
−9, and 17.73 × 10
−9 mol cm
−2, respectively. The surface concentration of electroactive Hb in the films could be calculated according to the equation
Γ =
Q/(
nFA), where
Q is the charge involved in the reaction,
n is the electron transfer number,
F is the Faraday constant, and
A is the geometric area of the GCE. The amounts of electroactive Hb molecules were estimated to be 6.543 × 10
−10, 7.499 × 10
−10, 9.338 × 10
−10, and 9.350 × 10
−10 mol cm
−2 in each film, respectively. The ration of electroactive Hb to total Hb on the electrode was 13.8, 7.91, 6.53, and 5.27%. The ration of electroactive Hb increased with increasing
n, from 1 to 3, and tended to level off when
n was more than 3. This may be due to the fourth layer being too far away from the Au electrode to perform direct electrochemistry. Consequently, the concentration of electroactive Hb in the third and fourth films was almost equal. Therefore, {Au NPs/Hb–Au NCs}
3 were chosen as the optimal layer-by-layer experimental conditions for further investigation.
 |
| | Fig. 2 (A) Cyclic voltammograms of {Au NPs/Hb–Au NCs}n films immobilized on CS-modified GCEs at 100 mV s−1 in pH 7.0 PBS (n = 1–5; scan potential from −0.9 to 0.3 V). (B) Redox peak currents varied with number of layers (from 1 to 5). | |
As depicted in Fig. S4,† the electron transfer number (n) of {Au NPs/Hb–Au NCs}3 on the electrode was found to be 1, implying a single electron transfer reaction. These results demonstrated that Hb realized direct electron transfer, indicating mutual transmission between ferrous iron and ferric iron. The proportion of protons/electrons involved was estimated to be 1, as shown in Fig. S5.† Therefore, the electron-transfer reaction can be presented as:25
| | |
HbhemeFe3+ + H+ + e ⇌ HbhemeFe2+
| (2) |
When nΔEp < 200 mV, the electron transfer rate constant (ks) was estimated using Laviron's equation:26
| |
 | (3) |
where
α is the charge transfer coefficient.
10 When the scan rate was 100 mV s
−1 and Δ
Ep = 80 mV,
ks was calculated to be 0.95 s
−1, which was larger than the value of 0.49 s
−1 reported for an Au colloid–cysteamine-modified gold electrode.
4
3.3. Biosensors based on electrocatalysis of GCE-CS–{Au NPs/Hb–Au NCs}3
DPV is a measurement method that imposes a range of pulsed voltages (10–100 mV) on a direct current voltage, which can improve sensitivity.27,28 Therefore, we selected DPV for analyte determination instead of CV. The electrocatalysis of H2O2 using GCE-CS–{Au NPs/Hb–Au NCs}3 was investigated with the successive addition of H2O2 to pH 7.0 PBS under a nitrogen atmosphere, and an obvious increase in the cathodic peak current at approximately −0.328 V was observed (Fig. 3). The more H2O2 added to the continuously stirred deoxygenated buffer solution, the larger the cathodic peak current became, indicating that Hb incorporated in the composite film acted as an effective catalyst for H2O2 reduction. The inset shows the calibration curve of the amperometric current and H2O2 concentration. Excellent linear relationships were obtained in the concentration range 9.70 × 10−9 to 2.14 × 10−5 M (a–n) (R2 = 0.9990, n = 8). This relationship can be described as IA = 3.68 × 10−7 + 3.52CH2O2, M. The limit of detection (LOD) of this biosensor for H2O2 was estimated as 3.55 × 10−9 M at a signal-to-noise ratio of 3 (S/N = 3). The mechanism can be expressed using the following equation:29| | |
HbhemeFe2+ + H2O2 + e → HbhemeFe3+ + H2O
| (4) |
 |
| | Fig. 3 Differential pulse voltammetry (DPV) signals measured at room temperature (scan potential, −0.9 to 0.3 V; pulse amplitude, 0.05 V; pulse width, 0.1 s, sensitivity (A/V), 1 × 10−6) for GCE-CS–{Au NPs/Hb–Au NCs}3 in PBS (0.10 M, pH 7.0) within the H2O2 concentration range 1.00 × 10−8 (curve a) to 1.76 × 10−7 M (curve n) at a scan rate of 100 mV s−1. Inset: plot of current vs. H2O2 concentration. | |
TCA is an important chemical intermediate in the synthesis of organic compounds and a by-product in drinking water disinfected with chlorine.30 The electrocatalytic reduction of TCA by GCE-CS–{Au NPs/Hb–Au NCs}3 is shown in Fig. 4.
 |
| | Fig. 4 DPV signals measured at room temperature (scan potential, −0.9 to 0.3 V; pulse amplitude, 0.05 V; pulse width, 0.1 s, sensitivity (A/V), 1 × 10−6) for GCE-CS–{Au NPs/Hb–Au NCs}3 in PBS (0.10 M, pH 7.0) within the TCA concentration range 1.00 × 10−11 (curve a) to 8.10 × 10−10 M (curve l) at a scan rate of 100 mV s−1. Inset: plot of current vs. square root of TCA concentration. | |
When different concentrations of TCA were added to pH 7.0 PBS under a nitrogen atmosphere, an obvious reduction peak at −0.340 V was observed. The catalytic reduction peak current increased linearly with the square root of TCA concentration in the solution. The linear regression equation was IA = 2.7279 × 10−8 + 1.32843 × 10−4CTCA1/2, M (R2 = 0.9993, n = 8). The linear relationship was obtained in the concentration range 5.00 × 10−11 to 8.10 × 10−6 M with an LOD of 3.07 × 10−11 M. The bioelectrocatalytic reduction mechanism of TCA by the sensor can be expressed as:31
| | |
2HbhemeFe2+ + CCl3COOH + H+ + e → 2HbhemeFe3+ + Cl2HCCOOH + Cl−
| (5) |
GCE-CS–{Au NPs/Hb–Au NCs}3 was then studied as a biosensor for O2. By continuously injecting an amount of pure O2 into thoroughly-deoxygenated PBS (5.0 mL, pH 7.0) under a nitrogen atmosphere, the electrocatalytic current at about −0.332 V increased with increasing O2 concentration, indicating that O2 was reduced by GCE-CS–{Au NPs/Hb–Au NCs}3. The peak current increased linearly with increasing oxygen concentration, as shown in Fig. 5. The biosensor exhibited an excellent oxygen sensing ability within the range 9.28 × 10−6 to 6.96 × 10−4 M (a–m). The linear equation was IA = 5.57711 × 10−8 + 1.06592 × 10−4CO2, M (R2 = 0.9995, n = 8), and the LOD was calculated as 3.10 × 10−6 M (S/N = 3). The bioelectrocatalytic reduction mechanism of O2 by the modified biosensor can be expressed by the following equation:25
| | |
2HbhemeFe2+ + 4H+ + O2 + 2e → 2HbhemeFe3+ + 2H2O
| (6) |
 |
| | Fig. 5 DPV signals measured at room temperature (scan potential, −0.9 to 0.3 V; pulse amplitude, 0.05 V; pulse width, 0.1 s, sensitivity (A/V), 1 × 10−6) for GCE-CS–{Au NPs/Hb–Au NCs}3 in PBS (0.10 M, pH 7.0) within O2 concentration range 9.28 × 10−6 (curve a) to 6.96 × 10−4 M (curve m) at a scan rate of 100 mV s−1. Inset: plot of current vs. O2 concentration. | |
Table 1 shows the determination of H2O2, TCA, and O2 by different sensors. The results show that the linear range and detection limits achieved in this work were better than those in some previous reports.
Table 1 Comparison of detection and quantification limits obtained in this work with those reported in the literature
| Analyte |
Sensor |
Line range |
LOD |
Reference |
| O2 |
Red blood cell on Au–NP/GCE |
2.0 × 10−3 to 8.0 × 10−3 M |
2.0 × 10−3 M |
32 |
| TiO2 and Nafion film modified GCE |
0.3–10 mg L−1 |
0.12 mg L−1 |
33 |
| GCE/nano Pd-multiwalled carbon nanotubes |
0.8 × 10−4 to 0.9 × 10−3 M |
0.2 × 10−4 M |
4 |
| GCE-CS–{Au NPs/Hb–Au NCs}3 |
9.28 × 10−6 to 6.96 × 10−4 M |
3.1 × 10−6 M |
This work |
| H2O2 |
Hb on Au NPs |
3.6 × 10−7 to 8.6 × 10−3 M |
1.2 × 10−7 M |
4 |
| Hb on Fe3O4@Pt NPs |
2.8 × 10−6 to 1.4 × 10−3 M |
4.8 × 10−8 M |
34 |
| Hb on Fe3O4@CS NPs |
5.0 × 10−6 to 9.0 × 10−4 M |
5 × 10−7 M |
35 |
| GCE-CS–{Au NPs/Hb–Au NCs}3 |
9.70 × 10−9 to 2.14 × 10−5 M |
3.55 × 10−9 M |
This work |
| TCA |
Horseradish peroxidase on gemini surfactant–polyvinyl alcohol composite film |
2.0 × 10−5 to 1.0 × 10−2 M |
5.7 × 10−3 M |
36 |
| Fe3O4@Au–CS modified GCE |
1.60 × 10−6 to 4.80 × 10−3 M |
1.0 × 10−6 M |
25 |
| Hb on multi-walled carbon nanotubes |
5.0 × 10−4 to 1.2 × 10−2 M |
1.0 × 10−4 M |
37 |
| GCE-CS–{Au NPs/Hb–Au NCs}3 |
5.0 × 10−11 to 8.1 × 10−6 M |
3.07 × 10−11 M |
This work |
3.4. Application of GCE-CS–{Au NPs/Hb–Au NCs}3 to real sample analysis
H2O2, applied in food anticorrosion, and is widely used in food production, processing, and packaging materials to kill harmful microbes and improve the storage period. H2O2 reportedly accelerates the aging and cancer by damaging cells. Some foods contain illegally added H2O2.38 The results of detecting H2O2 in milk powder are listed in Table 2, which clearly showed that the consequences of the proposed biosensor and the Chinese national standard were similar, indicating the excellent reliability of the biosensor. The recovery experiment concentration of TCA and O2 was carried out in tap water as a real sample. The recoveries of the biosensor for TCA and O2 were calculated to be 96.29–104.97% and 99.27–103.24%, respectively. The results showed that the detection method for H2O2, O2, and TCA presented in this work can be valid for analysis of real samples.
Table 2 Analytical application of GCE-CS–{Au NPs/Hb–Au NCs}3 to real samples
| |
DPV at the GCE-CS–{Au NPs/Hb–Au NCs}3 |
Standard method |
| Amount added |
Amount found |
Average recovery (%) |
RSD% (n = 3) |
Averaged value |
RSD% (n = 3) |
| Colorimetry. Chromatography. Iodine titrimetry. |
| TCA |
8.6 × 10−11 |
8.28 × 10−11 |
96.29 |
6.22 |
8.70 × 10−11a |
3.28 |
| 1.86 × 10−10 |
1.95 × 10−10 |
104.97 |
4.34 |
1.79 × 10−10a |
4.22 |
| 5.9 × 10−9 |
5.71 × 10−9 |
96.84 |
4.56 |
6.00 × 10−10a |
4.55 |
| O2 |
0 |
9.74 × 10−5 |
N |
5.77 |
9.23 × 10−5b |
2.55 |
| 3.62 × 10−4 |
3.74 × 10−4 |
103.24 |
6.23 |
3.61 × 10−4b |
2.10 |
| 6.03 × 10−4 |
5.99 × 10−4 |
99.27 |
4.35 |
6.01 × 10−4b |
1.99 |
| H2O2 |
8 × 10−10 |
7.76 × 10−10 |
97.01 |
3.36 |
7.99 × 10−10c |
5.87 |
| 3.4 × 10−9 |
3.28 × 10−9 |
96.36 |
4.12 |
3.23 × 10−9c |
6.10 |
| 1.31 × 10−8 |
1.33 × 10−8 |
101.45 |
4.23 |
1.33 × 10−8c |
5.91 |
4. Conclusions
We have successfully constructed a biosensor based on Hb–Au NCs using a layer-by-layer technique. Immobilized Hb on the electrode showed well-maintained bioactivity and enhanced electron transfer kinetics. The prepared biosensor achieved rapid and sensitive electrochemical responses to H2O2, TCA, and O2 without the addition of electron mediators. This strategy could potentially become a general and promising alternative to other bioactive molecules in various biosensor designs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant numbers: 21475070; 21175075; 21375066 and 81202249), the Natural Science Foundation of Jiangsu Province (Grant number: BK2011047, BK20151267), a Project Funded by “Six Kinds of Talents” in Jiangsu Province (2015-YY-008), a project funded by “College Students' Innovative Training Program” in Jiangsu Province (201610304035Z), and a project funded by China Environmental Protection Foundation.
Notes and references
- K. Charradi, C. Gondran, A. B. H. Amara, V. Prevot and C. Mousty, Electrochim. Acta, 2009, 54, 4237–4244 CrossRef CAS.
- N. Q. Jia, Q. Lian, Z. Y. Wang and H. B. Shen, Sens. Actuators, B, 2009, 137, 230–234 CrossRef.
- Y. Liu, Q. Y. Jiang, S. Y. Lu, Y. Zhang and H. Y. Gu, Appl. Biochem. Biotechnol., 2009, 152, 418–427 CrossRef CAS PubMed.
- H. Y. Gu, A. M. Yu and H. Y. Chen, J. Electroanal. Chem., 2001, 516, 119–126 CrossRef CAS.
- S. M. Chen and C. C. Tseng, Electrochim. Acta, 2004, 49, 1903–1914 CrossRef CAS.
- Y. S. Wang, R. Guo and J. Q. Xi, J. Colloid Interface Sci., 2009, 331, 470–475 CrossRef CAS PubMed.
- O. Y. Nadzhafova, V. N. Zaitsev, M. V. Drozdova, A. Vaze and J. F. Rusling, Electrochem. Commun., 2004, 6, 205–209 CrossRef CAS.
- J. M. Xu, W. Li, Q. F. Yin, H. Zhong, Y. L. Zhu and L. T. Jin, J. Colloid Interface Sci., 2007, 315, 170–176 CrossRef CAS PubMed.
- C. L. Xiang, Y. J. Zou, L. X. Sun and F. Xu, Sens. Actuators, B, 2009, 136, 158–162 CrossRef CAS.
- J. M. Gong and X. G. Lin, Microchem. J., 2003, 75, 51–57 CrossRef CAS.
- Y. Hu, H. Sun and N. F. Hu, J. Colloid Interface Sci., 2007, 314, 131–140 CrossRef CAS PubMed.
- I. Taurino, G. Sanzò, R. Antiochia, C. Tortolini, F. Mazzei, G. Favero, G. D. Micheli and S. Carrara, Anal. Chem., 2016, 79, 151–159 CAS.
- A. K. M. Kafi, A. Ahmadalinezhad, J. P. Wang, D. F. Thomas and A. C. Chen, Biosens. Bioelectron., 2010, 25, 2458–2463 CrossRef CAS PubMed.
- Y. Wang, X. L. Ma, Y. Wen, G. P. Duan, W. Ren, Z. R. Zhang and H. F. Yang, Microchim. Acta, 2009, 166, 283–288 CrossRef CAS.
- M. A. Rahman, H. B. Noh and Y. B. Shim, Anal. Chem., 2008, 80, 8020–8027 CrossRef CAS PubMed.
- Y. Li, J. Li, X. H. Xia and S. Q. Liu, Talanta, 2010, 82, 1164–1169 CrossRef CAS PubMed.
- M. C. Prieto-Blanco, M. F. Alpendurada, P. López-Mahía, S. Muniategui-Lorenzo, D. Prada-Rodríguez, S. Machado and C. Gonçalves, Talanta, 2012, 94, 90–98 CrossRef CAS PubMed.
- M. J. Cardador and M. Gallego, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 1824–1830 CrossRef CAS PubMed.
- P. S. Simone Jr, P. L. Ranaivo, G. Geme, M. A. Brown and G. L. Emmert, Anal. Chim. Acta, 2009, 654, 133–140 CrossRef PubMed.
- C. M. Yu, L. Wang, W. B. Li, C. Zhu, N. Bao and H. Y. Gu, Sens. Actuators, B, 2015, 211, 17–24 CrossRef CAS.
- Z. Q. Pan, C. G. Shi, H. Fan, N. Bao, C. M. Yu, Y. Liu, R. Lu, Q. H. Zhang and H. Y. Gu, Sens. Actuators, B, 2012, 174, 421–426 CrossRef CAS.
- M. J. Song, S. W. Hwang and D. Whang, Talanta, 2010, 80, 1648–1652 CrossRef CAS PubMed.
- L. G. Yu, G. M. Zhang, Y. Wu, X. Bai and D. Z. Guo, J. Cryst. Growth, 2008, 310, 3125–3130 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Chichester Brisbane Toronto, New York, 1980, ch. 9 Search PubMed.
- Y. Liu, T. Han, C. Chen, N. Bao, C. M. Yu and H. Y. Gu, Electrochim. Acta, 2011, 56, 3238–3247 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS.
- W. Lu, J. Ge, L. Tao, X. Cao, J. Dong and W. Qian, Electrochim. Acta, 2014, 130, 335–343 CrossRef CAS.
- M. K. Bojdi, M. H. Mashhadizadeh, M. Behbahani, A. Farahani, S. S. H. Davarani and A. Bagheri, Electrochim. Acta, 2014, 136, 59–65 CrossRef CAS.
- A. E. F. Nassar, Z. Zhang, V. Chynwat, H. A. Frank and J. F. Rusling, J. Phys. Chem., 1995, 99, 11013–11017 CrossRef CAS.
- J. Dojlido, E. Zbieć and R. Ŝwietlik, Water Res., 1999, 33, 3111–3118 CrossRef CAS.
- J. W. Shie, U. Yogeswaran and S. M. Chen, Talanta, 2009, 78, 896–902 CrossRef CAS PubMed.
- X. D. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666–3761 RSC.
- Z. Lin, Y. Liu and G. Chen, Electrochem. Commun., 2008, 10, 1629–1632 CrossRef CAS.
- H. Fan, Z. Q. Pan and H. Y. Gu, Microchim. Acta, 2010, 168, 239 CrossRef CAS.
- X. C. Tan, J. L. Zhang, S. W. Tan, D. D. Zhao, Z. W. Huang, Y. Mi and Z. Y. Huang, Sensors, 2009, 9, 6185 CrossRef CAS PubMed.
- W. Sun, X. Q. Li, Y. Wang, X. Li, C. Zhao and K. Jiao, Bioelectrochemistry, 2009, 75, 170 CrossRef CAS PubMed.
- K. Charradi, C. Gondran, A. Amara, V. Prevot and C. Mousty, Electrochim. Acta, 2009, 54, 4237 CrossRef CAS.
- H. Sies, Redox Biol., 2017, 11, 613–619 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05808j |
| ‡ Both authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Haiying Gu
* and
Haiying Gu