
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Generation of reactive cobalt oxo oxamate radical species for biomimetic oxidation of contaminants†
Nan Li *,
Yun Zheng,
Xuemei Jiang,
Ran Zhang and
Wenxing Chen*
*,
Yun Zheng,
Xuemei Jiang,
Ran Zhang and
Wenxing Chen*
National Engineering Lab for Textile Fiber Materials & Processing Technology (Zhejiang), Zhejiang Sci-Tech University, Hangzhou 310018, China. E-mail: linan@zstu.edu.cn; wxchen@zstu.edu.cn
First published on 5th September 2017
Abstract
The non-heme oxamate anionic cobalt(III) complexes [CoIII(opbaX)]− (opbaX = 4-X-o-phenylenebis(oxamate), X = H, NO2, CH3) with different substituents were synthesized and applied to targeted micropollutant degradation. Typical radical scavengers (isopropanol and chlorine anions) have no negative effect on the catalytic oxidation of substrates, and no DMPO–˙OH or DMPO–˙OOH (DMPO = 5,5-dimethyl-pyrroline-oxide) signal was detected by electron paramagnetic resonance spin-trap technique in [CoIII(opbaX)]−/H2O2 system, suggesting that the non-hydroxyl radical biomimetic catalytic mechanism was dominant in the oxidation process. The results of high-definition ESI-MS pronounced the presence of cobalt-oxo intermediates which played a key role in the catalytic oxidation of substrates. Furthermore, density functional theory calculations were used to evaluate the viability of such cobalt-oxo species and it demonstrated an optimizing electromer with a formulation of [CoIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O˙]− or [CoIII–OH]˙. The calculations explained that the catalytic activity of [CoIII(opbaX)]− was significantly enhanced by introducing an electron-withdrawing substituent which could change the coordination environment of cobalt to generate more electron-deficient cobalt-oxo species with stronger oxidizing power. This paper made a progress in verifying bio-mimic high oxidation state species and applied it to water purification, which provided deep insight into the properties of transition-metal centers in aqueous catalysis.
O˙]− or [CoIII–OH]˙. The calculations explained that the catalytic activity of [CoIII(opbaX)]− was significantly enhanced by introducing an electron-withdrawing substituent which could change the coordination environment of cobalt to generate more electron-deficient cobalt-oxo species with stronger oxidizing power. This paper made a progress in verifying bio-mimic high oxidation state species and applied it to water purification, which provided deep insight into the properties of transition-metal centers in aqueous catalysis.
Introduction
The widespread sewage containing a mass of industrial and natural chemical substances has been one of the most serious international distribution problems afflicting human health and aquatic ecosystems.1,2 Although most contaminants are present at trace concentrations, a number of them pose considerable toxicological hazards particularly when present as components of complex mixtures.3,4 Hence, the development of green and cost-effective processes aiming to eliminate critical pollutants, coupled with the search for ecosystem benign products, is a great challenge for environmental researchers.5 In recent years, many advanced oxidation technologies such as Fenton or Fenton-like systems,6,7 TiO2/H2O2 system,8 and photocatalytic processes,9,10 have been developed for this purpose. However, these predominantly catalytic mechanisms are mainly dominated by the hydroxyl radical (˙OH) process, which is inhibited due to its unselectivity in the presence of some inorganic or/and organic background constituents.11,12 Hence, it's necessary to look for efficient and sustainable catalysts with specificity.In nature, bioenzymes like cytochrome P450 and horseradish peroxidase (HRP) utilizes O2 or H2O2 to convert recalcitrant compounds under mild conditions with unexpected catalytic activities.13–16 However, these enzymes generate only in organisms and are perishable in practical application. So some researchers begin to distract their focus onto establishing artificial enzymes to imitate the essential and general principles of natural enzymes using stable and low-cost materials for a wide range of applications. It has been found that bioenzymes' selective oxidation of substrates attributes to the limited oxidation potential of high-valent metal-oxo active species.17 To date, Fe, Mn, Co, etc., metal complexes have been synthesized for bio-mimic targeted redoxes.18–20 For example, iron(III) porphyrin complexes can epoxide cyclohexene in protic solvent;21 and cobalt(III) complexes of bis-N,N′-disubstituted oxamides are able to catalyze secondary alcohols to the corresponding ketones in acetonitrile and to epoxide olefin in fluorobenzene.22,23 However, successful applications of these selective catalysts to the global issue of serious water pollution have been seldom reported.
As known, P450 and HRP both work with the same FeIVO active center in aqueous, while their different oxidant activation power may mainly attributed to the different electron nature of center Fe which is directly induced by the electronegativity of the fifth ligands and substituents on macrocyclic ligand.15,24–26 Generally, the generation of high-valent metal-oxo active species is determined by two competitive processes, heterolytic and homolytic cleavage of the O–O bond.21,27 Based on this theory, the transformed O–O bond cleavage has been achieved in the catalytic system with cobalt phthalocyanine through introducing axial fifth ligands in our previous studies, and the results indicated that the introduction of the fifth ligand changed the local environment around the cobalt cation and hence transformed the cleavage of peroxide O–O from homolysis to heterolysis.27,28 Here, we investigated the cleavage tendency of O–O bond to see if it was possible to change the catalyst's oxidation power by introducing various substituents on benzene ring of Co macrocyclic ligand. Besides, few researchers have presented direct evidence proving the existence of cobalt-oxo active species, so it's necessary to focus on the depiction of the functioning active intermediates.
In this study, we synthesized square-planar phenylenebis(oxamate) (opba) cobalt(III) complexes with related ligand derivatives (Scheme 1) and emphasized H2O2 activation mechanism through analysing the possible electron transport and substituents' effects on the catalysis. The effect of environmental factors such as pH, temperature, initial H2O2 carried out under mild conditions. The results of high-definition ESI-MS pronounced the presence of cobalt-oxo intermediates furthermore, the density functional theory calculations were used to evaluate the viability of such cobalt-oxo species and it demonstrated an optimizing electromer with a formulation of [CoIVO˙]− or [CoIII–OH]˙. In conclusion, the biologically inspired subject addressed in this work illuminated cobalt-oxo active species in water and broadened the designing strategy for synthesizing novel bioenzyme alternatives for wider applications.
 | ||
| Scheme 1 The [CoIII(opbaX)]− activators used in this work. | ||
Results and discussions
Characterization of cobalt complexes
The square-planar cobalt(III) complexes anions, [CoIII(opbaX)]−, were synthesized from the reaction between a Co(II) salt and the proligands in methanol solution by means of oxidizing the corresponding cobalt(II) complexes with dioxygen in situ. The solution state structure [CoIII(opbaX)]− and the oxidation state of Co was identified by Synapt G2-S HDMS (ESI) and XPS. The accurate masses of anions [CoIII(opbaX)]− and fragment ions were shown in Q-TOF MS and MS/MS spectra. In Q-TOF MS, this complex showed an ion at m/z 306.9402 (Fig. 1a), the mass was within 0.1 mDa deviating the theoretical mass (m/z 306.9401) for cobaltic complex anion [CoIII(opba)]− rather than cobaltous complex dianion ([CoII(opba)]2−, m/z 153.4701). Furthermore, the typical fragment ion masses were observed (see Fig. 1b and Table S1 for ESI†), which matched the theoretical masses within 0.5 mDa error. Likewise, the accurate masses of methyl and nitro substituent cobalt complex anions and fragment ions conformed to the expectation of calculated value (Tables S2, S3 and Fig. S1, S2 see ESI†). | ||
| Fig. 1 Synapt G2-S HDMS (ESI) in negative mode of complex 1 in MeCN aqueous solution for (a) Q-TOF MS and (b) MS/MS. | ||
Moreover, the oxidation state of Co in the complex is supported by XPS signals of the complex. The spin–orbit splitting of the Co2p level (ΔE) of complex 1 is shown in Fig. S3,† in which both the binding energy level splitting of Co2p1/2–Co2p3/2 (ΔE = 14.31 eV) and two weak satellite peaks demonstrated the generation of oxidation state 3+ of Co in the surface region.29,30 We guess that the strong electron-donating property of the N2–O2 coordination environment contributed to the generation of CoIII.31
To further prove the chemical structure of our catalysts, 1H NMR (D2O) spectra was employed. As shown in Fig. S4,† the sharp singlet assigned at δ 2.23 ppm was attributed to the Ar–methyl protons of complex 3.32,33 In the spectra of complex 1, 2 and 3, the multiplets observed in the low field region were the characteristic of benzene ring protons, while chemical shift to higher and lower magnetic field are ascribed to the contribution of electron-withdrawing nitro and electron-donating methyl respectively. In conclusion, the result of LC-MS and 1H NMR clearly described the chemical structure of our catalysts.
Therefore, it's evident that the synthesized compounds correspond to cobaltic complex anions [CoIII(opbaX)]−.
Catalytic oxidation of C.I. acid red 1 (AR1) and p-chlorophenol
In order to comparatively evaluate the effect of substituent ligands on the catalytic degradation of organic pollutants, the azo dye AR1 and p-chlorophenol (two typical aromatic pollutants) were selected as model substrates. As shown in Fig. 2, AR1 is barely degraded in the presence of the cobalt complexes alone (curve a–c, Fig. 2) and is the same when H2O2 is present alone (curve d, Fig. 2). When both the cobalt complex and 2 mM H2O2 was presented, the concentration of AR1 declined rapidly with increasing reaction time (curve e–g, Fig. 2) with an degradation rate order of [CoIII(opbaNO2)]− (2) > [CoIII(opba)]− (1) > [CoIII(opbaCH3)]− (3). In addition, the catalytic elimination of p-chlorophenol was carried out, and the removal rate was around 48%, 57% and 2% in the first 10 min in the presence of complex 1, 2 and 3 respectively (Fig. S5†). Hence, we hypothesize a correlation between the catalytic activity and the electron donor capacity of ligands to account for the phenomenon: the electron-withdrawing nitro substituent promoted a higher effective positive charge at cobalt compared with electron-donating methyl substituent, which was analogous to the ligand effect on Fe(III)–TAML (tetraamido macrocycle ligand),34 which indicated that the presence of electron-withdrawing substituent facilitated the generation of active intermediates.21,35,36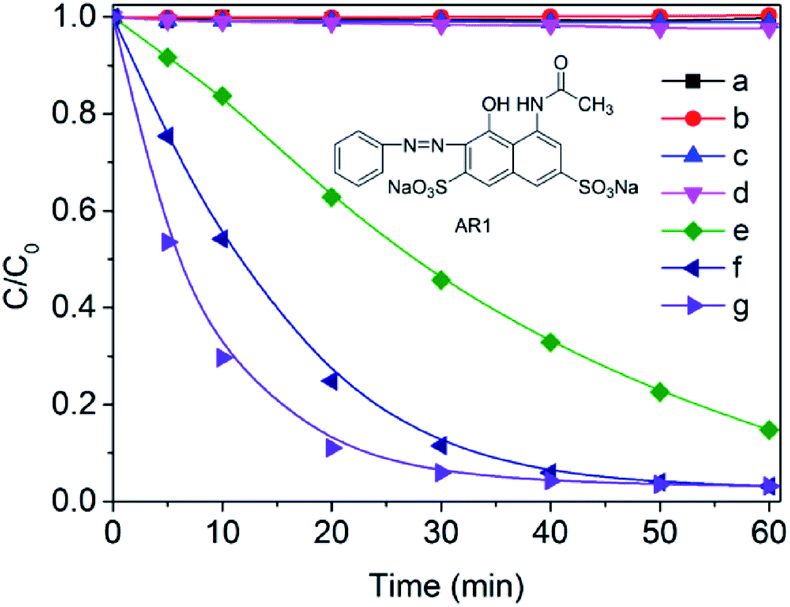 | ||
| Fig. 2 Bleaching of AR1 (0.05 mM) under different conditions: (a) complex 3, (b) complex 1 and (c) complex 2 without H2O2; (d) H2O2; (e) complex 3, (f) complex 1 and (g) complex 2 with H2O2; [H2O2] = 2 mM, [complexes 1–3] = 0.02 mM, T = 25 °C, pH 9.0. | ||
Effect of pH and temperature
The investigation of pH and temperature effect on 1-catalyzed deposition of AR1 activated by H2O2 is shown in Fig. 3. Complex 1 can efficiently activate H2O2 in the range of alkalinity (pH 8–11) which is consistent with the environmental conditions.37 Moreover, AR1 removal in initial 10 min increased from 37.3% to 83.6% as the temperature increased from 15 to 45 °C at pH 9.0 (Fig. 3a). An Ea value of 34.61 KJ mol−1 was obtained by plotting kobs versus 1/T (Fig. S6†). And the regression coefficient (R2) was close to 0.95, which indicated that the degradation kinetics of AR1 decomposed by 1/H2O2 followed pseudo first-order reaction kinetics well in the initial reaction stage where the concentration of H2O2 was enough.38 So we concluded that the decomposition of AR1 has less dependence on temperature from 15 to 45 °C because of the relatively small value of Ea which could be easily achieved in comparison with ordinary thermal reactions.39 | ||
| Fig. 3 (a) Effect of the temperature on catalyst oxidation of AR1 (0.05 mM) at pH 9.0; (b) effect of pH on the apparent rate constant kobs for the oxidation of AR1 by H2O2 catalyzed by 1 at different temperatures. The inset shows pH profiles of the removal rates of AR1 in the presence of 1 and H2O2 after reaction for 60 min. | ||
Furthermore, the pseudo-first-order rate constants (kobs) increased at the beginning and then decreased with the increase of pH values, finally the maximum rates occur at pH 9.0–9.5 (Fig. 3b). Hence, the result showed that the catalytic reaction kinetics were affected significantly by pH, which is interpreted as follows: the hydrolysis of the complex somewhat occurs and the dissociation of H2O2 was slower when pH moved toward neutral, which caused the limited coordination between HOO− and the central cobalt ions because the formation of active species was restrained. And when the pH of the solution was high, HOO−, the conjugate anion of H2O2, reacted with a non-dissociated molecule of H2O2 and ultimately resulted in the generation of oxygen and water.40,41
Effect of H2O2 concentration
The relationship between the degradation rate of AR1 and initial H2O2 concentrations were investigated and shown in Fig. S7,† which indicates that the AR1 can be effectively removed even at a very low concentration of H2O2 (0.5 mM), and even more than 90% of AR1 can be removed at such a low concentration of H2O2 in 1 hour. So this reaction system could significantly reduce the disproportionate amount of H2O2 that presented in some ˙OH-involving reaction systems, because in high backgrounds of complicated background, ˙OH inevitably react with easily handled constituents or the free coordination catalysts in homogeneous system.34,42Sustaining catalytic ability of the complexes
One of the most significant distinguishes between bio-enzymes and artificial catalyst is stability. To checkout if the catalysts were durable in water purification, representative 1 was examined by subsequent additions of dye and H2O2 at pH 9.0. As seen in Fig. 4, the removal of AR1 after 60 min was about 96% by the end of the first cycle, after eight cycles with repeated additions of the same amount of original dye and 1/3 times as much as the original amount of H2O2, the removal rate of AR1 was still maintained at more than 90%, which suggested that there existed no suicidal inactivation in our reaction system. | ||
| Fig. 4 Cyclic catalyst oxidation of AR1 (0.05 mM), [complex 1] = 0.02 mM, [H2O2] = 2 mM, T = 25 °C, pH 9.0. | ||
Detection of active species
As the oxidation mechanism of organic pollutants with H2O2 catalyzed by [CoIII(opbaX)]− in aqueous solution has not been previously reported, it is necessary to present clear insights into the function mechanisms of these catalysts. As we know, NaCl is a prevailing accelerant widely used in the dye industry and plays a vital role in the dyeing process for the transfer of dye stuff to fabric. However, various studies reported the retardation effect of NaCl on degradation of pollutants especially on the ˙OH mechanism.43,44 Hence, it is important to evaluate the impact of inorganic salts on the catalytic oxidation systems. As expected, the degradation efficiency of 1 displayed no significant change in the presence of NaCl compared with the control experiment (Fig. 5). This meant that this catalytic system has potential and practical application value in treating salt-containing wastewater and that there were no free ˙OH generated in this system. | ||
| Fig. 5 Effect of NaCl on catalytic oxidation of AR1 (0.05 mM) in the presence of 1 (0.02 mM) and H2O2 (2 mM), T = 25 °C, pH 9.0. Error bars represent the standard error from three independent experiments. | ||
In order to investigate the intermediate active species which generate based on the cleavage of O–O bond in hydrogen peroxide, isopropanol (IPA), a typical ˙OH scavenger,45,46 was employed to determine whether ˙OH dominated the reactions or not. As a represent, the catalysis of 1 in the presence of IPA was carried out. As shown in Fig. 6a, the degradation rate of 1 presented in IPA (its concentration was 5000 times as 1) was significantly in good agreement with the control experiment, which favored the non-˙OH mechanism. Moreover, EPR spin-trap technique (using DMPO (DMPO = 5,5-dimethyl-pyrroline-oxide) as the trapping reagent) was employed to detect the possibility that free radicals were produced in the reactions described above.47 As a result shown in Fig. 6b, no DMPO–˙OH signal or other free radicals showed up, indicating that ˙OH did not form in the catalytic reaction, which supported the positive results of isopropanol and NaCl experiments. An additional experiment conducted in methanol solution was conducted to probe whether there were any superoxide radicals (˙OOH) generated during the reaction process. As shown in Fig. 6c, no DMPO–˙OOH species were observed in 1/H2O2 system either, suggesting the catalytic mechanism belongs to non-hydroxyl radical pathway.
 | ||
| Fig. 6 (a) Concentration changes of AR1 (0.05 mM) in the presence of complexes 1–3 with or without isopropanol. Error bars represent the standard error from three independent experiments; (b) DMPO spin-trapping EPR spectra in AR1 (0.05 mM) aqueous solution in the presence of complex 1 and H2O2 (2 mM) with DMPO (20 mM); (c) DMPO spin-trapping EPR spectra in AR1 (0.05 mM) methanol solution in the presence of complex 1 and H2O2 (2 mM) with DMPO (20 mM). | ||
To obtain the evidence for the reactive intermediates, ESI-MS with a soft MS ionization technique, was employed for direct studies of catalytic intermediates, especially for the transient intermediates.48,49 The unsubstituted complex 1 was chosen for investigation and reacting with 2 equivalent m-chloroperbenzoic acid (mCPBA) playing as oxidant in the presence of trace NH3·H2O at −40 °C in CH3CN. The prominent ions at a mass/charge (m/z) ratio of 322.9379, as shown in Fig. 7, with a mass corresponding exactly to the formulation of [Co(O)(opba)]− within 2.8 mDa mass error. Hence, the electromer of high-valent cobalt-oxo ([CoIVO]− or [CoVO]−) and cobalt(III)-oxo ([CoIII–OH]) was presumed considering dehydrogenation of MS in the negative ion mode.
 | ||
| Fig. 7 (a) High-definition electrospray mass spectra of complex 1; (b) in the presence of 2 equivalents of mCPBA. | ||
Illuminating the valence state of the activated species is imperative for the study of electron transmission pathway. DFT calculations of Gaussian 09 program with functional B3LYP and basis set 6-31G method is potent.50 Since the related intermediate spin d6 square-planar cobalt(III) complexes have previously been reported, the electron spin density distributions were calculated with S = 0, S = 1, S = 2.51
Firstly we calculated high-valent models ([CoIVO]− or [CoVO]−) and chose the triplet (S = 1) spin ground state because it possessed the lowest energy of the optimized structures (Table S4†) in which we calculated the intermediates by placing an oxygen atom near the optimized [CoIII(opbaX)]− (Tables S5 and S6†). According to the calculated results, we concluded that high-valent cobalt-oxo species formed with a CoO bond length order of [CoN(O)(opbaCH3)]− > [CoN(O)(opba)]− > [CoN(O)(opbaNO2)]− (Fig. 8, Tables S7–S9,† N means the valence state of Co). In addition, the electron density on Co was decreased by electron-withdrawing –NO2 and the electrophilicity of Co was increased,52 while the electron-donating –CH3 functioned inversely.
 | ||
| Fig. 8 Unpaired electron spin distribution and geometrically optimized structure of [CoIII(opba)]− (a, b), [Co(O)(opbaCH3)]− (c, d), [Co(O)(opba)]− (e, f), [Co(O)(opbaNO2)]− (g, h), Co(OH)(opbaCH3) (i, j), Co(OH)(opba) (k, l), Co(OH)(opbaNO2) (m, n) based on DFT calculation. Red, oxygen; blue, nitrogen; purple, cobalt. Selected bond lengths (Å) of Co–O and the deviation height (Å) of Co are given. | ||
Moreover, a large unpaired electron spin density (∼1, neatly 50% of the total) located around the oxo moiety of CoO, which meant there existed an unpaired electron on the oxo moiety (Tables S10–S12†). And the spin density on excited Co reduced to 1.24 compared with the data calculated for ground state [CoIII(opba)]− (∼1.8), which meant Co lost a d-electron for the coordination to O after the O–O bond cleaved heterolytically. As a result, the CoIII was oxidized to CoIV. So the active intermediate can be described as [CoIVO˙]− species, like the radical species of OFeIV–AcO˙ reported by Yong Wang et al.53 Furthermore, the N2O2-plane deviation of Co empowered O to accept electron via oxidizing the substrates, especially the [˙OCo(opbaNO2)]− with a nitro substituent which has the shortest N2O2-plane deviation of Co (0.4078 Å).
To understand the origin of these properties, we have calculated the Frontier molecular orbitals (FMOs). According to the FMO theory, electrophiles require lowest unoccupied orbitals (LUMO) with high molecular orbital compositions on a reacting atom to achieve segistration with the FMOs on the nucleophilic substrate.54–56 In our calculations (Fig. 9a), LUMO and LUMO+1 of catalysts are mainly formed by the contributions of the O and Co d orbitals, which means the Co–O antibonding orbitals are the key FMOs; the O is the reacting atom, which interacts with the FMOs of the nucleophilic reagent. Moreover, the catalyst with the electron-donating methyl give highest energy levels of LUMO and singly occupied MO (SOMO), and that with electron-withdrawing cyano group has the lowest energy levels. The electron-withdrawing effect decreases the energy of LUMO intensively and the SOMO–LUMO gap is enhanced, which indicates that 3 might be more susceptible to oxidants after substituted by electron-withdrawing substituent while the electron of SOMO in 1 and 3 is more difficult to be excited. Thus the electron vacancies make the CoO unit possess significant desire for electron and electrophilic attack to the azo bond or naphthalene structure of dyes.57 This principle manifested well in the relative degradation efficiency that seemed to depend on the electron nature of the substituents (Fig. 2 and S5†).
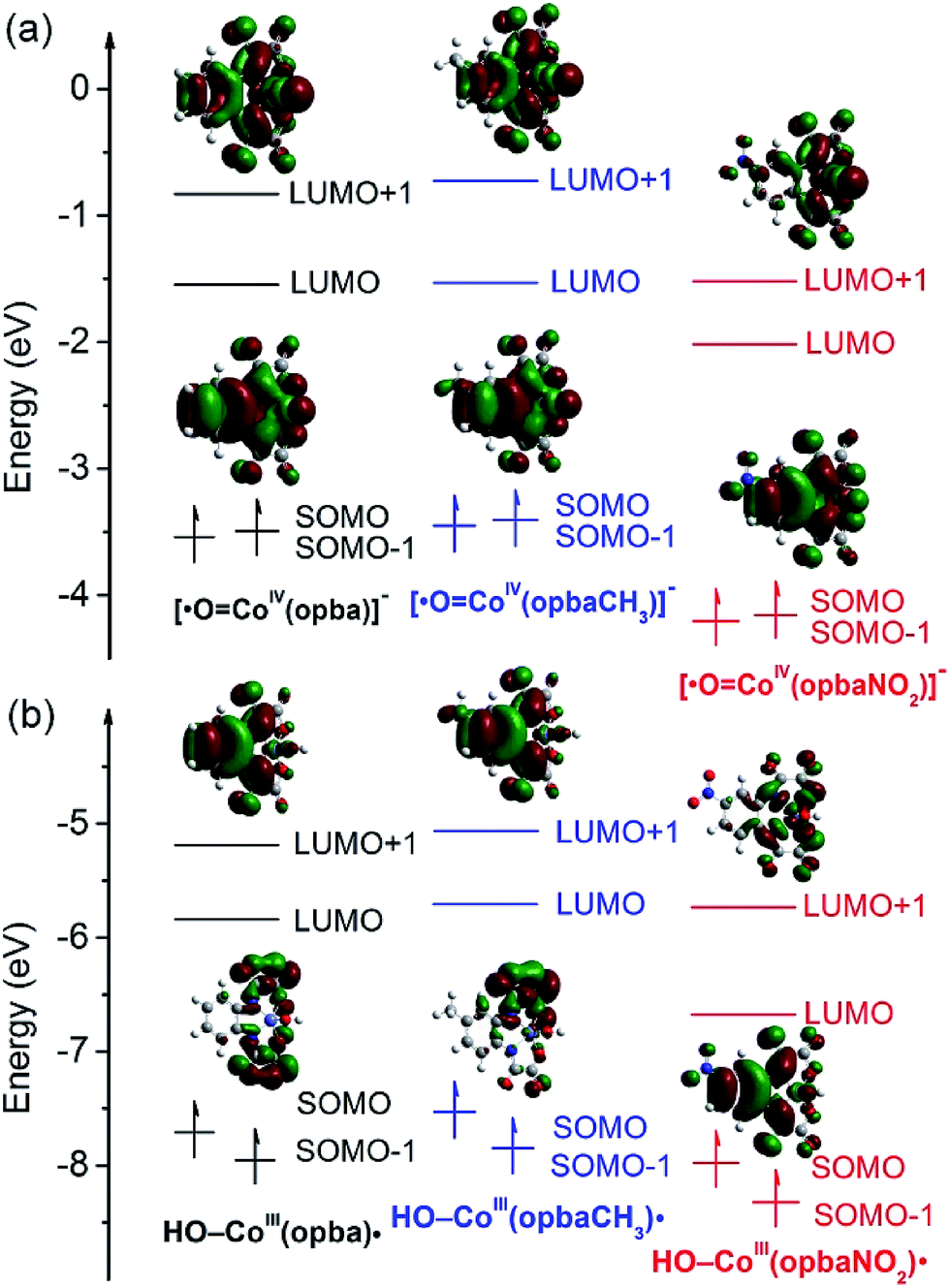 | ||
| Fig. 9 Frontier molecular orbitals energy level and molecular orbitals of [CoIVO˙]− and [CoIII–OH]˙. | ||
Secondly, the HO–CoIII(opbaX) patterns were also calculated by putting OH near to the optimized [CoIII(opbaX)]− in the same Gaussian 09 program (Fig. 9b). According to the total energy, we chose HO–CoIII(opba) and HO–CoIII(opbaCH3) models with S = 1, and HO–CoIII(opbaNO2) model with S = 2 (Table S13†). The different number of spinning electrons should ascribe to the extension effect of –NO2.58 The bond length between Co and O is around 1.8 Å, declaring the single bond between Co and O. Compared with high-valent models, these patterns possess smaller electron spin density distributed around the O atom (∼0.5), leaving near 1 spinning electron delocalized at the bis-benzoamido moiety (Tables S14–S16†) which manifested with the LUMO and SOMO orbitals (Fig. 9b). So the active intermediate can be depicted as [CoIII–OH]˙.59 Furthermore, the orders of Co–O bond length and orbital energy levels are the same as those of the high-valent models, which can also explain the oxidation power disparity of the three catalysts in contaminant removal. In conclusion, this catalytic system is proposed to proceed with either [CoIVO˙]− or [CoIII–OH]˙ reactive intermediates.
Accordingly, we depicted two generation pathway of reactive intermediates (Scheme 2). As known, the O–O bond of hydroperoxides cleaved either heterolytically or homolytically in aqueous, and the tendency of heterolysis was significantly affected by the electronic nature of oxamate complex.21,27 When coordinated with electron-withdrawing ligand, center Co was more affine to nucleophilic OOH moiety because of electron deficiency, which resulted to the rate acceleration in contaminant degradation.60,61 In pathway (a), electron-rich cobalt complexes showed a tendency to cleave the hydroperoxide O–O bond homolytically, whereas electron-deficient cobalt complexes show a tendency to cleave the heterolytically, in which the tendency was in the order of 2 > 1 > 3. Then the O–O cleavage followed due to the unstable structure of –CoIIIOOH2− with formations of OH− and CoIVO˙. Notably, this reactive radical was different from the results reported previously by Isabel Fernández et al., who proposed that oxamide cobalt complex played a key role in acetonitrile as CoIVO structure.22 Considering the Gray's “oxo-wall”,62 the pathway (b) is less exotic, where Co–O2H2 broke the O–O bond heterolytically and lost OH−, leaving electron deficient OH moiety.63 According to the electron spin density distribution result of HO–CoIII(opbaX), O atom obtained additional 0.5 spinning electron from Co, while the spin of Co did not essentially changed. This meant the bis-benzoamido moiety transferred electron to Co and Co transferred it to O, thus forming the ligand-based radical intermediate HO–CoIII(opbaX)˙. Electron-withdrawing substituent facilitated the O–O bond cleavage, which speeded up the formation of reactive intermediates and the degradation efficiency of pollutants.
 | ||
| Scheme 2 Proposed pathway and formation of cobalt-oxo electromer (L = bis-benzoamido). | ||
Experimental
Materials and reagents
The spin-trapping reagent, 5,5-dimethyl-pyrroline-oxide, (DMPO) was obtained from Tokyo Chemical Industry Co., Ltd. H2O2 (9.7 M), m-chloroperbenzoic acid (mCPBA, 85%), dyes including AR1, p-chlorophenol, and other chemicals used in the syntheses were of analytical grade from commercial suppliers and used as received. Ultrapure water (Millipore Milli-Q Advantage A10 Ultrapure water system) was used throughout the experimental process. The diethyl ester derivatives, 4-nitro-o-phenylenebis (oxamic acid) (H2Et2opbaNO2) and 4-methyl-o-phenylenebis (oxamic acid) (H2Et2opbaCH3), were synthesized by the corresponding commercially available o-phenylenediamine derivatives and ethyloxalyl chloride in tetrahydrofuran by standard condensation techniques in a similar preparation process as the unsubstituted analogue o-phenylenebis (oxamic acid) (H2Et2opba).Preparation of catalysts
The cobalt(III) complexes 1–3 were synthesized according to the steps reported previously.22,64 The Et2H2opbaX (X = H, NO2, CH3) (5 mmol) has been dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 mixture (100 mL) of methanol and water, and 50 mL NaOH (21 mmol) aqueous solution was added. This resulting mixture was stirred at 70 °C for 30 min to facilitate the hydrolysis of the ethyl ester groups. After the temperature of the reaction solution fell to room temperature, 5.0 mmol Co(NO3)2 (50 mL) was added dropwise with vigorous stirring under an oxygen atmosphere for 1 h. The resulting mixture was filtered and concentrated by evaporation of the solvent to about 10 mL, and then cooling in the presence of ethanol to obtain the dark-green product of complex 1 (ca. 1.6 g, 80% yield) after drying under vacuum. Similarly, the brick-red complex 2 (ca. 0.6 g, 30% yield) and dark-brown complex 3 (ca. 0.5 g, 25% yield) were synthesized respectively except for employing ether in place of ethanol.
:9 mixture (100 mL) of methanol and water, and 50 mL NaOH (21 mmol) aqueous solution was added. This resulting mixture was stirred at 70 °C for 30 min to facilitate the hydrolysis of the ethyl ester groups. After the temperature of the reaction solution fell to room temperature, 5.0 mmol Co(NO3)2 (50 mL) was added dropwise with vigorous stirring under an oxygen atmosphere for 1 h. The resulting mixture was filtered and concentrated by evaporation of the solvent to about 10 mL, and then cooling in the presence of ethanol to obtain the dark-green product of complex 1 (ca. 1.6 g, 80% yield) after drying under vacuum. Similarly, the brick-red complex 2 (ca. 0.6 g, 30% yield) and dark-brown complex 3 (ca. 0.5 g, 25% yield) were synthesized respectively except for employing ether in place of ethanol.
Catalytic experiments and analysis
The typical experimental procedure for the catalytic oxidation of AR1 and p-chlorophenol was conducted with complexes 1–3 respectively and carried out in a 40 mL glass beaker with a magnetic stirrer. The reaction temperature was maintained at 25 °C or 45 °C by means of a constant temperature water bath. The initial concentration of AR1 or p-chlorophenol in 0.01 M borate buffer was 0.05 mM with 0.02 mM cobalt complex, and then a certain amount of H2O2 was added to the reaction solution. At given time intervals, a small amount of reaction mixture of AR1 was frequently taken out for observing the concentration changes of substrates, and the concentration of dyes was determined using an ultraviolet-visible (UV-vis) spectrometer (Hitachi U-3010). The concentration changes of p-chlorophenol were analyzed by ultra-performance liquid chromatography (UPLC, Acquity BEH C18 column (1.7 μm, 2.1 × 50 mm), Waters). The removal rate was calculated by eqn (1), where C is the concentration of AR1 or p-chlorophenol at each reaction time and C0 is the initial concentration of the substrate.| Residual rate = C/C0 | (1) |
Assuming that the initial organic degradation constant fits the data to a pseudo-first-order reaction kinetic model when H2O2 is in considerable excess, we obtained the apparent rate constant kobs calculated from the slop of the plot ln(C/C0) versus to time.
Characterizations
The structure and cobalt oxidation state of the cobalt complexes 1–3 in MeCN aqueous solution were characterized by high-definition mass spectrometry in Q-TOF mode, which were performed on a Synapt G2-S HDMS (Waters) in negative mode equipped with electrospray ionization (ESI) source. The capillary voltage and cone voltage were set at 2.5 kV and 40 V respectively, and the desolvation gas was set to 900 L h−1 at 80 °C. The energies of collision ramp were controlled at 20–40 V for the MS/MS fragmentation information. The MS data were acquired using the lock spray to ensure mass accuracy and reproducibility with leucine-enkephalin (m/z 554.2615) in negative electrospray ionization mode. X-ray photoelectron spectroscopy (XPS) data were recorded on a Thermo Scientific K-Alpha spectrometer (monochromatic Al Kα, 1486.6 eV), and the binding energy peaks of the XPS spectra were calibrated by placing the principal C1s binding energy peak at 284.7 eV. 1H NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer. D2O was used as solvent and internal standard (δH = 4.71). The EPR signals were detected on a Bruker A300 spectrometer for the free radicals trapped by DMPO at room temperature. The parameter setting typically as follows: center field, 3520 G; microwave frequency, 9.77 GHz; modulation frequency, 100 kHz. We attempted to detect the residual p-chlorophenol using UPLC chromatographic separation with a BEH C18 column (1.7 μm, 2.1 × 100 mm) using 30% mobile phases A (acetonitrile) and 70% B (formic acid/water solution with a volume ratio of 1/1000). The flow rate was 0.4 mL min−1, and the column oven temperature was set at 30 °C.Detection of cobalt-oxo intermediates
The active intermediates were prepared from 0.02 mM solution of complex 1 in acetonitrile with slight ultrapure water at −40 °C (the pH value was adjusted to 9.5 with ammonia water previously) using 2 equivalents of mCPBA (85%), and the solution was infused directly into ion source of high-definition mass spectrometry with a syringe manually. In addition, to determine active intermediates, the source temperature was set at 40 °C to minimize the decomposition of intermediate species.Conclusions
Nature has developed various metalloenzymes such as heme-containing horseradish peroxidase and cytochrome P450; they are capable of stubborn substrate transformations. So there has long been significant interest in modelling such enzyme active sites and developing biomimetic catalysts for various purposes. Based on the same FeIV active species yet with different catalytic ability, here we designed [CoIII(opbaX)]−/H2O2 system which exhibited excellent catalytic performance in oxidizing targeted pollutants such as dyes and p-chlorophenol via non-hydroxyl radical process pathway under high backgrounds of constituents. The electron-drawing substituents have been proved to positively promote the H2O2 activation while electron-donating substituents were proved negatively. The results of radical trapping experiments and DFT calculations described [CoIVO˙]− and [CoIII–OH]˙ intermediates, which played as the key species in this oxidation processes. Accordingly, electron-withdrawing substituent enhanced the oxidizing power of [CoIII(opbaX)]− due to the electron environment change on cobalt-oxo moiety. This contribution provided further evidence for the formation of high-valent cobalt-oxo radical species, which are akin to the oxoiron(IV) complex proposed as the actual oxidant in the families of P450 and HRP. Meanwhile, future studies need to give more direct evidence and illumination of [CoIII–OH]˙ and [CoIVO˙]− in catalysis.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51703201), and Zhejiang Provincial Natural Science Foundation of China (No. LQ17E030003).Notes and references
- M. Šimšíková, M. Bartoš, J. Čechal and T. Šikola, Catal. Sci. Technol., 2016, 6, 3008–3017 Search PubMed.
- D. Chatterjee, S. Rothbart and R. van Eldik, RSC Adv., 2013, 3, 3606 RSC.
- S. Bhattacharya, S. Bala and R. Mondal, RSC Adv., 2016, 6, 25149–25158 RSC.
- S. Sengupta and R. Mondal, J. Mater. Chem. A, 2014, 2, 16373–16377 CAS.
- L. Xie, L. Zhou, T. Liu and X. Xu, RSC Adv., 2016, 6, 106935–106944 RSC.
- A. Y. Satoh, J. E. Trosko and S. J. Masten, Environ. Sci. Technol., 2007, 41, 2881–2887 CrossRef CAS PubMed.
- W. Huang, M. Brigante, F. Wu, C. Mousty, K. Hanna and G. Mailhot, Environ. Sci. Technol., 2013, 47, 1952–1959 CrossRef CAS PubMed.
- X. Li, C. Chen and J. Zhao, Langmuir, 2001, 17, 4118–4122 CrossRef CAS.
- X. Zhou, J. Lan, G. Liu, K. Deng, Y. Yang, G. Nie, J. Yu and L. Zhi, Angew. Chem., 2012, 124, 182–186 CrossRef.
- N. Guo, Y. Cao, Y. Rong and D. Jia, RSC Adv., 2016, 6, 106046–106053 RSC.
- E. M. Siedlecka, A. Więckowska and P. Stepnowski, J. Hazard. Mater., 2007, 147, 497–502 CrossRef CAS PubMed.
- H. Kim, W. Kim, Y. Mackeyev, G.-S. Lee, H.-J. Kim, T. Tachikawa, S. Hong, S. Lee, J. Kim and L. J. Wilson, Environ. Sci. Technol., 2012, 46, 9606–9613 CrossRef CAS PubMed.
- S. G. Sligar, Science, 2010, 330, 924 CrossRef CAS PubMed.
- P. R. O. De Montellano, Cytochrome P450: Structure, Mechanism, and Biochemistry, Springer Science & Business Media, 2005 Search PubMed.
- N. C. Veitch, Phytochemistry, 2004, 65, 249–259 CrossRef CAS PubMed.
- B. L. Allen, G. P. Kotchey, Y. Chen, N. V. Yanamala, J. Klein-Seetharaman, V. E. Kagan and A. Star, J. Am. Chem. Soc., 2009, 131, 17194–17205 CrossRef CAS PubMed.
- J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed.
- J. Bennett, Y. A. Miah, D. S. Varsani, E. Salvadori and T. S. Sheriff, RSC Adv., 2016, 6, 103372–103381 RSC.
- T. S. Sheriff, M. Watkinson, M. Motevalli and J. F. Lesin, Dalton Trans., 2010, 39, 53–55 RSC.
- S. Y. Shaban and R. van Eldik, Dalton Trans., 2011, 40, 287–294 RSC.
- W. Nam, H. J. Han, S.-Y. Oh, Y. J. Lee, M.-H. Choi, S.-Y. Han, C. Kim, S. K. Woo and W. Shin, J. Am. Chem. Soc., 2000, 122, 8677–8684 CrossRef CAS.
- I. Fernández, J. R. Pedro, A. L. Roselló, R. Ruiz, I. Castro, X. Ottenwaelder and Y. Journaux, Eur. J. Org. Chem., 2001, 2001, 1235–1247 CrossRef.
- J. Estrada, I. Fernández, J. Pedro, X. Ottenwaelder, R. Ruiz and Y. Journaux, Tetrahedron Lett., 1997, 38, 2377–2380 CrossRef CAS.
- N. A. Stephenson and A. T. Bell, J. Mol. Catal. A: Chem., 2007, 275, 54–62 CrossRef CAS.
- K. Auclair, P. Moënne-Loccoz and P. R. Ortiz de Montellano, J. Am. Chem. Soc., 2001, 123, 4877–4885 CrossRef CAS PubMed.
- D. S. Salnikov, R. Silaghi-Dumitrescu, S. V. Makarov, R. van Eldik and G. R. Boss, Dalton Trans., 2011, 40, 9831–9834 RSC.
- N. Li, W. Lu, K. Pei, Y. Yao and W. Chen, Appl. Catal., B, 2015, 163, 105–112 CrossRef CAS.
- N. Li, W. Lu, K. Pei, Y. Yao and W. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 5869–5876 CAS.
- L. I. Rodionova, A. V. Smirnov, N. E. Borisova, V. N. Khrustalev, A. A. Moiseeva and W. Grünert, Inorg. Chim. Acta, 2012, 392, 221–228 CrossRef CAS.
- T. Ivanova, A. Naumkin, A. Sidorov, I. Eremenko and M. Kiskin, J. Electron Spectrosc. Relat. Phenom., 2007, 156, 200–203 CrossRef.
- A. Mederos, S. Domínguez, R. Hernández-Molina, J. n. Sanchiz and F. Brito, Coord. Chem. Rev., 1999, 193, 857–911 CrossRef.
- R. Ruiz, C. Surville-Barland, A. Aukauloo, E. Anxolabehere-Mallart, Y. Journaux, J. Cano and M. C. Muñoz, J. Chem. Soc., Dalton Trans., 1997, 745–752 RSC.
- S. K. Sharma and R. Gupta, Inorg. Chim. Acta, 2011, 376, 95–104 CrossRef CAS.
- W. C. Ellis, C. T. Tran, R. Roy, M. Rusten, A. Fischer, A. D. Ryabov, B. Blumberg and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 9774–9781 CrossRef CAS PubMed.
- T. G. Traylor, C. Kim, J. L. Richards, F. Xu and C. L. Perrin, J. Am. Chem. Soc., 1995, 117, 3468–3474 CrossRef CAS.
- S. Roeser, F. Bozoglian, C. J. Richmond, A. B. League, M. Z. Ertem, L. Francàs, P. Miró, J. Benet-Buchholz, C. J. Cramer and A. Llobet, Catal. Sci. Technol., 2016, 6, 5088–5101 CAS.
- S. Kundu, A. Chanda, J. V. Thompson, G. Diabes, S. K. Khetan, A. D. Ryabov and T. J. Collins, Catal. Sci. Technol., 2015, 5, 1775–1782 CAS.
- A. Anushree, S. Kumar and C. Sharma, Catal. Sci. Technol., 2016, 6, 2101–2111 Search PubMed.
- J. Chen and L. Zhu, Catal. Today, 2007, 126, 463–470 CrossRef CAS.
- W. Lu, N. Li, S. Bao, W. Chen and Y. Yao, Carbon, 2011, 49, 1699–1709 CrossRef CAS.
- M. Kasiri, H. Aleboyeh and A. Aleboyeh, Appl. Catal., B, 2008, 84, 9–15 CrossRef CAS.
- S.-P. Sun, C.-J. Li, J.-H. Sun, S.-H. Shi, M.-H. Fan and Q. Zhou, J. Hazard. Mater., 2009, 161, 1052–1057 CrossRef CAS PubMed.
- H. Ghodbane and O. Hamdaoui, Chem. Eng. J., 2010, 160, 226–231 CrossRef CAS.
- J. Kiwi, A. Lopez and V. Nadtochenko, Environ. Sci. Technol., 2000, 34, 2162–2168 CrossRef CAS.
- D. Lawless, N. Serpone and D. Meisel, J. Phys. Chem., 1991, 95, 5166–5170 CrossRef CAS.
- L. Wang, M. Cao, Z. Ai and L. Zhang, Environ. Sci. Technol., 2014, 48, 3354–3362 CrossRef CAS PubMed.
- J. Sheng, X. Li and Y. Xu, ACS Catal., 2014, 4, 732–737 CrossRef CAS.
- M. R. Bukowski, K. D. Koehntop, A. Stubna, E. L. Bominaar, J. A. Halfen, E. Münck, W. Nam and L. Que, Science, 2005, 310, 1000–1002 CrossRef CAS PubMed.
- F. T. De Oliveira, A. Chanda, D. Banerjee, X. Shan, S. Mondal, L. Que, E. L. Bominaar, E. Münck and T. J. Collins, Science, 2007, 315, 835–838 CrossRef PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- S. Ozaki, H. Mimura, N. Yasuhara, M. Masui, Y. Yamagata, K. Tomita and T. J. Collins, J. Chem. Soc., Perkin Trans. 2, 1990, 353–360 RSC.
- C.-G. Liu, Z.-M. Su, W. Guan and L.-K. Yan, Inorg. Chem., 2008, 48, 541–548 CrossRef PubMed.
- Y. Wang, D. Janardanan, D. Usharani, K. Han, L. Que Jr and S. Shaik, ACS Catal., 2013, 3, 1334–1341 CrossRef CAS.
- S. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- R. A. Eikey and M. M. Abu-Omar, Coord. Chem. Rev., 2003, 243, 83–124 CrossRef CAS.
- R. Arumugaperumal, V. Srinivasadesikan, M.-C. Lin, M. Shellaiah, T. Shukla and H.-C. Lin, RSC Adv., 2016, 6, 106631–106640 RSC.
- H. Zhao and F. o. P. Gabbaï, Organometallics, 2012, 31, 2327–2335 CrossRef CAS.
- R. Flores, A. E. Torres and S. Fomine, RSC Adv., 2016, 6, 64285–64296 RSC.
- G. I. Berglund, G. H. Carlsson, A. T. Smith, H. Szöke, A. Henriksen and J. Hajdu, Nature, 2002, 417, 463–468 CrossRef CAS PubMed.
- W. Y. Tcho, B. Wang, Y. M. Lee, K. B. Cho, J. Shearer and W. Nam, Dalton Trans., 2016, 45, 14511–14515 RSC.
- D. Chatterjee, S. Rothbart and R. van Eldik, Dalton Trans., 2013, 42, 4725–4729 RSC.
- J. R. Winkler and H. B. Gray, Struct. Bonding, 2012, 142, 17–28 CrossRef CAS.
- A. I. Nguyen, R. G. Hadt, E. I. Solomon and T. D. Tilley, Chem. Sci., 2014, 5, 2874–2878 RSC.
- H. O. Stumpf, Y. Pei, O. Kahn, J. Sletten and J. P. Renard, J. Am. Chem. Soc., 1993, 115, 6738–6745 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08317c |
| This journal is © The Royal Society of Chemistry 2017 |