
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ni(II) and Co(II) complexes of an asymmetrical aroylhydrazone: synthesis, molecular structures, DNA binding, protein interaction, radical scavenging and cytotoxic activity†
Yueqin Li *a,
Zhiwei Yanga,
Minya Zhoua,
Yun Lia,
Jing Hea,
Xuehong Wanga and
Zhengfeng Linb
*a,
Zhiwei Yanga,
Minya Zhoua,
Yun Lia,
Jing Hea,
Xuehong Wanga and
Zhengfeng Linb
aSchool of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, P. R. China. E-mail: yueqinli@163.com; Fax: +86-025-85427024; Tel: +86-025-85427024
bInspection and Quarantine Technology Centre, Hainan Entry-exit Inspection and Quarantine Bureau, Haikou 570311, P. R. China
First published on 25th August 2017
Abstract
Two new nickel(II) and cobalt(II) benzhydrazone complexes having the general formula [ML2phen·3CH3OH] (where M = Ni and Co, L = 2-acetonaphthonebenzoylhydrazone, phen = 1,10-phenanthroline) have been synthesized via the reaction of Ni(OAc)2·4H2O and CoCl2·6H2O with 2 equivalents of HL and 1 equivalent of phen ligands in a MeOH/THF mixed medium. Both complexes have been characterized by single-crystal X-ray crystallography methods, which reveals a distorted octahedral coordination environment around the metal center with an MN4O2 chromophore [M = Ni(II) and Co(II)], with the hydrazone ligand acting as a monoanionic bidentate N,O-donor. These complexes exhibit quasi-reversible one-electron reduction responses (NiII–NiI, CoII–CoI) within the E1/2 at −0.808 and −0.767 V versus Ag/AgCl reference. The DNA-binding interactions of the complexes with herring sperm DNA have been investigated by UV-vis absorption, emission and viscosity measurements, which reveal that the complexes could interact with DNA via intercalation. The protein binding interactions of the complexes with BSA were investigated by UV-vis, fluorescence and synchronous fluorescence methods, which indicate strong binding of the complexes with BSA and a static quenching mechanism was observed. The Ni(II) complex displays higher DNA and BSA-binding affinity than the corresponding Co(II) analogue, which is expected of its smaller size. Moreover, the potential for free-radical scavenging of all the complexes was also determined using DPPH, hydroxyl and nitric oxide radicals under in vitro conditions, showing that the complexes are more effective in arresting the formation of hydroxyl than nitric oxide or DPPH radicals. Cytotoxicity experiments reveal that the two complexes both exhibit cytotoxic effects against human hepatocellular carcinoma cell line SMMC-7721 and human lung adenocarcinoma cell line A549.
Introduction
Cancer is one of the most fatal non-communicable diseases which causes a significant number of deaths every year. Chemotherapy is currently the most-utilized treatment for cancer treatment. cis-Platin and platinum drugs are some of the most widely used cytotoxic chemotherapeutic agents.1,2 Although they are active in the treatment of several types of cancer, the dose-limiting nephrotoxicity and the development of drug resistance prevent their potential efficacy.1–3 Therefore, in efforts to improve the efficacy and overcome the side effects, numerous transition metal complexes have been synthesized and tested for their anticancer activities.4,5 Several ruthenium-, gold-, gallium-, titanium-, and arsenic-based compounds have been investigated for their anticancer potential;6–11 however, none of these compounds have been approved clinically. It should be noted that reviews devoted to the general use of nickel and cobalt complexes in medicine have been published.12,13 A large body of evidence indicates that nickel and cobalt chelates are an effective method to inhibit tumor growth, and they have become promising agents in the treatment of cancer.14–25The effect of metal-based pharmaceuticals depends not only on the function of the metal but also on ligand moieties.26 Schiff base derivatives of hydrazones are an important class of ligands with interesting ligation properties due to the presence of several coordination sites. Moreover, the hydrazone unit offers a number of interesting features such as a degree of rigidity, a conjugated π-system and a NH unit that readily participates in hydrogen bonding and may be a site of protonation–deprotonation. It has been proved that hydrazone derivatives play vital role in certain pharmaceutical functions such as DNA binding and cleavage,18 antibacterial,27 and anticancer.28 In addition, the use of heterocyclic diimine as the co-ligand in ternary complexes is of considerable interest because some of the diimine containing nickel and cobalt complexes exhibit interesting biological as well as pharmacological properties.29 The pivotal role of diimine ligands in antitumour activities have been clarified by numerous literatures earlier.30,31 Overall, it has been demonstrated that physicochemical features, such as planarity, hydrophobicity, and size of the diimine, the nature of the co-ligand, and the coordination geometry of the metal complex, all played important roles in determining the binding mode of transition metal complexes to DNA.
DNA is the primary intracellular target of anti-cancer drugs, the interaction between small molecules and DNA can cause DNA damage in cancer cells, hence blocking the division of cancer cells and resulting in cell death. A study on the interaction of small molecules with DNA on a molecular level is very important in the development of novel chemotherapeutics and highly sensitive diagnostic agents.32,33 Small molecules can interact with DNA through the following three non-covalent modes: intercalation, groove binding and external static electronic effects.34 Both the planarity of ligand and the coordination geometry of the metal ion play important roles in deciding the intercalating ability of complexes to DNA.35,36 It has been proved that increasing the size of the substituent and planarity of ligands can enhance the DNA interaction and protein binding.17,37,38 Therefore, planar aromatic groups, such as benzyl and napthyl group were introduced as substituent to enhance planarity of the ligand. By varying the metal center, it is possible to modify the mode as well as the extent of interaction of the complex with nucleic acids and facilitate individual applications. On the other hand, serum albumins play an important and efficient role in drug delivery due to their remarkable binding properties. With the advantages of low cost and ready availability, bovine serum albumin (BSA) is a widely used serum albumin. The interaction of drug–protein may react in the formation of a stable protein complex which has important effect on the distribution, absorption, metabolism and properties of drugs. Such an interactions of drug compounds have aromatic rings are very important for protein sterilization and different regulation processes.39 Therefore, the activity of metal complexes towards DNA and protein BSA is useful in the design and synthesis of metal-based anticancer therapeutics.
Based on the above facts, we have been intense in studying the role of presence of aromatic rings, planarity and molecular composition of nickel and cobalt complexes on DNA and protein binding and cytotoxicity. Hence, our present investigation focuses on the synthesis of two new nickel and cobalt complexes containing 2-acetonaphthone benzoyl hydrazone ligand (HL) and 1,10-phenanthroline (phen) co-ligand. The structure of the new complexes has been characterized by various spectro-analytical and crystallographic techniques. All the synthesized compounds have been subjected to different experiments to assess their interacting ability with DNA/protien and antioxidant and cytotoxicity potential.
Experimental
Materials and instrumentation
2-Acetonaphthone benzoyl hydrazone (HL) has been synthesized and its molecular structure has been obtained previously by our group.40 1,10-Phenanthroline monohydrate, ethidium bromide (EB), herring sperm DNA, tris(hydroxymethyl) aminomethane hydrochloride (Tri–HCl), bovine serum albumin (BSA), 2,2-diphenyl-1-picrylhydrazyl (DPPH), cis-platin were purchased from Sigma-Aldrich Chemicals and used as received. Human hepatocellular carcinoma cell SMMC-7721 and human lung adenocarcinoma cell A549 were purchased from Shanghai Cell Bank, Chinese Academy of Sciences. SRB, fetal bovine serum and all other cell-culture reagents were obtained from Solarbio Science and Technology Co., Ltd. Beijing, China. All other chemicals and reagents used for biological studies were of high-quality biological grade.Elemental analyses for C, H and N were performed on a Perkin Elmer 240C elemental analyzer. ESI-MS spectra for the complexes were collected on a Thermo Fisher LTQ Orbitrap XL Electrospray Ionization Mass Spectrometer in a positive ion mode. FT-IR measurements (KBr pellets) were recorded in the range of 400–4000 cm−1 on the instrument of Thermo Nicolet Nexus 670 infrared spectrometer. Ultraviolet-visible (UV-vis) spectroscopy was obtained on a Varian Cary 300 UV-vis spectrophotometer. Fluorescence spectroscopy was measured with Perkin Elmer LS-55 spectrometer at room temperature.
Synthesis of NiL2phen·3CH3OH (1)
HL (0.576 g, 2 mmol) was dissolved in 10 mL MeOH/THF (V/V = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution, to which 2 mmol of triethylamine (0.201 g) was added. A solution of nickel acetate tetrahydrate (0.176 g, 1 mmol) in MeOH (10.0 mL) was added dropwise to the ligand solution, which was stirred at room temperature. After 1 h, a methanolic solution of 1,10-phenanthroline monohydrate (0.180 g, 1 mmol) was slowly added to the reaction mixture, resulting in a yellow green colour solution. The reaction mixture was allowed to stir for another 2 h, after which the precipitate that formed was filtered, washed successively with 2 mL of MeOH and 10 mL diethyl ether, and then dried at room temperature. X-ray quality single crystals, having a green colour and prime shape appeared on slow evaporation of the filtrate at room temperature. Yield 0.309 g (34%). Anal. calc for C53H50N6O5Ni (909.70 g mol−1): C 69.79, H 5.54, N 9.23%; found: C 69.70, H 5.57, N 9.31%. FTIR (KBr, cm−1): 1589 and 1523 s υ(C
:1) solution, to which 2 mmol of triethylamine (0.201 g) was added. A solution of nickel acetate tetrahydrate (0.176 g, 1 mmol) in MeOH (10.0 mL) was added dropwise to the ligand solution, which was stirred at room temperature. After 1 h, a methanolic solution of 1,10-phenanthroline monohydrate (0.180 g, 1 mmol) was slowly added to the reaction mixture, resulting in a yellow green colour solution. The reaction mixture was allowed to stir for another 2 h, after which the precipitate that formed was filtered, washed successively with 2 mL of MeOH and 10 mL diethyl ether, and then dried at room temperature. X-ray quality single crystals, having a green colour and prime shape appeared on slow evaporation of the filtrate at room temperature. Yield 0.309 g (34%). Anal. calc for C53H50N6O5Ni (909.70 g mol−1): C 69.79, H 5.54, N 9.23%; found: C 69.70, H 5.57, N 9.31%. FTIR (KBr, cm−1): 1589 and 1523 s υ(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NC), 1369 s υ(C–O). ESI-MS (MeOH): m/z = 813.25 [NiL2Phen + H]+ (calculated m/z = 812.24 for NiL2Phen+).
N–NC), 1369 s υ(C–O). ESI-MS (MeOH): m/z = 813.25 [NiL2Phen + H]+ (calculated m/z = 812.24 for NiL2Phen+).
Synthesis of CoL2phen·3CH3OH (2)
2-Acetonaphthone benzoyl hydrazone (0.576 g, 2 mmol) was dissolved in 10 mL MeOH/THF (V/V = 1:1) solution, to which 2 mmol of triethylamine (0.201 g) was added. A solution of cobalt chloride hexahydrate (0.129 g, 1 mmol) in MeOH (10.0 mL) was added dropwise to the ligand solution, which was stirred at room temperature. After one hour a methanolic solution of 1,10-phenanthroline monohydrate (0.180 g, 1 mmol) was slowly added to the reaction mixture, resulting in a deep brown colour solution. The reaction mixture was allowed to stir for another 2 h, after which the precipitate that formed was filtered, washed successively with 2 mL of MeOH and 10 mL diethyl ether, and then dried at room temperature. X-ray quality single crystals, having a brown colour and square shape appeared on slow evaporation of the filtrate at room temperature. Yield 0.529 g (58%). Anal. calc for C53H50N6O5Co (909.92 g mol−1): C 69.96, H 5.53, N 9.23%; found: C 69.90, H 5.57, N 9.31%. FTIR (KBr, cm−1): 1589 and 1517 s υ(CN–NC), 1367 s υ(C–O). ESI-MS (MeOH): m/z 846.24 [CoL2Phen·CH3OH + H]+, (calculated m/z = 845.26 for [CoL2Phen·CH3OH]+).
X-ray crystallography
The X-ray single-crystal data for complex 1 and 2 were recorded on a Brucker SMART Apex CCD detector diffractometer with graphite-monochromated MoKα radiation (λ = 0.71073 Å). The collected data were reduced using the SAINT program,41 and multi-scan absorption corrections were performed using the SADABS program.42 The structures were solved by direct methods and refined by full-matrix least squares on F2 using SHELXL-97.43 All non-hydrogen atoms were refined anisotropically and the hydrogen atoms in these structures were located via the difference Fourier map and constrained to ideal positions in the refinement procedure. Experimental details for X-ray data collection complexes are presented in Table 1.| Compound | 1 | 2 |
|---|---|---|
| Formula | C53H50N6O5Ni | C53H50N6O5Co |
| FW | 909.70 | 909.92 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c |
| a (Å) | 10.334(2) | 10.360(6) |
| b (Å) | 15.835(3) | 15.843(9) |
| c (Å) | 28.718(6) | 28.627(15) |
| α (°) | 90 | 90 |
| β (°) | 103.504(6) | 103.660 (18) |
| γ (°) | 90 | 90 |
| V (Å3) | 4569.5(16) | 4566(4) |
| Z | 4 | 4 |
| Dcalc (g cm−3) | 1.322 | 1.324 |
| μ (mm−1) | 0.48 | 0.43 |
| F (000) | 1912 | 1908 |
| Crystal size | 0.30 × 0.25 × 0.15 | 0.23 × 0.20 × 0.18 |
| Theta range (°) | 2.6–26.5 | 2.4–20.3 |
| Limiting indices | −10 ≤ h ≤ 12 | −11 ≤ h ≤ 12 |
| −15 ≤ k ≤ 18 | −18 ≤ k ≤ 18 | |
| −34 ≤ l ≤ 33 | −33 ≤ l ≤ 33 | |
| Reflections collected | 23095 |
30500 |
| Independent reflections | 8089 | 7997 |
| Data/restraints/parameters | 8089/0/594 | 7997/0/591 |
| T (K) | 296 | 293 |
| Goodness of fit on F2 | 1.05 | 0.97 |
| Final R indices[I > 2σ(I)] | R1 = 0.0384, wR2 = 0.1094 | R1 = 0.0726, wR2 = 0.1862 |
| R indices (all data) | R1 = 0.0597, wR2 = 0.1281 | R1 = 0.1513, wR2 = 0.2539 |
| Rint | 0.029 | 0.102 |
| ρmax, ρmin (e Å−3) | 0.36, −0.51 | 1.09, −1.35 |
Cyclic voltammetry
Cyclic voltammogram (CV) was recorded on a CHI 660C electrochemical work station using a three-electrode cell in which a platinum plate, a saturated Ag/AgCl and a platinum foil are used as the working, reference and auxiliary electrodes, respectively. A ferrocene/ferrocenium (Fc/Fc+) couple was used as an internal standard. Cyclic voltammograms of the complexes were recorded in DMF solutions at a scan rate of 100 mV s−1. Tetra-n-butylammonium perchlorate (TBAP, 0.1 M) was used as the supporting electrolytes. Prior to every electrochemical assay, the solutions were deoxygenated by purging them with nitrogen. All electrochemical measurements were performed at room temperature.DNA binding studies
Experiments involving the binding of compounds to herring sperm DNA were performed in double-distilled water with 5 mM Tris–HCl and 50 mM NaCl solution and the pH was adjusted to 7.2 with hydrochloric acid. DNA solution was prepared with 50 mM NaCl solution. The UV absorbance ratio at 260 and 280 nm (A260/A280) of 1.85 indicated that the DNA was sufficiently free of protein contamination.44 The concentrations of DNA were determined spectrophotometrically by assuming ε260 = 6600 M−1 cm−1,45 which was found to be 3.72 × 10−4 mol L−1 when the mass concentration was 200 μg mL−1. The ligand and complexes were dissolved in a combined solvent of 5% DMSO and 95% Tris–HCl buffer for all experiments. A stock solution of DNA was stored at 4 °C and used after no more than 4 days.UV-vis titration experiments were performed by keeping a fixed concentration of the metal complex constant (25 μM) and varying the nucleotide concentration (0–125 μM). However, when obtaining the absorption spectra, equal amounts of DNA were added to both complex and reference solutions to eliminate the absorbance of DNA itself. The complex-DNA solutions were allowed to incubate for 30 min before the absorption spectra were recorded.
Further support for the binding of complexes to DNA via intercalation was obtained using fluorescence spectral techniques in order to point out whether a complex can displace EB from a DNA–EB complex. EB displacement experiments were carried out by adding solutions of the complexes to a Tris–HCl buffer solution (pH 7.2) of a DNA/EB mixture. DNA was pre-treated with EB at a [DNA]/[EB] ratio of 10 for 30 min at room temperature, then a test solution was added to this mixture of EB–DNA and the change in fluorescence intensity was measured. The excitation wavelength was fixed at 545 nm for EB bound to DNA. Emissions were recorded with increasing concentrations of compounds and the emission range was adjusted before measurements. The ligand and complexes (0–50 mM) were then added to the mixture and their effect on the emission intensity was measured.
Viscosity experiments were carried on an Ubbelohde viscometer of 10 mL capacity, immersed in a thermostated water bath maintained at 37.0 ± 0.1 °C. The stock solutions of DNA (50 μM) and complexes (0–50 μM) were prepared in Tris–HCl/NaCl buffer. Mixing of the solutions was achieved by purging the nitrogen gas through viscometer. The flow time was measured three times for each sample with a digital stopwatch, and the mean flow time was calculated. Data were presented as (η/η0)1/3 versus binding ratio [complex]/[DNA], where η and η0 are the specific viscosity of DNA in the presence and absence of complex, respectively. The values of η and η0 were calculated from the relation:46 η0 = (t − tb)/tb, where tb is the flow time of buffer alone, and t is the observed flow time for DNA in the presence of complex. Relative viscosities for DNA were obtained from the relation, η/η0.
Protein binding studies
The protein-binding interactions of the ligand and complexes with BSA were investigated using both of fluorescence spectra and UV-vis absorption. Fluorescence spectra were obtained with an excitation wavelength of 280 nm and an emission wavelength of 345 nm corresponding to those of free BSA. The excitation and emission slit widths and scan rates were maintained at a constant for all experiments. Samples were thoroughly degassed using pure nitrogen gas for 15 minutes using quartz cells (4 × 1 × 1 cm) with high-vacuum Teflon stopcocks. Stock solutions of BSA were prepared in 50 mM phosphate buffer (pH 7.2) and stored in the dark at 4 °C for additional use. Concentrated stock solutions of metal complexes were prepared by dissolving them in DMSO:Tris–HCl buffer (5:95) and diluted suitably with phosphate buffer to obtain appropriate concentrations. 2.5 mL of BSA solution (1 μM) was titrated by successive additions of a 5 μL stock solution of complexes (10−4 M) using a micropipette. Synchronous fluorescence spectra were also obtained using the same concentrations of BSA and complexes as mentioned above with two different values of Δλ (difference between the excitation and emission wavelengths of BSA) such as 15 and 60 nm. For UV-vis absorption experiment, a 3 mL solution of BSA (1 μM) was titrated with various concentrations of the complexes. Equal solutions of complexes were added to the reference solutions to eliminate the absorbance of the complexes themselves. The UV-vis absorption spectra were measured from 200 to 400 nm.
Antioxidant activity
The DPPH (2,2-diphenyl-1-picrylhydrazyl) radical-scavenging activity of the compounds was measured according to the method described by Blois.47 The DPPH radical is a stable free radical having a λmax of 517 nm. Various concentrations (10–60 μM) of the test compounds were added to a solution of DPPH (125 mM, 2 mL) in methanol and the final volume was made up to 4 mL with double-distilled water. The solution was incubated at 37 °C for 30 min in the dark. The decrease in the absorbance of DPPH was measured at 517 nm. The same experiment carried out without the test compounds served as a control.The hydroxyl radical (OH˙) in aqueous media was generated by the Fenton system.48 The solution of the tested complex was prepared with DMF. The 5 mL assay mixture contained following reagents: safranin (20 μM), EDTA–Fe2+ (200 μM), H2O2 (196 mM), the tested compounds (1–6 μM) and a phosphate buffer (40 μM, pH = 7.4). The assay mixtures were incubated at 30 °C for 10 min in a water bath. The sample without the tested compound was used as the control. After that, the absorbance was measured at 520 nm.
Nitric oxide (NO) radical-scavenging activity was determined based on the reported method, in which sodium nitroprusside in an aqueous solution at physiological pH spontaneously generates nitric oxide, which interacts with oxygen to produce nitrate ions that can be estimated using the Griess reagent.49 For the experiment, sodium nitroprusside (10 mM) in phosphate buffered saline was mixed with a fixed concentration of the compound and incubated at room temperature for 150 min. After the incubation period, 0.5 mL of the Griess reagent containing 1% sulfanilamide, 2% H3PO4 and 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride was added. The reaction mixture without the sample but with equivalent amount of solvent served as a control. The absorbance of the chromophore formed was measured at 546 nm.
In the case of the above three assays, all of the tests were run in triplicate. All data are expressed as mean ± standard deviation (SD). Various concentrations of the compounds were used to fix a concentration at which the compounds showed in and around 50% of activity. The percentage of activity was calculated using the formula, % of activity = [(A0 − AC)/A0] × 100. A0 and AC are the absorbance in the absence and presence of the tested compounds respectively. The 50% activity (IC50) can be calculated using the percentage of activity results.
In vitro cytotoxic activity evaluation by SRB assays
In vitro cytotoxic activities of the two complexes and cis-platin were evaluated against two cancer cell lines including SMMC-7721 and A549 by using the Sulforhodamine B (SRB) assay. All cells were cultured in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum, 1% (w/v) penicillin (104 U mL−1) and 10 mg mL−1 streptomycin. Cell lines are maintained at 310 K in a 5% (v/v) CO2 atmosphere with 95% (v/v) humidity. Cultures were passaged weekly using trypsin–EDTA to detach the cells from their culture flasks. The two complexes and cisplatin were dissolved in DMSO and diluted to the required concentration with culture medium when used. The content of DMSO in the final concentrations did not exceed 0.1%. At this concentration, DMSO was found to be non-toxic to the cells tested. Rapidly growing cells were harvested, counted, and incubated at the appropriate concentration in 96-well micro plates for 24 h. The two complexes and cisplatin dissolved in culture medium were then applied to the culture wells to achieve final concentrations ranging from 10−4 to 102 μM. Control wells were prepared by addition of culture medium without cells. The plates were incubated at 37 °C in a 5% CO2 atmosphere for 48 h. Upon completion of the incubation, the cells were fixed with icecold 10% trichloroacetic acid (100 mL) for 1 h at 277 K, washed five times in distilled water and allowed to dry in the air and stained with 0.4% SRB in 1% acetic acid (100 mL) for 15 min. The cells were washed four times in 1% acetic acid and air-dried. The stain was solubilized in 10 mM unbuffered tris base (100 mL) and the OD of each well was measured at 540 nm on a microplate spectrophotometer. The IC50 values were calculated from the curves constructed by plotting cell survival (%) vs. the complexes concentration (μM). The medium without samples served as a control and triplicate measurements were made for all concentrations. All data are expressed as mean ± SD.Results and discussion
Benzhydrazone ligand derivative, as a pale yellow crystalline solid, were prepared in high yield by the condensation of 2-acetonaphthone with benzoylhydrazine in an equimolar ratio.40 It was then reacted with Ni(OAc)2·4H2O and CoCl2·6H2O in a 1:2 molar ratio, respectively, followed by 1,10-phen in the presence of triethylamine in a 2:1:1:2 molar proportion in methanol at room temperature (Scheme 1). After a few days, single crystals were obtained from the reaction mixture on slow evaporation at room temperature. The elemental analyses data, ESI-MS and FTIR of the complexes are consistent with their formulation as bis-hydrazone complexes of Ni(II) and Co(II) containing phen an ancillary ligand. The synthesized complexes were sparingly soluble in solvents such as ethanol, methanol or acetonitrile and readily soluble only in solvents such as DMF and DMSO, producing intensely orange-colored solutions.
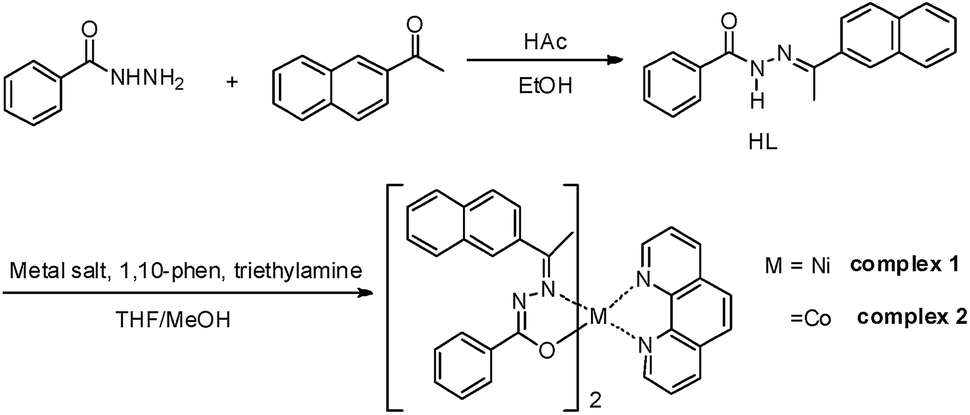 | ||
| Scheme 1 Synthesis of the ligand and complexes 1 and 2. | ||
The infrared spectra of the ligand and complexes 1 and 2 were showed in Fig. S1 (ESI†). The free ligand displayed a medium to strong band at 3348 cm−1, which is characteristic of the N–H functional group. The free ligands also displayed υCO absorption at 1666 cm−1 and υCN absorption at 1519 cm−1, which indicate that the ligands exist in the amide form in the solid state. However, bands that are due to υN–H and υCO stretching vibrations were not observed with the complexes, which indicates that the ligands underwent tautomerization and subsequent coordination of the imidolate enolate form during complexation. Coordination of the ligand to the Ni(II) and Co(II) ion through an azomethine nitrogen is expected to reduce the electron density in the azomethine link and thus lower the absorption frequency after complexation (1517–1589 cm−1), which indicates the coordination of azomethine nitrogen to the metal ions. The presence of a new band at 487 cm−1, which is due to υ(Ni–O) and υ(Co–O), is another indication of the involvement of the oxygen of the enolate group in coordination.50 The appearance of new additional bands in the regions 817 cm−1 and 711 cm−1 are due to phen ring Ar–H and CN stretching vibrations respectively.29
The UV-vis absorption spectra of the ligand and complexes 1 and 2 were obtained in methanol solution (10 μM) in the range of 200–800 nm at room temperature, as presented in Fig. 1. The spectrum of the hydrazone ligand displayed three absorption bands at 234, 278 and 306 nm. Generally, the bands appearing in the 230–280 nm range can be due to the π–π* transition within the aromatic rings, and the band at 306 nm can be due to the n–π* transitions within the >CN–N chromophore.15 In the case of the complexes, the bands appeared at 232, 270 and 303 nm. The slight shift in the absorption bands indicated the formation of the metal complexes and the fact that the azomethine nitrogen is involved in coordination.15,51 The band at 303 nm become broader in complexes than that in the HL, the reason could be assigned to ligand to metal (ion) charge transfer (LMCT) transitions.51 Unfortunately the expected weak d–d transition in the visible region for the one-electron paramagnetic complexes cannot be detected even with concentrated solutions. It may be lost in the low energy tail of the charge transfer transition.
 | ||
| Fig. 1 UV-vis absorption spectra of the ligand, and complexes 1 and 2 in MeOH solution. | ||
Single-crystal X-ray crystallography
All the synthesized compounds were analyzed by single crystals X-ray diffraction to confirm the molecular structures and geometry. The crystal structures of nickel and cobalt complexes are shown in Fig. 2 and slected bond distances (Å) and bond angles (°) are given in Table 2. The bond lengths and bond angles of HL can be found in our previous report.40 Both complexes are crystallized in the monoclinic space group P21/c with four molecules in the unit cell. The hexacoordinated Ni(II) and Co(II) center in 1 and 2 respectively, adopt a distorted octahedral geometry with NiN4O2 and CoN4O2 chromophores, the metal atom being coordinated by two N atoms and two O atoms of the two bidentate Schiff base ligands, along with two N atoms of phen. In complex 1, each of the two bidentate hydrazone ligands coordinates through imine nitrogen and enolate oxygen atoms. The two nitrogen atoms from the two hydrazone ligands are trans to each other and the corresponding enolate oxygen atoms are cis to each other. Each of the N atoms of phen is trans to an enolate oxygen atom. The bond lengths in the Ni(II) complex are: N1–N2 = 1.418(2) (L), N1–C1 = 1.314(3) (S), N2–C8 = 1.282(3) (S), O1–C1 = 1.271(3) (L), C1–C2 = 1.495 (S) Å, (L = longer, S = shorter) (Table 2), which are longer or shorter than those of the corresponding distances in the free ligand40 due to the bonding of the Ni(II) centre with the ligand. The Ni–O bond lengths (2.0020(16) and 2.0076(16) Å) are in the range of those expected for the Ni(II) ion in an octahedral environment.29 The Ni–N distances range from 2.123(2) to 2.170(2) Å, with the Ni–N(phen) distances being about 0.03 Å shorter than the Ni–N(imine) distances. In octahedral Ni(II) complexes of aroylhydrazones, it is generally found that the Ni–O bond distance is longer than the Ni–N distance. However, in the present case a reverse trend is observed. In fact, the Ni–N distance in this complex is comparatively longer than that normally observed for octahedral Ni(II) complexes of aroylhydrazones,28,52 but it is very similar to that reported for an anthracene aldehyde Schiff base complex of Ni(II),29 and the steric bulk of the naphthalene moiety may be responsible for this relatively longer Ni–N bond. The N6–Ni1–N5 angle (bite angle of phen) is 77.81(8)°, whereas for the hydrazone ligand, the bite angles lie in the range 77.00(6) to 77.88(6)°. The distortion, from a regular octahedral geometry is evident from the N2–Ni1–N4 trans-bond angle value of 162.33(7)°, while the other two trans angles, O1–Ni1–N6 and O2–Ni1–N5 are 170.73(7) and 168.07(7)°, respectively, which are closer to that expected for an octahedral geometry.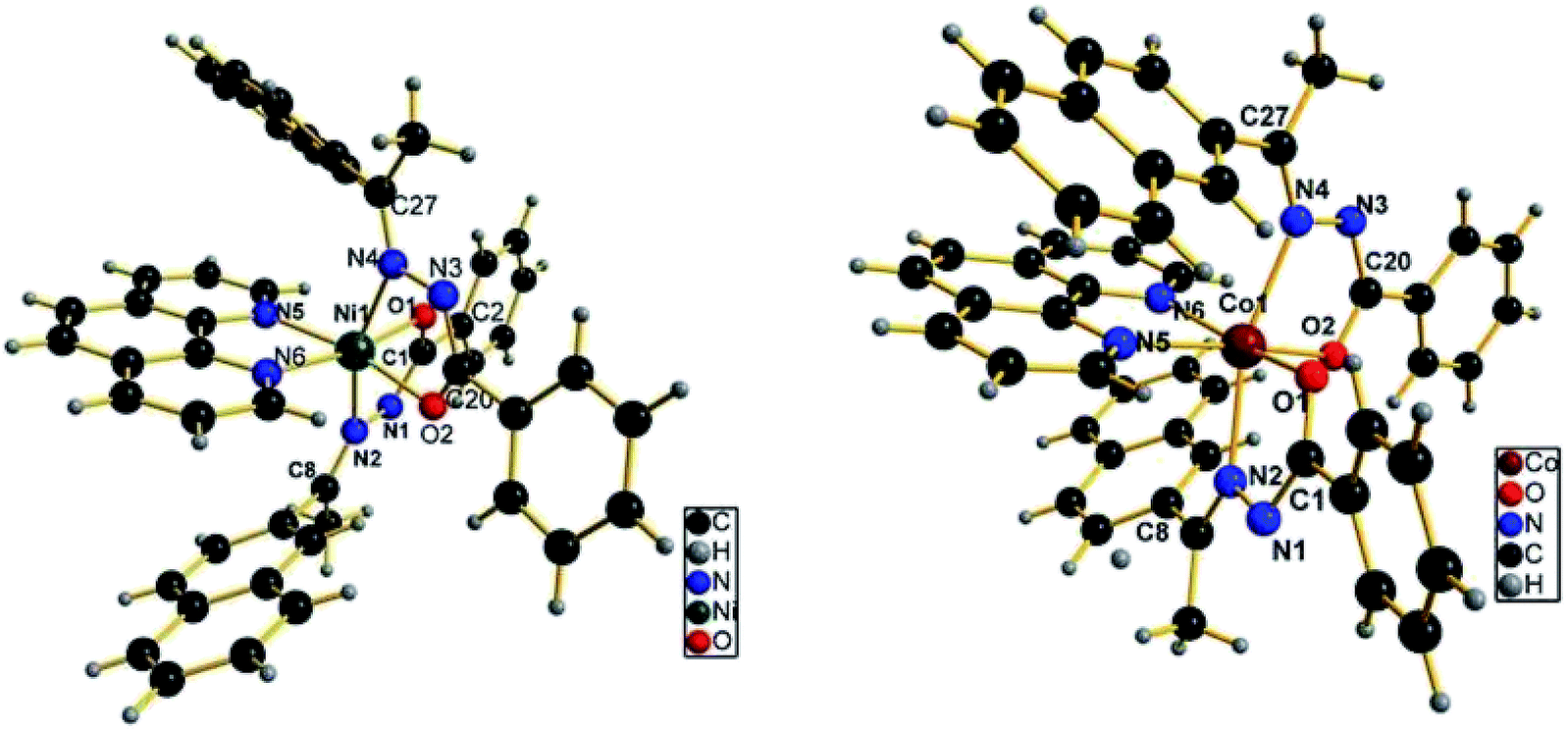 | ||
| Fig. 2 The coordination environment of Ni(II) and Co(II) complexes. | ||
| C53H50N6O5Ni (1) | C53H50N6O5Co (2) | ||
|---|---|---|---|
| Bond lengths (Å) | Bond angles (°) | Bond lengths (Å) | Bond angles (°) |
| N1–N2 1.418(2) | O1–Ni1–N2 77.00(6) | N1–N2 1.412(5) | O1–Co1–N2 76.90(13) |
| N3–N4 1.417(2) | O1–Ni1–N4 89.62(6) | N3–N4 1.393(6) | O1–Co1–N4 92.95(13) |
| N1–C1 1.134(3) | O1–Ni1–N5 95.82(7) | N1–C1 1.131(6) | O1–Co1–N5 92.82(17) |
| N2–C8 1.282(3) | O1–Ni1–N6 170.73(7) | N2–C8 1.287(6) | O1–Co1–N6 164.32(17) |
| O1–C1 1.271(3) | O1–Ni1–O2 93.90(7) | O1–C1 1.264(6) | O1–Co1–O2 97.13(17) |
| C1–C2 1.495(3) | O2–Ni1–N2 91.40(6) | C1–C2 1.505(6) | O2–Co1–N2 91.32(16) |
| Ni1–N2 2.170(2) | O2–Ni1–N4 77.88(6) | Co1–N2 2.205(4) | O2–Co1–N4 75.63(16) |
| Ni–N4 2.1611(19) | O2–Ni1–N5 168.07(7) | Co1–N4 2.219(4) | O2–Co1–N5 168.22(14) |
| Ni1–N5 2.1247(19) | O2–Ni1–N6 93.30(7) | Co1–N5 2.193(5) | O2–Co1–N6 96.44(18) |
| Ni1–N6 2.123(2) | N2–Ni1–N4 162.33(7) | Co1–N6 2.178(4) | N2–Co1–N4 162.49(17) |
| Ni1–O1 2.0020(16) | N5–Ni1–N2 84.09(7) | Co1–O1 2.019(3) | N5–Co1–N2 84.79(17) |
| Ni1–O2 2.0076(16) | N5–Ni1–N4 109.03(7) | Co1–O2 2.007(4) | N5–Co1–N4 110.24(17) |
| N6–Ni1–N2 108.67(7) | N6–Co1–N2 110.61(15) | ||
| N6–Ni1–N4 86.18(6) | N6–Co1–N4 82.84(15) | ||
| N6–Ni1–N5 77.81(8) | N6–Co1–N5 74.69(18) | ||
In complex 2, the Co(II) ion is also in a distorted octahedral environment, having a cis-, trans-, cis-arrangement of the O2, N2 (hydrazone), N2 donor atoms, with each hydrazone ligand acting as a monoanionic bidentate donor, coordinating through the azomethine N, deprotonated amide O atoms and bidentate N,N-donor of phen ligand. The Co–N distances range from 2.178(4) to 2.205(4) Å. Like the previously described Ni(II) complex, in the Co(II) complex, the Co–N(phen) distances are also about 0.03 Å shorter than the Co–N(imine) distances. The bond distances for Co–O1 and Co–O2 are 2.019(3) and 2.007(4) Å, respectively, which are comparable to the bond lengths reported for octahedral Co(II) complexes of aroylhydrazone.16 The bite angle of phen (N5–Co1–N6 angle) is 74.69(18)°, whereas for the hydrazone ligand, the bite angles vary between 75.63(16) and 76.70(13)°. The distortion, from a regular octahedral geometry is evident from the N2–Co1–N4 trans-bond angle value of 162.49(17)°, while the other two trans angles, O1–Co1–N6 and O2–Co1–N5 are 164.32(17) and 168.22(14)°, respectively. When compared the corresponding bond lengths of the Ni(II) complex with that of the Co(II) complex, it was found that the distances of Ni–N and Ni–O are both shorter than the corresponding Co–N and Co–O bond distances. In addition, and the bite angles of the Co(II) complex were more deviate from the ideal bond angles of 90° and 180° than that of the Ni(II) complex, which indicate that Co(II) complex has a more distorted octahedral geometry. X-ray determination confirms the structure that was proposed on the basis of spectroscopic data, which is consistent with the bivalency of the metal and the monoionic nature of the ligand in the complexes.
Electrochemical studies
Electrochemical studies were carried out for Ni(II) and Co(II) benzhydrazone complexes in DMF solution under an atmosphere of nitrogen. Tetrabutylammonium perchlorate (TBAP) (0.05 M) was used as the supporting electrolyte and the concentration of the complexes was 500 μM. The cyclic voltammograms of complexes are shown in Fig. S4 and Fig. S5 (ESI†), respectively. Both complexes were redox active and exhibit quasi reversible redox wave assignable to M2+/M+ couple. For complex 1, the cathodic and anodic peak potential was −0.935 and −0.681 V, respectively. While for complex 2, the corresponding value was −0.788 and −0.747 V, respectively. The separation of the cathodic and anodic peak potential, ΔEp was 0.254 and 0.041 V for 1 and 2, respectively. The formal potential E0′ (or voltammetric E1/2), taken as the average of Epc and Epa was −0.808 and −0.767 V for 1 and 2, respectively. Between −1.5 to −2.0 V, reductive waves are observed, which correspond to reductions of the phen ligand.29 The single-electron nature of the voltammograms was confirmed by a comparison of the current heights for the complexes and that for a simple [NiL2] complex (where L = thiophene aldehyde benzhydrazone) under identical conditions.15 The redox potentials were virtually independent of the scan rates, which indicates quasi-reversibility.53 In general, the reason for quasi-reversible electron transfer in the above complexes may either be due to slow electron transfer or the deposition of the complex on the electrode surface.DNA-binding studies
DNA is the primary pharmacological target of many antitumor compounds. DNA–metal complex interactions are of paramount importance in understanding the mechanism of tumour inhibition in the treatment of cancer. The interaction of the ligand HL and complexes 1 and 2 with DNA was studied by a number of techniques, such as absorption spectral titration, fluorescence spectroscopy and viscosity measurements. | ||
| Fig. 3 Electronic absorption spectra of HL (a) and complex 1 (b) and complex 2 (c) (25 μM) with increasing concentrations of DNA (0, 5, 10, 15, 20, 25, 50, 75, 100, 125 μM). Arrows show the changes in absorbance with respect to an increase in the DNA concentration (insert plots of [DNA] versus [DNA]/(εa − εf)). | ||
In order to compare quantitatively the binding strength of the compounds, the intrinsic binding constants (Kb) of them with CT-DNA were determined from the following eqn (1):57
| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εb − εf) | (1) |
| I0/I = 1 + Kq[Q] | (2) |
 | ||
| Fig. 4 Emission spectra of DNA–EB (50 μM), in the presence of 0, 10, 20, 40, 50, 60, 80 and 100 μM of compounds. The arrow indicates the changes in the emission intensity as a function of complex concentration (inset: Stern–Volmer plot of the fluorescence titration data). | ||
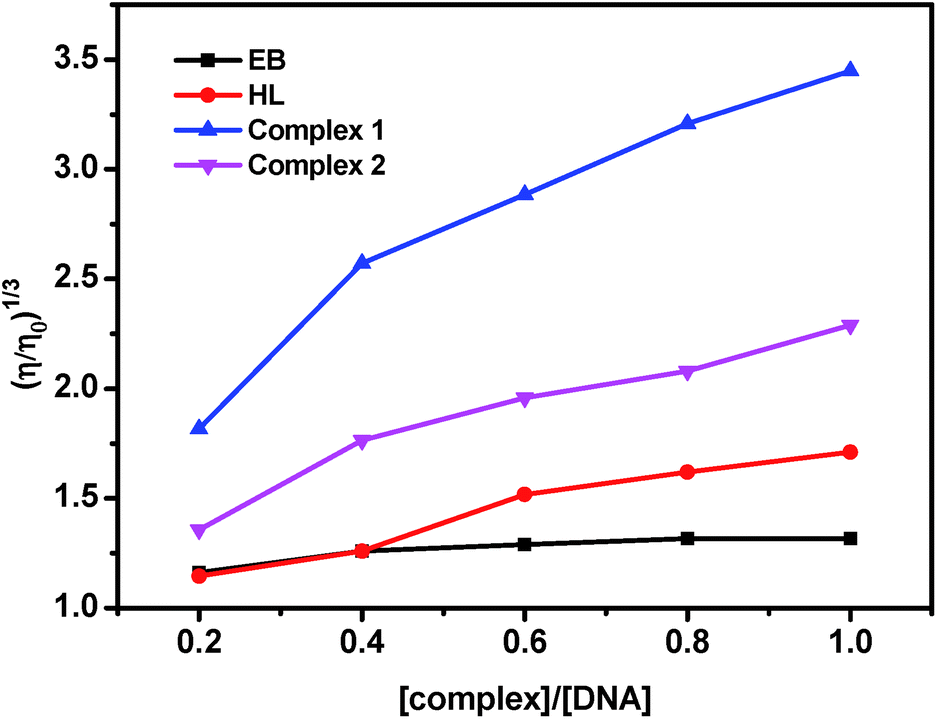 | ||
| Fig. 5 Effect of increasing amounts of EB, HL and complex 1 and 2 on the relative viscosity of DNA at 37.0 ± 0.1 °C. [DNA] = 50 μM and [compound] = 0–50 μM. | ||
Protein binding studies
 | ||
| Fig. 6 Absorption spectra of BSA (10 μM) in the present of the ligand and complexes 1 and 2 (10 μM). | ||
 | ||
| Fig. 7 (a–c) Emission spectra of BSA (1 μM; λex = 280 nm; λem = 345 nm) as a function of concentration of the compounds (0, 2, 4, 6, 8 and 10 μM). Arrow indicates the effect of the compound on the fluorescence emission of BSA (inset: plot of [Q] vs. I0/I). (d) Scatchard plots of the fluorescence titration of the ligand and the complexes with BSA. | ||
|
log[(I0 − I)/I] = logKbin + nlog[Q]
| (3) |
| Compound | Kq (M−1) | Kbin (M−1) | n |
|---|---|---|---|
| HL | 3.67(±0.13) × 104 | 1.35(±0.06) × 103 | 0.71(±0.04) |
| Complex 1 | 2.73(±0.10) × 105 | 4.30(±0.15) × 105 | 0.95(±0.05) |
| Complex 2 | 1.54(±0.04) × 105 | 1.49(±0.11) × 105 | 1.09(±0.07) |
 | ||
| Fig. 8 The synchronous spectra of BSA (1 μM) in the presence of increasing amounts of the ligand and complexes (0, 2, 4, 6, 8 and 10 μM) at a wavelength difference of Δλ = 15 nm and Δλ = 60 nm. The arrow shows the emission intensity changes upon an increase in the concentration of the compounds. | ||
Antioxidant activities
Since the experiments conducted so far revealed that Ni(II) and Co(II) complexes exhibit good DNA and protein binding affinity, it is considered worthwhile to study the antioxidant activity of these compounds. Many Ni(II) and Co(II) complexes bearing hydrazone ligands are known to have radical scavenging properties.15,16,71 The radical scavenging activities of our compounds in a cell free system have been examined with reference to hydroxyl radical (OH˙), DPPH radical (DPPH˙) and nitric oxide (NO˙) radical and their corresponding IC50 values have been tabulated in Table 4. The inhibitory effect of the hydrazone ligand and complexes on radicals was also depicted in Fig. 9, showing the suppression ratio increases with increasing concentration in the range of tested concentration. The order of the suppression ratio for DPPH˙ and OH˙ is complex 1 > complex 2, while for the NO radicals, the scavenging ability is very close to each other. Even though the DPPH˙ scavenging ability of complexes were lower than the standards, such as butylated hydroxyanisole (BHA) and butylated hydroxytoluene (BHT) studied in references,16 the ability was excellent for the DPPH and OH radicals when compared to the standards radicals (Table 4). The IC50 values (Table 4) indicated that the various radical scavenging activities of the complexes are in the order of OH˙ > DPPH˙ > NO˙. It is to be noted that no significant radical scavenging activities were observed in all the experiments carried out with Ni(OAc)2·4H2O and CoCl2·6H2O, even up to 1.0 mmol of concentration under the same experimental conditions. It can be concluded that the scavenging effects of the free ligand is significantly less when compared to that of its corresponding Ni(II) and Co(II) complexes, which is mainly due to the chelation of the organic ligand with the Ni(II) and Co(II) ions. Although the IC50 values observed in NO scavenging assay show much lower NO scavenging ability than those of the reported nickel(II) benzhydrazone complexes15,25 and cobalt(II) hydrazone complexes,25,30 the IC50 values observed in DPPH and HO scavenging assay do demonstrate that both complexes have a comparable effective in arresting the formation DPPH radicals as well as an obvious stronger OH scavenging potential. Therefore, the complexes have significant potential to be applied as scavengers to eliminate the DPPH and HO radicals. | ||
| Fig. 9 Trends in the inhibition of DPPH, OH and NO radicals by complexes at various concentrations. | ||
Cytotoxic activities
In vitro cytotoxic activities of the two complexes and cisplatin against two cancer cell lines human hepatocellular carcinoma cell line SMMC-7721 and human lung adenocarcinoma cell line A549 were conducted using a SRB assay in our study. The results were presented by means of cell viability curves and expressed as values in the studied concentration range from 0.1 to 100 μM. It is noteworthy that the IC50 values of complex 1 for the SMMC-7721 and A549 cell lines are 46.7 ± 1.2 and 39.5 ± 1.0 μM, and those of complex 2 are 66.6 ± 1.4 and 58.9 ± 1.2 μM, respectively. It is to be noted that the precursor and ligand did not display any inhibition of cell growth, even at concentrations of up to 100 mM, which clearly indicates that chelation of the ligand with the metal ion is responsible for the observed cytotoxic properties of the complexes. The results of SRB assays revealed that the complexes exhibited notable activity against both SMMC-7721 and A549 cell lines. The cytotoxic activity of Ni(II) complex was found to be better when compared with the Co(II) complex, which may be attributed to the extended planar structure induced by the π → π conjugation resulting from the chelating of the Ni(II) with aroylhydrazone primary ligand and phen diimine co-ligand and the cationic nature of the complex.17,39,72 Although the abovementioned complexes were active against the cell lines in vitro cytotoxicity experiments, none of the complexes could attain the effectiveness shown by the standard drug cis-platin (IC50 values of 18.0 and 25.3 μM, respectively).66 The order of their in vitro anticancer activities is in agreement with their DNA-binding abilities, which supports the conclusion that DNA may be the primarily molecular target of these complexes. The IC50 values are much higher than those previously reported for other nickel complex containing a coordinated hydrazone with planar napthyl group, which is responsible for the selectivity and the potential inhibition against the tumour cells.17 These findings suggest a strategy that increases the synergistic planarity and the cationic nature in mononuclear systems for the development of transition metal-based chemotherapeutic agents.Conclusions
Two new Ni(II) and Co(II) complexes containing 2-acetonaphthone benzoyl hydrazone (HL) and 1,10-phenanthroline have been successfully synthesized and characterized by various spectral and analytical techniques. The molecular structure of the complexes was confirmed by single crystal X-ray diffraction studies. The DNA-binding interaction of the compounds has been evaluated by UV-vis and fluorescence spectroscopy which revealed that both complexes can bind to DNA via intercalation in the order of HL < Co(II) complex < Ni(II) complex. Binding affinity of the compounds with BSA was significant and the synchronous spectral studies showed that the complexes bound with BSA in both tyrosine and tryptophan residues. From the above results we observed that Ni(II) complex exhibited higher DNA and BSA binding affinity that Co(II) complex, which could be ascribed to the smaller size of Ni(II) complex to fit well into the DNA/BSA grooves than the bulkier cation of Co(II) complex. Also, the antioxidant and cytotoxicity properties of the newly synthesised complexes showed that they can be investigated in detail as potential drugs. In addition, both complexes exhibited significant cytotoxicity against SMMC-7721 and A549 cell lines. The present study highlighted that synergistic planarity of aroylhydrazone primary ligand and diimine co-ligand, as well as the cationic nature of the complex may play an important role in biological activities, suggesting that the DNA/BSA-binding activities as well as the cytotoxicity can be tuned by changing the ligands and the geometry in these nickel and cobalt systems. This is a step toward enabling the rational design of novel metallodrugs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully thanked the facilities support from Jiangsu Key Lab for the Chemistry & Utilization of Agricultural and Forest Biomass and supported by the Natural Science Foundation of Hainan Province (Grant No. 214035).References
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed.
- R. A. Alderden, M. D. Hall and T. W. Hambley, J. Chem. Educ., 2006, 83, 728 CrossRef CAS.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS PubMed.
- W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC.
- C. R. Munteanu and K. Suntharalingam, Dalton Trans., 2015, 44, 13796–13808 RSC.
- S. H. van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 14, 1089–1097 CrossRef CAS PubMed.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817–7823 RSC.
- C. Chitambar, Future Med. Chem., 2012, 4, 1257–1272 CrossRef CAS PubMed.
- I. Ott, Coord. Chem. Rev., 2009, 253, 1670–1681 CrossRef CAS.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- M. C. Heffern, N. Yamamoto, R. J. Holbrook, A. L. Eckermann and T. J. Meade, Curr. Opin. Chem. Biol., 2013, 17, 189–196 CrossRef CAS PubMed.
- G. Barone, A. Terenzi, A. Lauria, A. M. Almerico, J. M. Leal, N. Busto and B. García, Coord. Chem. Rev., 2013, 257, 2848–2862 CrossRef CAS.
- M. D. Hall, T. W. Failes, N. Yamamoto and T. W. Hambley, Dalton Trans., 2007, 3983–3990 RSC.
- R. Raj Kumar and R. Ramesh, RSC Adv., 2015, 5, 101932–101948 RSC.
- D. S. Raja, N. S. P. Bhuvanesh and K. Natarajan, Dalton Trans., 2012, 41, 4365–4377 RSC.
- P. Sathyadevi, P. Krishnamoorthy, R. R. Butorac, A. H. Cowley, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2011, 40, 9690–9702 RSC.
- P. Krishnamoorthy, P. Sathyadevi, A. H. Cowley, R. R. Butorac and N. Dharmaraj, Eur. J. Med. Chem., 2011, 46, 3376–3387 CrossRef CAS PubMed.
- M. P. Sathisha, U. N. Shetti, V. K. Revankar and K. S. R. Pai, Eur. J. Med. Chem., 2008, 43, 2338–2346 CrossRef CAS PubMed.
- P. Krishnamoorthy, P. Sathyadevi, R. R. Butorac, A. H. Cowley, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2012, 41, 6842–6854 RSC.
- N. R. Filipovic, S. Bjelogrlic, T. R. Todorovic, V. A. Blagojevic, C. D. Muller, A. Marinkovic, M. Vujcic, B. Janovic, A. S. Malesevic, N. Begovic, M. Sencanski and D. M. Minic, RSC Adv., 2016, 6, 108726–108740 RSC.
- S. Tsiliou, L.-A. Kefala, F. Perdih, I. Turel, D. P. Kessissoglou and G. Psomas, Eur. J. Med. Chem., 2012, 48, 132–142 CrossRef CAS PubMed.
- F. Dimiza, A. N. Papadopoulos, V. Tangoulis, V. Psycharis, C. P. Raptopoulou, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2012, 107, 54–64 CrossRef CAS PubMed.
- J. Haribabu, K. Jeyalakshmi, Y. Arun, N. S. P. Bhuvanesh, P. T. Perumal and R. Karvembu, RSC Adv., 2015, 5, 46031–46049 RSC.
- P. Krishnamoorthy, P. Sathyadevi, P. T. Muthiah and N. Dharmaraj, RSC Adv., 2012, 2, 12190–12203 RSC.
- T. W. Hambley, Dalton Trans., 2007, 4929–4937 RSC.
- M. V. Angelusiu, S.-F. Barbuceanu, C. Draghici and G. L. Almajan, Eur. J. Med. Chem., 2010, 45, 2055–2062 CrossRef CAS PubMed.
- V. P. Singh, S. Singh, D. P. Singh, P. Singh, K. Tiwari, M. Mishra and R. J. Butcher, Polyhedron, 2013, 56, 71–81 CrossRef CAS.
- S. Mondal, B. Pakhira, A. J. Blake, M. G. B. Drew and S. K. Chattopadhyay, Polyhedron, 2016, 117, 327–337 CrossRef CAS.
- S. Ramakrishnan, V. Rajendiran, M. Palaniandavar, V. S. Periasamy, B. S. Srinag, H. Krishnamurthy and M. A. Akbarsha, Inorg. Chem., 2009, 48, 1309–1322 CrossRef CAS PubMed.
- A. N. Wein, A. T. Stockhausen, K. I. Hardcastle, M. R. Saadein, S. Peng, D. Wang, D. M. Shin, Z. Chen and J. F. Eichler, J. Inorg. Biochem., 2011, 105, 663–668 CrossRef CAS PubMed.
- S. Anbu, R. Ravishankaran, A. A. Karande and M. Kandaswamy, Dalton Trans., 2012, 41, 12970–12983 RSC.
- A. Basu, D. Thiyagarajan, C. Kar, A. Ramesh and G. Das, RSC Adv., 2013, 3, 14088–14098 RSC.
- A. Erdem and M. Ozsoz, Electroanalysis, 2002, 14, 965–974 CrossRef CAS.
- H. Xu, K.-C. Zheng, Y. Chen, Y.-Z. Li, L.-J. Lin, H. Li, P.-X. Zhang and L.-N. Ji, Dalton Trans., 2003, 2260–2268 RSC.
- M. Asadi, E. Safaei, B. Ranjbar and L. Hasani, New J. Chem., 2004, 28, 1227–1234 RSC.
- E. Ramachandran, D. S. Raja, J. L. Mike, T. R. Wagner, M. Zeller and K. Natarajan, RSC Adv., 2012, 2, 8515–8525 RSC.
- S. Ramakrishnan, E. Suresh, A. Riyasdeen, M. A. Akbarsha and M. Palaniandavar, Dalton Trans., 2011, 40, 3245–3256 RSC.
- G. Ayyannan, M. Mohanraj, G. Raja, N. Bhuvanesh, R. Nandhakumar and C. Jayabalakrishnan, Inorg. Chim. Acta, 2016, 453, 562–573 CrossRef CAS.
- Y. Li, Z. Yang, B. Song, H. Xia and Z. Wang, Inorg. Nano-Met. Chem., 2017, 47, 966–972 CrossRef CAS.
- Bruker SMART and SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2002 Search PubMed.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector, University of Gottingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- G. Felsenfeld and S. Z. Hirschman, J. Mol. Biol., 1965, 13, 407–427 CrossRef CAS PubMed.
- S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Biochemistry, 1992, 31, 9319–9324 CrossRef CAS PubMed.
- M. S. Blois, Nature, 1958, 181, 1199–1200 CrossRef CAS.
- C. C. Winterbourn, Biochem. J., 1981, 198, 125–131 CrossRef CAS PubMed.
- L. C. Green, D. A. Wagner, J. Glogowski, P. L. Skipper, J. S. Wishnok and S. R. Tannenbaum, Anal. Biochem., 1982, 126, 131–138 CrossRef CAS PubMed.
- L.-H. Jia, R.-Y. Li, Z.-M. Duan, S.-D. Jiang, B.-W. Wang, Z.-M. Wang and S. Gao, Inorg. Chem., 2011, 50, 144–154 CrossRef CAS PubMed.
- P. Sathyadevi, P. Krishnamoorthy, R. R. Butorac, A. H. Cowley and N. Dharmaraj, Metallomics, 2012, 4, 498–511 RSC.
- S. Naskar, S. Naskar, R. J. Butcher and S. K. Chattopadhyay, Inorg. Chim. Acta, 2010, 363, 3641–3646 CrossRef CAS.
- M. S. Kryatova, O. V. Makhlynets, A. Y. Nazarenko and E. V. Rybak-Akimova, Inorg. Chim. Acta, 2012, 387, 74–80 CrossRef CAS.
- E. C. Long and J. K. Barton, Acc. Chem. Res., 1990, 23, 271–273 CrossRef CAS.
- M. Alagesan, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2013, 42, 7210–7223 RSC.
- M. Sirajuddin, S. Ali and A. Badshah, J. Photochem. Photobiol., B, 2013, 124, 1–19 CrossRef CAS PubMed.
- A. Wolfe, G. H. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- B.-d. Wang, Z.-Y. Yang, Q. Wang, T.-k. Cai and P. Crewdson, Bioorg. Med. Chem., 2006, 14, 1880–1888 CrossRef CAS PubMed.
- J. Ravichandran, P. Gurumoorthy, M. A. Imran Musthafa and A. Kalilur Rahiman, Spectrochim. Acta, Part A, 2014, 133, 785–793 CrossRef CAS PubMed.
- P. A. Vekariya, P. S. Karia, J. V. Vaghasiya, S. Soni, E. Suresh and M. N. Patel, Polyhedron, 2016, 110, 73–84 CrossRef CAS.
- Y.-J. Hu, Y. Liu, J.-B. Wang, X.-H. Xiao and S.-S. Qu, J. Pharm. Biomed. Anal., 2004, 36, 915–919 CrossRef CAS PubMed.
- P. Sathyadevi, P. Krishnamoorthy, E. Jayanthi, R. R. Butorac, A. H. Cowley and N. Dharmaraj, Inorg. Chim. Acta, 2012, 384, 83–96 CrossRef CAS.
- K. Karami, Z. Mehri Lighvan, A. M. Alizadeh, M. Poshteh-Shirani, T. Khayamian and J. Lipkowski, RSC Adv., 2016, 6, 78424–78435 RSC.
- A. S. Sharma, S. Anandakumar and M. Ilanchelian, RSC Adv., 2014, 4, 36267–36281 RSC.
- F. Samari, B. Hemmateenejad, M. Shamsipur, M. Rashidi and H. Samouei, Inorg. Chem., 2012, 51, 3454–3464 CrossRef CAS PubMed.
- X.-W. Li, X.-J. Li, Y.-T. Li, Z.-Y. Wu and C.-W. Yan, J. Photochem. Photobiol., B, 2013, 118, 22–32 CrossRef CAS PubMed.
- D. Senthil Raja, N. S. P. Bhuvanesh and K. Natarajan, Inorg. Chem., 2011, 50, 12852–12866 CrossRef PubMed.
- X.-Z. Feng, Z. Lin, L.-J. Yang, C. Wang and C.-l. Bai, Talanta, 1998, 47, 1223–1229 CrossRef CAS PubMed.
- E. U. Akusoba and J. N. Miller, Proc. Anal. Div. Chem. Soc., 1979, 16, 92–95 RSC.
- J. Tang, F. Luan and X. Chen, Bioorg. Med. Chem., 2006, 14, 3210–3217 CrossRef CAS PubMed.
- L. Tabrizi, P. McArdle, A. Erxleben and H. Chiniforoshan, Eur. J. Med. Chem., 2015, 103, 516–529 CrossRef CAS PubMed.
- F. A. Beckford, J. M. Shaloski, G. Leblanc, J. Thessing, L. C. Lewis-Alleyne, A. A. Holder, L. Li and N. P. Seeram, Dalton Trans., 2009, 10757–10764 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR, ESI-mass and cyclic voltammograms of the complexes. CCDC 1511152 and 1518021. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra05504h |
| This journal is © The Royal Society of Chemistry 2017 |