
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoenzymatic one-pot reaction of noncompatible catalysts: combining enzymatic ester hydrolysis with Cu(I)/bipyridine catalyzed oxidation in aqueous medium†
Henning Sand and
Ralf Weberskirch *
*
Faculty of Chemistry and Chemical Biology, TU Dortmund, Otto-Hahn Str. 6, D 44227 Dortmund, Germany. E-mail: Ralf.Weberskirch@tu-dortmund.de
First published on 3rd July 2017
Abstract
The combination of chemical catalysts and biocatalysts in a one-pot reaction has attracted considerable interest in the past years. However, since each catalyst requires very different reaction conditions, chemoenzymatic one-pot reactions in aqueous media remain challenging and are limited today to metal-catalysts that display high activity in aqueous media. Here, we report the first combination of two incompatible catalytic systems, a lipase based ester hydrolysis with a water-sensitive Cu/bipyridine catalyzed oxidation reaction, in a one-pot reaction in aqueous medium (PBS buffer). Key to the solution was the compartmentalization of the Cu/bipyridine catalyst in a core–shell like nanoparticle. We show the synthesis and characterization of the Cu/bipyridine functionalized nanoparticles and the application in the oxidation of allylic and benzylic alcohols in aqueous media. Furthermore, the work demonstrates the implementation of a one-pot reaction process with optimized reaction conditions involving a lipase (CAL-B) to hydrolyze various acetate ester substrates in the first step, followed by oxidation of the resulting alcohols to the corresponding aldehydes under aerobic conditions in aqueous media.
Introduction
The design and implementation of one-pot tandem reactions has received significant attention in organic chemistry in the past years as an important tool to carry out multi-step reactions without the need to isolate and purify intermediates.1–3 Of particular interest is the combination of enzymes with metal–organic catalysts.4,5 However, owing to the fact that the majority of enzymes are only active in aqueous medium and most metal–organic catalysts prefer organic solvents, only a few examples of such reactions have been reported so far.6–12One of the first examples is the dynamic kinetic resolution (DKR) reported by Williams and co-workers based on a lipase catalyzed resolution of alcohols under substrate-racemizing conditions catalyzed by a rhodium catalyst [Rh2(OAc)4].6 The chemo-enzymatic reaction was carried out by using the enzyme Pseudomonas fluorescens lipase and 2 mol% of the rhodium catalyst [Rh2(OAc)4] simultaneously in an organic solvent. The Pd-catalyzed Suzuki coupling followed by an enzyme catalyzed asymmetric reduction in aqueous medium was one of the first examples reported by Gröger and coworker. Initial attempts required adjustment of the reaction temperature, solvent composition and pH after the Pd-catalyzed Suzuki reaction before the enzymatic reduction was carried out.7 In a subsequent work, the conditions for the first step were modified and both reactions could be conducted at r.t. in a 2-PrOH/H2O solvent mixture that required only a pH adjustment before the enzymatic reaction step.8 An very impressive example has been recently reported by Gröger and coworker who described the one-pot reaction of the CuCl/PdCl2 catalyzed Wacker oxidation followed by an enzymatic reduction. To avoid any enzyme inhibition by Cu-ions, the Wacker oxidation with CuCl/PdCl2 was conducted in the interior of a polydimethylsiloxane thimble, which enables diffusion of organic substrate and product into the exterior aqueous enzyme phase while Cu ions are withheld.9
In most cases, the chemoenzymatic one-pot reaction of an enzyme with a metal organic catalyst was only possible after extensive optimization of the reaction conditions, by using aqueous solvent mixtures,7–10 adjusting reaction temperature or pH,7–9 addition of scavengers,12 by consecutive addition of the two catalysts to the one-pot system10,12,13 or by using a fed-batch continuous-flow process with compartmentalized combination of catalysts.14
As a result, each catalytic reaction is conducted under non-optimal conditions in favor of the one-pot reaction and overall activity is often quite low. Another challenge is that all chemoenzymatic one-pot reactions described so far require a metal- or organo catalyst that displays activity in aqueous media, water-sensitive reactions such as the L-proline catalyzed aldol reaction in combination with an asymmetric reduction catalyzed by the alcohol dehydrogenase (ADH) had to be conducted in organic media.15
A reaction that attracted our attention recently, is the oxidation of a broad range of primary alcohols to the corresponding aldehydes by using a Cu(I)/bipyridine/nitroxyl co-catalyst system by Stahl and coworker.16,17 Compared to other oxidation procedure, the catalyst systems works at room temperature under aerobic conditions. Furthermore, catalyst activity can be significantly enhanced by using ABNO instead of TEMPO as the N-oxyl source enabling also the oxidation of secondary alcohols under mild reaction conditions.18 However, extensive testing of different solvents showed that the best activity results were achieved using acetonitrile as a solvent whereas the presence of water reduced the product yield significantly.16
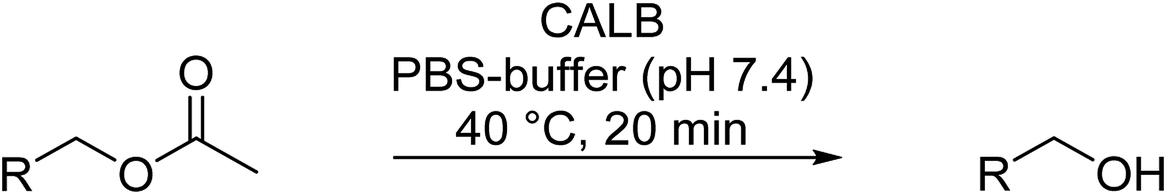
An important reaction in organic chemistry is the selective synthesis of an aldehyde from the corresponding ester.19 The most common reducing agent involve the use of diisobutylaluminum hydride (DIBALH), which is commercially available but gives moderate to high yields (48–90%) and requires very low temperature (−70 °C) in organic solvents, such as hexane.20 A second method is based on two steps via reduction to the alcohol with LiAlH4 followed by oxidation to the aldehyde with Pyridinium Chlorochromate (PCC) or Pyridinium Dichromate (PDC) in THF (see Scheme 1) giving high yields with >90%.21 However, the latter reaction uses chromium compounds that are category 1 heavy metals according to the International agency for Research on Cancer.22 Therefore, the application of catalytic reaction steps under environmentally friendly reaction conditions (e.g. aqueous medium and moderate temperature) is of great interest for the sustainable production of chemicals.23,24
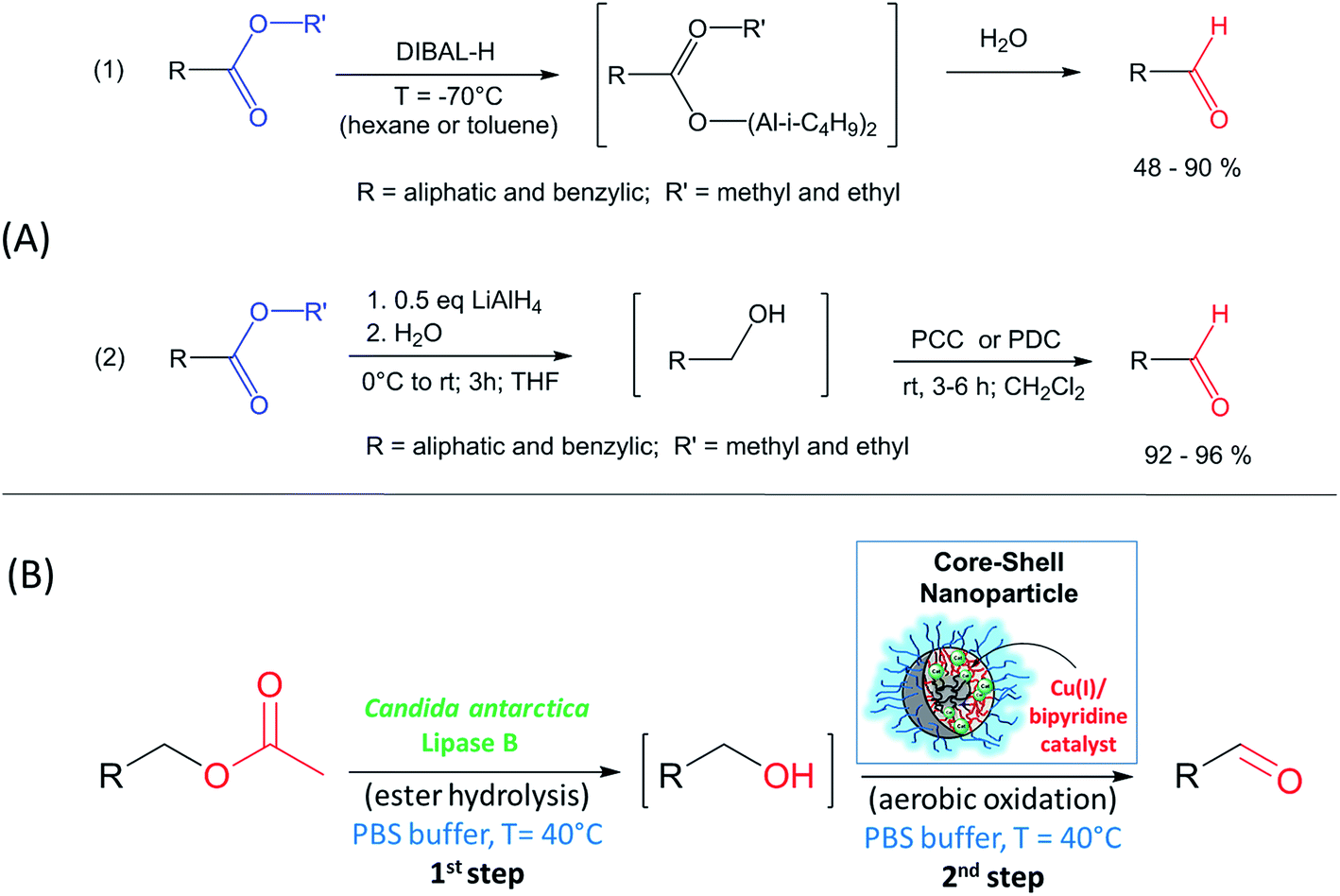 | ||
| Scheme 1 (A) Most common methods in organic chemistry to synthesize aldehydes from the corresponding aliphatic or benzylic esters. (B) Alternative approach by the combination of enzymatic hydrolysis and Cu(I)/bipyridine mediated oxidation in a one-pot process in PBS buffer. | ||
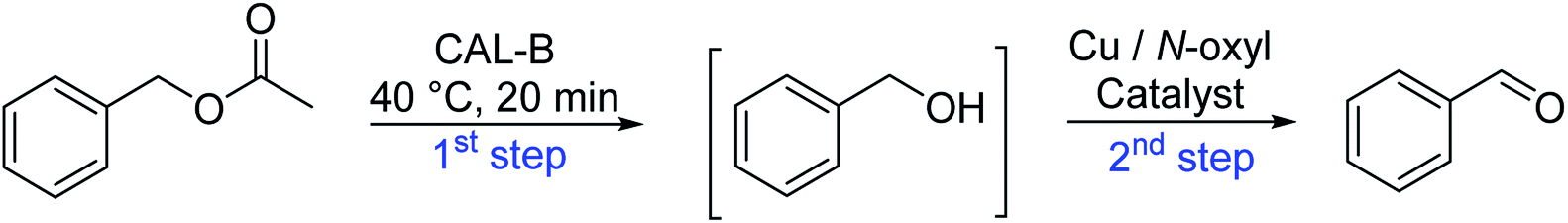
Herein, we report the first example to prepare aldehydes from esters by a combination of a lipase catalyzed ester cleavage with a Cu(I)/bipyridine catalyzed oxidation reaction in a one-pot two-step reaction in aqueous medium (PBS buffer). The key to the combination of these two catalysts in a reaction sequence is the compartmentalization of the Cu(I)/bipyridine catalyst in the hydrophobic core of a polymeric nanoparticle thus avoiding any mutual inhibition with the commercially available enzyme lipase B from Candida antarctica (Immobead 150). At the same time, the amphiphilic nanoparticle structure makes the Cu(I)/bipyridine catalyst compatible with the aqueous reaction medium that is preferred by the enzyme (Scheme 1).
Results and discussion
The idea to combine a lipase based ester hydrolysis with the Cu(I)/bipyridine aerobic oxidation was based on several considerations. Firstly, the lipase B from Candida antarctica (CAL-B) is a well-known biocatalyst for ester hydrolysis and transesterification reactions.25–27 It exists in various commercial forms, such as the Immobead 150, where the lipase is immobilized on the surface of polymer microbeads as a carrier.28 Secondly, the very mild reaction conditions under which the enzyme operates makes it an ideal candidate to be combined with the Cu(I)/bipyridine based aerobic oxidation since both reactions work at moderate temperatures under aerobic conditions but differ in the best solvent for each reaction, acetonitrile for the aerobic oxidation reaction and water or PBS buffer for the enzymatic ester hydrolysis.Therefore, initial experiments focused on studying enzyme activity with a variety of ester substrates. We synthesized various acetyl esters, including aromatic, allylic, aliphatic and heterocyclic esters (ESI†) and studied their enzymatic hydrolysis by using the commercially available lipase B from Candida antarctica (CAL-B) immobilized on polyacrylate beads (Immobead 150). In a control experiment, we checked the hydrolytic stability of benzyl acetate in PBS buffer at 40 °C for 20 min and no hydrolysis was observed. In the presence of the enzyme CALB, benzylic esters were converted efficiently after 20 min at 40 °C to the corresponding alcohols in 87–93% yield (entry 1–4, Table 1). The allylic ester showed similar enzymatic activities with 67–89% yield (entry 5–7, Table 1), whereas lowest enzyme activity was observed for the aliphatic substrate with 59% yield (entry 8, Table 1). However, when the reaction time was extended to 40 min the latter also gave quantitative yield. Furthermore, the immobilized enzyme could be isolated and separated from the reactants by simple filtration (ESI†).
|
|||
|---|---|---|---|
| Entry | Substrate | Conversiona (%) | Yieldb (%) |
| a Determined by 1H-NMR-spectroscopy.b Isolated yield.c Reaction time of 40 min leads to quantitative conversion. | |||
| 1 | 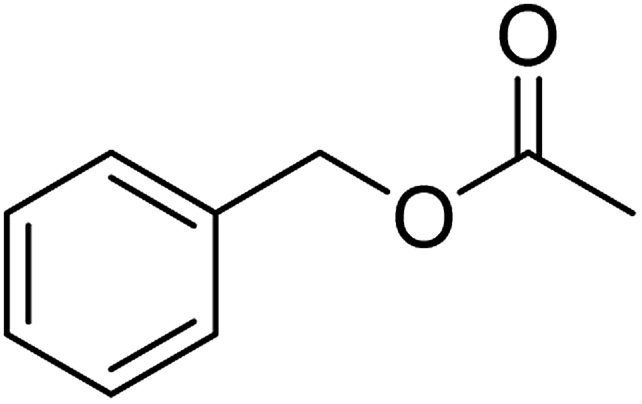 |
97 (±3) | 93 (±2) |
| 2 | 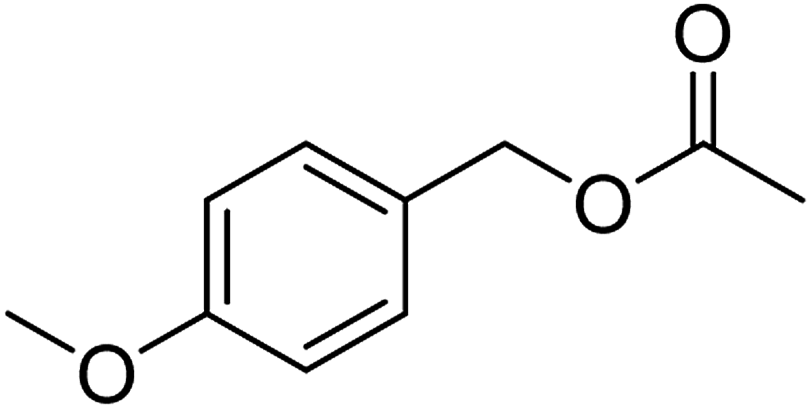 |
93 (±2) | 87 (±2) |
| 3 |  |
96 (±2) | 87 (±2) |
| 4 | 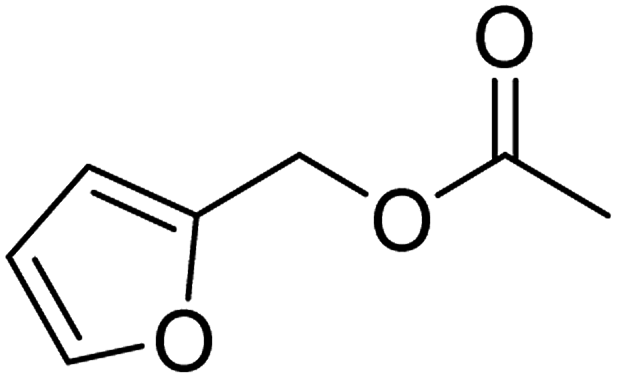 |
95 (±1) | 89 (±3) |
| 5 | 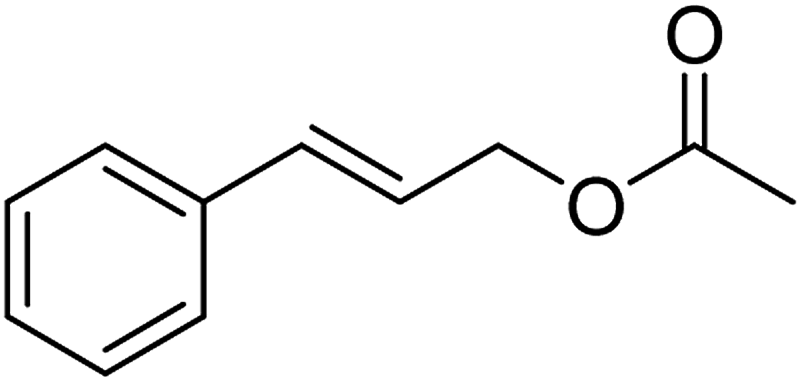 |
88 (±2) | 79 (±2) |
| 6 | 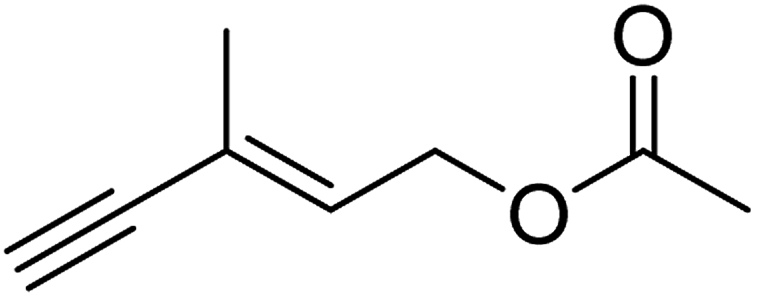 |
96 (±3) | 89 (±3) |
| 7c | 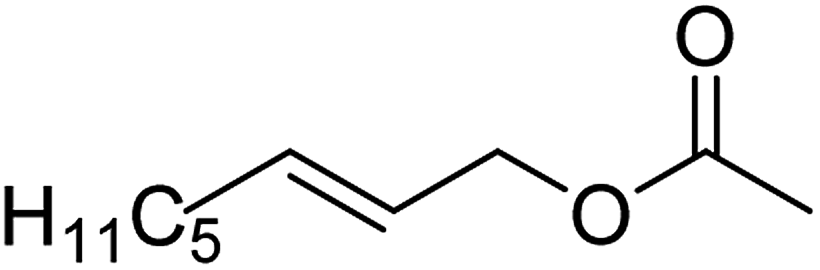 |
71 (±4) | 67 (±4) |
| 8c | 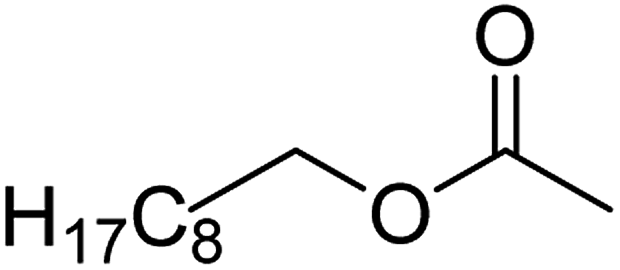 |
67 (±5) | 59 (±4) |
Although in the classical Cu(I)/bipyridine mediated oxidation of alcohols small amounts of water as co-solvent led to a drastic reduction of product yield,16 we were recently able to transfer this reaction to an aqueous medium by site-isolation of the Cu(I)/bipyridine catalyst in micelles prepared from polymeric amphiphiles.29 Oxidation of benzyl alcohol as a model compound was shown to be quantitative at room temperature after 3 h (with 5 mol% Cu(I)Br; 5 mol% polymer ligand, 5 mol% TEMPO and 10 mol% NMI as a base) in water as reaction medium, suggesting that the micellar core–shell like structure is able to protect the catalyst very efficiently from the aqueous phase. Therefore, our thought was to combine the enzymatic ester cleavage with the micellar catalytic oxidation in a one-pot two-step system by using benzyl acetate as a model substrate. Initial studies revealed, however, that the enzymatic ester cleavage was quantitative after product isolation, whereas the subsequent oxidation step of the benzyl alcohol showed only 9% yield. The low product yield indicated inhibition of the Cu(I)/bipyridine catalyst in the presence of the enzyme most likely due to the dynamic nature of micellar aggregates.
A strategy to overcome this limitation of micelles is to stabilize them by crosslinking the micellar core or shell and thus prevent the dynamic exchange of single polymer amphiphiles.30–33 The main focus to apply shell crosslinked nanoparticles has been drug delivery whereas few reports used them also for catalysis application.34,35 In contrast to medical application, where the focus is mainly the stabilization of micelles to prevent any premature drug release, the nanoparticles design for catalysis application should enable high accessibility of the catalyst and easy diffusion of the substrate(s)/product(s) in and out the nanoparticle. Therefore, we decided to use the microemulsion approach for the preparation of catalytically active nanoparticles.36,37 These nanoparticles are composed of a crosslinked core to prevent any interaction between the catalyst and the enzyme and an amphiphilic polymer shell which is compatible with the aqueous phase and provides an hydrophobic layer that contains the Cu(I)/bipyridine catalyst (Scheme 2).
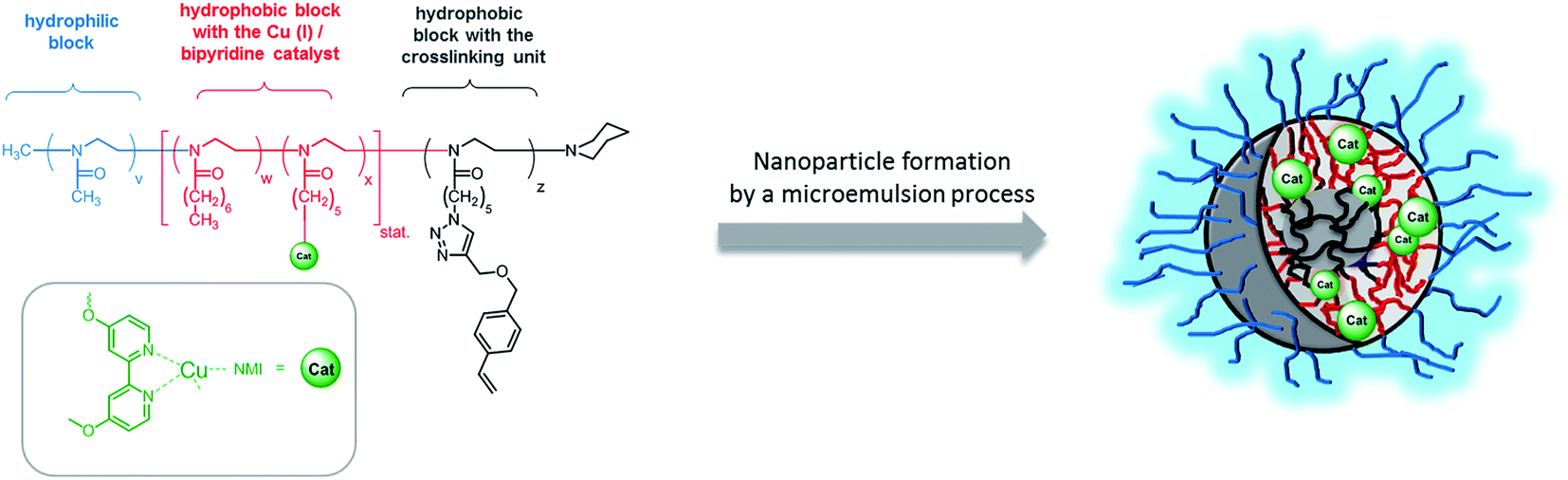 | ||
| Scheme 2 Schematic structure of the core-crosslinked nanoparticle NP1 prepared from the amphiphilic block copolymer P1. | ||
To synthesize the desired nanoparticles we needed to introduce a new 2-oxazoline monomer M1 with a styrene functionality for the nanoparticle crosslinking reaction. The monomer synthesis is depicted in Scheme 3 and is based on two fragments that are coupled via Click chemistry to yield M1. The 2-(5-azidopentyl)-2-oxazoline fragment 2 was synthesized from 6-bromohexanoic acid 1 according to Lav et al.38 in four steps with an overall yield of 43%. In the first step 1 was reacted with NaN3 in a nucleophilic substitution reaction. The α,ω-azido hexanoic acid was then converted to the corresponding active ester with N-hydroxyl succinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) as a coupling agent. The corresponding NHS-ester was then reacted with 2-chlorethylamine before the 2-oxazoline ring was formed in the presence of a KOH as a base. The second fragment 1-(prop-2-yn-1-yloxy)methyl)-4-vinylbenzene 4 was obtained by etherification of p-vinyl benzyl chloride 3 with propargyl alcohol PA in the presence of tetrabutylammonium iodide (TBAI) and sodium hydride (NaH) in 98% yield. Both fragments 2 and 4 were finally coupled by Cu-mediated click chemistry to give the desired monomer M1. The structure and purity of the monomer M1 were confirmed by 1H NMR (ESI†).
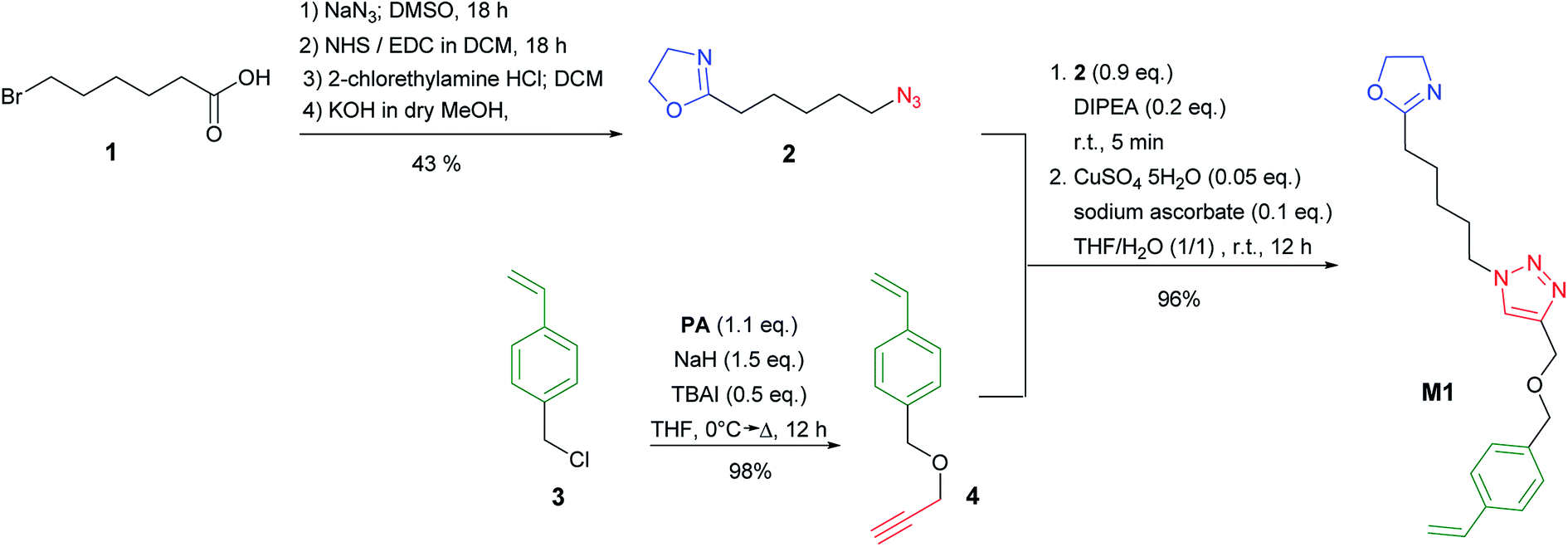 | ||
| Scheme 3 Synthesis scheme of the 2-oxazoline monomer M1. | ||
The synthesis of the amphiphilic bipyridine functionalized block copolymers was then carried out by a cationic ring-opening polymerisation mechanism (Scheme 4). The living nature of the polymerisation allows the preparation of tailor-made block copolymers of defined composition and molecular weight.39–41 Based on this approach, we now extended the amphiphilic diblock copolymer structure that is suitable for micellar catalysis by another polymer block composed of the styrene-functionalized 2-oxazoline M1 without an additional spacer (three block model, P1) or with a short poly(2-n-heptyl-2-oxazoline) block (four block model, P2). The bipyridine ligand was introduced into the polymers P1 and P2 in a polymer analogous reaction after the polymerisation has been finished. The structure, composition and molecular weight of the resulting polymers P1 and P2 were analysed by 1H NMR spectroscopy and size exclusion chromatography. Table 2 summarizes the analytical data.
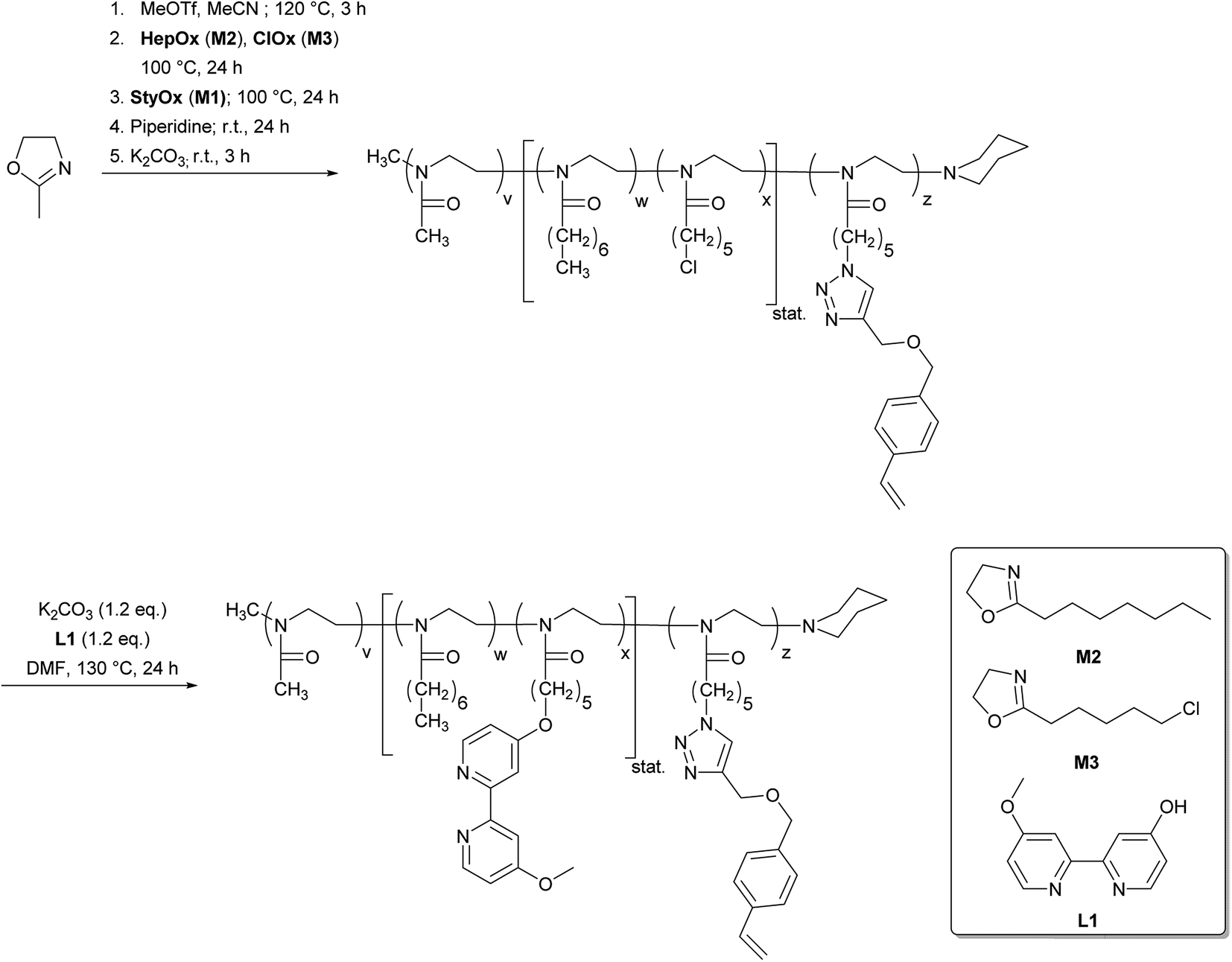 | ||
| Scheme 4 Synthesis of the amphiphilic, bipyridine-functionalized block copolymer P1. | ||
| No. | Polymer compositiona | Mnb (g mol−1) | Mnc (g mol−1) | Đc | cmcd (mol l−1) | dhe (in H2O) (nm) | dhe (in MeOH) (nm) |
|---|---|---|---|---|---|---|---|
| a Polymer composition as determined by 1H-NMR analysis; Me = 2-methyl-2-oxazolin; Hep = 2-n-heptyl-2-oxazolin; BiPy = 4-methoxy-4-pentoxy-2,2-bipyiridine units; Sty = styrene-oxazoline M1.b Determined by 1H-NMR end group analysis in CDCl3.c Determined by SEC analysis in DMF/5 g l−1 LiBr with linear PS standards.d With pyrene (0.1 mM in MeOH).e By DLS measurements in water (1 mM) at room temperature. | |||||||
| P1 | Me40(Hep5BiPy4)Sty1 | 6070 | 6860 | 1.18 | 1.2 × 10−6 | 15.2 (±0.51) | 2.8 (±0.16) |
| P2 | Me53(Hep4BiPy3)Hep3Sty1 | 7170 | 8940 | 1.50 | 5.0 × 10−7 | 18.1 (±0.43) | 3.8 (±0.27) |
The critical micelle concentrations (cmc = 0.5–1.2 µmol l−1) and the hydrodynamic radii of the micelles with 15.2 to 18.1 nm matched the typical range for amphiphilic poly(2-oxazoline)s.42–44 In the last step, P1 and P2 were employed in a microemulsion procedure using hexanediol dimethacrylate (HDDMA) as a crosslinking monomer and AIBN as the radical initiator. This resulted in stable core-crosslinked nanoparticles NP1 and NP2 with sizes in a range of 23–27 nm in water, which is slightly larger than the pure micelles. This is in good agreement with recent data.36,37 Successful core-crosslinking and nanoparticle formation was confirmed in methanol as a non-selective solvent, showing similar values for NP1 and NP2 compared to water (see Table 3). Furthermore, nanoparticle formation was confirmed by TEM measurements, which showed spherical particles for both nanoparticles NP1 and NP2 (Fig. 1).
 | ||
| Fig. 1 TEM images of NP1 (a, b) and NP2 (c, d) in water on a 100 nm and 50 nm scale. | ||
Application of the nanoparticles NP1/NP2 in catalysis
In the first set of experiments, the catalytic activity of the nanoparticles NP1/NP2 in the aerobic oxidation of benzyl alcohol as a test substrate was investigated and compared with the micellar catalytic approach based on P1/P2. As can be seen from Table 4, no difference in catalytic activity was observed when using polymer micelles based on P1/P2 or polymer nanoparticles NP1/NP2 with a crosslinked core. This result supported our hypothesis that core-crosslinked nanoparticles do not show a reduced catalytic activity compared to polymer micelles. Larger differences are observed when using different N-oxyl sources, TEMPO versus ABNO, which is in agreement with literature data.18
|
||||
|---|---|---|---|---|
| Entry | Polymer | N-Oxyl | t (h) | Conversionb (%) |
| a 40 mol of CuBr, 5 mM solution of P1/NP1 and P2/NP2.b Average conversion (2 runs) after work-up and isolation determined by 1H-NMR-spectroscopy. | ||||
| 1a | P1 | ABNO | 2 | 95 (±3) |
| 1b | P1 | TEMPO | 3 | 99 (±0) |
| 2a | P2 | ABNO | 2 | 98 (±2) |
| 2b | P2 | TEMPO | 3 | 96 (±2) |
| 4a | NP1 | ABNO | 2 | 96 (±1) |
| 4b | NP1 | TEMPO | 3 | 95 (±1) |
| 5a | NP2 | ABNO | 2 | 96 (±0) |
| 5b | NP2 | TEMPO | 3 | 95 (±1) |
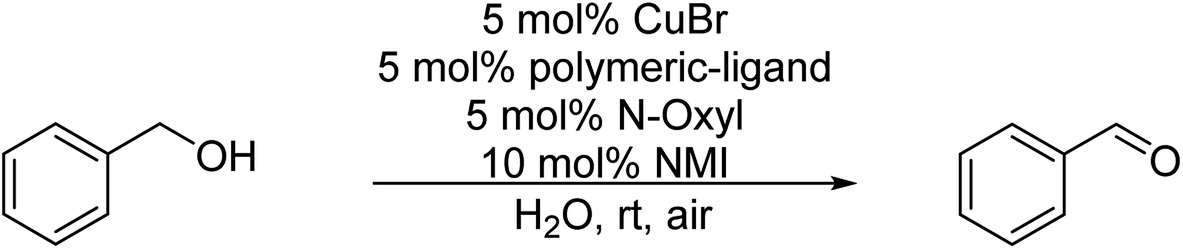
Since we did not observe any difference in catalytic activity between NP1 (three block copolymer) and NP2 (four block copolymer), we continued to work with NP1 to investigate the substrate scope of the polymeric nanoparticles. Table 5 summarizes the results of representative alcohols in the Cu(I)/N-oxyl catalysed aerobic oxidation with NP1 as polymeric ligand.
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | N-Oxyl | t (h) | Conversionb (%) | Isolated yieldc (%) |
| a 40 mol of CuBr, 5 mM solution of NP1.b Average conversion (3 runs) after work-up and isolation determined by 1H-NMR-spectroscopy.c Average isolated yield (3 runs) after purification. | |||||
| 1a | 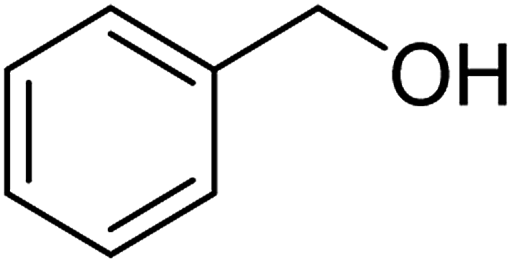 |
ABNO | 2 | 96 (±1) | 94 |
| 1b | TEMPO | 3 | 95 (±1) | 91 (±3) | |
| 2a | 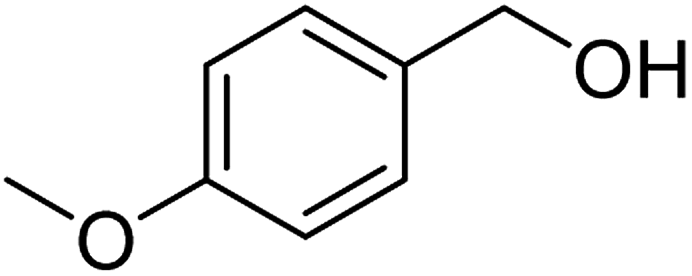 |
ABNO | 2 | 98 (±1) | 95 (±1) |
| 2b | TEMPO | 3 | 98 (±1) | 95 (±2) | |
| 3a | 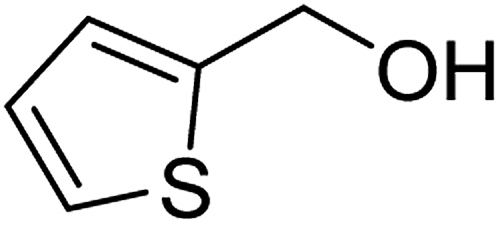 |
ABNO | 2 | 98 (±1) | 93 (±2) |
| 3b | TEMPO | 3 | 99 (±0) | 96 (±1) | |
| 4a | 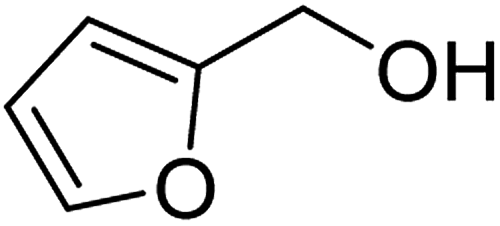 |
ABNO | 2 | 86 (±2) | 78 (±3) |
| 4b | TEMPO | 3 | 64 (±1) | 58 (±1) | |
| 5a | 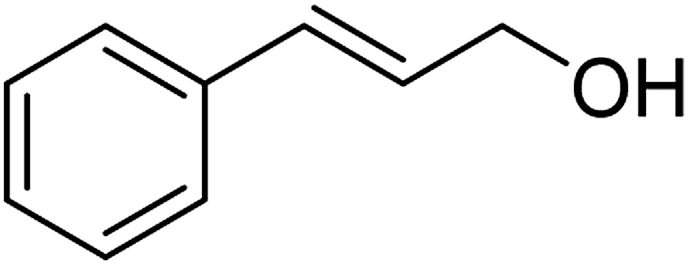 |
ABNO | 2 | 98 (±1) | 96 (±2) |
| 5b | TEMPO | 3 | 99 (±0) | 96 (±2) | |
| 6a | 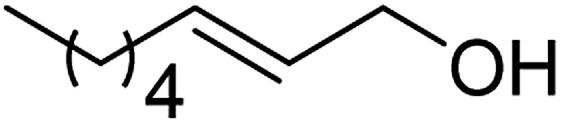 |
ABNO | 2 | 84 (±1) | 79 (±3) |
| 6b | TEMPO | 3 | 57 (±1) | 49 (±3) | |
| 7a |  |
ABNO | 2 | 46 (±2) | 39 (±3) |
| 7b | TEMPO | 3 | 2 (±0) | n. d. | |
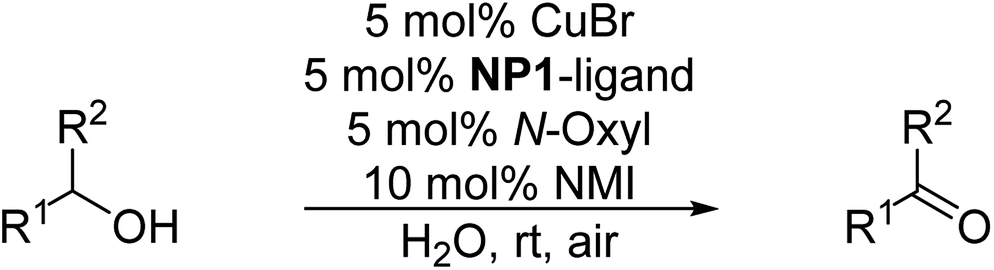
As can be seen from Table 5, aromatic alcohols gave excellent results with high yields (entry 1–5, Table 5), only the allylic and aliphatic substrates led to lower product yield due to their lower reactivity (entry 6, 7 Table 5). Importantly, for all tested alcohols there was no significant difference in activity between the micellar catalytic approach and the nanoparticle system (see ESI, Table S1†).
In the next set of experiments, we compared the recycling properties of the polymeric nanoparticles with the micellar catalytic approach using either TEMPO or ABNO as the N-oxyl source. Therefore, the oxidation of benzyl alcohol was examined in five consecutive runs for the amphiphilic polymer P1 and the nanoparticle system NP1 (Fig. 2).
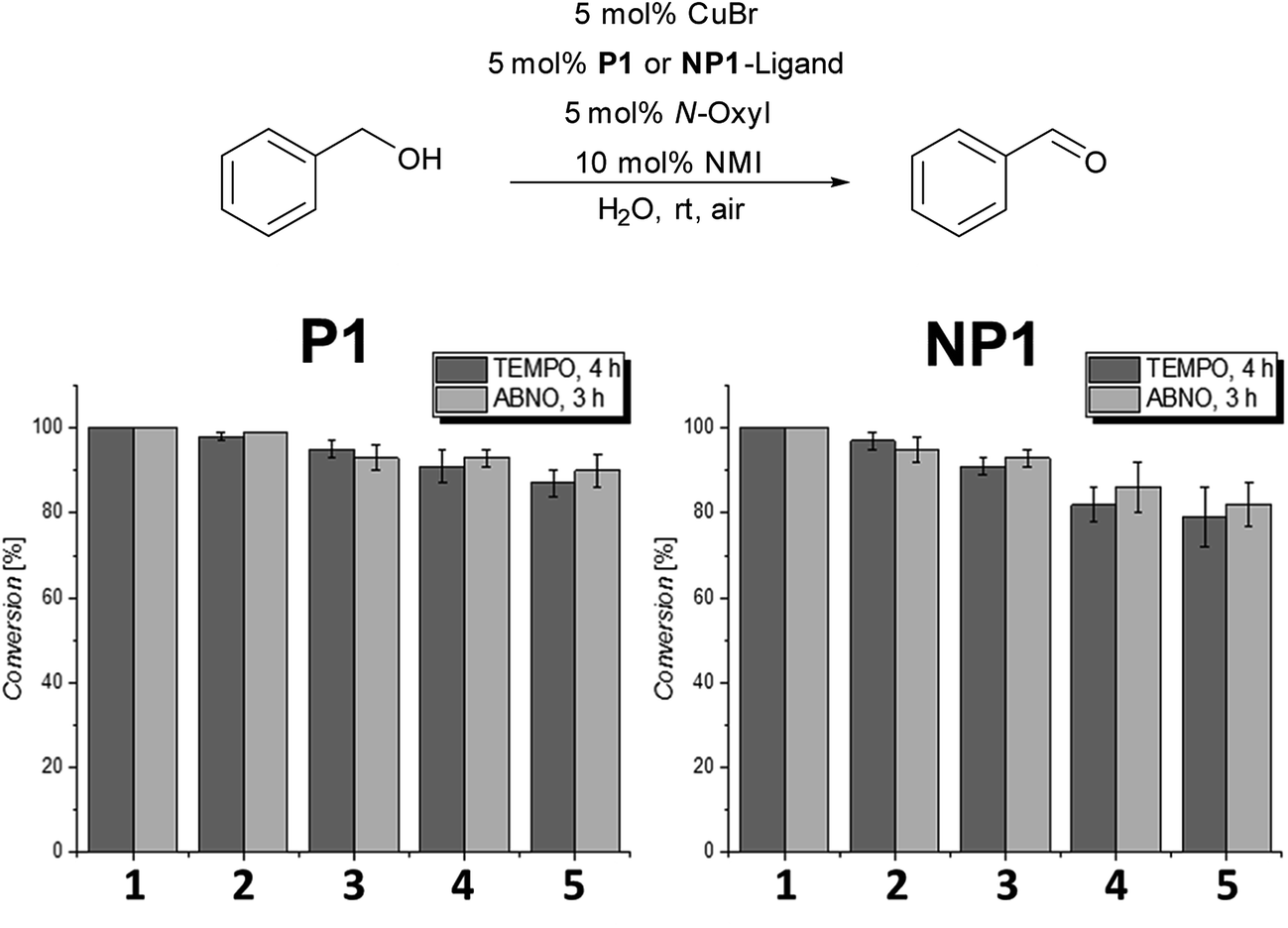 | ||
| Fig. 2 Recycling experiments in 5 consecutive runs with P1 (micellar catalytic approach) and NP1 (nanoparticle approach); conversions were determined by 1H-NMR-spectroscopy. | ||
As can be seen in Fig. 2 there is no significant difference in catalyst activity when comparing the micellar catalytic system based on P1 with the corresponding nanoparticle system NP1. The P1-system showed still around 90% conversion after the 5th run for both ABNO and TEMPO whereas the NP1-system achieved only around 80 to 85% after the 5th cycle for TEMPO and ABNO.
Multi-step-catalysis
In the last set of experiments we studied the combination of the enzymatic ester hydrolysis which resulted in the formation of alcohols followed by the Cu(I)/bipyridine based oxidation of these alcohols to their corresponding aldehydes. To realize such a two-step-reaction, various aspects had to be considered.In the initial experiments, we conducted the tandem reaction as a one-pot two-step reaction and isolated the intermediary product benzyl alcohol. After 2 h at room temperature, we obtained a 97% conversion in PBS buffer versus 81% in water, clearly favouring the PBS buffer for the enzymatic step (Table 6, entry 1–2). However, the second step, involving the aerobic oxidation of benzyl alcohol to the corresponding benzaldehyde gave only unsatisfactory conversion of 20–26% in PBS buffer, even after extending the reaction time from 2 h to 24 h (Table 6, entry 3).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | T (°C) | t (h) | DIPEA (eq.) | Conversionb step 1 (%) | Conversionb step 2 (%) |
| a First reaction step: CAL-B, 40 °C, 20 min; second step: 5 mol% CuBr, 5 mol% NP1-ligand, 5 mol% ABNO, 10 mol% NMI.b Average conversion (3 runs) after work-up and isolation determined by 1H-NMR-spectroscopy.c DIPEA (1 eq.), 40 °C, 20 min. | ||||||
| 1 | H2O | rt | 2 | 0 | 81 (±2) | 4 (±1) |
| 2 | PBS | rt | 2 | 0 | 97 (±3) | 20 (±3) |
| 3 | PBS | rt | 24 | 0 | 97 (±3) | 26 (±2) |
| 4c | PBS | rt | 4 | 1 | ≥99 | 82 (±3) |
| 5c | PBS | 40 | 4 | 1 | ≥99 | 96 (±1) |
No catalyst inhibition was expected by the side product acetic acid, due to its hydrophilicity which should prevent it from being solubilized in the Cu(I)/bipyridine nanoparticle. However, the acetic acid led to a shift in pH of the aqueous medium to around 5 that was assumed to be the reason for the significantly lower Cu(I)/bipyridine activity. A possibility to overcome the problem was to add a base such as N,N-diisopropyl ethylamine (DIPEA) to the aqueous medium after the ester hydrolysis and thus neutralize the acetic acid. Consequently, the protocol for the one-pot two-step reaction was changed and 1 eq. DIPEA was added after the enzymatic hydrolysis and allowed to stir for 20 min before the aerobic oxidation for the benzyl alcohol was carried out in the presence of NP1. The results obtained now showed quantitative conversion of the ester hydrolysis step after 20 min at 40 °C and a 82 to 96% conversion for the aerobic oxidation step at r.t. and 40 °C, respectively (Table 6, entry 4, 5). In an attempt to assess the scope and utility of this one-pot two-step system, a small series of aromatic/allylic alcohols with various functional groups was examined (Table 6). The results highlight the efficiency of the consecutive enzymatic ester hydrolysis and aerobic oxidation reaction with yields of 73 to 93% of the aldehydes. By using 20 times the amount of catalysts (NP1, CALB) and reagents two upscale experiments were carried out for the two substrates benzyl acetate and p-methoxy benzyl acetate (Table 7, entry 6, 7). The results for the isolated yields of the corresponding aldehydes were 93 and 95%, respectively and were in excellent agreement with the previous reported product yields of the small scale experiments (Table 7, entry 1, 2).
|
||||
|---|---|---|---|---|
| Entry | Substrate | Conversionb step 1 (%) | Conversionb step 2 (%) | Isolated yieldc (%) |
| a Reactions were carried out in PBS-buffer (pH 7.4) at 40 °C; (1) CAL-B, 20 min; (2) DIPEA (1 eq.) 20 min; (3) 5 mol% CuBr, 5 mol% NP1-ligand (12.14 mg, 2 µmol, 0.0125 eq.), 5 mol% ABNO, 10 mol% NMI, 4 h.b Average conversion (3 runs) after work-up and isolation determined by 1H-NMR-spectroscopy.c Average isolated yield of the aldehyde (3 runs).d 5 h reaction time lead to nearly quantitative conversion.e Scale-up-experiment: CuBr (86 mg, 600 µmol, 5 mol%), NP1-ligand (243 mg, 40 µmol, 5 mol%), ABNO (22.4 mg, 160 µmol, 5 mol%), NMI (25.6 µl, 320 µmol, 10 mol%), acetate (3.2 mmol, benzyl: 480.6 mg; 4-methoxybenzyl: 576.6 mg, 1 eq.), same reaction conditions as described in a. | ||||
| 1 |  |
≥99 | 96 (±1) | 93 |
| 2 | 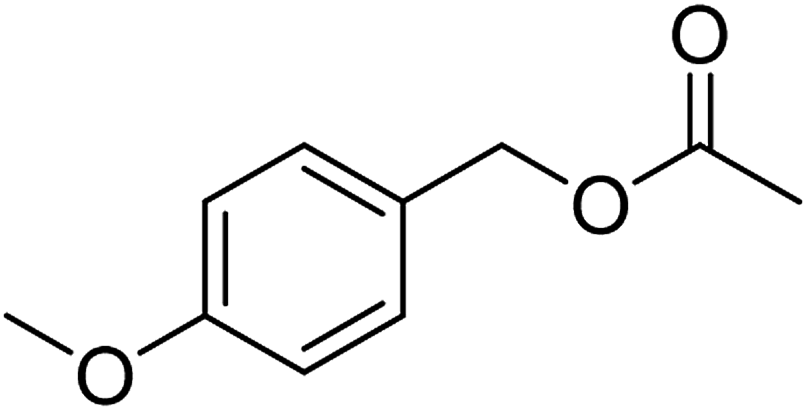 |
≥99 | 97 (±1) | 93 |
| 3d | 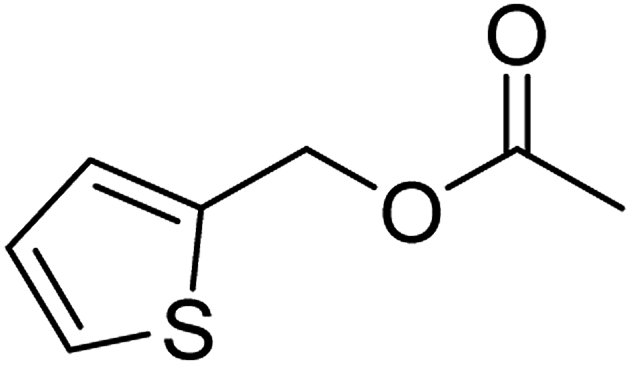 |
≥99 | 84 (±2) | 80 |
| 4d | 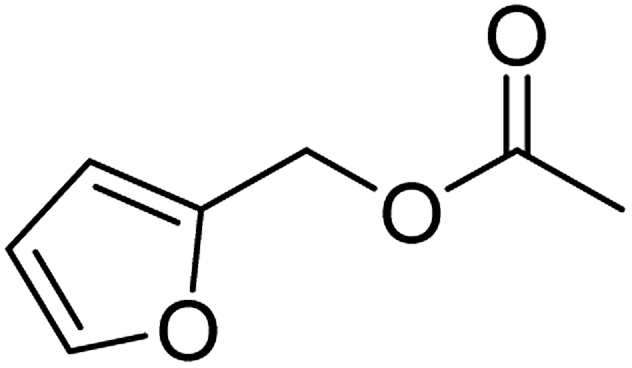 |
≥99 | 78 (±4) | 73 |
| 5 | 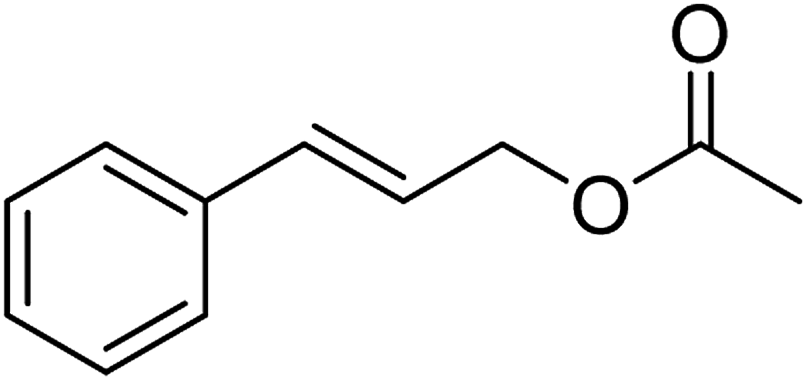 |
≥99 | 96 (±1) | 91 |
| 6e | 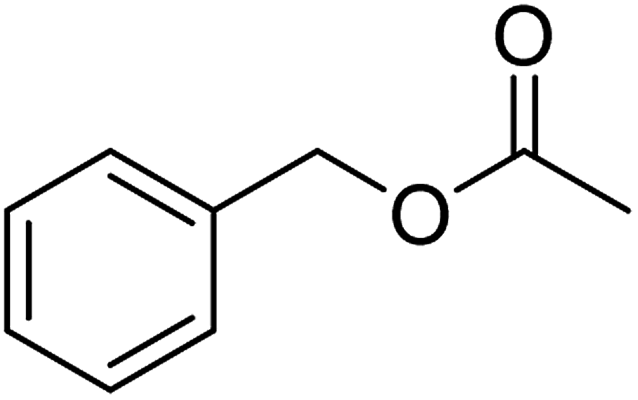 |
≥99 | 93 (±2) | 93 |
| 7e | 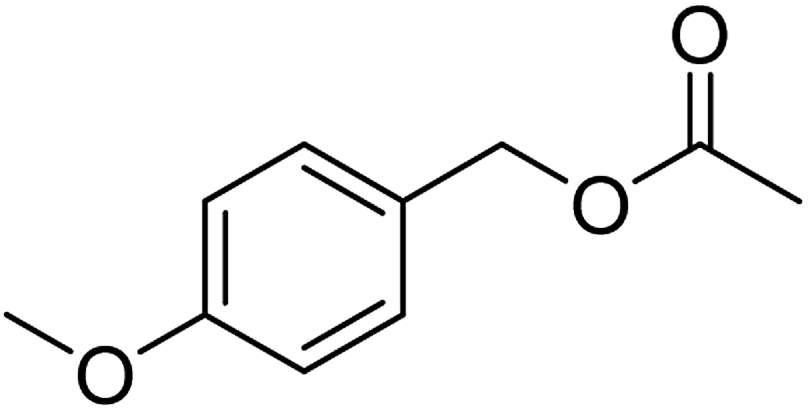 |
≥99 | 96 (±1) | 95 |
Conclusions
Herein, we presented the first successful one-pot two-stage reaction of an enzymatic ester hydrolysis followed by Cu(I)/bipyridine catalysed aerobic oxidation of the alcohol intermediate to the corresponding aldehyde. The key to the successful one-pot reaction in an aqueous medium was the site-isolation of the Cu(I)/bipyridine catalyst in core-crosslinked nanoparticles with a hydrophilic shell and a hydrophobic core which were prepared by a microemulsion process. The nanoparticle design efficiently prevents any interaction of the Cu/bipyridine catalyst with the enzyme that has been shown to lead to catalyst inhibition. Furthermore, it provides a hydrophobic nano-environment for the Cu/bipyridine system as a prerequisite for catalyst activity in the aerobic oxidation. The nanoparticles displayed no significant loss in activity compared to the micellar catalytic approach and could also be used in five consecutive runs still displaying a 80% substrate conversion in the 5th run. The one-pot tandem reaction was finally conducted as a two-step procedure, where the acetyl ester hydrolysis proceeded quantitatively after 20 min at 40 °C in the presence of the enzyme CAL-B followed by addition of DIPEA to neutralize the acetic acid side product. Then, the Cu(I)/bipyridine functionalized nanoparticle NP1 was added to the reaction mixture to carry out the aerobic oxidation of the intermediate alcohols to the corresponding aldehydes in high yields of 73–93% for various aromatic and allylic acetic esters to their corresponding aldehydes. We believe that this approach of metal-catalyst compartmentalization within core–shell nanoparticles should also be of interest to other catalyst combinations to prevent mutual catalyst inhibition on the one side and provide a hydrophobic reaction environment for the metal-catalyst in aqueous medium.Experimental
Measurements and materials
1H (500.13 MHz) and 13C NMR (100.63 MHz) spectra were recorded on a Bruker DRX 500 spectrometer. GC-EI-HRMS measurements were performed on a Thermo Electro at 160 °C and 70 eV, using perfluorokerosene as reference. Gel permeation chromatography (SEC) was carried out on a DMF (5 g l−1 LiBr) based SEC at 60 °C with PSS GRAM analytical 1000 A and 30 °A columns equipped with a Knauer RI detector Smartline 2300 using linear polystyrene standards for the poly(2-oxazoline). Dynamic light scattering was carried out on a Malvern Zetasizer Nano-Z5 with a HeNe-laser (λ = 633 nm). Fluorescence spectroscopy was recorded on a Hitachi F2700 using a wavelength of λ = 334 nm and a 0.1 mM solution of pyrene in methanol. The analyses were carried out with the program FLWinLab. TEM measurements were performed on a Philips CM200 using an Orius SC200 camera of Gatan. All commercially available chemicals were purchased from Sigma Aldrich, TCI Chemicals, ABCR and Acros Organics and used without further purification. CALB immobilized on Immobead 150 was also purchased from Sigma Aldrich.Monomer synthesis
1H-NMR (400 MHz, CDCl3): δ = 0.86 ppm (m, CH3), 1.30 (m, 4 × CH2), 1.61 (quin, 3J = 7.4 Hz, CH3CH2), 2.25 (t, 3J = 7.7 Hz, CCH2), 3.80 (t, 3J = 9.4 Hz, CH2N), 4.20 (t, 3J = 9.5 Hz, CH2O). 13C-NMR (100 MHz, CDCl3): δ = 14.0 ppm (CH3), 22.6 (CH3CH2), 25.9 (CH2CH2CO), 27.9 (CH2CH2CH2CH3), 28.9 (CH2CN), 29.2 (CH2CH2CH2CN), 31.6 (CH2CH2CH3), 54.3 (CH2N), 67.1 (CH2O), 168.6 (CO). HR-ESI-MS: Mcalculated = 169.1467 [M = C10H19NO]; Mmeasured = 170.1538 [M + H]+.
1H-NMR (400 MHz, CDCl3): δ = 1.47 pp (m, CH2CH2CH2Cl), 1.63 (quin, 3J = 7.6 Hz, CH2CH2C(O)N), 1.76 (quin, 3J = 7.2 Hz, CH2CH2O), 2.25 (t, 3J = 7.5 Hz, CH2C(O)N), 3.5 (t, 3J = 6.7 Hz, NCH2CH2O), 3.78 (t, 3J = 9.7 Hz, CH2Cl), 4.19 (t, 3J = 9.5 Hz, NCH2CH2O). 13C-NMR (100 MHz, CDCl3): δ = 25.1 ppm (CH2CH2C(O)N), 26.3 (CH2CH2CH2C(O)N), 27.6 (CH2CH2CH2Cl), 32.1 (CH2C(O)N), 44.7 (CH2N), 54.3 (CH2CH2CH2Cl), 67.1 (NCH2CH2O), 168.1 (C(N)O). HR-ESI-MS: Mcalculated = 175.0764 [M = C8H14ClNO]; Mmeasured = 176.0838 [M + H]+.
1H-NMR (500 MHz, CDCl3): δ = 1.28–1.42 ppm (m, CCH2CH2CH2), 1.48–1.64 (m, CCH2CH2CH2CH2CH2N3), 2.20 (td, J = 7.46/1.22 Hz, CCH2), 3.19 (td, J = 6.85/1.47 Hz, CH2N3), 3.66–3.82 (m, CH2O), 4.14 (td, J = 9.42/1.71 Hz, CH2N). 13C-NMR (125 MHz, CDCl3): δ = 25.3 ppm (CH2CH2C(O)N), 26.1 (CH2CH2CH2C(O)N), 27.6 (CH2CH2CH2N3), 28.4 (CH2C(O)N), 51.1 (CH2N), 54.2 (CH2CH2CH2N3), 67.0 (NCH2CH2O), 168.1 (C(O)N). HR-ESI-MS: Mcalculated = 182.1168 [M = C8H14N4O]; Mmeasured = 183.1246 [M + H]+.
1H-NMR (500 MHz, CDCl3): δ = 2.49 ppm (t, J = 2.2 Hz, CHC), 4.19 (d, J = 2.4, CHCCH2), 4.63 (s, OCH2Ar), 5.27 (d, J = 10.8 Hz, CH2CH), 5.77 (d, J = 17.6 Hz, CH2CH), 6.74 (dd, J = 17.6 Hz/11.2 Hz, CH2CH), 7.31–7.45 (m, Ar). 13C-NMR (125 MHz, CDCl3): δ = 56.9 (CH2CCH), 71.1 (OCH2Ar), 74.6 (CH2CCH), 79.5 (CH2CCH), 113.9 (CH2CH), 126.2 (2 × CHCCH), 128.3 (2 × CH2CCH), 136.4 (CH2CH), 136.7 (CH2C(Ar)), 137.2 (CHC(Ar)).
1H-NMR (500 MHz, CDCl3): δ = 1.32–1.42 (m, CCH2CH2CH2), 1.62–1.71 (m, CCH2CH2CH2CH2), 1.86–1.96 (m, CCH2CH2), 2.26 (t, J = 7.48 Hz, CCH2CH2), 3.79 (t, J = 9.46 Hz, OCH2CH2N), 4.18 (td, J = 9.42/1.71 Hz, OCH2CH2N), 4.28–4.38 (m, CH2NN), 4.58 (s, CHCCH2), 4.66 (s, OCH2Ar), 5.23 (d, J = 10.8 Hz, CH2CH), 5.74 (dd, J = 17.70, 0.61 Hz, CH2CH), 6.70 (dd, J = 17.6 Hz/11.2 Hz, CH2CH), 7.28–7.42 (m, Ar), 7.53 (s, CHN). 13C-NMR (125 MHz, CDCl3): δ = 25.1 ppm (CH2CH2C(O)N), 25.9 (CH2CH2CH2C(O)N), 27.5 (CH2CH2CH2N), 29.9 (CH2C(O)N), 50.0 (CH2N), 54.2 (CH2CH2CH2N), 63.6 (OCH2CN), 67.1 (NCH2CH2O), 72.2 (OCH2Ar), 113.8 (CH2CH), 122.3 (CHN), 126.2 (2 × CHCCH), 128.1 (2 × CH2CCH), 136.4 (CH2CH), 137.0 (CH2C(Ar)), 137.3 (CHC(Ar)), 145.0 (OCH2CN), 168.0 (C(O)N). HR-ESI-MS: Mcalculated = 354.2056 [M = C20H26N4O2]; Mmeasured = 355.2136 [M + H]+.
1H-NMR (400 MHz, CDCl3): δ = 3.91 ppm (s, CH3O), 6.53 (dd, 1J = 7.2 Hz, CHCHCOCH3), 6.88 (dd, 1J = 5.8 Hz, CHCHCOH), 7.11 (s, br, CCHCOH), 7.39 (s, CCHCOCH3), 7.71 (d, 1J = 7 Hz, CHCHCOH), 8.42 (d, 1J = 5.8 Hz, CHCHCOCH3). 13C-NMR (100 MHz, CDCl3): δ = 55.5 ppm (CH3O), 106.1 (2 × CCHCOR), 111.2 (2 × CHCHCOR), 150.2 (2 × CHCHCOR), 167.0 (2 × NCCH, 2 × COR). HR-ESI-MS: Mcalculated = 202.0742 [M = C11H10N2O2]; Mmeasured = 203.0816 [M + H]+.
Polymer synthesis
All polymerizations were carried out in Schlenk tubes under inert atmosphere using freshly distilled and dried solvents. A typical procedure for P1 was as follows:To 2-methyl-2-oxazoline (990.10 µl, 11.75 mmol, 35 eq.) in 10 ml acetonitrile was given methyltriflate (37.99 µl, 335.72 µmol, 1 eq.) at 0 °C. After stirring the mixture for 3 h at 120 °C, 2-heptyl-2-oxazoline (295.97 µl, 1.68 mmol, 5 eq.) and (2-(5-chloropentyl)-2-oxazoline) (294.85 mg, 1.68 mmol, 5 eq.) were added at room temperature. After stirring the mixture for 24 h at 100 °C M2 (356.99 mg, 1.01 mmol, 3 eq.) was added at room temperature. The mixture was stirred for 18 h at 100 °C and the reaction terminated at room temperature by addition of piperidine (36.54 µl, 369.28 µmol, 1.1 eq.) for 12 h. After removal of the solvent, the residue was dissolved in 15 ml of chloroform and stirred with K2CO3 for 3 h. After filtration, PP1 was purified by precipitation in ice cold diethyl ether and dried under reduced pressure.
1H-NMR (500 MHz, CDCl3): δ = 0.85 ppm (s(br), CH3(HepOx)), 1.26 (s(br), 4 × CH2(HepOx)), 1.45–1.90 (m, CH2(HepOx), 3 × CH2(ClOx), 2 × CH2(StyOx), 3 × CH2(Pip)), 1.90–2.06 (s(br), CH2(StyOx)), 2.06–2.19 (m, CH3(MeOx), 2 × CH2(Pip)), 2.19–2.50 (m, CH2(HepOx), CH2(ClOx), CH2(StyOx)), 2.94/3.01 (CH3(In)), 3.19–3.75 (m, CH2–CH2(backbone), CH2(ClOx)), 4.33 (s(br), CH2(StyOx)), 4.58–4.70 (m, 2 × CH2(StyOx)), 5.20–5.26 (m, CH(StyOx)), 5.69–5.79 (m, CH(StyOx)), 6.64–6.74 (m, CH(StyOx)), 7.28–7.42 (m, 4 × CHAr(StyOx)), 7.49–7.65 (s(br), CHTriazol(StyOx)).
L1 (18.23 mg, 0.90 mmol, 1.2 eq.) and K2CO3 (12.5 mg, 0.90 mmol, 1.2 eq.) were added to a solution of PP1 (1 g, 1 eq. related to Cl-functionality) in 20 ml N,N-dimethylformamide. After stirring the mixture for 24 h at 100 °C, the solvent was removed. The residue was dissolved in dichloromethane, filtered and the solvent was removed. The polymer was purified by precipitation in ice cold diethyl ether and dialyzed against ethanol (MWCO 1000) for 24 h. After removal of the solvent, the polymer P1 was again precipitated in ice cold diethyl ether. Drying yielded 0.83 g polymer P1 as a white powder in 82% yield.
1H-NMR (500 MHz, CDCl3): δ = 0.85 ppm (s(br), CH3(HepOx)), 1.26 (s(br), 4 × CH2(HepOx)), 1.45–1.90 (m, CH2(HepOx), 3 × CH2(BiPyOx), 2 × CH2(StyOx), 3 × CH2(Pip)), 1.90–2.06 (s(br), CH2(StyOx)), 2.06–2.19 (m, CH3(MeOx), 2 × CH2(Pip)), 2.19–2.50 (m, CH2(HepOx), CH2(BiPyOx), CH2(StyOx)), 2.94/3.01 (CH3(In)), 3.19–3.75 (m, CH2–CH2(backbone)), 3.93 (s(br), OCH3(BiPyOx)), 4.10 (s(br), OCH2(BiPyOx)), 4.33 (s(br), CH2(StyOx)), 4.58–4.70 (m, 2 × CH2(StyOx)), 5.20–5.26 (m, CH(StyOx)), 5.69–5.79 (m, CH(StyOx)), 6.64–6.74 (m, CH(StyOx)), 6.82 (s(br), 2 × CH(BiPyOx)), 7.28–7.42 (m, 4 × CHAr(StyOx)), 7.49–7.65 (s(br), CHTriazol(StyOx)), 7.94 (s(br), 2 × CH(BiPyOx)), 8.44 (s, 2 × CH(BiPyOx)).
P2: The synthesis of the precursor copolymer PP2 was similar to the one described for PP1 starting with the addition of methyltriflate (37.99 µl, 335.72 µmol, 1 eq.) to 2-methyl-2-oxazoline (990.10 µl, 11.75 mmol, 35 eq.) in 10 ml acetonitrile at 0 °C. After stirring the mixture for 3 h at 120 °C, 2-heptyl-2-oxazoline (295.97 µl, 1.68 mmol, 5 eq.) and (2-(5-chloropentyl)-2-oxazoline) (294.85 mg, 1.68 mmol, 5 eq.) were added at room temperature to form the second block for 24 h at 100 °C. Then 2-heptyl-2-oxazoline (295.97 µl, 1.68 mmol, 5 eq.) was added at room temperature and polymerization proceeded at 100 °C for another 24 h (3rd block) before M2 (356.99 mg, 1.01 mmol, 3 eq.) was added at room temperature to form the fourth block after 18 h at 100 °C. The polymerisation was terminated at room temperature by addition of piperidine (36.54 µl, 369.28 µmol, 1.1 eq.) for 12 h. After precipitation in diethyl ether PP2 as a white powder. Coupling reaction with the bipyridine ligand L1 was carried out as described before by using (1 g, 1 eq. related to Cl-functionality) in 20 ml N,N-dimethylformamide and addition of L1 (18.23 mg, 0.90 mmol, 1.2 eq.) and K2CO3 (12.5 mg, 0.90 mmol, 1.2 eq.). Drying yielded 0.87 g polymer P1 as a white powder in 86% yield. The 1H-NMR (500 MHz, CDCl3) for P2 shows the same peak signals as P1 (see above). Further analytical data of P1 and P2 are summarized in Table 1.
Nanoparticle synthesis
Micellar/nanoparticle catalysis
All catalytic oxidations were carried out in open Schlenk tubes under air atmosphere at 20 °C and constant stirring rates. For preparation of the catalytically active polymer solution only freshly distilled and dried acetonitrile was used. A typical procedure was as follows:CuBr (4.30 mg, 30 µmol, 0.05 eq.) was added to polymer P1 or nanoparticle NP1 (51.34 mg, 10 µmol, 0.017 eq.) in 2 ml acetonitrile under an inert atmosphere. After stirring for 1 h at room temperature, the solvent was removed and the polymer was dissolved in 2 ml water (5 mM solution). N-Methylimidazole (4.78 µl, 60 µmol, 0.1 eq.), N-oxyl-radical (ABNO 4.21 mg/TEMPO 4.69 mg, 30 µmol, 0.05 eq.) and benzyl alcohol (62.38 µml, 0.6 mmol, 1.0 eq.) were added to the solution. The solution was stirred at room temperature with a constant rate in an open flask for 2 h (ABNO) or 3 h (TEMPO). To isolate the product, the solution was extracted with diethyl ether (4 × 20 ml). The organic layers were dried with MgSO4 and removed under reduced pressure. The product was purified via column chromatography (cyclohexane/ethyl acetate 5/1) and yielded with 94%. The catalyst solution in water could be used for further runs by simply adding N-methylimidazole and N-oxyl radical again.
Enzymatic ester cleavage
All enzymatic ester cleavages were carried out under air atmosphere at 40 °C and constant stirring rates. A typical procedure was as follows: benzyl acetate (86.63 µl, 600 µmol) was added to 50 mg of the immobilized enzyme CALB (Immobead 150) in 1 ml of PBS-buffer. After stirring for 20 min at 40 °C, the solution was extracted with diethyl ether (3 × 10 ml). The organic layers were dried with MgSO4 and the solvent removed under reduced pressure. The product was purified via column chromatography (cyclohexane/ethyl acetate 5/1). Benzyl alcohol (60.3 mg, 557 µmol, 93%) was obtained as a colorless liquid.One-pot multi-step-reaction
All reactions were carried out in open Schlenk tubes under air atmosphere at 40 °C and constant stirring rates. For the preparation of the catalytically active nanoparticle solution only freshly distilled and dried acetonitrile was used. A typical procedure is described below:Acknowledgements
We thank Monika Meuris (Prof. J. C. Tiller, BCI, TU Dortmund) for preparing the TEM-images. Furthermore, we thank the working group of Prof. Tiller for providing some analytical devices.Notes and references
- L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed.
- J. C. Waslike, S. J. Obrey, R. T Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef PubMed.
- Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC.
- C. A. Denard, J. F. Hartwig and H. Zhao, ACS Catal., 2013, 3, 2856–2864 CrossRef CAS.
- H. Gröger and W. Hummel, Curr. Opin. Chem. Biol., 2014, 19, 171–179 CrossRef PubMed.
- P. M. Dinh, J. A Howarth, A. R Hudnott and J. M. J. Williams, Tetrahedron Lett., 1997, 37, 7623–7626 CrossRef.
- E. Burda, W. Hummel and H. Gröger, Angew. Chem., Int. Ed., 2008, 47, 9551–9554 CrossRef CAS PubMed.
- S. Borchert, E. Burda, J. Schatz, W. Hummel and H. Gröger, J. Mol. Catal. B: Enzym., 2012, 84, 89–93 CrossRef CAS.
- H. Sato, W. Hummel and H. Gröger, Angew. Chem., Int. Ed., 2015, 54, 4488–4492 CrossRef CAS PubMed.
- K. Tenbrink, M. Seßler, J. Schatz and H. Gröger, Adv. Synth. Catal., 2011, 353, 2363–2367 CrossRef CAS.
- N. Ríos-Lombardía, C. Vidal, E. Liardo, F. Morís, J. García-Álvarez and J. González-Sabín, Angew. Chem., Int. Ed., 2016, 55, 8691–8695 CrossRef PubMed.
- N. Ríos-Lombardı, C. Vidal, M. Cocina, F. Morıs, J. García-Alvarez and J. González-Sabin, Chem. Commun., 2015, 51, 10937–10940 RSC.
- J. S. Willemsen, R. P. Megens, G. Roelfes, J. C. M. van Hest and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2014, 14, 2892–2898 CrossRef.
- J. M. Sperl, J. M. Carsten, J.-K. Guterl, P. Lommes and V. Sieber, ACS Catal., 2016, 6, 6329–6334 CrossRef CAS.
- M. Heidlindemann, G. Rulli, A. Berkessel, W. Hummel and H. Gröger, ACS Catal., 2014, 4, 1099–1103 CrossRef CAS.
- J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901–16910 CrossRef CAS PubMed.
- J. M. Hoover, B. L. Rayland and S. S. Stahl, J. Am. Chem. Soc., 2011, 135, 2357–2367 CrossRef PubMed.
- J. E. Steves and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 15742–15745 CrossRef CAS PubMed.
- J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156–1184 CrossRef CAS.
- L. I. Zakharkin and I. M. Khorlina, Tetrahedron Lett., 1962, 3, 619–620 CrossRef.
- J. S. Cha, J. Hyun Chun, J. M. Kim, D. Y. Lee and S. Dong Cho, Bull. Korean Chem. Soc., 1999, 20, 1373–1374 CAS.
- H. S. Kim, Y. J. Kim and Y. R. Seo, Journal of Cancer Prevention, 2015, 20, 232–240 CrossRef PubMed.
- I. N. Francesco, F. Fontaine-Vive and S. Antoniotti, ChemCatChem, 2014, 6, 2784–2791 CrossRef.
- V. Polshettiwar and R. S. Varma, Acc. Chem. Res., 2008, 41, 629–639 CrossRef CAS PubMed.
- E. M. Anderson, M. Karin and O. Kirk, Biocatal. Biotransform., 1998, 16, 181–204 CrossRef CAS.
- P. Berglund, Biomol. Eng., 2001, 18, 13–22 CrossRef CAS PubMed.
- S. Lutz, Tetrahedron: Asymmetry, 2004, 15, 2743–2748 CrossRef CAS.
- B. Chen, J. Hu, E. M. Miller, W. Xie, M. Cai and R. A. Gross, Biomacromolecules, 2008, 9, 463–471 CrossRef CAS PubMed.
- H. Sand and R. Weberskirch, RSC Adv., 2015, 5, 38235–38242 RSC.
- K. B. Thurmond, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1997, 119, 6656–6665 CrossRef CAS.
- V. Butun, N. C. Billingham and S. P. Armes, J. Am. Chem. Soc., 1998, 120, 12135–12136 CrossRef.
- Y. Kim, M. H. Pourgholami, D. L. Morrisba and M. H. Stenzel, J. Mater. Chem., 2011, 21, 12777–12783 RSC.
- N. Engelhardt, A. Ernst, A.-L. Kampmanna and R. Weberskirch, Macromol. Chem. Phys., 2013, 214, 2783–2791 CrossRef CAS.
- L. C. Lee, J. Lu, M. Weck and C. W. Jones, ACS Catal., 2016, 6, 784–787 CrossRef CAS.
- J. Lu, J. Dimroth and M. Weck, J. Am. Chem. Soc., 2015, 137, 12984–12989 CrossRef CAS PubMed.
- A.-L. Kampmann, M. Luksin, I. Pretzer and R. Weberskirch, Macromol. Chem. Phys., 2016, 217, 1704–1711 CrossRef CAS.
- A.-L. Kampmann, T. Grabe, C. Jaworski and R. Weberskirch, RSC Adv., 2016, 6, 99752–99763 RSC.
- T.-X. Lav, P. Lemechko, E. Renard, C. Amiel, V. Langlois and G. Volet, React. Funct. Polym., 2013, 73, 1001–1008 CrossRef CAS.
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21, 151–208 CrossRef CAS.
- B. M. Culbertson, Prog. Polym. Sci., 2002, 27, 579–626 CrossRef CAS.
- S. Kobayashi and H. Uyama, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 192–209 CrossRef CAS.
- D. Schönfelder, K. Fischer, M. Schmidt, O. Nuyken and R. Weberskirch, Macromolecules, 2005, 38, 254–262 CrossRef.
- B. M. Rossbach, K. Leopold and R. Weberskirch, Angew. Chem., Int. Ed., 2006, 45, 1309–1312 CrossRef CAS PubMed.
- L. Lempke, A. Ernst, F. Kahl, R. Weberskirch and N. Krause, Adv. Synth. Catal., 2016, 358, 1491–1499 CrossRef CAS.
- H. Witte and W. Seeliger, Justus Liebigs Ann. Chem., 1974, 996–1009 CrossRef CAS.
- J. Kim and M. H. Litt, J. Polym. Sci., 1989, 27, 2711–2722 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05451c |
| This journal is © The Royal Society of Chemistry 2017 |