
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanostructured WO3 photoanodes for efficient water splitting via anodisation in citric acid†
Jifang Zhang a,
Ivette Sallesb,
Sam Peringc,
Petra J. Cameronc,
Davide Mattiaa and
Salvador Eslava*a
a,
Ivette Sallesb,
Sam Peringc,
Petra J. Cameronc,
Davide Mattiaa and
Salvador Eslava*a
aDepartment of Chemical Engineering, University of Bath, Bath, UK. E-mail: s.eslava@bath.ac.uk
bDepartament d'Enginyeria Química, Biològica i Ambiental, Universitat Autònoma de Barcelona, Bellaterra 08193, Spain
cDepartment of Chemistry, University of Bath, BA2 7AY, UK
First published on 13th July 2017
Abstract
In this work we report the production of nanostructured WO3 photoanodes for solar water splitting produced via anodisation using for the first time citric acid (CA), a safer and more environmentally friendly alternative to fluoride-based electrolytes. Photoelectrochemical solar water splitting has shown potential as a renewable method for hydrogen production, a key ingredient to advance the de-carbonisation of our economy. Many methods to produce WO3 photoanodes are time-consuming and require high temperatures and/or toxic chemicals, such as fluoride-based electrolytes. Here we report on a systematic investigation of the anodisation of tungsten using CA to establish a relation between (i) anodisation parameters (current, time and electrolyte), (ii) the resulting nanostructured morphology and (iii) its performance as a photoanode for water splitting. Characterisation was carried out by X-ray diffraction, scanning electron microscopy, linear sweep voltammetry, and ultraviolet-visible spectroscopy. After optimisation, the obtained WO3 photoanodes produced a photocurrent of 0.88 mA cm−2 at 1.0 V vs. Ag/AgCl in 0.5 M aqueous H2SO4 under AM1.5 solar irradiation. At low applied potentials (below 0.67 V vs. Ag/AgCl), closer to practical conditions, the photoanodes produced in CA outperformed a conventional counterpart made using a NH4F electrolyte. The CA-anodised photoanodes also showed higher stability, retaining 90% of their activity after 1 h of chopped solar illumination. This work demonstrates the promise of anodisation in citric acid as an efficient and more sustainable method for the production of WO3 photoanodes for solar water splitting.
Introduction
Photoelectrochemical (PEC) water splitting is a promising approach to produce hydrogen, a sustainable clean fuel, and significant progress has been made since its first advent.1 In a PEC cell, the choice of materials for electrodes is essential. Among different metal oxide semiconductors, tungsten trioxide (WO3) has received much attention due to its band gap of 2.7 eV within the visible-light region, the relatively long diffusion length (150 nm) of its hole carriers, and its excellent stability in acidic conditions.2 Many methods, including chemical vapour deposition, solvothermal, sol–gel and anodisation have been deployed to produce nanostructured WO3.3–10 Their optimised performance is associated with the production of larger active surface areas, better light harvesting capability, and more effective transport of charge carriers. However, many deposition methods such as chemical vapour deposition and flame deposition involve complex experimental setups or conditions which compromise the large scale production.3,4 Sol–gel and solvothermal methods are relatively simpler to carry out, and WO3 nanostructures can be formed too with careful selection of solvents and reaction conditions.5–8 Nevertheless, long hours at high temperatures are often required. Anodisation, compared to other preparation techniques for photoelectrodes, is inexpensive and simple, making it suitable for large scale fabrication. It consists of growing a natural oxide layer of a metal foil by electrolytic passivation, followed by crystallisation at adequate temperatures (typically 400 to 600 °C). As the metal oxide layer grown on top is tightly bound to the metal support, the efficacy of charge collection is high. However, there is one major disadvantage in the anodisation of tungsten. Current reports exclusively use electrolytes that contain one or more fluorides (e.g. NaF, HF or NH4F) as etching agents, which potentially bring serious safety issues due to the presence or formation of HF.9,11–13 Few F-free alternative electrolytes have been reported to successfully anodise tungsten, such as oxalic acid and NH4NO3, which have shown highly porous nanostructures.14,15 Therefore, it is meaningful to explore more F-free electrolytes and approaches for safer, faster and more scalable anodisation for the production of WO3 photoanodes.An ideal anodising electrolyte for the formation of photoanodes needs to assist the oxidation of the top surface of the precursor metal foil under an electric field and promote a morphology and porosity that eventually enhance the photoresponse. Citric acid (CA, C6H8O7) is known for its chelating properties and is widely used to soften water due to its ability to bind metals.16 It is also used as a structure-directing agent in solution-based synthesis methods.17,18
Here, we demonstrate for the first time that CA offers a sustainable, fast and effective replacement of fluoride-containing electrolytes for the anodisation of tungsten foil and the preparation of nanostructured WO3 photoanodes for solar water splitting. We report the results of different anodising conditions using CA and compare the performance of the resulting photoanodes with that using NH4F as electrolyte. The comparison is made by analyses of their structural as well as photoelectrochemical properties, including photocurrent density under solar simulation measured by linear sweep voltammetry (LSV), applied bias photon-to-current efficiency (ABPE), and incident photon-to-current conversion efficiency (IPCE). The stability of the photoanodes is also tested.
Experimental
Materials
Tungsten foil (0.1 mm, 99.95%) was purchased from Alfa Aesar. Citric acid (CA, 99%) and N-methylformamide (NMF, 99%) were purchased from Sigma-Aldrich. Ammonium fluoride (NH4F, 98+%) and sulfuric acid (H2SO4, 5 M) were supplied by Acros Organics and Fluka Analytical, respectively. Analytical acetone was obtained from VWR Chemicals and deionised water was used.Anodisation
Tungsten foil was cut into 15 × 30 mm rectangles and sonicated for 15 minutes in acetone. After sonication, the foil pieces were rinsed with H2O and dried under pressurised air. The anodisation was carried out by using the cleaned foil pieces as the anode of a two-electrode cell and a 3 mm-thick stainless steel piece as the cathode. Teflon and rubber templates were used to limit the anodisation to a circular area of 13 mm in diameter. The anode and cathode were held in parallel at a distance of 10 mm and immersed in electrolytes consisting of 0.1 M CA in either H2O or a solution of 80 vol% NMF and 20 vol% H2O. A jacketed beaker connected to a refrigerated circulating bath (DC-10, Thermo) kept the electrolyte at a constant temperature of 0 °C. Samples were anodised for 30 minutes at constant currents, controlled by a DC power supply (Agilent 6675A). One of the previously published procedures using fluoride electrolyte10 was repeated for comparison: tungsten pieces were anodised for 6 h under 40 V at 40 °C in NMF solution including 20 vol% H2O and 0.05 wt% NH4F. After anodisation, all samples were rinsed with H2O, dried in air, and calcined in air at 450 °C for 4 h.Physical characterisations
Morphology of photoanodes after fabrication processes was characterised by Field Emission Scanning Electron Microscopy (FESEM), JEOL 6301F, with an acceleration voltage of 5 keV. Film thicknesses were measured at ten different sites to calculate the mean value and standard deviation (quoted with a ± sign). Area fraction is measured using ImageJ. X-ray diffraction was performed with a BRUKER AXS D8 advance diffractometer using a Vantec-1 detector and CuKα radiation.Photoelectrochemical performance
Photoelectrochemical measurements were carried out in a three-electrode photoelectrochemical quartz cell using the prepared WO3 electrode as the working electrode, a Pt wire counter electrode, a Ag/AgCl (in 3.5 M KCl) reference electrode, and 0.5 M H2SO4 electrolyte. Solar simulated light on an 8 mm-diameter area was provided by a 300 W Xe Lamp (LOT Quantum Design) equipped with an AM1.5G filter. The irradiation intensity was set to 1 sun (100 mW cm−2). An external potential (provided by Ivium CompactStat) was linearly swept from 0 to 1.2 V vs. Ag/AgCl at a rate of 20 mV s−1. Applied bias photon-to-current efficiency (ABPE) was obtained using the following equation:where jph is the net photocurrent density measured at an applied bias Vb and Ptotal is the total solar incident irradiation.
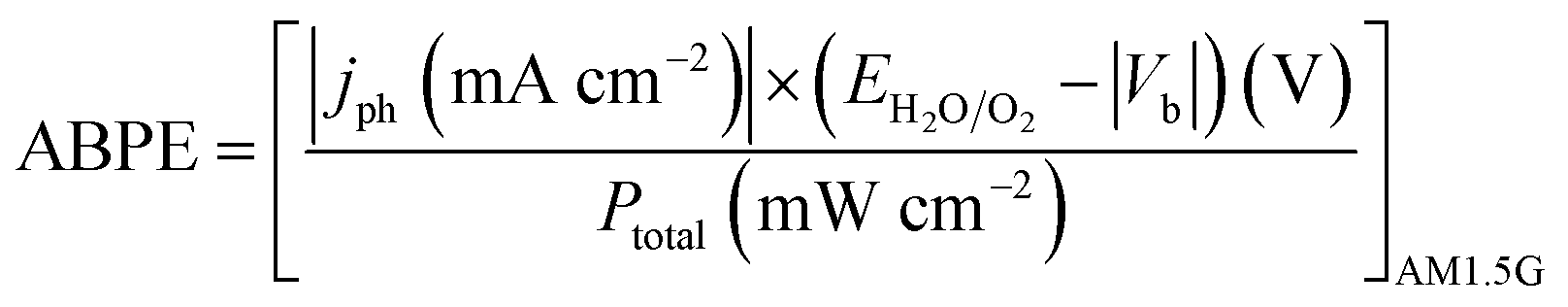
Incident photon-to-current efficiency (IPCE) measurements were performed from 300 to 500 nm with the same light source and a triple grating Czerny-Turner monochromator. The intensity of monochromatic light was measured at the working electrode position with a SEL033/U photodetector (International Light Technologies). The values of IPCE were then calculated using the formula below:
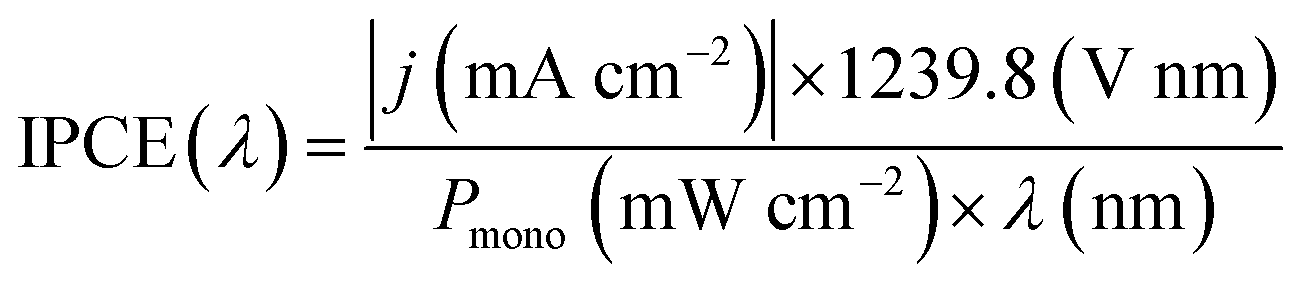
Photoelectrochemically active surface area (PECSA) values were calculated by measuring double layer capacitances with cyclic voltammetry. Potential was scanned at rates from 5 to 160 mV s−1 in a window of 0.2 V around open-circuit potential. The active surface area was then calculated using equation: PECSA = CDL/CS,19 where CDL is the slope for the fitted line plotted from measured currents against scanning rates, and CS (specific capacitance) took the value of the unit area CDL for a photoanode that has compact oxide layer produced from a controlled weak anodisation. The same electrochemical and light irradiation setup used for photocurrent density measurements was also used herein.
Results and discussion
Anodisation was carried out in two selected types of solvents: H2O and a mixture of NMF and H2O. NMF is added for its high dielectric constant which can favour higher charge density and assist oxide growth,20 and H2O is added as an oxygen donor and to assist the dissolution of CA, which is not soluble in NMF and unstable in other anodising solvents such as ethylene glycol or glycerol. Although it is sometimes argued that H2O concentration should be minimal for fast and deep anodisation and that H2O content in air is sufficient to act as oxygen source, adding H2O ensures the formation of a porous oxide.12,21 We used a mixture of H2O and NMF in a 20 and 80 vol% proportion, as this was found to be optimal by Tacca et al.10 The same solvents were used with NH4F for reference of the effectiveness of CA as etching agent.The morphology of WO3 films on the tungsten foil surface after anodisation was examined by FESEM. In the H2O solution, the CA-assisted anodisation etched the tungsten foil into a canyon-like nanostructure with valleys and corrugated WO3 walls and rods occupying approximately 70% of the area (Fig. 1a and b). This differs from the commonly seen WO3 mesoporous mesh-like structures observed after anodisation in fluoride-containing media.9,11–13 The height of the nanostructured valleys is estimated to be around 500 nm, below which there is a thin, compact layer of WO3, which adds up to a total thickness of ca. 3.7 ± 1.5 μm (Fig. S1a, ESI†). The width of the walls and rods are between 100 and 200 nm. It is worth noting that their corrugated features should allow for stronger light scattering and absorption, as has been demonstrated in WO3 photoanodes with helical nanostructure produced with oblique angle deposition.22
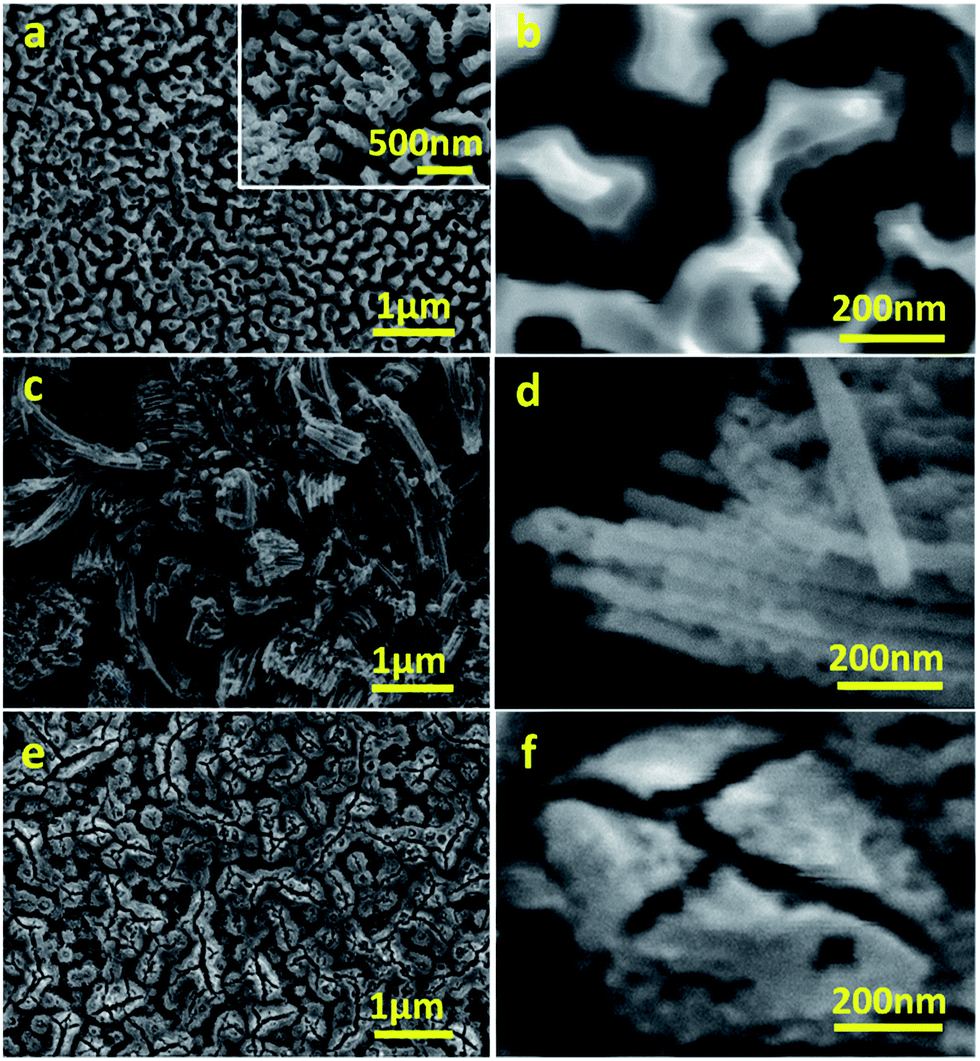 | ||
| Fig. 1 Representative FESEM micrographs of WO3 photoanodes anodised in CA/H2O at 0.1 A for 30 min (a, b), in CA/NMF/H2O at 0.015 A for 30 min (c, d), and in NH4F/NMF/H2O at 40 V for 6 h (e, f). | ||
The etching of the tungsten foil top surface during the anodisation in CA/H2O was investigated at different times by FESEM to understand the formation of the canyon-like nanostructure. First, a compact layer of tungsten oxide is formed under electrochemical oxidation, where cracks appear due to the difference in density of the oxide layer compared to tungsten, inducing strong local stresses (Fig. S2a†). Next, field-assisted dissolution starts to play an important role in the vicinity of the cracks, rendering the formation of holes of several hundred nanometres across (Fig. S2b†). The relatively compact oxide is then carved into nanowalls/nanorods (Fig. S2c†). The existence of nanorods is a result of horizontal dissolution as demonstrated by the “natural bridges” between two nanorods on the top right corner of Fig. 1a inset. Extended anodisation time can cause total dissolution of tungsten foil and hence a less effective working area. (Fig. S2d†). The morphological migration observed here is akin to the formation mechanism proposed by Chai and co-workers using oxalic acid anodisation, although they obtained a different porous structure consisting of spherical voids.23
The morphology can also be controlled by changing the anodisation current. Results for a series of specimens anodised at different current values (0.05, 0.08 and 0.10 A) for 30 min indicate further features in the formation of the WO3 layer (Fig. 2). When anodised at 0.05 A, cracks (deep valleys) dominate the morphology of the layer, which appears to be rougher and with a less uniform porosity (Fig. 2a). At 0.08 A, more shallow valleys spread over the majority of the surface, with relatively limited flat domains (Fig. 2b). Anodisation at 0.10 A obtains the most uniform porosity and finer features (Fig. 2c). Above 0.10 A, large cavities up to 100 μm arise due to pitting corrosion (Fig. 2d). The higher porosity gained from nanorods/nanowalls compared to other structures is beneficial for its photoresponse.
 | ||
| Fig. 2 FESEM micrographs of photoanodes after anodisation in CA/H2O for 30 min at 0.05 A (a), 0.08 A (b), 0.10 A (c) and 0.15 A (d). | ||
The morphology of CA-anodised films was found to depend also on the choice of solvents. When anodised in CA/NMF/H2O, a different morphology was obtained where nanowires of ca. 50 nm in diameter with different lengths and random orientation are predominant (Fig. 1c and d). This indicates that the high dielectric constant of NMF solvent compared to water has an effect on the final morphology.
For comparison to literature, we anodised the same tungsten foil in NH4F/NMF/H2O and obtained a worm-like porous morphology (Fig. 1e and f), as reported in the literature, different to the canyon-like nanostructure or the nanowire structures obtained with CA.10 Cross sectional FESEM micrographs show that the thicknesses of the films under study are ca. 5.8 ± 1.7 μm for CA/NMF/H2O anodised film and ca. 7.4 ± 2.7 μm for NH4F/NMF/H2O anodised film (Fig. S1b and c†). Therefore, CA has a similar etching capability with NH4F, despite the citrate ligand being bulkier.
Fig. 3 shows the XRD patterns of anodised films after calcination. The phases of the oxide layer in all cases are monoclinic (JCPDS no. 43-1035), which is the typical phase encountered upon various anodisation methods10,12–14,24 and has proved to be superior to other phases including orthorhombic and hexagonal in photocatalysis.25 Despite the fact that calcination temperature and dwell time are the same, the oxide formed using NH4F as electrolyte has preferential orientation along (−222)/(222), which was also observed elsewhere and found to be the most stable orientation during aging tests.10,26 Notably, the calcination step after anodisation is essential because for uncalcined photoanodes, the oxide formed has poor crystallinity and is not photoresponsive (see Fig. S3†).
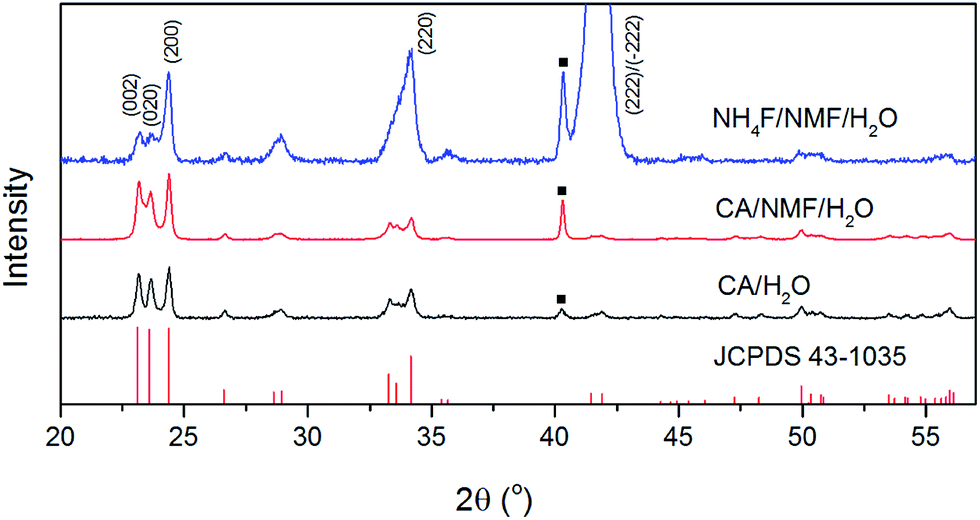 | ||
| Fig. 3 XRD patterns of calcined WO3 photoanodes anodised in CA/H2O at 0.1 A for 30 min (black), in CA/NMF/H2O at 0.015 A for 30 min (red), and in NH4F/NMF/H2O at 40 V for 6 h (blue) with reference to monoclinic WO3 (JCPDS no. 43-1035). Squares indicate diffraction from metallic tungsten (110) under WO3. | ||
Kubelka–Munk conversion of UV-visible diffuse reflectance spectroscopy of anodised films after calcination is shown in Fig. 4. All anodised films show the WO3 bandgap absorbing at wavelengths below 450 nm. For wavelengths above 450 nm, the NH4F/NMF/H2O anodised films absorb more light, which could be due to F doping originated from the electrolyte, as observed in F-doped TiO2 powders.27 Lower, but still evident is the absorption at those high wavelengths in CA/H2O and CA/NMF/H2O films, which could be due to minor carbon doping from CA or NMF. Although the Kubelka–Munk conversion compensates for specular reflectance, this cannot be completely ruled out in these measurements.
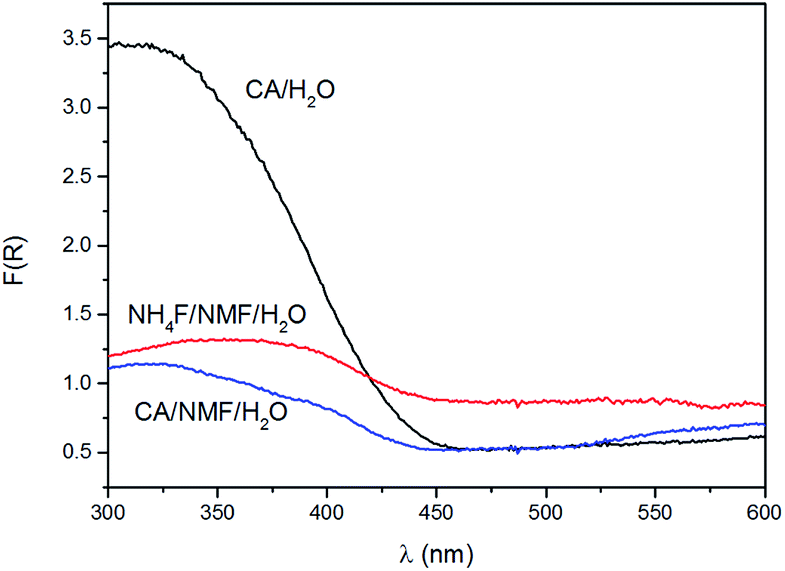 | ||
| Fig. 4 Kubelka–Munk function, F(R), of films anodised in CA/H2O at 0.1 A for 30 min (black), in CA/NMF/H2O at 0.015 A for 30 min (blue), and in NH4F/NMF/H2O at 40 V for 6 h (red) obtained from UV-visible diffuse reflectance spectroscopy. | ||
Anodised films were tested for photoelectrochemical water splitting. The photocurrent densities under solar illumination measured on films are found to be optimal when anodised at 0.1 A for 30 min in CA/H2O and 0.015 A for 30 min in CA/NMF/H2O (Fig. S4†). Fig. 5a shows representative LSV curves of these optimised WO3 films, with or without solar illumination in a three-electrode system. In spite of differences in WO3 thickness between the CA/H2O- and the CA/NMF/H2O-anodised photoanodes (3.7 ± 1.5 vs. 5.8 ± 1.7 μm, resp.), they show similar performance: the net photocurrent densities measured at 1.0 V vs. Ag/AgCl in 0.5 M H2SO4 are 0.88 mA cm−2 and 0.77 mA cm−2, respectively. This could be assigned to a stronger light scattering effect in the corrugated canyon-like nanostructure (formed in CA/H2O) compared to the thin nanowires (formed in CA/NMF/H2O). Another cause is that the ordered structure favours a more facile charge collection between the oxide and the metal basis than the randomly oriented nanowires. These values are comparable with published data for fluoride-based anodisations,12,13,24 while using a more benign electrolyte. The film prepared with NH4F/NMF/H2O, used here as a competitive benchmark, showed a higher photocurrent at 1 V vs. Ag/AgCl. Accordingly, ABPE values show higher efficiencies below 0.67 V vs. Ag/AgCl for CA-anodised samples (Fig. 5b). These differences can be ascribed to their different morphology and crystal orientation, observed by FESEM and XRD, which could affect their light absorption, charge transfer efficiency, and amount of surface states. One should note that a higher response at lower onset potential can be advantageous for tandem PEC cells working with little or no applied potentials.
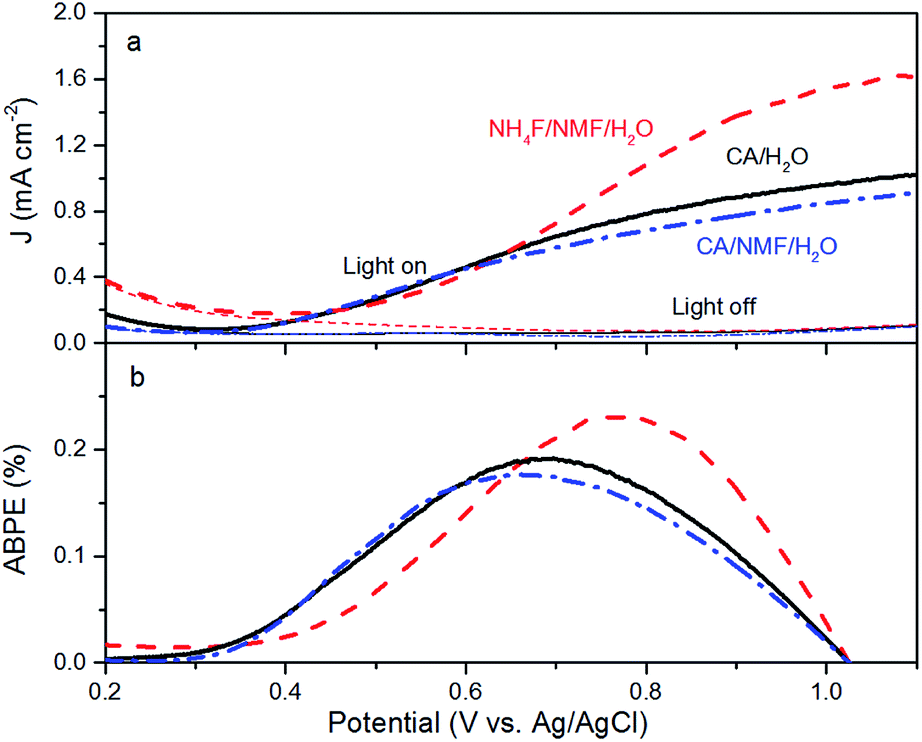 | ||
| Fig. 5 (a) Current densities of WO3 photoanodes in the presence and absence of AM1.5G illumination (100 mW cm−2). (b) Calculated ABPE of WO3 electrodes. Electrodes were prepared by anodising tungsten foils in CA/H2O at 0.1 A for 30 min (black solid line), in CA/NMF/H2O at 0.015 A for 30 min (blue dash-dot line), and in NH4F/NMF/H2O at 40 V for 6 h (red dashed line). | ||
The trends from LSV measurements were further confirmed by IPCE measured at 0.55 V and 1 V vs. Ag/AgCl (Fig. 6a). Photoanodes prepared using CA have similar performance at both conditions and at all wavelengths. When NH4F was used, the response was not as high as using CA at 0.55 V vs. Ag/AgCl, which is consistent with the photocurrent and ABPE results in Fig. 4. Tauc plot based on IPCE spectra at 1 V vs. Ag/AgCl shows estimated band gaps of about 2.7 eV for all three photoanodes (Fig. 6b). It is notable that at 1 V vs. Ag/AgCl there is an IPCE maximum between 350 and 420 nm, centred at 375 nm, for WO3 prepared in NH4F/NMF/H2O. This IPCE maximum is not present at 0.55 V vs. Ag/AgCl for the same film. This agrees with and confirms the higher photocurrent density observed in LSV measurements at higher applied potentials.
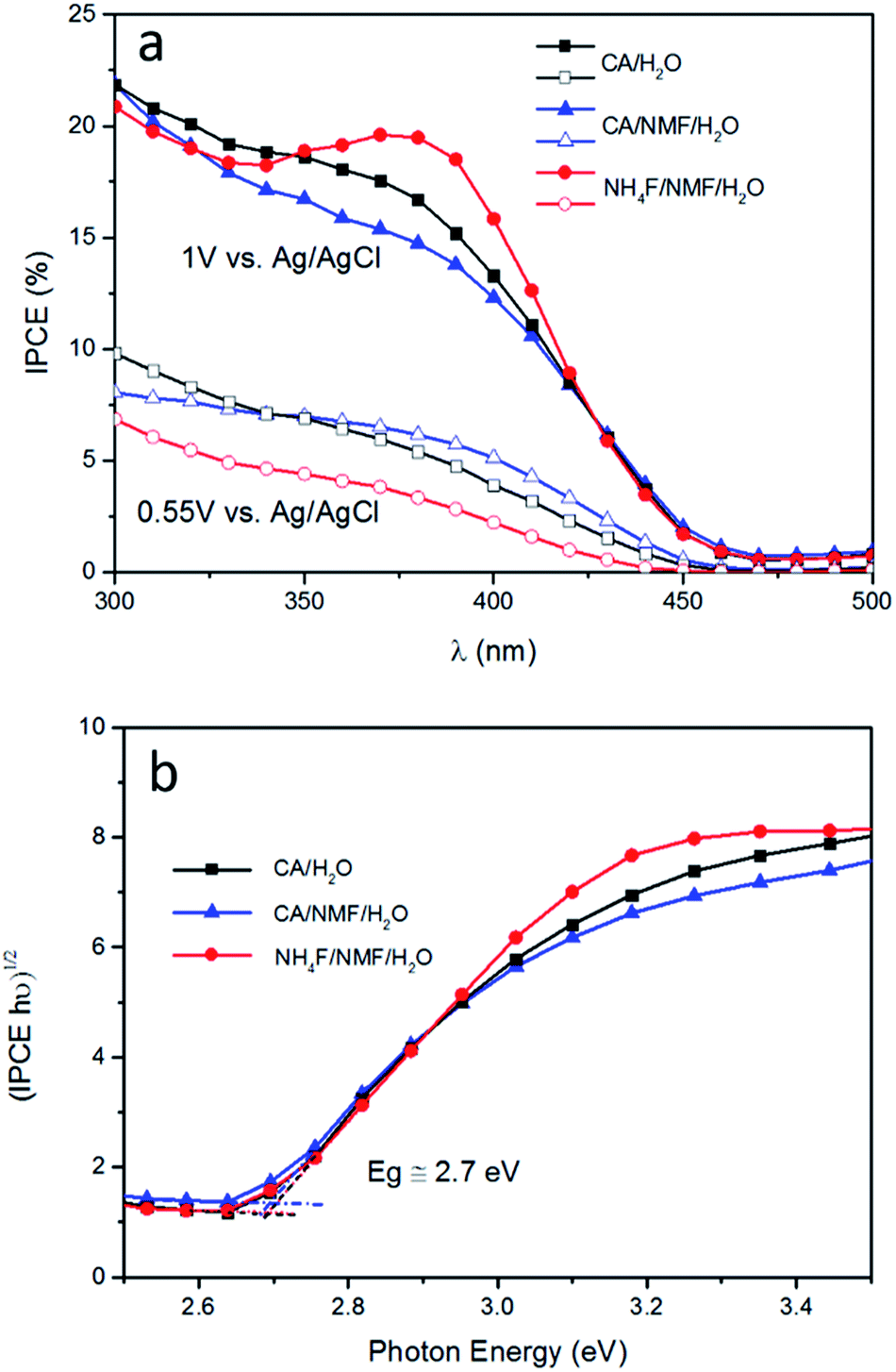 | ||
| Fig. 6 (a) Incident photon-to-current conversion efficiency (IPCE) results for photoanodes subjected to anodisations in CA/H2O at 0.1 A for 30 min (black squares), in CA/NMF/H2O at 0.015 A for 30 min (blue triangles), and in NH4F/NMF/H2O at 40 V for 6 h (red circles). Filled shapes indicate measurements at an applied bias of 1 V and hollow shapes at 0.55 V vs. Ag/AgCl. Both measurements were carried out in 0.5 M H2SO4. (b) Band gaps for WO3 on all photoanodes are estimated to be ∼2.7 eV based on IPCE spectra at 1 V vs. Ag/AgCl. | ||
The stability of WO3 photoanodes was tested at 1 V vs. Ag/AgCl in 0.5 M H2SO4 solution for 60 min with 6 min cycles of light chopping (Fig. 7). The photoanode anodised in CA/H2O shows a decay of 0.14 mA cm−2 in photocurrent density within the first two cycles, which can be attributed to photoelectrochemical instability caused by corrosion from accumulating holes at electrode–electrolyte interface.27 After two cycles, the decay slows down and, overall, 69% of its initial stability is retained. For the CA/NMF/H2O anodised sample, 90% of its initial photoresponse is maintained after the test. We attribute this superior result to its morphology consisting of thin nanowires having a higher photoelectrochemically active surface area (3.8 cm2 vs. 1.7 cm2), which is in favour of faster charge transfer. This leads to less accumulation of holes at the semiconductor–liquid junction and thereby higher photoelectrochemical stability.28 On the other hand, for the sample anodised in NH4F/NMF/H2O, poorer stability is observed. The photocurrent density suffers from gradual decline throughout the amount of time being tested, meaning a low photoelectrochemical stability. In addition, the photocurrent densities cannot reach its previous level after each dark period, which represents poor chemical stability. The photocurrent density is almost halved (53%) after one hour. Therefore, the stability test shows a clear advantage of CA-anodised photoanode over the fluoride-anodised photoanode.
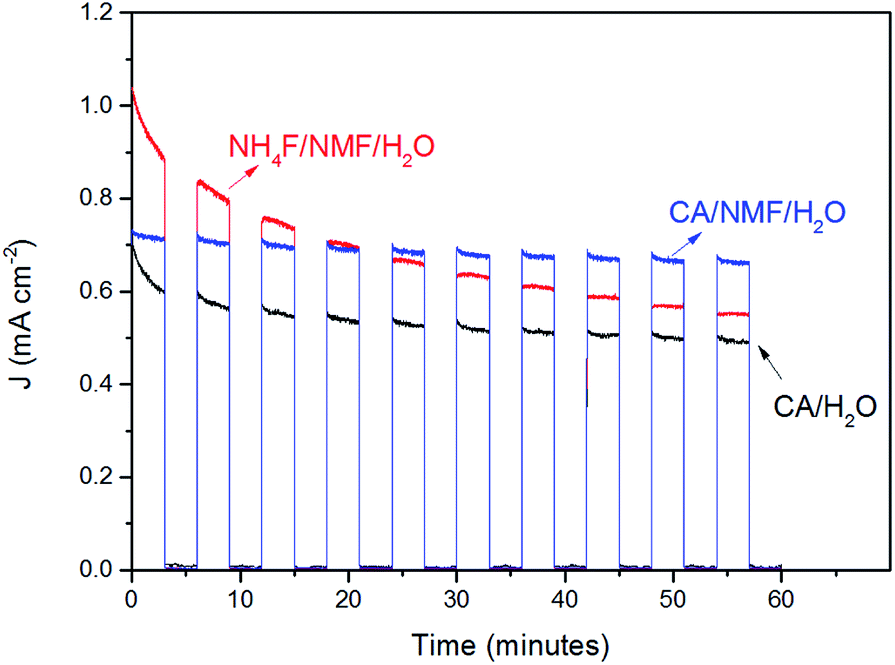 | ||
| Fig. 7 Stability test of WO3 electrodes prepared using CA/H2O at 0.1 A for 30 min (black), in CA/NMF/H2O at 0.015 A for 30 min (blue), and in NH4F/NMF/H2O at 40 V for 6 h (red) at 1 V vs. Ag/AgCl in 0.5 M H2SO4 under AM1.5G illumination (100 mW cm−2). Light was chopped with 3 min intervals. | ||
The performance of several porous anodic WO3 films is listed in Table 1 along with their synthesis and characterisation conditions. The nanowalls/nanorods structures formed here by CA anodisation in aqueous solution bear much resemblance to Ng's work where corrugated nanorods of 450 nm in height were obtained using Na2SO4 and NaF as an electrolyte.30 Moreover, the reported formation process is similar to that observed herein, which proves the success of CA as an effective electrolyte to replace fluorides. Fluoride-free anodisations have been explored in other works, with the formation of WO3 layers with different morphologies and dimensions. For example, a slow growth of a “nanosponge” was achieved in a 10 wt% K2HPO4/glycerol electrolyte.15 The thickness was up to 8.7 μm for a long 26 h anodisation. A 7.5 μm film of WO3 nanotubes was reported for 0.2 M NH4NO3/ethylene glycol by Wei et al.14 The plateau photocurrent density under solar irradiation was 2.5 mA cm−2 in the presence of HCOONa as a hole scavenger. More ordered and higher-aspect-ratio nanotubes were also recently formed anodising in molten H3PO4, although no photoresponse was reported.31 To the best of our knowledge, the present work is the first to report the formation of WO3 nanostructures using CA.
| Anodising media | Anodising voltage and time | Morphology | Phase | Electrolyte | J (mA cm−2) at 1.23VRHE | Onset (V) vs. RHE | Light source and intensity |
|---|---|---|---|---|---|---|---|
| a Photocurrent densities and onset potentials listed were approximatively read on figures from references. | |||||||
| 80 v% NMF/20 v% H2O/0.05 wt% NH4F10 | 40 V, 6 h | Crispy surface crust | Monoclinic | 1 M H2SO4 | 3.6 | 0.5 | AM1.5, 300 mW cm−2 |
| 1.5 M HNO3/40 mg NH4F24 | 30 V, 4 h | Triple-layered | Monoclinic | 0.5 M Na2SO4 | 0.9 | 0.4 | Xe lamp, 100 mW cm−2 |
| 0.1 M NaF12 | 60 V, 24 h | Mesh-like | Monoclinic | 0.1 M HCl | 0.75 | 0.45 | AM1.5, 100 mW cm−2 |
| 1 M Na2SO4/0.5 wt% NaF29 | 50 V, 30 min | Mesh-like | n.d. | 0.5 M H2SO4 | 1.0 | 0.6 | Xe lamp λ > 400 nm, 100 mW cm−2 |
| 0.1 M Na2SO4/0.5 wt% NaF13 | 50 V, 30 min | Mesh-like | Monoclinic | 0.1 M KH2PO4 + KOH (pH 7) | 0.9 | 0.7 | AM1.5, 100 mW cm−2 |
| 0.2 M NH4NO3/ethylene glycol14 | 10 V, 35 min | Nanochannel | Monoclinic | 0.5 M Na2SO4 + 0.1 M HCOONa | 2.5 | 0.52 | AM1.5, 100 mW cm−2 |
| 0.1 M CA (this work) | 0.1 A 30 min | Nanowalls/nano-rods | Monoclinic | 0.5 M H2SO4 | 0.88 | 0.5 | AM1.5, 100 mW cm−2 |
Conclusion
In this work, citric acid (CA) was studied for the anodisation of tungsten foil as an alternative to fluoride-based electrolytes such as NH4F. A systematic investigation of anodisation process parameters (time, currents and solvents) was performed to establish a relation between the obtained morphology and its performance in solar water splitting. We demonstrated, for the first time, that a CA/H2O electrolyte can produce porous canyon-like nanostructure consisting of WO3 nanowalls and nanorods. Using a mixture of CA, H2O and NMF, a different porous nanostructure was obtained consisting of randomly oriented ∼50 nm wide WO3 nanowires. These morphologies differ from the classic mesh-like porous nanostructures obtained with NH4F as an anodising electrolyte. The performance of these anodised films was tested for photoelectrochemical water splitting using a three-electrode system and simulated sunlight. The CA-anodised films obtained photocurrent densities around 0.8 mA at 1 V vs. Ag/AgCl in 0.5 M H2SO4. These CA-anodised films outperformed NH4F-anodised films at low applied potentials (below 0.67 V vs. Ag/AgCl) and showed much better photoelectrochemical stability. As such, CA represents a safer and more environmentally friendly alternative to fluoride-based electrolytes for the production of nanostructured WO3 photoanodes with comparable or superior performance. This work also opens an avenue for sustainable anodisation procedures for the production of other nanostructured metal oxides as photoelectrodes using CA.Acknowledgements
SE would like to acknowledge the financial support from EPSRC (EP/P008097/1).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- J. Lin, P. Hu, Y. Zhang, M. Fan, Z. He, C. K. Ngaw, J. S. C. Loo, D. Liao and T. T. Y. Tan, RSC Adv., 2013, 3, 9330 RSC.
- J. Ding and K. Kim, J. Nanosci. Nanotechnol., 2016, 16, 1578 CrossRef CAS PubMed.
- V. Chakrapani, J. Thangala and M. K. Sunkara, Int. J. Hydrogen Energy, 2009, 34, 9050 CrossRef CAS.
- C. Santato, M. Odziemkowski, M. Ulmann and J. Augustynski, J. Am. Chem. Soc., 2001, 123, 10639 CrossRef CAS PubMed.
- J. Su, X. Feng, J. D. Sloppy, L. Guo and C. A. Grimes, Nano Lett., 2011, 11, 203 CrossRef CAS PubMed.
- D. Qin, C. Tao, S. A. Friesen, T. Wang, O. K. Varghese, N. Bao, Z. Yang, T. E. Mallouk and C. A. Grimes, Chem. Commun., 2012, 48, 729 RSC.
- Y. Hou, F. Zuo, A. P. Dagg, J. Liu and P. Feng, Adv. Mater., 2014, 26, 5043 CrossRef CAS PubMed.
- A. Watcharenwong, W. Chanmanee, N. R. de Tacconi, C. R. Chenthamarakshan, P. Kajitvichyanukul and K. Rajeshwar, J. Electroanal. Chem., 2008, 612, 112 CrossRef CAS.
- A. Tacca, L. Meda, G. Marra, A. Savoini, S. Caramori, V. Cristino, C. A. Bignozzi, V. Gonzalez Pedro, P. P. Boix, S. Gimenez and J. Bisquert, ChemPhysChem, 2012, 13, 3025 CrossRef CAS PubMed.
- N. R. De Tacconi, C. R. Chenthamarakshan, G. Yogeeswaran, A. Watcharenwong, R. S. De Zoysa, N. A. Basit and K. Rajeshwar, J. Phys. Chem. B, 2006, 110, 25347 CrossRef CAS PubMed.
- K. R. Reyes-Gil, C. Wiggenhorn, B. S. Brunschwig and N. S. Lewis, J. Phys. Chem. C, 2013, 117, 14947 CAS.
- Q. Liu, Q. Chen, J. Bai, J. Li, J. Li and B. Zhou, J. Solid State Electrochem., 2014, 18, 157 CrossRef CAS.
- W. Wei, S. Shaw, K. Lee and P. Schmuki, Chem.–Eur. J., 2012, 18, 14622 CrossRef CAS PubMed.
- W. Lee, D. Kim, K. Lee, P. Roy and P. Schmuki, Electrochim. Acta, 2010, 56, 828 CrossRef CAS.
- K. Kirimura, Y. Honda and T. Hattori, Compr. Biotechnol., 2011, 3, 135 Search PubMed.
- Q. Han, X. Wu, Y. Liao, D. Li, R. Yue, H. Liu and Y. Chen, Mater. Lett., 2013, 95, 9 CrossRef CAS.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91 RSC.
- S. Trasatti and O. A. Petrii, Pure Appl. Chem., 1991, 63, 711 CrossRef CAS.
- V. Cristino, S. Caramori, R. Argazzi, L. Meda, G. L. Marra and C. A. Bignozzi, Langmuir, 2011, 27, 7276 CrossRef CAS PubMed.
- M. Paulose, K. Shankar, S. Yoriya, H. E. Prakasam, O. K. Varghese, G. K. Mor, T. A. Latempa, A. Fitzgerald and C. A. Grimes, J. Phys. Chem. B, 2006, 110, 16179 CrossRef CAS PubMed.
- X. Shi, I. Y. Choi, K. Zhang, J. Kwon, D. Y. Kim, J. K. Lee, S. H. Oh, J. K. Kim and J. H. Park, Nat. Commun., 2014, 5, 4775 CrossRef CAS PubMed.
- Y. Chai, C. W. Tam, K. P. Beh, F. K. Yam and Z. Hassan, J. Porous Mater., 2013, 20, 997 CrossRef CAS.
- H. Qi, J. Wolfe, D. Wang, H. J. Fan, D. Fichou and Z. Chen, Nanoscale, 2014, 6, 13457 RSC.
- S. S. Kalanur, Y. J. Hwang, S. Y. Chae and O. S. Joo, J. Mater. Chem. A, 2013, 1, 3479 CAS.
- P. Dias, T. Lopes, L. Meda, L. Andrade and A. Mendes, Phys. Chem. Chem. Phys., 2016, 18, 5232 RSC.
- T. W. Kim and K. S. Choi, J. Phys. Chem. Lett., 2016, 7, 447 CrossRef CAS PubMed.
- D. Bae, B. Seger, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, Chem. Soc. Rev., 2017, 46, 1933 RSC.
- W. Li, J. Li, X. Wang, S. Luo, J. Xiao and Q. Chen, Electrochim. Acta, 2010, 56, 620 CrossRef CAS.
- C. Y. Ng, K. Abdul Razak and Z. Lockman, J. Porous Mater., 2015, 22, 537 CrossRef CAS.
- M. Altomare, O. Pfoch, A. Tighineanu, R. Kirchgeorg, K. Lee, E. Selli and P. Schmuki, J. Am. Chem. Soc., 2015, 137, 5646 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FESEM micrographs, XRD patterns and photocurrent density measurements. See DOI: 10.1039/c7ra05342h |
| This journal is © The Royal Society of Chemistry 2017 |