
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper doped BaMnO3 perovskite catalysts for NO oxidation and NO2-assisted diesel soot removal†
Verónica Torregrosa-Rivero,
Vicente Albaladejo-Fuentes,
María-Salvadora Sánchez-Adsuar and
María-José Illán-Gómez *
*
Carbon Materials and Environment Research Group, Department of Inorganic Chemistry, Faculty of Science, Universidad de Alicante, Alicante, Spain. E-mail: illan@ua.es; Tel: +34 965903975
First published on 13th July 2017
Abstract
The activity for NO oxidation and for NO2-assisted diesel soot removal of a BaMn1−xCuxO3 (x = 0, 0.1, 0.2, 0.3) perovskite-type catalyst has been tested by Temperature Programmed Reaction (TPR) and isothermal experiments at 450 °C. Fresh and used catalyst characterization by ICP-OES, N2 adsorption, XRD, XPS, IR spectroscopy and H2-TPR was performed. Results showed that: (i) manganese is partially substituted by copper in the perovskite structure leading to the formation of a manganese-deficient perovskite with a new hexagonal structure, (ii) in BaMn1−xCuxO3 catalysts, manganese seems to be mainly Mn(III) and, as a consequence, the amount of oxygen vacancies increases gradually with the copper content and (iii) the presence of copper into the perovskite structure enhances the reducibility of the catalyst and increases the mobility of lattice oxygen. BaMn0.7Cu0.3O3 is the most active catalyst for NO2 generation and, consequently, shows the lowest T50% value, the highest CO2 selectivity, the best performance during TPR cyclic experiments, and the highest soot oxidation rate at 450 °C. This behavior is a result of the enhancement of the redox properties of the catalyst due to the replacement of Mn(III)/Mn(IV) by Cu(II) in the perovskite structure.
1. Introduction
Perovskites are ceramic mixed oxides with ABO3 structural type formula, wherein A is the larger ionic radius cation (around three times larger than B) and 12-oxygen coordinated, while B is a smaller and 6-oxygen coordinated cation.1 The ideal structure of these mixed oxides is cubic, following the rA + rO = √2(rB + rO) ionic radius relationship. However, most of the solids with perovskite structure do not follow this equation, and for this reason, Goldschmidt2,3 introduced the tolerance factor t parameter, which is defined as: t = rA + rO/√2(rB + rO). The t parameter establishes that the perovskite structure is allowed when tolerance factor is between 0.8 and 1.1, although a distorted or non-ideal structure is formed when the t parameter is different than 1.Magnetic and electrical properties of perovskites have been widely reported2 but also, catalytic properties have been explored.4,5 Up to the present, it is accepted that the catalytic activity of perovskite oxides depends on the nature of the B cation.6,7 Thus, the fact that 90% of metallic elements can form stable perovskite oxides but also the possibility of modulating the physicochemical properties of the solid, such as oxygen mobility, specific surface area, redox behavior and so on, by selection and/or partial substitution of the A and B cations, justifies the wide use of perovskite as a catalyst in the redox processes.8
The first perovskite catalytic studies in the 1970s were focused on NO and CO oxidation.3 Afterward, the research of perovskite-type oxides as a catalyst was expanded to other processes as hydrogenations, methane reforming, oxidation of hydrocarbons, photocatalysis and also pollution control reactions.9–17 Libby18 suggested that the LaCoO3 activity for hydrocarbon oxidation pointed out perovskite-type catalysts as a promising alternative to noble metal based catalyst for automotive emissions control. In spite of the huge number of reports focused on this topic, perovskite oxides activity has been always lower than those obtained for the commercial catalysts based on noble metals. However, Kim and co-workers19 lately showed that similar NO oxidation activity as a Pt-based catalyst can be achieved by lanthanum manganese perovskites. Considering these results, the application of perovskites for automotive emissions control aroused a great interest again, including NO, CO, and hydrocarbons oxidation and NOx and soot removal.20–25
Diesel engines work with a high air/fuel ratio so, they show higher energy efficiency and lower pollutants emissions than gasoline engines.26 However, in diesel engines fuel and air do not mix as thoroughly as in gasoline engines, and this promotes the formation of NOx and soot particles, being these the main pollutants emitted by diesel engines.27 It has been shown that soot particles are directly related to many human health and environmental problems and, for this reason, the reduction of automotive soot emissions are mandatory. Removal of soot from diesel engines is mainly carried out using diesel particulate filters (DPF).28 This system retains soot, which has to be burned increasing the temperature to avoid the filter collapse. As NO2 promotes soot oxidation, coating the filter surface with a catalyst able to oxidize NO to NO2, is an interesting methodology for reducing the soot combustion temperature.29
In 1995, Teraoka et al.30 compared the performance of lanthanum-based perovskites and simple oxides for simultaneous NOx and soot removal. These authors concluded that perovskites featured higher NOx reduction activity and soot combustion rate than simple oxides and also, that the lowest ignition temperature was obtained for the Mn- and Co-perovskites. Recently, Chen et al.20 have demonstrated that LaCoO3 perovskites show higher NO oxidation activity, a key step for NO2 assisted soot oxidation, and stability against hydrothermal aging than those prepared with Mn or Fe.
Based on these results, perovskites oxides are being considered as an alternative to noble-metal catalysts in DPF. Thus, over the last decade, it has been demonstrated that partial substitution of A and B cations in perovskites can enhance the activity for soot oxidation.31 On the one hand, lanthanum substitution by potassium or strontium allows obtaining soot oxidation activity close to that showed for platinum catalysts under loose contact,32 tight contact33 and also for both modeling or realistic conditions.34 On the other hand, partial substitution of manganese by cobalt in LaMn0.9Co0.1O3 oxide reduces soot combustion temperature35 due to the higher oxygen surface concentration of this catalyst compared with the non-substituted LaMnO3 perovskite. In this sense, after copper addition to the La0.8K0.2MnO3 catalyst, a higher stability is achieved, but the catalyst activity is not enhanced.36
In spite of the extensive number of studies published related with the use of lanthanum-based perovskites for soot removal, rare reports of barium based perovskites for this application have been found. Doggali et al.37 reported that barium doping of a LaCoO3 enhanced CO oxidation activity due to a change of the catalyst basicity. Milt and co-workers21 tested BaCoO3 perovskite for NOx adsorption and soot combustion, concluding that the mixed oxide is active for both processes, however, the relationship between physicochemical properties and catalytic activity was not established in detail. Gao et al.38 proved that BaMnO3 formation, by the aging of a Ba/MnOx–CeO2 catalyst improves the catalyst stability because the perovskite phase inhibits cerium oxide sintering, being also active for soot oxidation.
Considering all this information, the aim of this paper is to correlate physicochemical properties of BaMnO3 perovskites with their catalytic activity for NO and soot oxidation under loose contact conditions. In addition, considering that perovskite doping with copper has previously shown interesting results,39,40 it has also been determined the effect of partial substitution of manganese by copper on the catalysts performance.
2. Experimental section
2.1 Catalyst preparation
BaMn1−xCuxO3 catalysts (x = 0, 0.1, 0.2, 0.3) were prepared by a citrate sol–gel method. Ba(CH3COOH)2 (Sigma-Aldrich, 99%), Mn(NO3)2·4H2O (Sigma-Aldrich, 97%), Cu(NO3)2·3H2O (Panreac, 99%) were used as metal precursors. In brief, a 0.1 M citric acid solution with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio with respect to barium was prepared and heated up to 60 °C. The pH of this solution was increased to 8.5 by drop wise ammonia addition. Afterward, the stoichiometric amounts of barium, manganese, and copper precursors were added to the solution in this order. Once the precursor salts were dissolved, the pH value was readjusted to 8.5 with ammonia. The solution was kept at 65 °C for 5 h and later dried at 90 °C for 48 h. The dried gel was calcined at 150 °C for 1 h and then, at 850 °C for 6 h. The final powder was ground using an agate mortar and pestle. The nomenclature used for the catalysts prepared is included in Table 1.
:2 molar ratio with respect to barium was prepared and heated up to 60 °C. The pH of this solution was increased to 8.5 by drop wise ammonia addition. Afterward, the stoichiometric amounts of barium, manganese, and copper precursors were added to the solution in this order. Once the precursor salts were dissolved, the pH value was readjusted to 8.5 with ammonia. The solution was kept at 65 °C for 5 h and later dried at 90 °C for 48 h. The dried gel was calcined at 150 °C for 1 h and then, at 850 °C for 6 h. The final powder was ground using an agate mortar and pestle. The nomenclature used for the catalysts prepared is included in Table 1.
| Catalyst | Molecular composition | Nominal Cu (wt%) | ICP-OES Cu (wt%) | SBET (m2 g−1) |
|---|---|---|---|---|
| BMC0 | BaMnO3 | — | — | 5.0 |
| BMC1 | BaMn0.9Cu0.1O3 | 2.6 | 2.4 | 2.2 |
| BMC2 | BaMn0.8Cu0.2O3 | 5.2 | 4.8 | 3.9 |
| BMC3 | BaMn0.7Cu0.3O3 | 7.8 | 7.6 | 3.7 |
2.2 Characterization
A Perkin-Elmer device, model Optima 4300 DV, was used for measuring copper content by ICP-OES. For this analysis, copper was extracted by solving the samples with magnetic stirring in 8 M HCl solution at room temperature. The BET surface area was determined by N2 adsorption at −196 °C in an Autosorb-6B instrument from Quantachrome. Samples were previously degasified at 250 °C for 4 h.XRD diffractograms were recorded between 20–80° 2θ angles with a step rate of 1.5°/2 min and using Cu Kα (0.15418 nm) radiation in a Bruker D8-Advance device.
The morphology of catalysts was analyzed using a ZEISS Merlin VP Compact Field Emission Scanning Electron Microscopy (FESEM) equipment.
The chemical surface properties of the samples were studied by XPS. XPS spectra were registered using a K-Alpha Photoelectron Spectrometer by Thermo-Scientific with an Al Kα (1486.6 eV) radiation source. This analysis was done with a 5 × 10−10 mbar pressure in the chamber. Binding energy (BE) and kinetic energy (KE) scales were adjusted by setting C 1s transition at 284.6 eV, and the BE and KE values were then determined with the peak-fit software of the spectrophotometer.
Reducibility of the catalysts was tested by Temperature Programmed Reduction with H2 (H2-TPR). These experiments were carried out in a Pulse Chemisorb 2705 device from Micromeritics fitted with a TCD to determine the outlet gas composition changes. 20 mg of the sample were heated at 10 °C min−1 from room temperature to 1000 °C in 5% H2/Ar atmosphere (40 mL min−1, Pt = 1 atm). The H2 consumption was quantified using a CuO sample supplied by Micromeritics.
Infrared spectroscopy analysis of the fresh and used samples was done using a JASCO FT/IR 4100 spectrometer with and MTC detector and fitted with a Harrick's Praying Mantis diffuse reflection accessory. DRIFTS spectra were recorded in the 5000–500 cm−1 wavenumber interval with a 4 cm−1 resolution and as an average of 40 scans.
2.3 Activity tests
The activity of the catalysts for NO and soot oxidation was determined by Temperature Programmed Reaction (TPR) using an atmosphere containing 500 ppm NOx, 5% O2 and balance with N2, and a gas flow of 500 mL min−1. For NO oxidation experiments, 80 mg of the sample were diluted with SiC in a mass ratio 1:4, and heated up from 25 to 800 °C, at 10 °C min−1 in a quartz fixed-bed reactor. Soot oxidation activity was analyzed by adding 20 mg of Printex U (a carbon black used as model soot), in loose contact in the reactor. For the most active catalysts, isothermal soot oxidation reactions at 450 °C were also carried out. The gas composition as a function of temperature was monitored by specific NDIR-UV gas analyzers for NO, NO2, CO, CO2 and O2 (Rosemount Analytical Model BINOS 1001, 1004 and 100).
The NO2 generation, soot conversion and CO2 selectivity percentages were calculated using eqn (1)–(3) respectively:
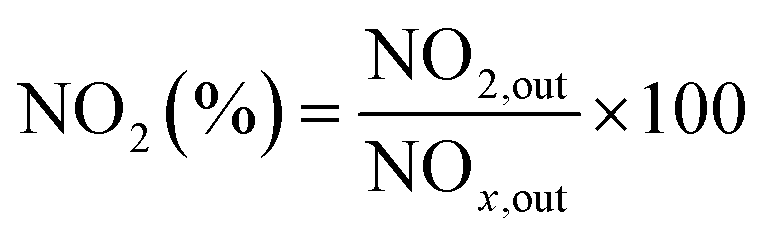 | (1) |
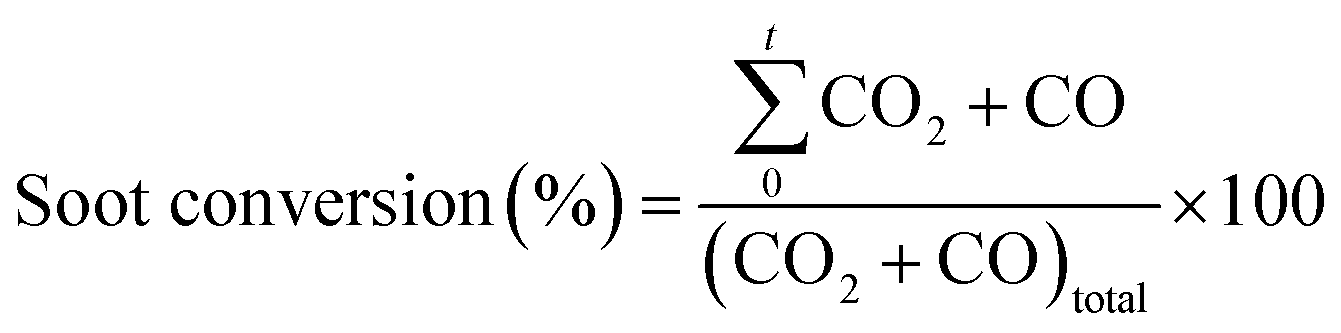 | (2) |
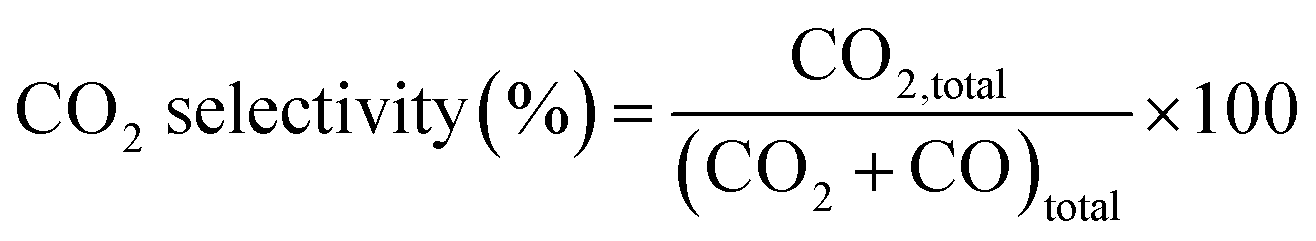 | (3) |
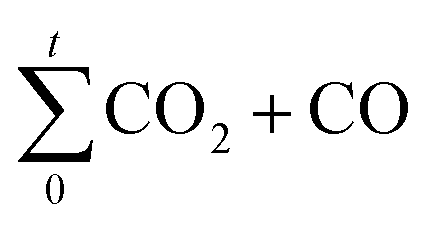 is the amount of CO2 and CO evolved at a time t and CO2,total and (CO2 + CO)total are the CO and CO2 + CO evolved during all the experiment time.
is the amount of CO2 and CO evolved at a time t and CO2,total and (CO2 + CO)total are the CO and CO2 + CO evolved during all the experiment time.
3. Results and discussion
3.1 Characterization of the fresh catalysts
Concerning morphological properties, FESEM images (Fig. S1 of ESI† shows the FESEM images of BMC0 and BMC3 as examples) indicate that highly agglomerated irregular grains in the range of micrometer size are formed for all catalysts, either without (BMC0) or with (BMC1, BMC2 and BMC3) copper. Thus, it seems that the presence of copper does not appreciably modify the morphology of the perovskites.
XRD diffractograms obtained for the BaMn1−xCuxO3 catalysts are shown in Fig. 1a and b. Besides, to facilitate the understanding of the results, all the phases identified in the catalysts are also indicated in Table 2. Thus, BMC0 XRD pattern exactly fits with that previously reported by Cussen et al.41 for a 2H-BaMnO3 perovskite with hexagonal structure, with main peaks at 26°, 26.5°, 27.5°, 31.5°, 41°, 41.5°, 49.5°, 52.5°, 53.2° and 56° 2θ values. Additionally, Ba3Mn2O3 oxide (only its main XRD peak is indicated in Fig. 1a)22 is also identified as a minority phase. The incorporation of copper modifies appreciably the XRD patterns of the BMC1, BMC2, and BMC3 catalysts compared with that obtained for BMC0. First, for the catalysts with higher copper content (BMC2 and BMC3), a very small amount of CuO is identified as a segregated phase. In addition, as the copper content increases, peaks corresponding to the original 2H-BaMnO3 hexagonal structure progressively disappear and they are replaced by new diffraction peaks at 27°, 31°, 41.5°, 52.7° and 55° 2θ values, being these peaks the unique ones observed for BMC3 catalyst. Shinoda and co-workers43 noticed a similar trend for a BaCo1−xMnxO3 oxides series, concluding that by increasing the manganese content from x = 0 to x = 0.3, the original 2H-type structure changed to a 10H-type structure through an intermediate state in which a mixture of the former and a 12H-type structure are identified, belonging all of them to the hexagonal crystal structure. These authors pointed out that, by cobalt substitution the amount of BO6 octahedrons sharing corners was promoted and, for hence, a new hexagonal structure type was formed. Considering Shinoda results, it could be suggested that, in the BaMn1−xCuxO3 catalysts, copper is partially incorporated into the perovskite structure and, as a consequence, the original 2H-BaMnO3 perovskite structure is progressively modified, thus generating a new BO6 octahedrons order which leads to a new copper-perovskite structure that becomes the unique phase for BMC3 (x = 0.3).
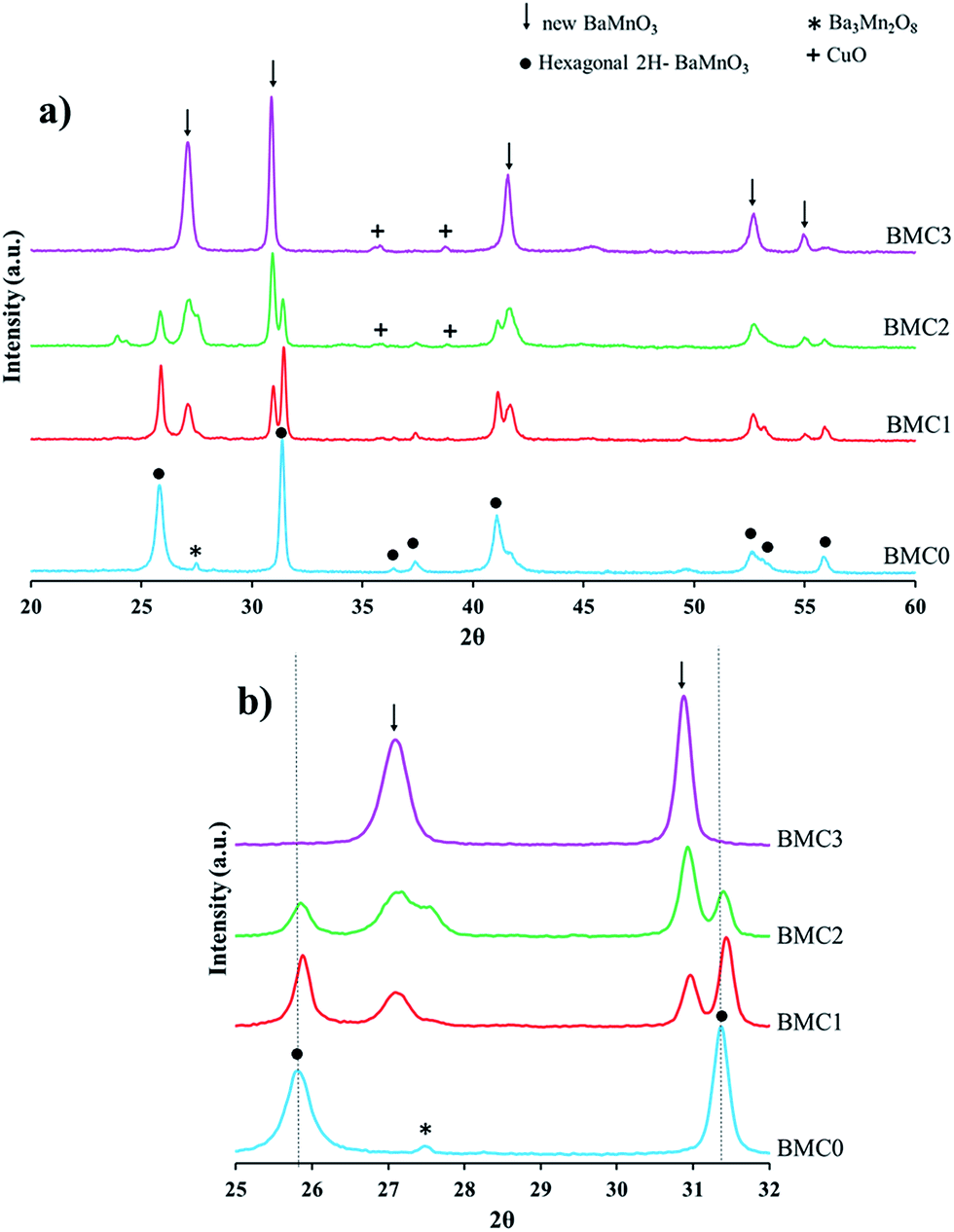 | ||
| Fig. 1 (a) XRD patterns of the BaMn1−xCuxO3 catalysts. (b) Magnification of the XRD patterns to identify the shift of the main peaks. | ||
| Catalyst | XRD phase identification | Average crystal sizea (nm) | aa (Å) | ca (Å) |
|---|---|---|---|---|
| a In brackets the crystallographic data calculated for the new BaMnO3 phase are shown. | ||||
| BMC0 | 2H-BaMnO3, Ba3Mn2O8 | 31.8 | 5.703 | 4.831 |
| BMC1 | 2H-BaMnO3, new BaMnO3 | 38.4 (38.3) | 5.698 (5.774) | 4.808 (4.364) |
| BMC2 | 2H-BaMnO3, new BaMnO3, CuO | 43.2 (35.9) | 5.695 (5.784) | 4.811 (4.365) |
| BMC3 | New BaMnO3, CuO | — (33.2) | — (5.792) | — (4.368) |
Moreover, from the XRD diffractograms, it is also concluded that copper is partially substituting manganese into the perovskite structure. In Fig. 1b, where an amplified area of the diffractograms is shown, it is clearly observed that the position of the two main peaks of the BaMnO3 hexagonal structure, at 26° and 31.5°, shifts to higher diffraction angle as the amount of copper increases. A similar analysis has been previously reported in a wide number of studies,46–48 and consequently, the shift of the diffraction peaks is generally accepted as an evidence of the copper introduction (totally or partially) into the lattice. Thus, it is confirmed that total or partial incorporation of copper into the mixed oxide structure is achieved, and this fact generates the distortion of the original hexagonal phase due to the different anionic radius of Cu(II) (73 pm)44 and Mn(III) (65 pm)/Mn(IV) (53 pm).45
In a previous report from Negas and Roth,49 where the structural properties of different BaMnO3−x phases were analyzed, the diffraction peaks of a new phase in the XRD pattern of the 4H-type BaMnO3 are included. However, even though the authors did not present specific observations about this new phase, they suggested that it could correspond with a manganese-deficient perovskite. Comparing the pattern of the Negas and Roth phase with the new phase detected for BMC1, BMC2, and BMC3 catalysts, it is concluded that the new structure corresponds with the proposed manganese-deficient perovskite. Thus, using the crystallographic data included in the Negas and Roth paper49- and assuming that both structures have hexagonal symmetry − a and c unit cell parameters, for the two structures identified in the XRD patterns, were calculated, being the data included in Table 2. It is observed that lattice parameters a and c for the 2H-BaMnO3 structure (which is the phase identified in BMC0, BMC1 and BMC2 catalysts) decrease as the copper content in the catalyst increases, whilst the two parameters for the new BaMnO3 phase (observed in the BMC1, BMC2 and BMC3 XRD patterns) increase with the amount of copper. A similar effect was found for the average crystal size of the two structures, which was calculated using Scherrer equation (Table 2):50 an increase of the 2H-BaMnO3 structure average crystal size is observed as the copper content increases, while for the new phase (manganese-deficient perovskite), a decrease of this value is featured as this structure becomes predominant.
Thus, from XRD characterization results, it could be concluded that copper is partially substituting manganese into the lattice, causing the distortion of the perovskite structure. This distortion increases with the copper content and, as a consequence, a new structure corresponding to a manganese-deficient perovskite is formed, which becomes to be the main crystalline phase for the highest copper content (BMC3) catalyst. Additionally, the presence of very low intense peaks of CuO is observed for the two highest copper content catalysts (BMC2 and BMC3).
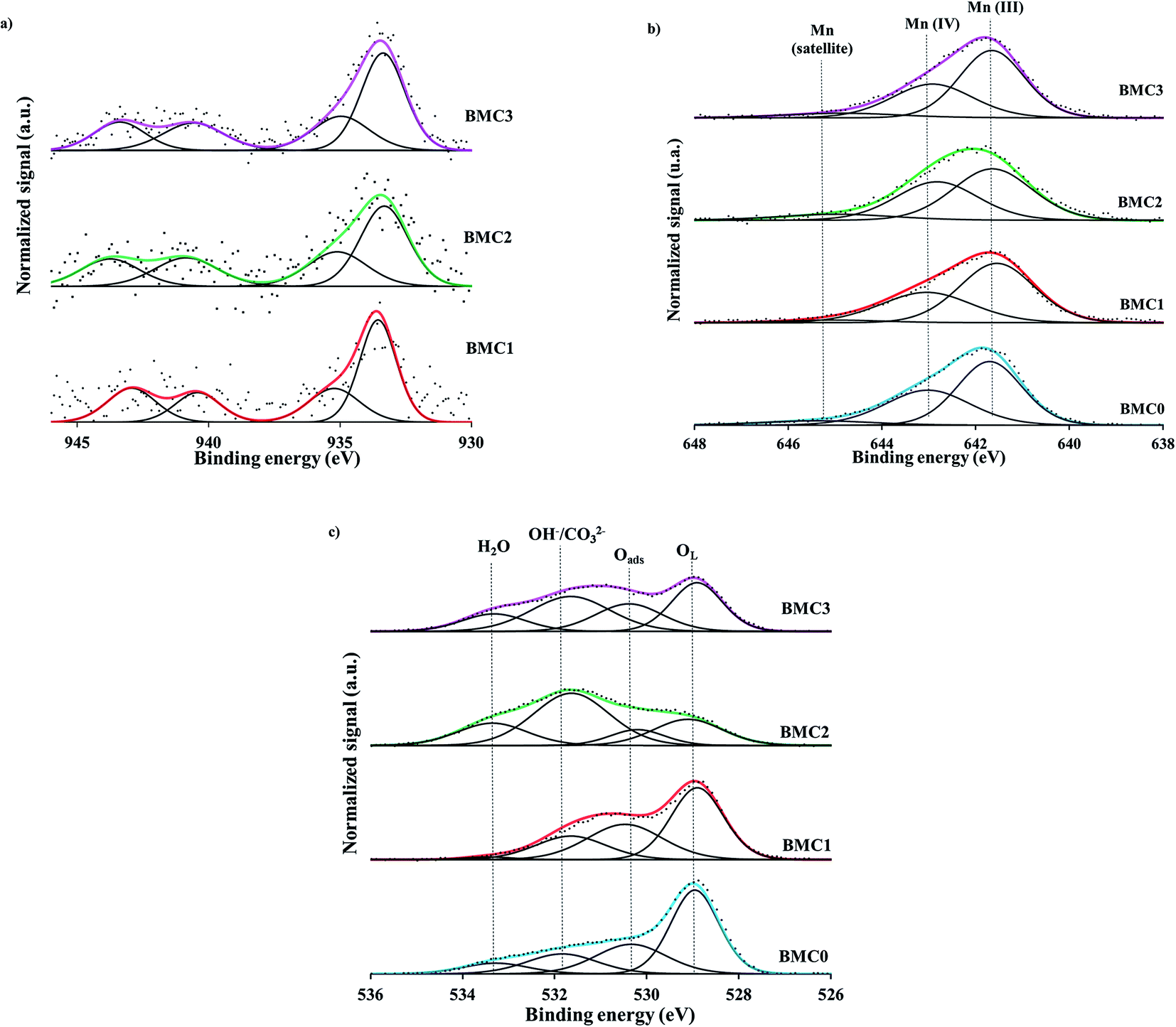 | ||
| Fig. 2 XPS spectra of the (a) Cu 2p3/2 transition, (b) Mn 2p3/2 transition and (c) O 1 s transition for the BaMn1−xCuxO3 catalysts. | ||
In agreement with previous results,42 the two contributions observed in the Cu 2p3/2 XPS spectra can be assigned to two different Cu(II) species: (i) near surface located copper species (CuS) with weaker electronic interaction with the perovskite matrix or supported copper oxide, that appears at lower BE (933.5 eV), and (ii) copper with a strong interaction with the perovskite structure and located in a deeper position into the lattice (CuL), which corresponds with the peak at higher BE (935.0 eV). Additionally, in Table 3 the Cu/(Ba + Mn + Cu) ratio calculated from the atomic percentages determined by XPS (Table S1, in ESI†) and the corresponding nominal ratio (calculated from the stoichiometric composition of the BaMn1−xCuxO3 catalysts) are compared. For copper catalysts, the XPS ratio is equal (BMC1) or lower (BMC2 and BMC3) than the nominal one, being the difference larger as the copper content increases (0.01 for BMC2 and 0.04 for BMC3). Considering that by XPS only the surface of the solid is analyzed, this result supports that, as it was previously observed for BaTi1−xCuxO3 catalysts,40,42 copper is being partially introduced into the perovskite structure, agreeing with the conclusion drawn from XRD data.
| Catalyst | Cu/Ba + Mn + Cu (nominal) | Cu/Ba + Mn + Cu (XPS) | Mn(IV)/Mn(III) (XPS) | OL/Ba + Mn + Cu (XPS) |
|---|---|---|---|---|
| BMC0 | — | — | 0.70 | 1.50 |
| BMC1 | 0.05 | 0.05 | 0.90 | 1.40 |
| BMC2 | 0.1 | 0.09 | 0.80 | 1.30 |
| BMC3 | 0.15 | 0.11 | 0.60 | 1.20 |
For all BaMn1−xCuxO3 catalysts, the XPS spectra of the Mn 2p3/2 transition, shown in Fig. 2b, feature a BE maximum at 641.8 eV approximately. It is well known that XPS is a useful tool to determine the oxidation state of an element, as the BE maximum value is straightly related to the oxidation states. Nevertheless, the assignment of the manganese oxidation states by XPS is a difficult task, as it has been featured by Dicastro and Polzonetti54 and also, by Nelson et al.55 Thus, the peak maximum of the Mn 2p3/2 transition suggests the presence of Mn(III) and Mn(IV) in all the catalysts. Based on the Ponce et al. procedure56 and, on the asymmetric shape of these spectra, the band has been deconvoluted considering three contributions: (i) the first peak at ∼641.7 eV corresponds to Mn(III), (ii) the peak at ∼643 eV is assigned to Mn(IV) and, (iii) the peak at 645 eV is a Mn(III) satellite peak. From the area of the corresponding peaks, the ratio Mn(IV)/Mn(III) was calculated and values, lower than 1 for all the catalysts, indicate that Mn(III) is the main oxidation state for all the BaMn1−xCuxO3 perovskites. Note also that the Mn(IV)/Mn(III) ratio decreases with the copper content, suggesting that copper promotes an increase of Mn(III) amount. Based on the BaMnO3 stoichiometric formula, Mn(IV) has to be the predominant Mn oxidation state in the catalysts, and an increase of the Mn(IV) amount would be expected to compensate the additional defect of positive charge due to manganese substitution by copper. However, XPS results reveal just the opposite effect as the Mn(III) amount increases with the copper content and, consequently, the electroneutrality must be achieved by the generation of oxygen vacancies, that is, by the formation of a non-stoichiometric (oxygen deficient) perovskite BaMn1−xCuxO3−δ.57 Thus, the highest amount of oxygen vacancies must be generated in the BMC3 catalyst as it shows the lowest Mn(IV)/Mn(III) ratio.
The analysis of the XPS O 1s transition brings valuable data about oxygen species present in the BaMn1−xCuxO3 catalysts series. Thus, the O 1s transition spectra recorded for all the catalysts, shown in Fig. 2c, reveal four contributions assigned, from lower to higher binding energies, to: lattice oxygen (OL), adsorbed oxygen (O*), hydroxyls and carbonates groups (OH−/CO32−) and adsorbed water (H2O).42 In these spectra, it is clearly observed that copper introduction promotes the generation of surface oxygen species, such as adsorbed oxygen (O*) and hydroxyls/carbonates species (OH−/CO32−), since the area corresponding to these species increases from BMC0 to BMC3 catalyst. As a consequence, the OL/(Ba + Mn + Cu) ratio (Table 3), calculated from the atomic percentages obtained by XPS (see Table S1 in ESI†) and the area of OL peak, decreases gradually as the amount of copper increases, showing the lowest value for BMC3 catalyst. In fact, the amount of oxygen calculated from XPS data is larger than the nominal one and increases with the copper content. Li et al.58 observed by XPS that A and B cations substitution in La1−xKxCo1−yCuyO3 catalysts led to an increase of the surface oxygen groups associated with the generation of oxygen vacancies. Also, Zhan and co-workers59 concluded that the increase of the Oads/OL ratio, due to the lanthanum substitution in the LaMn0.5Cu0.5O3 perovskite, indicates that a higher amount of oxygen defects exists in the catalysts. Thus, the decrease of the OL/(Ba + Mn + Cu) ratio and the increase of the surface oxygen peak areas observed for the BaMn1−xCuxO3 catalysts allow concluding that the incorporation of copper into the structure leads to an increase of the oxygen vacancies42,60 and that the BMC3 catalyst is the perovskite with the highest number of structural defects as it is expected based on the analysis of the Mn 2p3/2 spectra. Additionally, during a set of O2-TPD experiments (Fig. S2 in ESI†), desorption of oxygen (α-O2) at low temperature (350–650 °C) was observed for all the catalysts. However, it is worth mentioning that it cannot be drawn any trend from these profiles due to the low amount of α-O2 desorbed (ascribed to the low specific surface area of these catalysts). Therefore, these results support that a larger amount of structural defects have been formed in copper containing catalysts, as it is widely reported61,62 that α-O2 can be associated to the desorption of oxygen adsorbed in the vacancies of the structure.
In summary, XPS data reveal that Mn(IV) and Mn(III) oxidation states coexist in the BaMn1−xCuxO3 catalysts. Besides, the amount of Mn(III) species increases with the copper content and, consequently, a higher amount of oxygen defects are created in the structure to balance out the deficient positive charge generated.
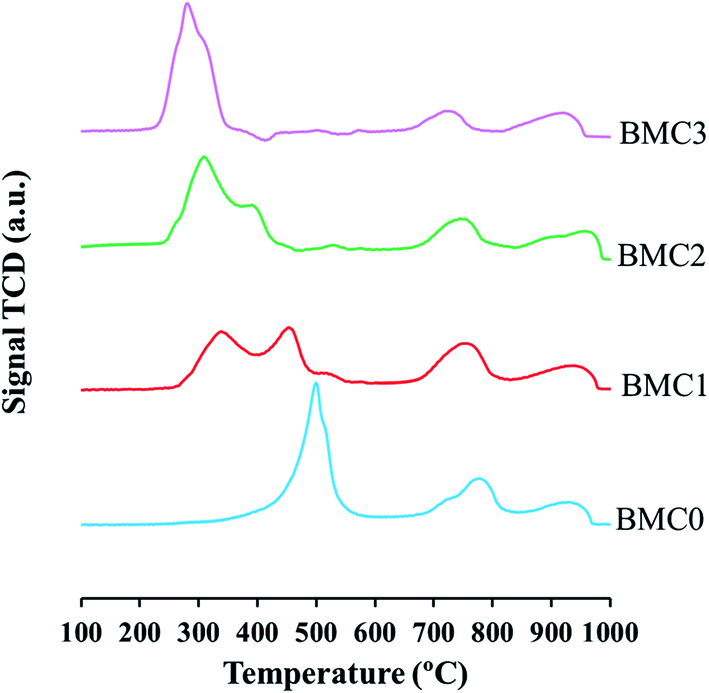 | ||
| Fig. 3 H2-TPR profiles of the BaMn1−xCuxO3 catalysts. | ||
Based on this assignment, the most relevant information related to the redox properties of the catalysts is located in the first temperature region. BMC0 profile shows only one well-defined peak with a maximum at 500 °C. Kapteijn et al.64 studied the reduction of different manganese oxides and concluded that MnO2 reduction mechanism involves two reduction processes: firstly, MnO2 is reduced to Mn3O4 and this oxide is finally reduced to MnO. The asymmetric peak observed in the BMC0 H2-TPR profile suggests that Mn(IV)/Mn(III) reduction also occurs in two different steps: below 450 °C, the reduction of Mn(IV) to Mn(III) takes place, and then, Mn(III) is reduced to Mn(II).
In the H2 consumption profiles of BMC1 and BMC2, two peaks are identified. The peak with maximum at 350 °C is ascribed to the Cu(II) to Cu(0) reduction and appears at a lower temperature than that obtained for a CuO used as a reference.42 This peak shows a shoulder at low temperature that could reveal that copper reduction also takes place in two steps (Cu(II) to Cu(I) and then, Cu(I) to Cu(0)).65,66 The peak at 450 °C for the BMC1 catalyst and at 400 °C for the BMC2 corresponds to the Mn(III)/Mn(IV) reduction which, for both catalysts, takes place at a lower temperature than for the BMC0 catalyst. In the H2-TPR profile of the BMC3 catalyst, it is not observed a clear separation of copper and manganese reduction processes since only one H2 consumption peak is shown. Rojas et al.60 observed similar results for LaMn1−xCuxO3 perovskites, for which the reduction of copper and manganese appears overlapped in the H2-TPR profiles. Furthermore, for BMC3 catalyst the manganese reduction temperature decreases by 200 °C respect to the BMC0 catalyst.
Thus, from H2-TPR results it can be concluded that copper incorporation promotes the reduction of manganese since the reduction temperature for Mn(IV)/Mn(III) decreases as the copper content increases. Tien-Thao and coworkers67 also observed this synergic effect in the reduction of Co and Cu in LaCo1−xCuxO3 mixed oxides. These authors justified the decrease in the reduction temperature of cobalt in the presence of copper by the formation of metallic copper (which is more easily reduced than cobalt) during the reduction experiment which can move toward the catalyst surface and promotes the reduction of lattice cobalt.
Despite the fact that O2-TPD experiments do not allow to determine the reducibility of catalysts by itself, it can be used to obtain additional information about the redox properties. It is generally accepted61,62,68,69 that desorption of oxygen at high temperature (beyond 650 °C and called β-O2) comes from the perovskite structure and it is related with the reduction of high oxidation state B cations. Therefore, the amount of β-O2 can be related with the catalyst reducibility. The desorption profiles recorded for all the catalysts (Fig. S2 in ESI†) clearly show a high temperature (650–950 °C) oxygen desorption peak that comes from the release of oxygen from the lattice (β-O2). The intensity of this peak becomes higher as the copper content increases, suggesting that copper introduction promotes the oxygen mobility in the perovskite lattice, but also that the copper improves the reducibility of the manganese. In Fig. 4, the nominal H2 consumption (mL of H2 per gram of catalysts) expected if manganese is Mn(III) or Mn(IV) in the catalyst is compared with the experimental H2 consumption obtained from the H2-TPR profiles. Firstly, it is noticed that the experimental H2 consumption is in between both nominal values confirming that, as it was previously deduced from XPS results, both Mn(IV) and Mn(III) oxidation states exist in the BaMn1−xCuxO3 catalysts. Additionally, it is noticeably that the experimental H2 consumption follows the same trend than the Mn(IV)/Mn(III) ratio obtained by XPS, thus pointing out that Mn(III) becomes the predominant oxidation state as copper content increases.
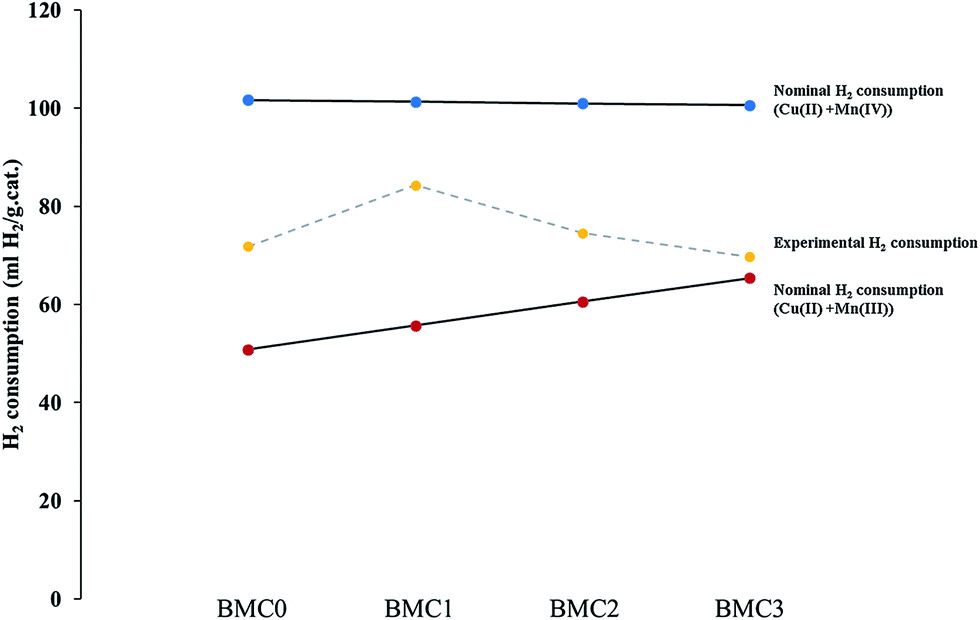 | ||
| Fig. 4 H2 consumption (mL g−1 of catalyst) of the BaMn1−xCuxO3 catalysts. | ||
In summary, the characterization data indicate that manganese is partially substituted by copper in the perovskite structure, leading to a new structural order of the BO6 octahedrons which originates the formation of a manganese-deficient perovskite with a new hexagonal structure. Even though Mn(III) and Mn(IV) coexist in BaMn1−xCuxO3 catalysts, manganese seems to be mainly as Mn(III) and, as a consequence, oxygen vacancies exist to balance out the deficient positive charge generated by the copper substitution. Both, the amount of Mn(III) and of oxygen vacancies increase with the copper content. Finally, the replacement of manganese by copper significantly facilitates the reducibility of the catalyst and increases the mobility of the lattice oxygen of the perovskite.
3.2 Catalytic activity tests
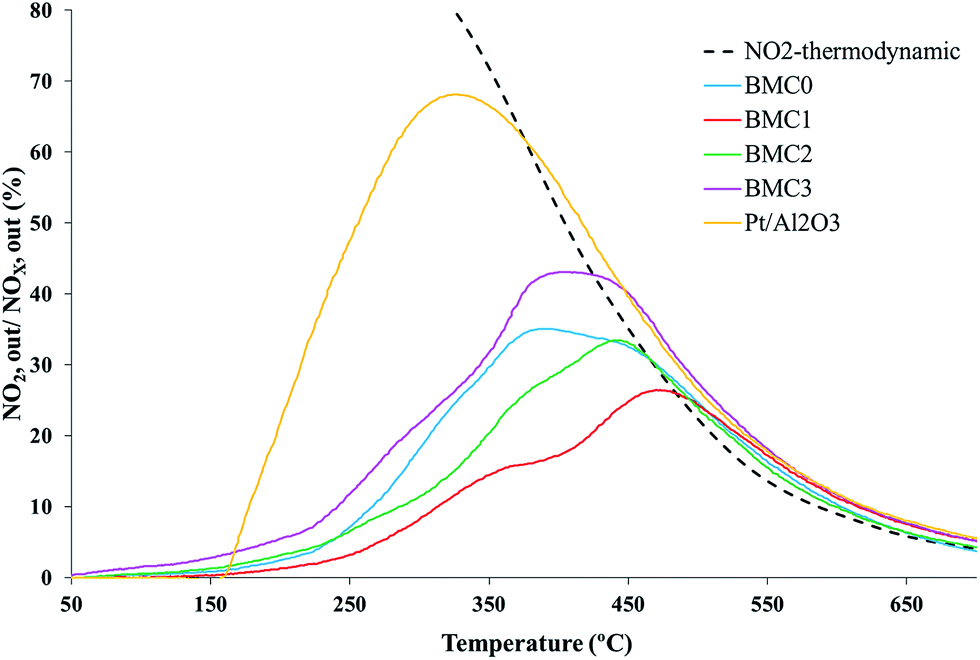 | ||
| Fig. 5 TPR NO2 generation profiles of the BaMn1−xCuxO3 and platinum-based reference catalysts. | ||
These profiles reveal that BMC0 and BMC3 catalysts show the highest activity for NO to NO2 oxidation among the perovskites prepared, with NO2 generation maximum values of 35% at 350 °C and 43% at 400 °C, respectively. It is remarkable that these catalysts showed only one phase in the XRD patterns and a higher Mn(III) amount than the other copper catalysts (BMC1 and BMC2). The better performance of the BMC3 catalyst respect to BMC0, seems to be related with the enhanced redox properties (higher reducibility and O2 mobility) of the former catalyst due to copper introduction in the perovskite structure.39 Anyway, the NO to NO2 oxidation activity of BMC3 catalyst is still far away from the activity shown by the 1% Pt/Al2O3 catalyst used as a reference.
On the other hand, the activity of the BMC1 and BMC2 catalysts for NO oxidation is even lower than that observed for the copper-free catalyst BMC0. For BMC1 and BMC2 two different BaMnO3 structures have been identified by XRD. Additionally, their amount of Mn(III) is lower than for BMC0 and BMC3 and, consequently, a lower amount of oxygen vacancies are created on the catalysts.
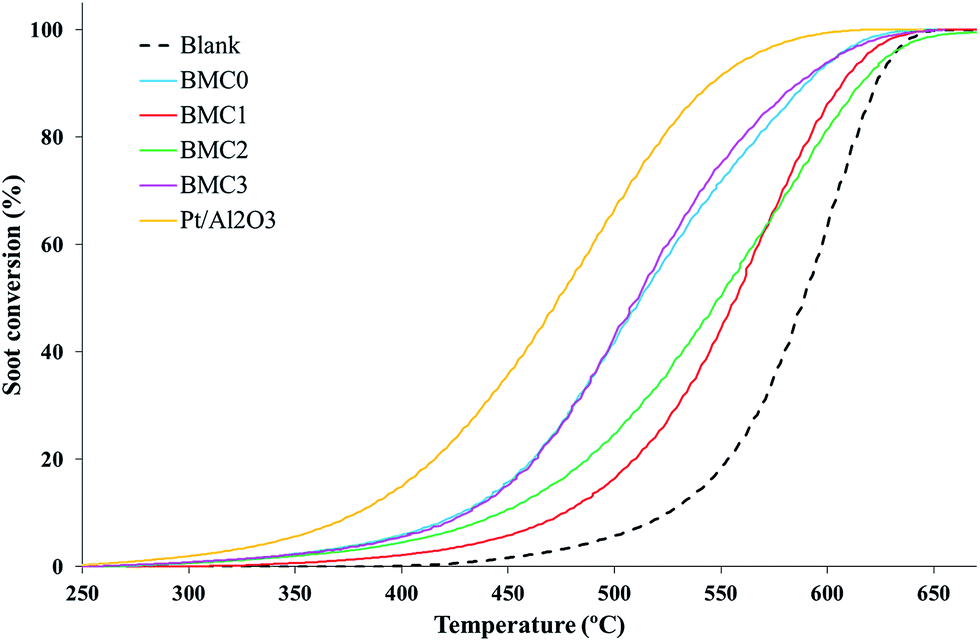 | ||
| Fig. 6 TPR soot conversion profiles of the BaMn1−xCuxO3 and platinum-based reference catalysts in NO/O2 atmosphere. | ||
To prove that the catalytic activity is directly related to the NO2 generation, O2-soot combustion experiments under temperature programmed conditions, using a 5% O2 in N2 atmosphere, were carried out. The soot oxidation profiles obtained (Fig. S3 in ESI†) do not reveal any significant decrease of the T5% and the T50% values respect to the blank experiment, confirming that the activity of the catalysts for soot oxidation is due to their ability to accelerate the NO2 generation. In Table 4, the CO2 selectivity data have also been included. As it was expected,39,40 all the catalysts show a high CO2 selectivity, even though only BMC0 and BMC3 perovskites are as selective as the 1% Pt/Al2O3 reference. BMC1 and BMC2 catalyst, indeed, show a lower CO to CO2 oxidation activity than the other catalysts. This result was also observed during the NO to NO2 activity tests, where it was related with the lower amount of surface oxygen vacancies and with the mixture of phases observed in XRD patterns.
| Catalyst | T5% (°C) | T50% (°C) | CO2 selectivity (%) |
|---|---|---|---|
| Blank | 485 | 589 | 45 |
| BMC0 | 390 | 512 | 98 |
| BMC1 | 443 | 557 | 79 |
| BMC2 | 406 | 548 | 88 |
| BMC3 | 394 | 511 | 97 |
| 1% Pt/Al2O3 | 344 | 474 | 100 |
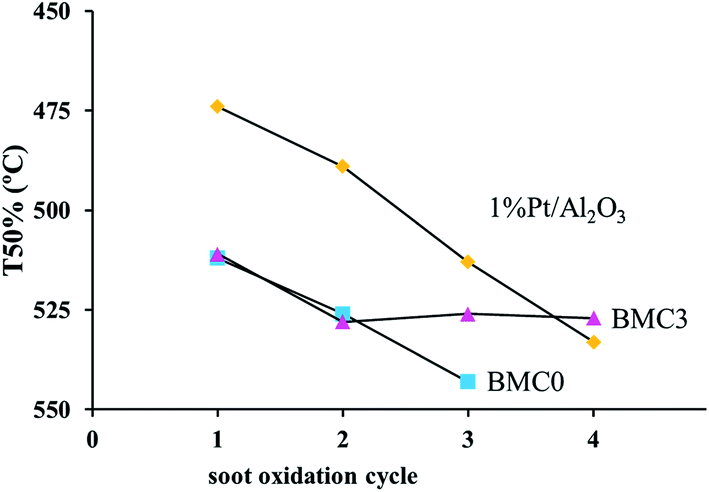
In order to further analyze the performance of the BMC0 and BMC3 catalysts, consecutive TPR soot oxidation cycles were carried out using the same parcel of catalyst (the TPR profiles for the consecutive cycles are shown in Fig. S4 in ESI†). In Fig. 7 the T50% values in the consecutive TPR cycles are plotted and compared with the results obtained for the 1% Pt/Al2O3 catalyst used as a reference. As it is observed, the value for BMC0 catalyst progressively decreases, indicating the catalyst suffers a deactivation. On the other hand, the T50 value for BMC3 catalyst slightly drops after the first cycle, but remains constant in the 2nd and 3rd cycle, and even during an additional 4th cycle experiment carried out for the two most active catalysts (BMC3 and 1% Pt/Al2O3). Finally, the platinum catalyst used as a reference also features deactivation during consecutive TPR cycles due to a very likely sintering process, as a consequence of the high reaction temperature achieved during TPR.71,72 Note that after the 4th cycle, the T50 value for BMC3 is close to that of 1% Pt/Al2O3.
 | ||
| Fig. 7 T50% values for consecutive soot oxidation TPR cycles. | ||
Thus, these results indicate that due to the incorporation of copper into the structure, as in BMC3, the catalyst is not deactivated during consecutives TPR soot oxidation cycles, whilst BMC0 shows a loss of activity. Therefore, the presence of copper in the perovskite, in agreement with literature studies of copper catalysts for the NO2-assisted soot combustion,39 improves the general performance of the catalysts.
In order to explain the effect of copper on the BMC3 catalysts performance for soot combustion, the two used BMC0 and BMC3 catalysts were deeply characterized after the third soot oxidation cycle by XRD, ICP-OES, XPS and infrared spectroscopy. The XRD patterns of the fresh and used samples (compared in Fig. 8) do not shown significant changes, so, it is confirmed that the structures of the catalysts are stable after consecutive TPR cycles, and consequently, the deactivation of the BMC0 catalyst is not due to a modification of the perovskite structure. Considering the conditions of TPR experiment, a very likely shift of copper species towards catalyst surface could be a feasible explanation.31 To analyze this possibility, the copper content in the used BMC0 and BMC3 catalysts was determined by ICP-OES and, also, the surface composition was analyzed by XPS, being the results shown in Table 5. The metal content and the surface chemical composition of BMC0 and BMC3 catalysts are almost identical to that of the fresh catalysts, indicating that copper remains incorporated into the perovskite structure and there is not migration of copper species towards the surface. Fig. 9 shows the infrared spectra recorded for the fresh and used BMC0 and BMC3 catalysts. The comparison of both spectra reveals the presence of small bands assigned to C–O bonds of surface oxygen species formed on the carbon deposit,73 corresponding to un-burnt soot, which are present in the used catalysts. These carbon deposits remain at the end of the experiment due to the high thermal stability of the carbon black used as model soot.73 Note that the intensity of these bands is higher for BMC0 catalyst than for BMC3 catalyst, indicating a larger accumulation in BMC0 due to the absence of copper. To remove the carbon deposit, the used samples were calcined at 900 °C for 2 h in a furnace; however, only a decrease in the intensity of the bands is observed supporting the high thermal resistance of the carbon black (Fig. 9). Thus, according to this IR results, it seems that the BMC0 deactivation is due to the accumulation of carbon deposits on the catalyst surface, being this effect less significant for BMC3 as this catalyst, due to the presence of copper, shows the highest soot oxidation rate, which minimizes the accumulation of un-burnt soot on the surface.
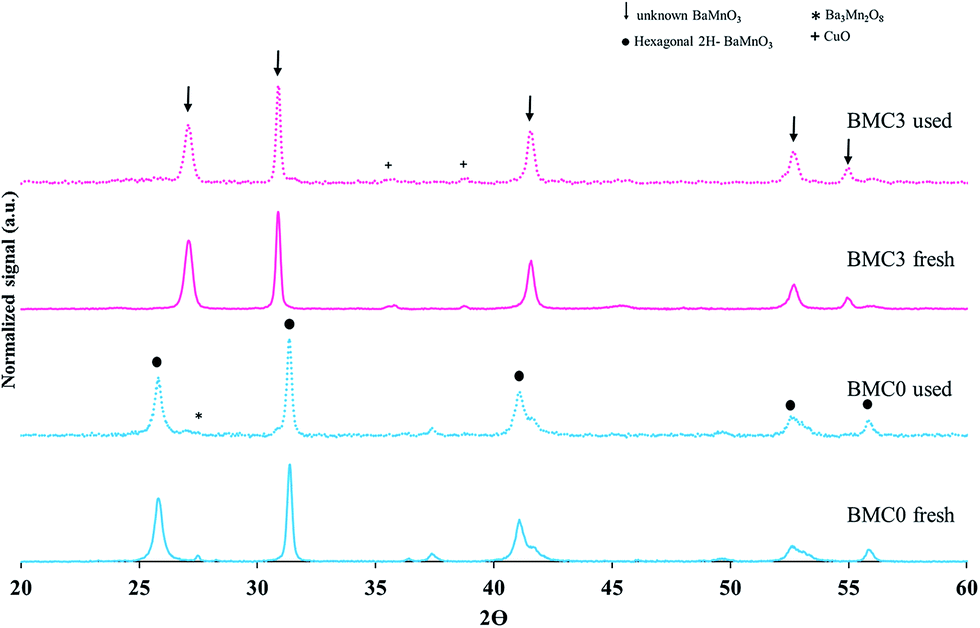 | ||
| Fig. 8 XRD patterns of the fresh and used (a) BaMnO3 (BMC0) and (b) BaMn0.7Cu0.3O3 (BMC3) catalysts. | ||
| Catalyst | ICP-OES Cu (wt%) | Cu/Ba + Mn + Cu (XPS) | Mn(IV)/Mn(III) (XPS) | OL/Ba + Mn + Cu (XPS) |
|---|---|---|---|---|
| BMC0_used | — | — | 0.70 | 1.50 |
| BMC3_used | 7.5 | 0.10 | 0.50 | 1.20 |
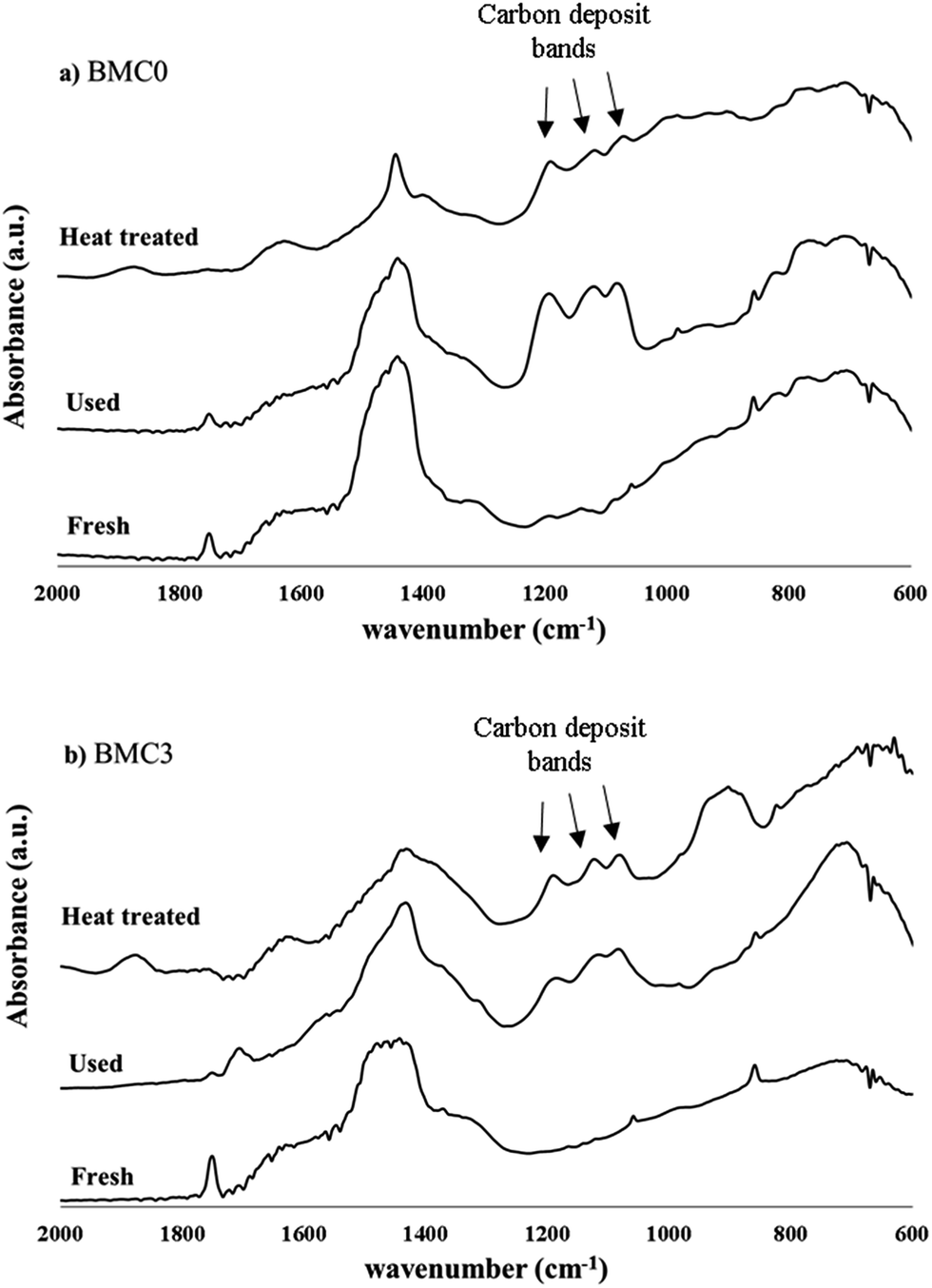 | ||
| Fig. 9 IR spectra for the fresh, used and heat treated BaMnO3 (BMC0) and BaMn0.7Cu0.3O3 (BMC3) catalysts. | ||
Therefore, all the characterization results allow concluding that the incorporation of copper in the BMC3 catalyst leads to the formation of a manganese-deficient perovskite, with a new hexagonal structure, which seems to be more active for NO to NO2 and soot oxidation than the copper-free catalyst (BMC0). Furthermore, this perovskite does not lose activity during four consecutive TPR cycles of soot oxidation as copper remains into the lattice of the perovskite.
Finally, to complete the analysis of the catalytic activity of the BMC0 and BMC3 catalysts and, in order to determine the soot oxidation rate under more realistic isothermal conditions, soot oxidation experiments at 450 °C were carried out. The soot oxidation profiles at 450 °C, featured in Fig. 10, reveal that BMC3 catalyst shows higher activity than BMC0 at 450 °C, that is, the copper content catalyst is able to oxidize soot faster than the copper-free perovskite. Thus, these profiles confirm that the addition of copper improves the NO2 generation activity and, consequently, BMC3 catalyst is more active for soot removal under isothermal conditions than BMC0. In fact, the t80% value (time to achieve the 80% of soot conversion) obtained from BMC3 soot conversion profile is even lower than the values obtained for other copper based perovskites catalysts.74
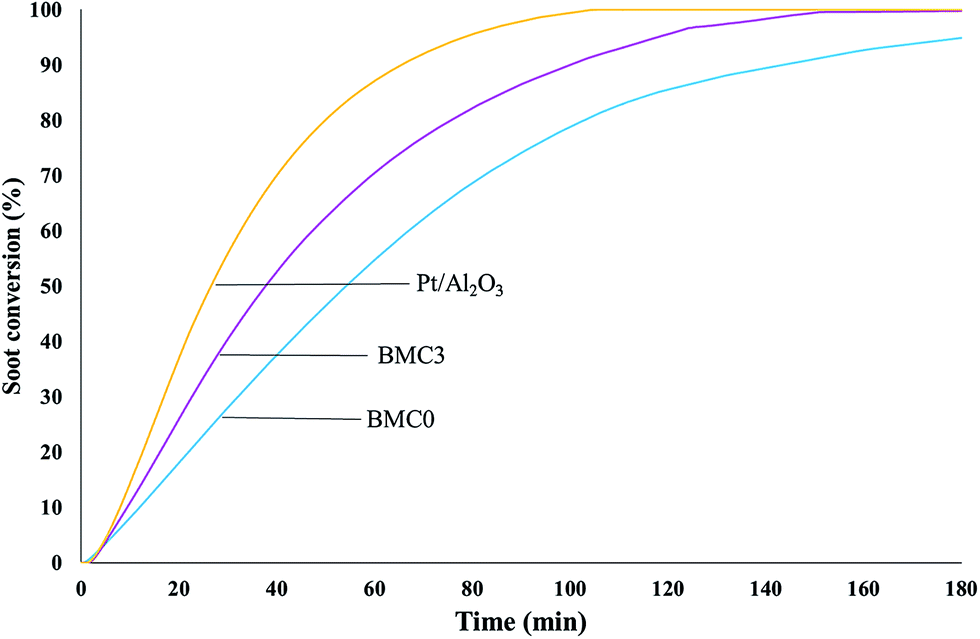 | ||
| Fig. 10 Soot conversion profiles, at 450 °C, for BaMnO3 (BMC0), BaMn0.7Cu0.3O3 (BMC3) and platinum-based catalysts. | ||
4. Conclusions
In this work a series of BaMn1−xCuxO3 (x = 0, 0.1, 0.2 and 0.3) catalysts have been synthesized and the effect of copper substitution on NO to NO2 oxidation and on NO2-assisted soot removal activity have been analyzed. From the characterization and activity test results the following conclusions have been drawn:- Partial substitution of manganese by copper in the perovskite structure of a BaMnO3 mixed oxide promotes a new structural order of the BO6 octahedrons and as a result, a new hexagonal manganese deficient perovskite structure is formed. This new structure is the only phase for the BMC3 (x = 0.3) catalyst whilst for BMC1 (x = 0.1) and BMC2 (x = 0.2) a mixture of two phases is observed.
- In the BaMn1−xCuxO3 catalysts, Mn(IV) and Mn(III) oxidation states coexist. Besides, the amount of Mn(III) species increases with the copper content and a higher amount of oxygen defects are created in the structure to balance out the deficient positive charge generated. Additionally, the presence of copper in the catalyst enhances the reducibility of the manganese species and increases the mobility of the lattice oxygen of the perovskite.
- BaMn0.7Cu0.3O3 is the most active catalyst for NO2 generation and, consequently, presents the lowest T50% value, the highest CO2 selectivity, and the best performance during TPR cyclic experiments, but also, the highest soot oxidation rate at 450 °C. This behavior is a result of the enhancement of the redox properties of the catalyst due to the partial substitution of Mn(III)/Mn(IV) by Cu(II) in the perovskite structure.
Acknowledgements
The authors thank Spanish Government (MINECO project CTQ2015-64801-R), the European Union (FEDER) and the Generalitat Valenciana (Project PROMETEOII/2014/10) for the financial support. Vicente Albaladejo-Fuentes thanks the University of Alicante for his Ph.D. grant and Veronica Torregrosa-Rivero thanks SECAT for her “Introduction to Research” grant.References
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2017 CrossRef.
- A. S. Bhalla, R. Guo and R. Roy, Mater. Res. Innovations, 2000, 4, 3–26 CrossRef CAS.
- L. G. Tejuca, J. L. G. Fierro and J. M. D. Tascón, Adv. Catal., 1989, 36, 237–328 CAS.
- S. Royer, D. Duprez, F. Can, X. Courtois, C. Batiot-Dupeyrat, S. Laassiri and H. Alamdari, Chem. Rev., 2014, 114, 10292–10368 CrossRef CAS PubMed.
- J. Zhu, H. Li, L. Zhong, P. Xiao, X. Xu, X. Yang, Z. Zhao and J. Li, ACS Catal., 2014, 4, 2917–2940 CrossRef CAS.
- H. Zhu, P. Zhang and S. Dai, ACS Catal., 2015, 5, 6370–6385 CrossRef CAS.
- H. Tanaka and M. Misono, Curr. Opin. Solid State Mater. Sci., 2001, 5, 381–387 CrossRef CAS.
- N. Yamazoe and Y. Teraoka, Catal. Today, 1990, 8, 175–199 CrossRef CAS.
- G. Pecchi, P. Reyes, R. Zamora, L. E. Cadús and J. L. G. Fierro, J. Solid State Chem., 2008, 181, 905–912 CrossRef CAS.
- S. Rousseau, S. Loridant, P. Delichere, A. Boreave, J. P. Deloume and P. Vernoux, Appl. Catal., B, 2009, 88, 438–447 CrossRef CAS.
- J. A. Gómez-Cuaspud, C. A. Perez and M. Schmal, Catal. Lett., 2016, 1–12 Search PubMed.
- F. Bidrawn, G. Kim, N. Aramrueang, J. M. Vohs and R. J. Gorte, J. Power Sources, 2010, 195, 720–728 CrossRef CAS.
- Z.-X. Wei, C.-M. Xiao, W.-W. Zeng and J.-P. Liu, J. Mol. Catal. A: Chem., 2013, 370, 35–43 CrossRef CAS.
- N. Tien-Thao, H. Alamdari and S. Kaliaguine, J. Solid State Chem., 2008, 181, 2006–2019 CrossRef.
- F. N. Agüero, M. R. Morales, S. Larrégola, E. M. Izurieta, E. Lopez and L. E. Cadús, Int. J. Hydrogen Energy, 2015, 40, 15510–15520 CrossRef.
- L. Jia, J. Gao, W. Fang and Q. Li, J. Rare Earths, 2010, 28, 747–751 CrossRef CAS.
- E. Grabowska, Appl. Catal., B, 2016, 186, 97–126 CrossRef CAS.
- W. F. Libby, Science, 1971, 171, 500–501 Search PubMed.
- C. H. Kim, G. Qi, K. Dahlberg and W. Li, Science, 2010, 327, 1624–1627 CrossRef CAS PubMed.
- J. Chen, M. Shen, X. Wang, J. Wang, Y. Su and Z. Zhao, Catal. Commun., 2013, 37, 105–108 CrossRef CAS.
- V. G. Milt, M. A. Ulla and E. E. Miró, Appl. Catal., B, 2005, 57, 13–21 CrossRef CAS.
- H. Iwakuni, Y. Shinmyou, H. Yano, H. Matsumoto and T. Ishihara, Appl. Catal., B, 2007, 74, 299–306 CrossRef CAS.
- W. Xu, J. Cai, J. Zhou, Y. Ou, W. Long, Z. You and Y. Luo, ChemCatChem, 2016, 8, 417–425 CrossRef CAS.
- J. Zhu and A. Thomas, Appl. Catal., B, 2009, 92, 225–233 CrossRef CAS.
- W. Y. Hernández, M. N. Tsampas, C. Zhao, A. Boreave, F. Bosselet and P. Vernoux, Catal. Today, 2015, 258, 525–534 CrossRef.
- L. L. Ferreira Squaiella, C. A. Martins and P. T. Lacava, Fuel, 2013, 104, 183–193 CrossRef.
- M. V. Twigg, Appl. Catal., B, 2007, 70, 2–15 CrossRef CAS.
- B. Guan, R. Zhan, H. Lin and Z. Huang, J. Environ. Manage., 2015, 154, 225–258 CrossRef CAS PubMed.
- W. F. Shangguan, Y. Teraoka and S. Kagawa, Appl. Catal., B, 1997, 12, 237–247 CrossRef CAS.
- Y. Teraoka, K. Nakano, S. Kagawa and W. F. Shangguan, Appl. Catal., B, 1995, 5, L181–L185 CrossRef CAS.
- F. E. López-Suárez, A. Bueno-López, M. J. Illán-Gómez, A. Adamski, B. Ura and J. Trawczynski, Environ. Sci. Technol., 2008, 42, 7670–7675 CrossRef.
- H. Wang, Z. Zhao, C. M. Xu and J. Liu, Catal. Lett., 2005, 102, 251–256 CrossRef CAS.
- H. Shimokawa, H. Kusaba, H. Einaga and Y. Teraoka, Catal. Today, 2008, 139, 8–14 CrossRef CAS.
- L. Li, X. Shen, P. Wang, X. Meng and F. Song, Appl. Surf. Sci., 2011, 257, 9519–9524 CrossRef CAS.
- J. Wang, Y. Su, X. Wang, J. Chen, Z. Zhao and M. Shen, Catal. Commun., 2012, 25, 106–109 CrossRef CAS.
- X. S. Peng, H. Lin, W. F. Shangguan and Z. Huang, Chem. Eng. Technol., 2007, 30, 99–104 CrossRef CAS.
- P. Doggali, S. Kusaba, Y. Teraoka, P. Chankapure, S. Rayalu and N. Labhsetwar, Catal. Commun., 2010, 11, 665–669 CrossRef CAS.
- Y. Gao, X. Wu, S. Liu, D. Weng, H. Zhang and R. Ran, Catal. Today, 2015, 253, 83–88 CrossRef CAS.
- F. E. López-Suárez, S. Parres-Esclapez, A. Bueno-López, M. J. Illán-Gómez, B. Ura and J. Trawczynski, Appl. Catal., B, 2009, 93, 82–89 CrossRef.
- V. Albaladejo-Fuentes, F. E. López-Suárez, M. S. Sánchez-Adsuar and M. J. Illán-Gómez, Appl. Catal., A, 2016, 519, 7–15 CrossRef CAS.
- E. J. Cussen and P. D. Battle, Chem. Mater., 2000, 12, 831–838 CrossRef CAS.
- V. Albaladejo-Fuentes, F. E. López-Suárez, M. S. Sánchez-Adsuar and M. J. Illán-Gómez, Appl. Catal., A, 2014, 488, 189–199 CrossRef CAS.
- R. Shinoda, A. Iwase and T. Matsui, J. Appl. Phys., 2014, 116, 6 CrossRef.
- F. Parrino, E. García-López, G. Marcì, L. Palmisano, V. Felice, I. N. Sora and L. Armelao, J. Alloys Compd., 2016, 682, 686–694 CrossRef CAS.
- D. Cordischi, M. Faticanti, G. Minelli, M. Occhiuzzi and P. Porta, Phys. Chem. Chem. Phys., 2003, 5, 1467–1473 RSC.
- A. Tarjomannejad, A. Niaei, A. Farzi, D. Salari and P. R. Zonouz, Catal. Lett., 2016, 146, 1544–1551 CrossRef CAS.
- N. A. Merino, B. P. Barbero, P. Ruiz and L. E. Cadús, J. Catal., 2006, 240, 245–257 CrossRef CAS.
- V. C. Coletta, F. C. F. Marcos, F. G. E. Nogueira, M. I. B. Bernardi, A. Michalowicz, R. V. Goncalves, E. M. Assaf and V. R. Mastelaro, Phys. Chem. Chem. Phys., 2016, 18, 2070–2079 RSC.
- T. Negas and R. S. Roth, J. Solid State Chem., 1971, 3, 323–339 CrossRef CAS.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- J. Giménez-Mañogil, A. Bueno-López and A. García-García, Appl. Catal., B, 2014, 152–153, 99–107 CrossRef.
- L. Dong, L. Zhang, C. Sun, W. Yu, J. Zhu, L. Liu, B. Liu, Y. Hu, F. Gao, L. Dong and Y. Chen, ACS Catal., 2011, 1, 468–480 CrossRef CAS.
- J. Ghijsen, L. Tjeng, J. van Elp, H. Eskes, J. Westerink, G. Sawatzky and M. Czyzyk, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 11322–11330 CrossRef CAS.
- V. Di Castro and G. Polzonetti, J. Electron Spectrosc. Relat. Phenom., 1989, 48, 117–123 CrossRef CAS.
- A. J. Nelson, J. G. Reynolds and J. W. Roos, J. Vac. Sci. Technol., A, 2000, 18, 1072 CAS.
- S. Ponce, M. Peña and J. L. Fierro, Appl. Catal., B, 2000, 24, 193–205 CrossRef CAS.
- K. Tabata, Y. Hirano and E. Suzuki, Appl. Catal., A, 1998, 170, 245–254 CrossRef CAS.
- Z. Li, M. Meng, Y. Zha, F. Dai, T. Hu, Y. Xie and J. Zhang, Appl. Catal., B, 2012, 121–122, 65–74 CrossRef CAS.
- H. Zhan, F. Li, P. Gao, N. Zhao, F. Xiao, W. Wei and Y. Sun, RSC Adv., 2014, 4, 48888–48896 RSC.
- M. L. Rojas, J. L. G. Fierro, L. G. Tejuca and A. T. Bell, J. Catal., 1990, 124, 41–51 CrossRef CAS.
- T. Seiyama, Catal. Rev.: Sci. Eng., 1992, 34, 281–300 CAS.
- D. Fino, N. Russo, G. Saracco and V. Specchia, J. Catal., 2003, 217, 367–375 CrossRef CAS.
- R. Zhang, A. Villanueva, H. Alamdari and S. Kaliaguine, Appl. Catal., A, 2006, 307, 85–97 CrossRef CAS.
- F. Kapteijn, L. Singoredjo, A. Andreini and J. A. Moulijn, Appl. Catal., B, 1994, 3, 173–189 CrossRef CAS.
- A. Glisenti, M. Pacella, M. Guiotto, M. M. Natile and P. Canu, Appl. Catal., B, 2016, 180, 94–105 CrossRef CAS.
- F. Patcas, F. C. Buciuman and J. Zsako, Thermochim. Acta, 2000, 360, 71–76 CrossRef CAS.
- N. Tien-Thao, H. Alamdari, M. H. Zahedi-Niaki and S. Kaliaguine, Appl. Catal., A, 2006, 311, 204–212 CrossRef CAS.
- I. Rossetti and L. Forni, Appl. Catal., B, 2001, 33, 345–352 CrossRef CAS.
- N. T. Thao and N. T. Thuan, Journal of Science, Natural Sciences and Technology, 2009, 25, 112–122 Search PubMed.
- B. Van Setten, M. Makkee and J. Moulijn, Catal. Rev., 2001, 43, 489–564 CAS.
- S. Liu, X. Wu, D. Weng, M. Li and H. R. Lee, Chem. Eng. J., 2012, 203, 25–35 CrossRef CAS.
- M. H. Wiebenga, C. H. Kim, S. J. Schmieg, S. H. Oh, D. B. Brown, D. H. Kim, J. H. Lee and C. H. F. Peden, Catal. Today, 2012, 184, 197–204 CrossRef CAS.
- S. Mahamulkar, K. Yin, P. K. Agrawal, R. J. Davis, C. W. Jones, A. Malek and H. Shibata, Ind. Eng. Chem. Res., 2016, 55, 9760–9818 CrossRef CAS.
- F. E. López-Suárez, A. Bueno-López, M. J. Illán-Gómez and J. Trawczynski, Appl. Catal., A, 2014, 485, 214–221 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04980c |
| This journal is © The Royal Society of Chemistry 2017 |