
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of molecularly imprinted polymers (MIPs) as a solid phase extraction (SPE) sorbent for the determination of ibuprofen in water†
Y. A. Olcerab,
M. Demirkurta,
M. M. Demirb and
A. E. Eroglu *a
*a
aDepartment of Chemistry, İzmir Institute of Technology, Urla 35430, İzmir, Turkey. E-mail: ahmeteroglu@iyte.edu.tr
bDepartment of Materials Science and Engineering, Izmir Institute of Technology, Urla 35430, İzmir, Turkey
First published on 20th June 2017
Abstract
Ibuprofen is a well-known endocrine disrupter. In this study, highly selective molecularly imprinted polymers (MIPs) with different morphologies were synthesized via precipitation and bulk polymerization of methacrylic acid (MAA) and trimethylolpropane trimethacrylate (TRIM) in the presence of ibuprofen as a template. Non-imprinted polymers (NIPs) were also synthesized via the same procedure in the absence of ibuprofen. Spherical and monolithic MIPs were obtained using different experimental conditions, and the spherical MIP was shown to have better sorption towards ibuprofen. The optimum sample pH, sorbent amount, sample volume, and sorption time were determined to be 8.0, 25.0 mg, 10.0 mL, and 30.0 min, respectively. A methanol water mixture (MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O, 80:20, pH 3.0) was employed as an eluent with >97% (±0.8, n = 3) desorption. The MIP demonstrated high selectivity towards ibuprofen in the presence of naproxen and ketoprofen. The validity of the proposed method was checked via spike recovery tests using drinking and tap water samples. The method worked efficiently for both water types, resulting in the recoveries of 97.2% (±0.3, n = 3) and 97.7% (±0.2, n = 3).
:H2O, 80:20, pH 3.0) was employed as an eluent with >97% (±0.8, n = 3) desorption. The MIP demonstrated high selectivity towards ibuprofen in the presence of naproxen and ketoprofen. The validity of the proposed method was checked via spike recovery tests using drinking and tap water samples. The method worked efficiently for both water types, resulting in the recoveries of 97.2% (±0.3, n = 3) and 97.7% (±0.2, n = 3).
1. Introduction
Endocrine disrupting chemicals (EDCs) as man-made or natural exogenous compounds can interfere with processes that are controlled by hormones in the body.1 These compounds inhibit a group of enzymes called cyclooxygenases (COXs) that catalyze the synthesis of prostaglandin (PG).2,3 In this way, they show adverse effects such as feminization of fish, sex transposition, and hormone-related cancers.Ibuprofen is an example of an EDC. It is a propionic acid derivative of nonsteroidal anti-inflammatory drugs (NSAIDs) used in mild analgesics. As an EDC, IBU causes deficiency of one or both testicles (cryptorchidism) in the male foetus by interfering with COXs during testis development (dysgenesis). The descent would have the risk of poor semen quality and testicular germ cancer because of the disruption. The risk increases when IBU is used during the second trimester of the gestation period and further increases by the simultaneous usage of more than one type of endocrine disrupting mild analgesics.4 Hence, the timing and amount of exposure are important parameters for the distruption.5
Another issue that should be taken care of while dealing with EDCs is that they have non-monotonic dose response curves. These curves are completely different from the view point of the dose makes poison (in monotonic curves).6 Even very small amounts of these chemicals may create huge disruption. Thus, low detection limits or pre-concentration methods are required for the instrumental determination of these compounds.
Ibuprofen disruption is not only caused by the usage of drugs. This compound can be detected in wastewater, surface and ground waters, and even in drinking waters. Pollution due to IBU can arise from the sewage systems of both domestic and industrial regions. In addition, EDCs can contaminate irrigation waters produced in the wastewater treatment plants, and the products irrigated with this water.7
There are several solid phase extraction (SPE) methods in literature to determine ibuprofen using commercial SPE cartridges prior to instrumental determinations.3,8–11 Furthermore, there are research groups that prefer to synthesize their own sorbents for preconcentration.12–15
Molecular imprinting is inspired from the superiority of natural receptors that can specifically recognize only one type of substances among other species in the living body.16 MIP synthesis is achieved via the creation of cavities with special binding sites during polymer knitting via a crosslinking agent. Imprinting sites are initially created via interaction between a monomer and template molecule during pre-polymerization.16,17 These interactions provide the specific name of the total process, semi-covalent imprinting or non-covalent imprinting. The non-covalent approach is based on polar interactions, whereas semi-covalent imprinting occurs from covalent bonding.18 In covalent imprinting, a clear structure of the cavities and free polymerization conditions are provided. However, non-covalent imprinting is preferred because of the fast removal, rebinding, and release of the template.16
Various polymerization methods can be used for the synthesis of MIPs. Bulk and precipitation polymerization have been commonly used. The main distinction between these two polymerization methods is the usage of solvent in the reaction mixture.19 While the former method is carried out in the presence of small amounts of solvent, the latter method involves large amounts of solvent in the polymerization mixture. The MIPs prepared via bulk polymerization have a monolithic morphology. If the monoliths cannot be directly used, such as in capillary columns, they need to be crushed, ground, and sieved to have microparticles prior to the sorption process.20 This step is considered to be tedious and time-consuming. On the other hand, MIPs prepared via precipitation polymerization present spherical and monodispersed beads with a nano/sub micrometer diameter. They are suitable for use as SPE sorbents in traditional analytical applications.19 Since polymerization is carried out in the presence of a template molecule of interest, the molecules are surrounded by polymeric network chains. The removal of the template by solvent leaves behind cavities with a well-defined size, shape, and active surface sites.
Processes in which MIP is used as a sorbent in the SPE is called molecular-imprinted solid-phase extraction (MISPE).21 MISPE takes place as a sample preparation and pre-concentration method. It exhibits same advantages, such as stability, flexibility, and activity under various conditions, better wetting characteristics, high surface area, and better retention of analyte, with polymeric sorbents compared to the conventional SPE sorbents.22 However, selective and strong binding sites for the analyte under aqueous conditions and robust, rapid, and cost-effective synthesis of MIP make it a favorable sorbent as compared to other polymeric sorbents.16 In our study, we generated an MISPE method by synthesizing morphologically different MIPs for the selective and sensitive determination of ibuprofen in water samples prior to HPLC-DAD determination.
2. Experimental
2.1 Materials and methods
All the chemicals were of analytical reagent grade. Deionized ultrapure water (UPW) (18.2 MΩ) was used throughout the study. Glassware and plastic containers were cleaned using a soapy solution, rinsed with deionized water, and dried with acetone prior to use. Stock standard solutions of ibuprofen, ketoprofen, and naproxen, 500.0 mg L−1, were prepared by dissolving ibuprofen (Sigma Aldrich, product code: I4883, CAS no. [15687-27-1]), ketoprofen (product code: K1751, CAS no. [22071-15-4]), and naproxen (product code: J63103, CAS no. [26159-34-2]), respectively, in methanol (MeOH). Lower concentration standards were prepared daily by appropriate dilution from their stock standards before use.2.2 Instrumentation
HPLC analyses were performed using an Agilent 1200 series HPLC equipped with a diode array detector (DAD) (Agilent Technologies, USA). A Supelco C18 (Lichrosphere RP 18-5, 25 cm × 4.6 mm) column was used for separation. From the stock solution of ibuprofen, 0.025, 0.050, 0.10, 0.25, 0.50, 1.0, and 5.0 mg L−1 standards were prepared. MeOH:H2O mixtures in different compositions, several column temperatures, and flowrates were tried. The limit of detection (LOD) and limit of quantification (LOQ) were calculated.
2.3 Synthesis of MIP and NIP
In the precipitation polymerization strategy of MIP100, first, 0.0687 g ibuprofen, 226.0 μL methacrylic acid (MAA), and 100.0 mL acetonitrile were mixed in an amber reaction bottle and stirred for 1.0 hour for pre-polymerization. This MIP was given the name MIP100 as 100 mL of solvent, acetonitrile, was used in the synthesis. Then, 2.1 mL of trimethylolpropane trimethacrylate (TRIM) was added to the reaction mixture to obtain a molar ratio of 1:8:20 (template:monomer:crosslinker). 4,4-Azobis(4-cyanovaleric acid) (AIVN) was used as the initiator in a percentage of 2.0 (mole AIVN/total mole excluding ibuprofen) and was carefully added under Ar gas. Polymerization was carried out in an oil bath at 60 °C for 8 hours.
After polymerization, the template molecule was removed using two different solutions; MeOH and a MeOH:H2O (acetic acid, pH 3) (80:20) mixture. After complete removal of ibuprofen, MIPs were dried in an oven at 60.0 °C. The preparation of NIP100 was the same as that of MIP100, except for the addition of ibuprofen. The schematic of MIP synthesis is given in Fig. 1.
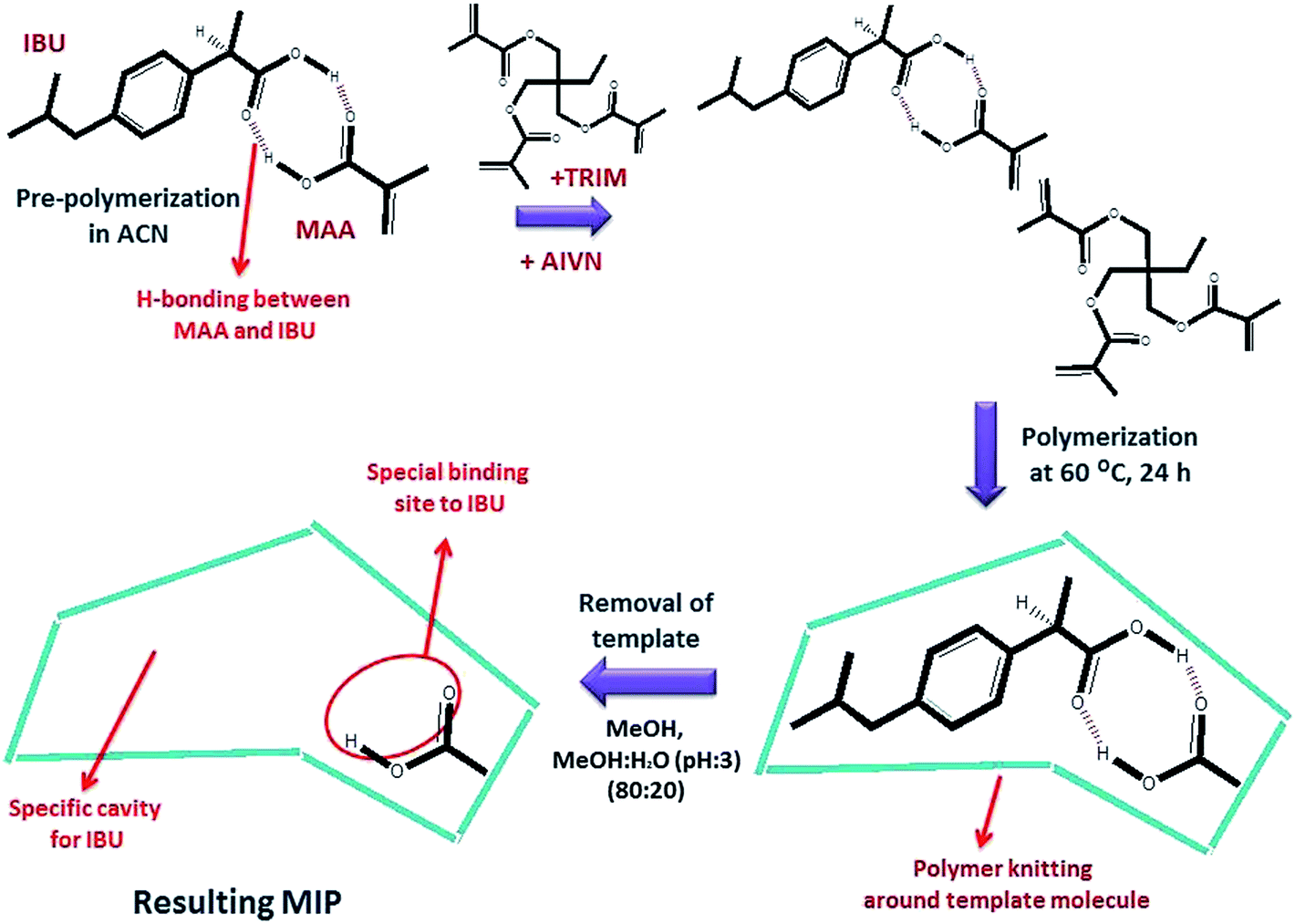 | ||
| Fig. 1 Schematic of the synthesis of an MIP via co-polymerization of MAA and TRIM. | ||
MIP10 and NIP10 were synthesized according to the abovementioned synthesis of MIP100 and NIP100, but 10 mL acetonitrile was used instead.
2.4 Characterization
Sorption performances of MIPs and NIPs were compared to prove the presence of cavities in MIPs. For this purpose, first, the binding characteristic assay was applied. Sample solutions were prepared from the stock solution of ibuprofen at 1.0, 5.0, 10.0, 20.0, 50.0, 100.0, and 250.0 mg L−1. MIPs and/or NIPs (25.0 mg) were added to ten milliliters of these solutions. Mixtures were shaken at 480 rpm using an orbital shaker for 8 hours.To demonstrate the selectivity of MIP100 towards ibuprofen, the sorption procedure was performed in the presence of structurally related compounds such that 10.0 mL mixture of ibuprofen, naproxen, and ketoprofen (50.0 mg L−1 each) were prepared and shaken with 25.0 mg of MIP100 as explained before.
2.5 Optimization of parameters
Herein, all the sorption procedures were realized using an orbital shaker at 480 rpm. Effluents and eluates were analyzed using HPLC-DAD. First, the effect of solution pH was examined using the values between 7.0 and 10.0 (adjusted with nitric acid and sodium hydroxide). Ibuprofen concentration was 1.0 mg L−1 (10.0 mL solution) and the amount of sorbent was 25.0 mg. In a separate experiment, the sorbent (MIP100) amount was also investigated by changing it from 5.0 to 100.0 mg for again 1.0 mg L−1 of 10.0 mL ibuprofen solution. For the determination of the optimum sample volume, 5.0, 10.0, 25.0, and 50.0 mL of sample solutions were mixed with 25.0 mg of sorbents for 1.0 mg L−1 of 10.0 mL ibuprofen. To find the optimum sorption time with the other parameters kept constant, the sorption procedure was carried out for different durations for 1.0 mg L−1 of 10.0 mL ibuprofen solution and 25.0 mg of sorbent.Analysis of eluates is also an important part of the MISPE process to prove that the analyte species retained by MIP are eluted with an appropriate eluent. MeOH and MeOH:H2O mixture (acetic acid, pH 3.0) (80:20) were used as eluents for the desorption of 1.0 mg L−1.
Reusability of the sorbent was checked via repetitive sorption/desorption of 1.0 mg L−1 ibuprofen with MIP100 for 30 minutes. After loading, the sorbent was washed with 10 mL MeOH:H2O (acetic acid, pH 3.0) (80:20) solution, dried in an oven, and reused in the next sorption cycle. This procedure was repeated ten times.
2.6 Method validation
Sorption efficiency of the sorbents was investigated by analyzing 10.0 mL aliquots of the 100.0 μg L−1 ibuprofen-spiked samples of ultrapure, drinking, and tap water using the optimum parameters for the MISPE process.3. Results and discussion
3.1 Instrumentation
Optimum parameters for mobile phase composition, flow rate, and column temperature were determined as 80:20 MeOH:H2O (acetic acid, pH 3.0), 0.8 mL min−1, and 30 °C, respectively. The LOD was 0.023 mg L−1 and LOQ was 0.075 mg L−1.
3.2 Synthesis of the MIP and NIP
The SEM images of MIPs and NIPs are shown in Fig. 2. Fig. 2a and b illustrate the controlled aggregation of small polymeric droplets with monolithic appearance of the resulting polymers because the polymerization reaction contains only small volume of the solvent and mostly monomer units and ibuprofen. This monolithic morphology can also be explained by a pseudo diagram that explains the morphology of the resulting polymer with respect to the composition of the cross-linker and the volume of the solvent. High amounts of solvent cause the MIP/NIP to be more smaller in shape (micro-sizes).23 It has also been established that the ratio of the total monomer to the porogen (w/v%, total monomer/porogen) determines the polymerization method as well as the morphology. For instance, for values bigger than 5%, polymer formation is thought to be in accordance with the bulk polymerization route.24 For MIP10 and NIP10, this ratio is 24.5%. | ||
| Fig. 2 SEM images of MIPs: (a) MIP10, (b) NIP10, (c) MIP100, and (d) NIP100. | ||
On the other hand, when the polymer mixture contains higher amounts of solvent, the polymer droplets become evident (Fig. 2c and d). Not surprisingly, the fabrication method falls into precipitation polymerization such that MIP100 and NIP100 yield uniform spheres. Aggregation does not occur due to the large amount of solvent, meaning that they have sufficient area to escape aggregation. Ratios smaller than 5% indicate that the polymers will be synthesized via precipitation polymerization, which results in spherical individual polymer particles with a homogenous binding site distribution.24 The total monomer to porogen ratio is calculated to be 2.5% for MIP100 and NIP100. In addition, there is no remarkable difference between MIP100 and NIP100 in terms of morphology. The template molecule is too small to be differentiated from the SEM images.
MIPs are crosslinked network structures. The development of the network is a diffusion-limited process, meaning that small crosslinked domains have much lower diffusion capability as compared to smaller linear chains. Therefore, a complete conversion of monomers into the crosslinked structure cannot be achieved.
After the synthesis of the MIP, the template (ibuprofen) was removed by washing out with MeOH. A total of ten consecutive washing cycles were found to be sufficient for the complete removal (Fig. S-1†).
3.3 Characterization
Sorption capacities of MIP100/NIP100 and MIP10/NIP10 particles as a function of ibuprofen concentration are shown in Fig. 3. The ordinate Q shows the ratio of mmol of ibuprofen to 1.0 g of MIP or NIP. At low concentrations, there is no remarkable difference between MIPs and NIPs. When the concentration is increased, difference between the sorption capacities of MIPs and NIPs starts to appear.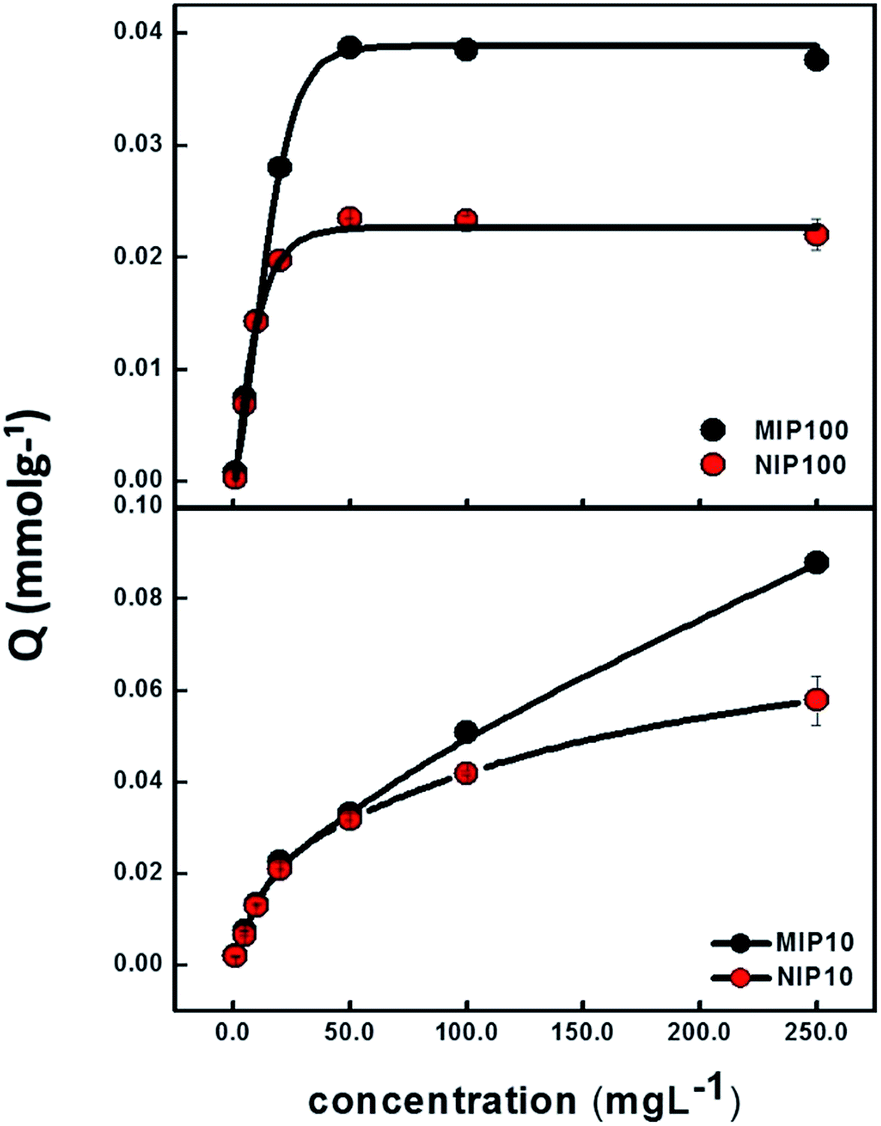 | ||
| Fig. 3 Binding characteristic assay showing the sorption capacities of MIP100/NIP100 (above) and MIP10/NIP10 (below). (Agilent 1200 Series HPLC-DAD system, Supelco C18 (Lichrosphere RP 18-5, 25 cm × 4.6 mm) column, 80:20 MeOH:H2O (pH 3.0) mobile phase, 0.8 mL min−1 flow rate, 220 nm, and n = 3). | ||
Above 50 mg L−1 of ibuprofen, the sorption capacities of MIP100 and NIP100 remain constant at the Q values of 0.0387 and 0.0235, respectively. This nearly two-fold capacity of MIP100 as compared to that of NIP100 also validates the presence of specific cavities for ibuprofen. The difference in the sorption capacity of MIP10 and NIP10 becomes evident above 50.0 mg L−1. However, a further increase in the concentration does not result in saturation although their sorption capacities seem to be much higher than those of MIP100 and NIP100. The investigation of this effect is a topic of further studies.
Cavities in MIPs are developed both on the surface and in the interiors of the structure. Since adsorption is an interface phenomenon, the main contribution to sorption performance may stem from the selective sites on the surface. The spherical MIPs have much more specific surface areas than the monolithic MIPs; thus, there are more cavities on their surface as compared to MIP10. Ibuprofen species retained by MIP10 and NIP10 may be thought to be more on the polymer network and not in the cavities. Thus, it could be said that MIP100 showed better (more specific) sorption performance. Hence, the rest of the experiments were performed using MIP100 as the sorbent.
Fig. 4 shows the MIP100 and NIP100 sensitivity/selectivity to ibuprofen in the presence of structurally related compounds, namely, naproxen and ketoprofen. For ibuprofen, MIP100 and NIP100 showed a remarkable difference in the sorption capacity. On the other hand, the difference in the sorption of imprinted and non-imprinted sorbents towards both naproxen and ketoprofen was indistinguishable from each other. This could be additional proof for the presence of specific binding sites in MIP100 against ibuprofen.
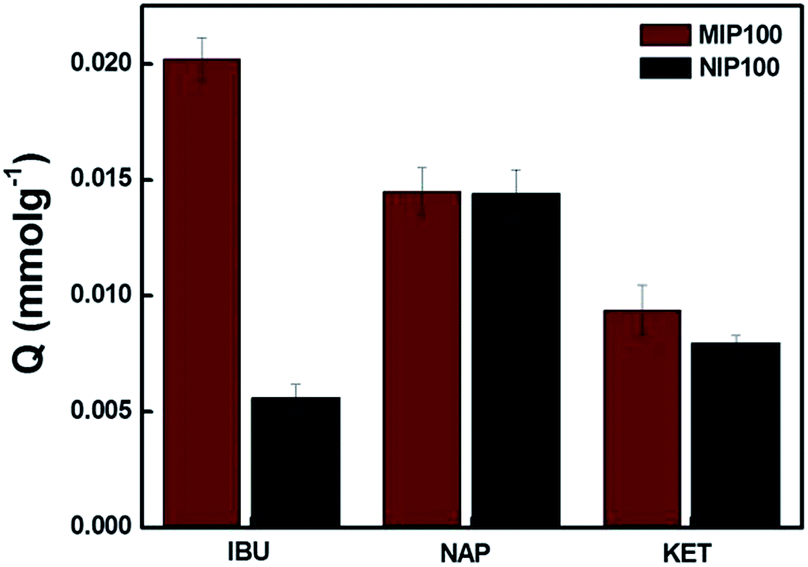 | ||
| Fig. 4 Cross sensitivity of MIP100 and NIP100. (Agilent 1200 Series HPLC-DAD system, Supelco C18 (Lichrosphere RP 18-5, 25 cm × 4.6 mm) column, 80:20 MeOH:H2O (pH 3.0) mobile phase, 0.8 mL min−1 flow rate, 220 nm, and n = 3). | ||
The Q value of MIP100 in the sorption of ibuprofen decreased to 0.0202, which was 0.0387 in the binding characteristics assay. Ibuprofen may not only be sorbed in the cavities but also retained by the surface of the polymer. The decrease in the sorption by MIP100 may be the result of the inhibition of ibuprofen by naproxen and ketoprofen during surface sorption. Like MIP100, NIP100 also shows a decrease in the sorption of ibuprofen because of the same reason.
3.4 Optimization of the working parameters
Sorption percentages of both MIP100 and NIP100 in the pH range from 7.0 to 10.0 are shown in Fig. 5. MIP100 showed quantitative sorption (99.0%, ± 0.3, n = 3) at pH 7.0 and 8.0, and then, the sorption capacity decreased with the increasing pH for both MIP100 and NIP100. A decrease in the sorption may be related to the charges of both ibuprofen and polymeric particles during sorption. Ibuprofen has a pKa value of 5.2, meaning that it is negatively charged in the pH range from 7.0 to 10.0. On the other hand, it can be said that a further increase in pH makes the functional groups, both in the cavities and on the surface, of MIP100 and NIP100 deprotonate and hence less available for sorption. This situation may be arising from the point of zero charge of the synthesized MIPs.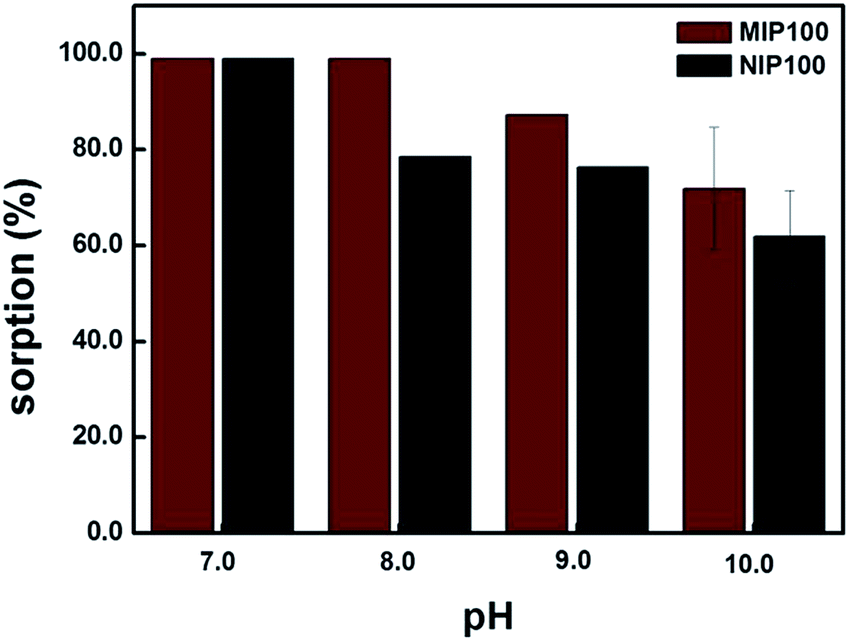 | ||
| Fig. 5 Effect of pH on sorption. (Agilent 1200 Series HPLC-DAD system, Supelco C18 (Lichrosphere RP 18-5, 25 cm × 4.6 mm) column, 80:20 MeOH:H2O (pH 3.0) mobile phase, 0.8 mLmin−1 flow rate, 220 nm, and n = 3). | ||
At pH 7.0, there is no remarkable difference in the sorption capacities of MIP100 and NIP100 at 1.0 mg L−1, as observed in the binding characteristic assay. Herein, note that regardless of the initial pH of the mixtures, the pH value of the mixtures changed to 7.0 after the mixtures were shaken with the sorbents.
For pH 8.0, MIP100 also exhibited quantitative sorption, but the sorption percentage of NIP100 decreased to 78.2% (±0.4, n = 3). The reason could be the rapid access of –OH to the surface of the NIP100 for deprotonation, whereas the cavities of MIP100 could not be accessed. Note that the good sorption ability of NIPs results in high sorption performance of the MIPs.25 Therefore, around 80% sorption by NIP under the specified conditions can also be considered in light of this thought.
At pH 9.0 and 10.0, there is a remarkable decrease in the sorption percentage of both MIP100 and NIP100. It may be said that –OH is now able to reach the specific binding sites in MIP100. In conclusion, the remaining experiments were carried out at pH 8.0, where a significant difference in the sorption was observed.
MISPE procedure was also applied using different amounts of MIP100. There is a linear increase in sorption with respect to the sorbent amount up to 25.0 mg of MIP100 (Fig. S-2†). Then, sorption levels off and remains unchanged (98.5%, ±0.7251, n = 3). It is clear that values smaller than 25.0 mg are not sufficient to have a nearly complete sorption of 1.0 mg L−1 of ibuprofen. Therefore, it was decided to use 25.0 mg sorbent in the remaining experiments.
The effect of the sample volume on the sorption percentage of MIP100 was examined with all the other parameters held constant. Ibuprofen solutions of 5.0 and 10.0 mL gave a high sorption percentage (98.6% (±3.5), n = 3 and 98.5% (±3.3), n = 3, respectively) and the percentage sorption immediately decreased after a further increase in the sample volume (Fig. S-3†). This might have been caused by two reasons: first, the total number of moles of ibuprofen was higher than the available sorption sites and second, the mixing was inefficient when the volume was increased beyond 10.0 mL. The number of cavities in 25.0 mg of MIP100 may not be sufficient for this increase. To guarantee quantitative sorption, 10.0 mL of 1.0 mg L−1 ibuprofen solution was used in the remaining experiments.
Using the predetermined parameters, the effect of shaking time on sorption was examined. The interaction time was not critical for sorption. Even 1 min was found to be sufficient for quantitative sorption (Fig. S-4†). The reason may be the ability of the high diffusion rate of spherical MIP100 because of the homogenous binding sites. To ensure that quantitative sorption was achieved, 30 min was chosen as the sorption time in the remaining experiments.
Desorption is an equally important part of the SPE process as sorption. Therefore, the analyte sorbed by MIP should be recovered from the sorbent using a proper eluent, and the desorption percentage should be calculated to understand the analyte concentration in an unknown sample. Herein, two different eluents, MeOH and MeOH:H2O (acetic acid, pH 3) (80:20), were tried for the desorption process. Both solutions have the ability of making hydrogen bonding stronger than the analyte molecule ibuprofen. These solvents disturb the hydrogen bonding between the analyte and the solid sorbent. Although both eluents gave >97% (97.2%, ±0.8, n = 3) desorption (Fig. S-5†), MeOH:H2O (acetic acid, pH 3) (80:20) solution was chosen to be used in the remaining experiments since it was the mobile phase employed in the HPLC-DAD determinations.
Fig. 6 shows that up to fifth sorption cycle, MIP100 shows greater than 90% sorption. The decrease in the sorption capacity may be explained by the disruption of specific cavities during the extraction process. Thus, it can be concluded that the sorbent MIP100 can be used efficiently up to five times under the experimental conditions applied. This is obviously an important point to study when a competitive sorbent is going to be synthesized.
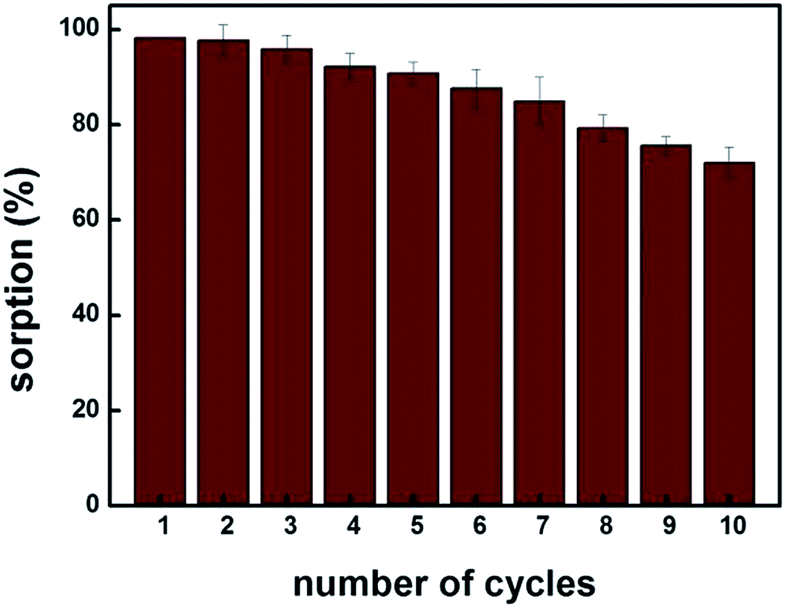 | ||
| Fig. 6 Reusability of the sorbent. (Agilent 1200 Series HPLC-DAD system, Supelco C18 (Lichrosphere RP 18-5, 25 cm × 4.6 mm) column, 80:20 MeOH:H2O (pH 3.0) mobile phase, 0.8 mL min−1 flow rate, 220 nm, and n = 3). | ||
3.5 Method validation
MIP100 was used in the sorption of ibuprofen in various types of spiked water samples. For all the samples, the sorption percentages were greater than 97% for n = 3, 97.4% (±0.3), 97.2% (±0.3), and 97.7% (±0.2) for ultrapure, drinking, and tap water, respectively (Fig. 7). The identical results of spiked drinking and tap water samples with those of ultrapure water clearly demonstrate that the proposed methodology can be applied in real samples.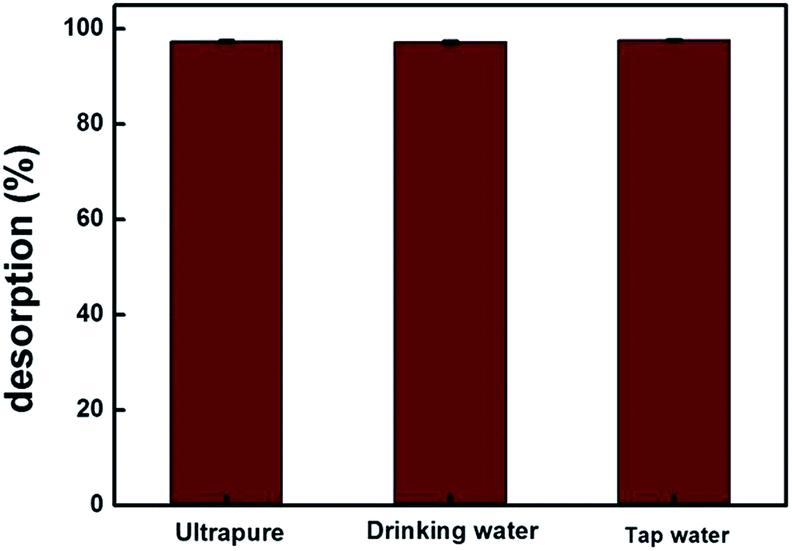 | ||
| Fig. 7 Validation of the proposed method with spiked water samples. (Agilent 1200 Series HPLC-DAD system, Supelco C18 (Lichrosphere RP 18-5, 25 cm × 4.6 mm) column, 80:20 MeOH:H2O (pH 3.0) mobile phase, 0.8 mL min−1 flow rate, 220 nm, and n = 3). | ||
4. Conclusion
MIPs are crosslinked network structures. Herein, two different types of molecularly imprinted polymers were prepared successfully for the specific recognition of ibuprofen prior to its determination by HPLC-DAD (so-called MIP100 for the material prepared via precipitation polymerization and MIP10 prepared via bulk polymerization route). The MIP prepared via precipitation polymerization was employed for the MISPE optimization steps because of its better sorption performance.The selectivity of MIP100 to ibuprofen was also demonstrated in the presence of structurally related compounds, namely, naproxen and ketoprofen. MIP showed better sorption performance than NIP and was found to be specific to ibuprofen. After the generation of the MISPE method by optimizing experimental parameters, the validity of the method was checked via spike recovery experiments with different types of water samples (drinking and tap waters). The method has been shown to be efficient and applicable to both water types.
The MIP particles synthesized herein can be used in many applications such as in the form of chromatographic materials in the preparation of packed and open-tubular columns and SPME fiber coating.
Acknowledgements
The authors thank The Scientific and Technological Research Council of Turkey (TUBITAK) for the research project coded 114Z777.References
- WHO/UNEP, World Health Organization, 2012.
- F. E. Bloom, Analgesic, http://www.global.britannica.com/science/analgesic.
- H. B. Lee, T. E. Peart and M. L. Svoboda, J. Chromatogr. A, 2005, 1094, 122–129 CrossRef CAS PubMed
.
- D. M. Kristensen, U. Hass, L. Lesne, G. Lottrup, P. R. Jacobsen, C. Desdoits-Lethimonier, J. Boberg, J. H. Petersen, J. Toppari, T. K. Jensen, S. Brunak, N. E. Skakkebaek, C. Nellemann, K. M. Main, B. Jegou and H. Leffers, Hum. Reprod., 2011, 26, 235–244 CrossRef CAS PubMed
- T. Damstra, S. Barlow, A. Bergman, R. Kavlock and G. V. D. Kraak, Global Assessment of the State-of-the-Science of Endocrine Disruptors, International Programme on Chemical Safety. WHO/PCS/EDC/02.2, World Health Organization, Geneva, Switzerland, 2002 Search PubMed
- EPA, Endocrine Disruption Research: Testing for Potential Low-Dose Effects, 2015, http://www.2.epa.gov/chemical-research/endocrine-disruption-research-testing-potential-low-dose-effects.
- D. Löffler and T. A. Ternes, J. Chromatogr. A, 2003, 1021, 133–144 CrossRef
- M. J. Gomez, M. Petrovic, A. R. Fernandez-Alba and D. Barcelo, J. Chromatogr. A, 2006, 1114, 224–233 CrossRef CAS PubMed
- M. Gros, M. Petrovic and D. Barcelo, Talanta, 2006, 70, 678–690 CrossRef CAS PubMed
- J. L. Santos, I. Aparicio, E. Alonso and M. Callejon, Anal. Chim. Acta, 2005, 550, 116–122 CrossRef CAS
- A. Kot-Wasik, J. Debska, A. Wasik and J. Namiesnik, Chromatographia, 2006, 64, 13–21 CAS
- M. Abd Rahim, W. A. W. Ibrahim, Z. Ramli, M. M. Sanagi and H. Y. Aboul-Enein, Chromatographia, 2016, 79, 421–429 CAS
- A. Sarafraz-Yazdi, A. Amiri, G. Rounaghi and H. Eshtiagh-Hosseini, Anal. Chim. Acta, 2012, 720, 134–141 CrossRef CAS PubMed
- K. Aguilar-Arteaga, J. A. Rodriguez, J. M. Miranda, J. Medina and E. Barrado, Talanta, 2010, 80, 1152–1157 CrossRef CAS PubMed
- C. Y. Hung, Y. T. Huang, H. H. Huang and C. C. Hwang, J. Chin. Chem. Soc., 2006, 53, 1173–1180 CrossRef CAS
- M. Komiyama, T. Takeuchi, T. Mukawa and H. Asanuma, Molecular Imprinted From Fundamentals to Applications, 2003 Search PubMed
- K. Farrington and F. Regan, Biosens. Bioelectron., 2007, 22, 1138–1146 CrossRef CAS PubMed
- N. Fontanals, R. M. Marce and F. Borrull, Contrib. Sci., 2010, 2, 199–213 Search PubMed
- J. Haginaka, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 866, 3–13 CrossRef CAS PubMed
- B. Sellergren, Man-Made Mimics of Antibodies and their Application in Analytical Chemistry, Elsevier Science, Amsterdam, 2001 Search PubMed
- J. O. Mahony, K. Nolan, M. R. Smyth and B. Mizaikoff, Anal. Chim. Acta, 2005, 534, 31–39 CrossRef CAS
- S. Weigel, R. Kallenborn and H. Huhnerfuss, J. Chromatogr. A, 2004, 1023, 183–195 CrossRef CAS PubMed
- P. A. G. Cormack and A. Z. Elorza, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 804, 173–182 CrossRef CAS PubMed
- L. Nune, E. Turiel, A. Martin-Esteban and J. L. Tadeo, Talanta, 2010, 80, 1782–1788 CrossRef PubMed
- C. Baggiani, C. Giovannoli, L. Anfossi, C. Passini, P. Baravalle and G. Giraudi, J. Am. Chem. Soc., 2012, 134, 1513–1518 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05254e |
| This journal is © The Royal Society of Chemistry 2017 |