DOI:
10.1039/C7RA04175F
(Paper)
RSC Adv., 2017,
7, 31448-31456
Magnetically separable AgI–BiOI/CoFe2O4 hybrid composites for Hg0 removal: characterization, activity and mechanism
Received
12th April 2017
, Accepted 14th June 2017
First published on 20th June 2017
Abstract
A series of magnetically separable AgI–BiOI/CoFe2O4 hybrid composites were successfully synthesized via a solvothermal and subsequent coprecipitation method. The microstructure and magnetism of the materials were characterized by X-ray diffraction (XRD), N2 adsorption–desorption, scanning electron microscopy (SEM), transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), X-ray photoelectron spectroscopy (XPS), UV-vis diffuse reflectance spectroscopy (DRS), photocurrent test, electron spin resonance (ESR) and vibrating sample magnetometer (VSM). The photocatalytic performance of AgI–BiOI/CoFe2O4 composites on Hg0 removal from simulated flue gas was carefully designed and evaluated under fluorescent light (FSL) irradiation. The results showed that AgI–BiOI/CoFe2O4 composites displayed superior photocatalytic activities because of the synergistic effects between AgI, BiOI, and CoFe2O4 under FSL irradiation. The optimal weight ratio between AgI and the total weight of AgI–BiOI/CoFe2O4 photocatalyst was 0.3. The presence of a small amount of SO2 had a dramatic inhibition on Hg0 removal, while the inhibitory effect of NO on Hg0 removal could only be observed at a higher NO concentration. The trapping experiments indicated that photoinduced holes (h+) and superoxide radicals (˙O2−) were the primary active substances in the AgI–BiOI/CoFe2O4 photocatalytic oxidation system. According to the experimental and characterization results, one plausible mechanism for enhanced Hg0 removal performance over AgI–BiOI/CoFe2O4 composites was proposed.
1. Introduction
Mercury has received great attention owing to its toxicity, persistence and bioaccumulation in recent years.1,2 Coal-fired power plants are thought to be responsible for most of the anthropogenic mercury emissions into the environment.3,4 In 2011, the State Environmental Protection Administration of China issued a new national standard to specify the emission limits of mercury and its compounds from coal-fired flue gas.5 In China, coal accounts for about 70% of the total primary energy consumption.6 Mercury from flue gases often exists as elemental mercury (Hg0), oxidized mercury (Hg2+) and particulate mercury (Hgp).7 Compared with Hg2+ and Hgp, Hg0 is hardly removed owing to its volatility and indissolubility.8 To meet the increasingly severe emission standards, it is urgent to effectively remove Hg0 from coal-fired power plants.
In recent years, wet catalytic oxidation technology including UV/fenton,9 UV/H2O2,10 H2O2/Na2S2O8,11 NaClO2,12 and gas-phase photocatalytic oxidation with TiO2,13 SiO2–TiO2,14 CuO/TiO2,15 TiO2–aluminum silicate fiber,16 and BiOIO3,17 as photocatalysts have been explored extensively to remove Hg0 from flue gas. Most of these technologies can effectively remove Hg0 with the aid of UV energy.9,13–17 Under UV light irradiation, the produced reactive species such as hydroxyl radicals (˙OH) and hydroperoxyl radicals (˙HO2) possessing higher oxidability would oxidize Hg0 into Hg2+. This technique is promising since it could be carried out at low temperatures. However, the method needs lots of UV light irradiation and H2O2 or with the assistance of TiO2. Moreover, TiO2 itself has a large band gap (about 3.2 eV), which could only be excited under UV light to produce some useful reactive substances. The high cost of supplying successive UV light in coal-fired power plant limits its practical application. To solve this problem, it is urgent to fabricate a novel photocatalyst that can generate reactive species under visible light or even sunlight irradiation.
To date, a lot of novel Ag-based and Bi-based photocatalytic materials have been employed as efficient visible-light-responsive photocatalysts, such as Ag2CO3,18 Ag3PO4,19 Ag–AgI/Al2O3,20 BiOX (X = Cl, Br, I),21 AgI/BiOI,22 and AgI/Bi5O7I.23 During the photocatalytic oxidation processes, large amounts of reactive substances such as holes (h+), superoxide radicals (˙O2−) and ˙OH were generated. Hg0 could be possibly oxidized by these radicals due to their superior oxidation ability. To realize the above inference, the activity of Hg0 removal using visible-light-driven Ag/AgI–Ag2CO3 photocatalyst was investigated tentatively in our previous studies.24 The results indicated that this photocatalytic oxidation technology can effectively remove Hg0 under fluorescent light. Moreover, this technology can combine with wet flue gas desulfurization system (WFGD) to simultaneously remove Hg0 and SO2 at relatively low temperature, which brings out us a new insight into Hg0 removal from flue gas.
Although photocatalysis is efficient under fluorescent light, however, the applications of this type of powder photocatalysts also face a major bottleneck such as separation and cyclic utilization.25,26 In consideration of the easy separation of powder photocatalysts from reaction solution, a useful strategy to integrate Ag-based or Bi-based composites with magnetic nanoparticles such as Fe3O4,27 CoFe2O4,28 ZnFe2O4,29 NiFe2O4,30 and SrFe12O19,31 was proposed. Among the magnetic materials, CoFe2O4 has been widely employed in photocatalytic degradation of organic pollutants due to its small band gap energy and moderate saturation magnetization.28,32 Moreover, it also was reported that there were significant synergistic effects of individual components in CoFe2O4-based nanocomposites. For example, when active components such as BiOBr,33 BiVO4,34 ZnO,35 CdS,36 and Ag/Ag3VO4,37 were combined with CoFe2O4 respectively and employed in photocatalytic degradation of organic pollutants, the hybrid composites all showed enhanced photocatalytic activities than single component under visible light irradiation, and more importantly, they could be used for many times and recovered easily by an external magnetic force.
Thus, to reduce the cost of photocatalyst and obtain better photocatalytic activity with easy recovery, in this work, a series of novel magnetically separable AgI–BiOI/CoFe2O4 hybrids with lower silver content were synthesized via a solvothermal and subsequent coprecipitation method to remove Hg0 under fluorescent light (FSL) irradiation, which has not yet been researched in the literatures. As was expected, the as-synthesized ternary AgI–BiOI/CoFe2O4 composites displayed higher Hg0 removal activity than single component CoFe2O4 and BiOI under fluorescent light. The cyclic experiment showed that the optimal hybrid had stable photocatalytic activity and was relatively stable in crystal structures in the photocatalytic process. The synthesized ternary composite proved to be promising for wet photocatalytic oxidation removal of Hg0 from flue gas.
2. Experimental
2.1. Synthesis of CoFe2O4 nanoparticles
The CoFe2O4 composites were synthesized via a solvothermal method.37 All chemical agents employed were of analytical grade. Typically, 1.24 g of Co(NO3)2·6H2O and 3.44 g of Fe(NO3)3·9H2O were dissolved into 40 mL deionized water to obtain a mixed solution with magnetic stirring for about 60 min. Then, 30 mL of 1 M NaOH solution was added dropwise into the above mixed solution with stirring for another 60 min. Subsequently, the suspension was placed into a Teflon-lined autoclave (100 mL) and heated at 180 °C for 12 h. The resultant product was allowed to cool down to ambient temperature and then washed with distilled water several times. Finally, the obtained CoFe2O4 (abbreviated as CFO) composites were dried under vacuum at 60 °C for 12 h.
2.2. Synthesis of AgI–BiOI/CoFe2O4 microparticles
AgI–BiOI/CoFe2O4 samples were fabricated via a modified coprecipitation method,38,39 in which the mass ratio of CoFe2O4 (CFO) and total mass of the photocatalyst was fixed at 20%. Firstly, 4.82 g of Bi(NO3)3·5H2O and 0.36 g of AgNO3 were dissolved into 200 mL aqueous solution containing 20 mL acetic acid. Secondly, 1.00 g of as-prepared CFO powder was added into the above solvent with continuous stirring for 60 min. Then, 100 mL of 0.12 M KI solution was added to the above suspension. Finally, after stirring for another 60 min and placed for 12 h, the resulting precipitate was washed with ethanol water mixture for 3–5 times, filtered and dried at 60 °C for 24 h, respectively. The obtained composites were designated as AgI0.1BiOI/CFO for simplicity, where the value of 0.1 was the weight ratio between AgI and the total weight of the photocatalyst. Equally, other different photocatalysts were synthesized via the same procedure and named as BiOI/CFO, AgI/CFO, AgI0.2BiOI/CFO, AgI0.3BiOI/CFO, AgI0.4BiOI/CFO, and AgI0.5BiOI/CFO. For comparison, pure AgI and BiOI were also synthesized via the same method without the addition of CoFe2O4.
2.3. Characterization of photocatalysts
X-ray powder diffraction (XRD) analyses were performed on a D8 advance diffractometer (Bruker, Germany). N2 adsorption–desorption tests were carried out on an Autosorb iQ surface area and porosity analyzer (Quantachrome, USA) and the photocatalysts were degassed at 150 °C in vacuum for 8 h before the nitrogen adsorption isotherm was constructed. Scanning electron microscope (SEM) was carried out on a field emission scanning electron microscope (NoVa™ Nano SEM 430, FEI Company). Transmission electron microscope (TEM) and high-resolution transmission electron microscope (HRTEM) analyses were conducted on a Tecnai G20 microscopy (FEI, USA). X-ray photoelectron spectroscopy (XPS) experiments were performed on an Escalab 250xi spectrometer (Thermo Fisher, USA). UV-vis diffuse reflection spectra (DRS) were recorded on an UV2401 spectrophotometer (Shimadzu, Japan). The free radicals were detected on an ER200-SRC electron spin resonance spectrometer (ESR, Bruker, Germany). The magnetism of the composite was analyzed on a MPMS XL-7 magnetometer (Quantum Design, USA).
2.4. Photoelectrochemical measurement
The photoelectrochemical performances of the composites were conducted on a CHI 760E electrochemical work-station (Chenhua, China) in a standard three electrode system using fabricated photo-anode as the working electrode. A Pt wire and a saturated calomel electrode were respectively utilized as the counter and reference electrode. The 0.5 M Na2SO4 solution (pH = 5.8) purged with N2 was utilized as the electrolyte. A 300 W PLS-SXE300 Xe illuminator with a UV-cutoff filter (λ ≥ 420 nm) was used as the light source.
2.5. Photocatalytic activity test
The photocatalytic performances of as-prepared composites were implemented on a laboratory-scale wet bubbling photocatalytic reactor at ambient temperature. A detailed description about the experimental setup and process was provided in our previous work.24 In the experiment, the simulated flue gas was composed of 6% of O2, 12% of CO2, 55 μg m−3 of Hg0 vapor, and balance N2. The total flow rate was approximately 1.50 L min−1. 0.2 g of photocatalyst and 1 L of deionized water were uniformly mixed as the reaction solution and the reaction temperature was set as 45 °C. During the test, Hg0 concentration data was collected by an online VM-3000 mercury analysis instrument. The Hg0 removal efficiency η (%) was calculated according to the following equation:| | |
η = (1 − Cout/Cin) × 100
| (1) |
where Cout and Cin represent the outlet and inlet Hg0 concentrations (μg m−3), respectively.
3. Results and discussion
3.1. Photocatalytic activity
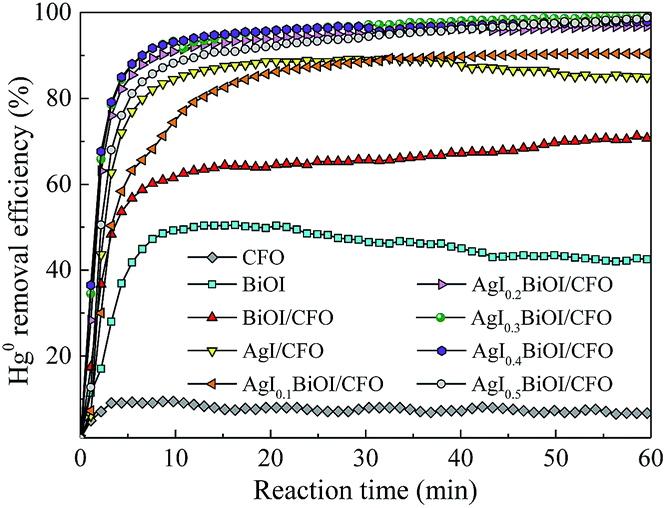
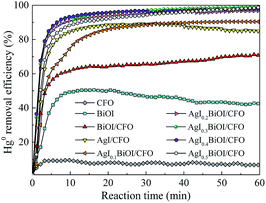
3.1.1. Effect of AgI content. The photocatalytic performances of Hg0 by as-prepared materials were studied under fluorescent light (FSL) irradiation and the corresponding results were shown in Fig. 1. It can be observed that under FSL irradiation, pure CFO and BiOI could only remove about 6% and 42% of Hg0 in 60 min, respectively, whereas approximately 68% of Hg0 was removed by BiOI/CFO. This comparison suggested that BiOI had relatively poor Hg0 removal ability, but the introduction of magnetic CFO onto BiOI could efficiently enhance its photocatalytic performance. The above phenomena can be explained that because of a small band gap of CoFe2O4 and a high e−–h+ pair recombination rate, pure CFO can hardly oxidize Hg0 to Hg2+. However, when BiOI was combined with CFO, a more efficient charge separation from BiOI/CFO can be achieved since the introduction of CFO can enhance the photoluminescent intensity of metal oxides.33,40
 |
| | Fig. 1 Effect of AgI content on Hg0 removal efficiency. | |
Furthermore, when different amounts of AgI were doped with BiOI/CFO, all ternary AgI–BiOI/CFO composites displayed much higher Hg0 removal efficiency than BiOI/CFO and the photocatalytic performances of ternary AgI–BiOI/CFO composites depended on AgI content. The AgI0.3BiOI/CFO exhibited the highest Hg0 removal efficiency (99%) when the weight ratio between AgI and AgI–BiOI/CFO was increased from 10% to 50%. It was reported that the composites with multifunctional components could combine individual advantages to achieve a high photocatalytic performance.41 Thus, a much better Hg0 removal efficiency could be obtained for AgI–BiOI/CFO hybrids.
3.1.2. Effect of FSL illumination. Fig. 2 shows the effect of FSL illumination on Hg0 removal efficiency. It was clear that only about 5% and 35% of Hg0 removal efficiencies were observed only with FSL irradiation and only with AgI0.3BiOI/CFO, respectively, indicating that Hg0 cannot be easily removed without FSL irradiation or photocatalyst. However, once both AgI0.3BiOI/CFO composite and FSL irradiation were employed simultaneously in the reaction system, Hg0 removal efficiency dramatically increased to about 99%. The enhanced Hg0 removal activity could be evidently due to the synergistic effect of AgI0.3BiOI/CFO and FSL irradiation.
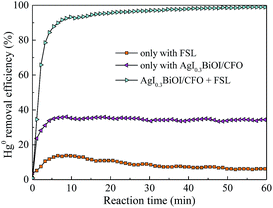 |
| | Fig. 2 Effect of FSL irradiation on Hg0 removal efficiency. | |
3.1.3. Effect of flue gas composition. In the coal-fired power plants, actual exhaust gas inevitably contained some concentrations of SO2 and NO, which may exert inhibitory or promotional impacts on Hg0 removal. Fig. 3 shows the effects of SO2 and NO concentrations on Hg0 removal. When 200 ppm of SO2 was introduced into the reaction stream in the ranges of 30–60 and 90–120 min, greater inhibitory effects on Hg0 removal appeared. However, when 100 and 200 ppm of NO were added, there was almost no loss in Hg0 removal efficiency, but a decrement of Hg0 removal efficiency from 93% to 75% was observed when NO concentration further increased to 600 ppm. Moreover, once SO2 and NO were cut off, the removal efficiency of Hg0 restored to about its initial value, suggesting that the structure of the photocatalyst had not been destroyed by SO2 and NO. According to previous reports,9,42 the inhibition could be ascribed to the consumption of photogenerated reactive species by SO2 and NO.
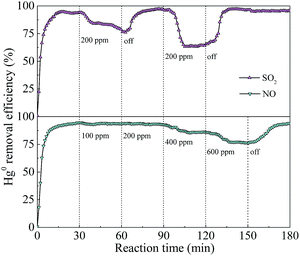 |
| | Fig. 3 Effect of SO2 and NO on Hg0 removal efficiency. | |
3.2. Photocatalyst characterization
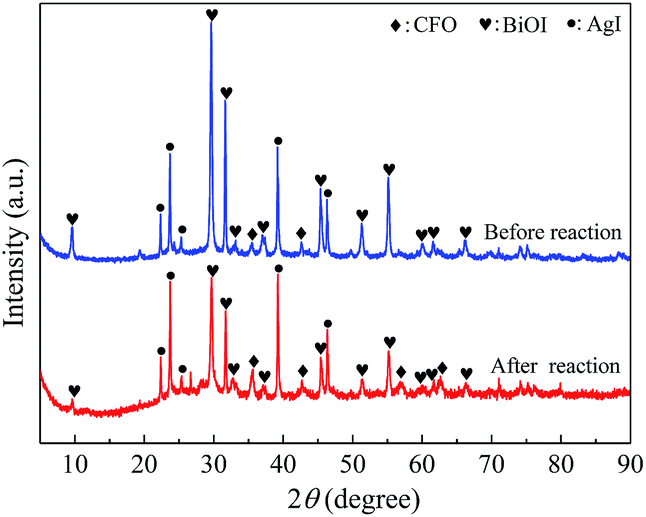
3.2.1. XRD and N2 adsorption–desorption. The XRD patterns of CoFe2O4 (CFO), BiOI, AgI, and AgI–BiOI/CoFe2O4 samples with differing AgI content were shown in Fig. 4. The diffraction peaks shown in Fig. 4a–c were assigned to the signal phase of CoFe2O4 (JCPDS 22-1086), tetragonal BiOI (JCPDS 73-2062) and hexagonal AgI (JCPDS 09-0374), respectively.22,43,44 By comparison, the diffraction intensities of BiOI in BiOI/CFO (Fig. 4d) and AgI in AgI/CFO (Fig. 4e) greatly decreased, suggesting the presence of CoFe2O4. It was clear that the diffraction patterns of AgI intensified gradually with the increasing of AgI/(AgI–BiOI/CFO) weight ratio (Fig. 4f–j), and the peak intensities of BiOI accordingly weakened. Moreover, two weak diffraction peaks of CoFe2O4 located at 2θ = 35.51° and 42.67° were observed from XRD patterns of Fig. 4f–j, indicating the coexistence of AgI, BiOI, and CoFe2O4 in AgI–BiOI/CoFe2O4 samples.
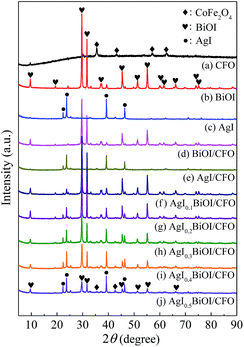 |
| | Fig. 4 XRD patterns of the photocatalysts. | |
Table 1 summarizes the physical features of selected composites. Obviously, CFO showed the largest BET surface area, total pore volume, and average pore diameter among the three samples. When BiOI itself or both BiOI and AgI were combined with CFO, the BET surface areas and total pore volumes of BiOI/CFO and AgI0.3BiOI/CFO were greatly decreased, which indicated that BiOI and AgI were successfully doped with CFO.
Table 1 Physical features of the selected composites
| Sample |
BET surface area (m2 g−1) |
Total pore volume (cm3 g−1) |
Average pore diameter (nm) |
| CFO |
55.07 |
2.61 × 10−1 |
20.52 |
| BiOI/CFO |
21.28 |
7.85 × 10−2 |
20.49 |
| AgI0.3BiOI/CFO |
20.74 |
6.56 × 10−2 |
17.28 |
3.2.2. SEM, TEM and HRTEM. The morphology features of selective composites were analyzed by SEM, TEM and HRTEM. As shown in Fig. 5, CFO exhibited a rough irregular surface, while when AgI was combined with CoFe2O4, many AgI granules with smooth surface (see red circular marks) appeared. As for BiOI/CFO, the petal-like BiOI particles (see blue circular mark) were observed (Fig. 5c). From Fig. 5d, it can be seen that the petal-like BiOI particles in AgI0.3BiOI/CFO composites became dispersed and somehow smaller than BiOI/CFO after the introduction of AgI. To provide the evidence that AgI and BiOI particles were successfully mixed with magnetic CFO, the TEM and HRTEM images of CFO and AgI0.3BiOI/CFO were investigated. The TEM images shown in Fig. 5e and f indicated that the CoFe2O4 grains were uniformly distributed on the surface of AgI0.3BiOI/CFO photocatalyst. The lattice spacing belonged to (311) plane of CFO was observed (Fig. 5g), while as for AgI0.3BiOI/CFO (Fig. 5f), three different lattice spacings were observed. The lattice spacings of 0.25, 0.27 and 0.30 nm corresponded to the (311) plane of CFO, (102) plane of AgI and (102) plane of BiOI, respectively, which further indicated that BiOI and AgI were successfully doped with CFO.36,45
 |
| | Fig. 5 SEM images of (a) CFO, (b) AgI/CFO, (c) BiOI/CFO and (d) AgI0.3BiOI/CFO, TEM images of (e) CFO and (f) AgI0.3BiOI/CFO, and HRTEM images of (g) CFO and (h) AgI0.3BiOI/CFO. | |
3.2.3. XPS. To obtain the surface compositions and chemical status of the as-prepared samples, XPS technique was employed and the corresponding results were displayed in Fig. 6. Fig. 6a exhibits the XPS spectra of Bi 4f. For BiOI/CFO, the peaks located at 164.49 and 159.15 eV were belonged to Bi 4f5/2 and Bi 4f7/2,46 while the peaks of Bi 4f5/2 and Bi 4f7/2 from AgI0.3BiOI/CFO shifted to higher binding energy. Fig. 6b shows the XPS spectra of I 3d. The peaks in BiOI/CFO centered at 630.33 and 618.93 eV corresponded to the spectrums of I 3d3/2 and I 3d5/2,47 while for AgI/CFO and AgI0.3BiOI/CFO, their peaks of I 3d3/2 and I 3d5/2 all shifted to higher binding energies. The peaks belonged to Ag 3d3/2 and Ag 3d5/2 was observed in Fig. 6c. It was noted that the peaks of Ag 3d in AgI0.3BiOI/CFO have a red-shift to higher binding energy compared with these of AgI/CFO.
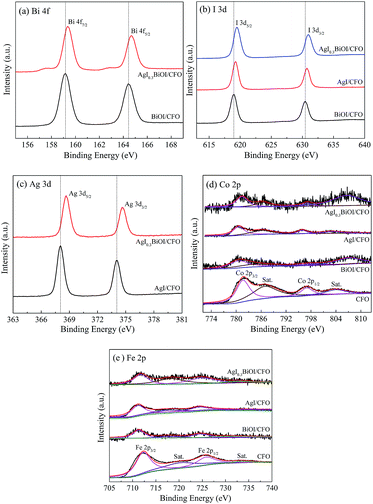 |
| | Fig. 6 XPS spectra of the photocatalysts: (a) Bi 4f, (b) I 3d, (c) Ag 3d, (d) Co 2p, and (e) Fe 2p. | |
The XPS spectra of Co 2p and Fe 2p were displayed in Fig. 6d and e, respectively. The peaks of both Co 2p and Fe 2p in BiOI/CFO, AgI/CFO and AgI0.3BiOI/CFO were weaker than that of CFO due to the interposition of CFO into the composites and relatively lower content of CFO. In Fig. 6d, the peaks centered at 797.0 eV (with a satellite peak at 804.50 eV) and 781.8 eV (with a satellite peak at 787.1 eV) were ascribed to Co 2p1/2 and Co 2p3/2, which corresponded to the characteristic peaks of Co2+ ions in CoFe2O4.37 Also, it was observed that the satellite peak of Co 2p1/2 in BiOI/CFO and AgI0.3BiOI/CFO shifted greatly to higher binding energies. In Fig. 6e, the photoionization region of Fe 2p yielded Fe 2p3/2 binding energies of 712.32 eV and Fe 2p1/2 binding energies of 725.84 eV, which is consistent with the Fe 2p binding energy for CoFe2O4.37,43 According to previous works,48,49 all the shifts of binding energy in the XPS spectra can be ascribed to the intense interaction between closely contacted phases of AgI, BiOI and CFO, suggesting the successful syntheses of AgI/CFO, BiOI/CFO and AgI0.3BiOI/CFO.
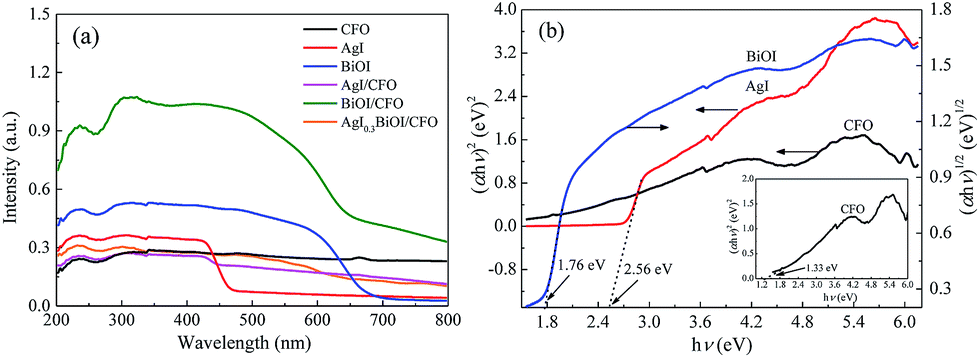
3.2.4. DRS. The optical performances of selective composites were performed on a UV-vis spectrophotometer. From Fig. 7a, it was observed that CFO with black surface displayed an almost constant absorption capacity in the wavelength range of 300–800 nm, while pure AgI and BiOI exhibited the absorption edges of around 475 and 670 nm, respectively. When CFO was coupled with AgI and BiOI, the absorption edges of AgI/CFO and BiOI/CFO all shifted to shorter wavelengths than those of pure AgI and BiOI. However, the absorption strengths of AgI/CFO and BiOI/CFO increased in visible region in comparison to AgI and BiOI, indicating the synergistic effect between CoFe2O4 and AgI or BiOI. When BiOI/CFO was decorated with AgI, the absorption intensity of AgI0.3BiOI/CFO decreased greatly compared to BiOI/CFO because the band gap of AgI was larger than BiOI.22 In the photocatalytic process, the component that can absorb more energy was beneficial for the photocatalytic activity of Hg0 removal to some extent, but the main factor could be the electron–hole (e−–h+) pairs separation efficiency of the component.50,51
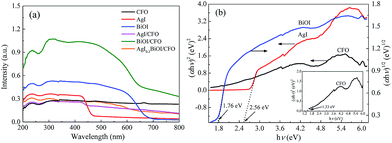 |
| | Fig. 7 (a) UV-vis diffuse reflectance spectra of selective samples; (b) plots of (αhν)2 versus hν for AgI, CFO and plot of (αhν)2 versus hν for BiOI. | |
According to the absorption spectra in Fig. 7a, the band gap values of the semiconductors can be measured by the equation as follows:
where
α,
h,
ν,
A, and
Eg are the absorption coefficient, Planck constant, light frequency, a proportionality constant, and the band gap, respectively. The value of
n depended on the types of photon transition in semiconductors (
n = 1 for direct transition and
n = 4 for indirect transition). Thus, the value of
n is 1 for direct gap semiconductor AgI and CoFe
2O
4 while 4 for indirect gap semiconductor BiOI.
23,37 Hence, as
Fig. 7b shows, the band gap energies (
Eg) of AgI, BiOI and CoFe
2O
4 were estimated to be 2.56, 1.76 and 1.33 eV, respectively. The conduction band (CB) and valence band (VB) values of a semiconductor were measured by the following empirical equations:
22where
ECB and
EVB represent the CB and VB potentials, respectively;
X is the electronegativity (the
X values for AgI, BiOI and CoFe
2O
4 are 5.354, 5.941, and 5.815 eV, respectively
22,37);
EC represents the energy of free electrons on the hydrogen scale (4.5 eV) and
Eg refers to the band gap energy of the semiconductor. Thus, the
ECB and
EVB of the composites were accordingly calculated and summarized in
Table 2.
Table 2 Band gap, conduction and valence band of the samples
| Sample |
Band gap, Eg (eV) |
Conduction band, ECB (eV) |
Valence band, EVB (eV) |
| AgI |
2.56 |
−0.43 |
2.13 |
| BiOI |
1.76 |
0.56 |
2.32 |
| CoFe2O4 |
1.33 |
0.65 |
1.98 |
3.2.5. Photocurrent analysis and ESR. The above DRS analysis can be further supported by prompt photocurrent response of the composites under visible light irradiation. In general, the higher the e−–h+ pairs separation efficiency, the higher the photocurrent produced, thereby leading to a much higher photocatalytic activity. Fig. 8 compares the prompt photocurrent responses of different samples and a good reproducibility of the photocurrent was observed in several on/off cycling tests. The photocurrent of BiOI/CFO was steadier than those of AgI/CFO and AgI0.3BiOI/CFO, implying that there was little recombination process within the whole irradiation period. In comparison to BiOI/CFO and AgI/CFO, AgI0.3BiOI/CFO photocatalyst exhibited much higher photocurrents density. This suggested that the e−–h+ pairs separation performance in the photo-electrochemical process of AgI0.3BiOI/CFO had been promoted, which could be ascribed to the appropriate energy band matching between AgI, BiOI and CoFe2O4.
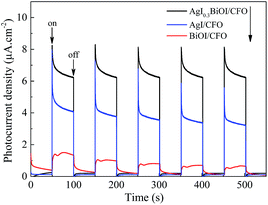 |
| | Fig. 8 Prompt photocurrent responses of the samples under visible light irradiation. | |
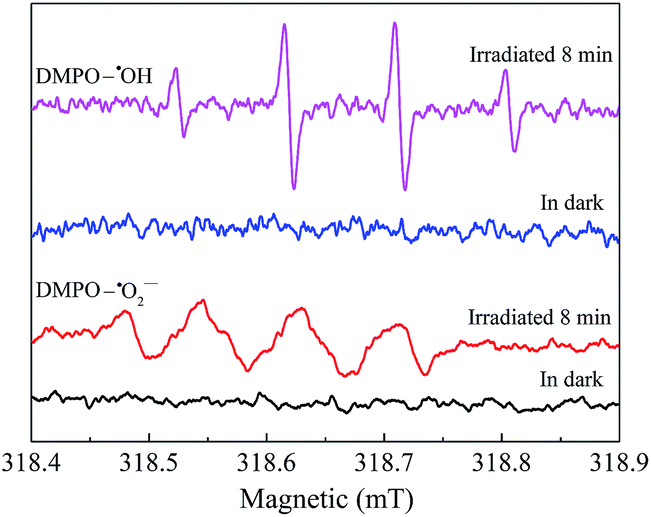
To explore the reactive substances produced during the photocatalytic oxidation process, ESR technology was utilized and the corresponding result was shown in Fig. 9. Under visible light irradiation, the characteristic peaks of DMPO–˙OH were detected in the suspension of AgI0.3BiOI/CFO, while no such signal appeared in the dark.52 Furthermore, when AgI0.3BiOI/CFO was dispersed in methanol, DMPO–˙O2− signals also only appeared under visible light. In the photocatalytic process, ˙OH, ˙O2− and holes played vital roles in pollutant removal.24,53–55 Thus, the ESR results provided a strong indication that the photogenerated carriers in AgI0.3BiOI/CFO possibly had not only strong redox ability but also survived long enough to react with the absorbed O2 and OH− or H2O to generate reactive ˙O2− and ˙OH.55
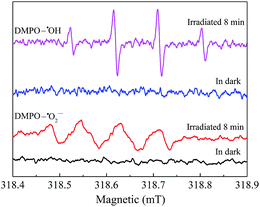 |
| | Fig. 9 ESR spectra for DMPO–˙OH and DMPO–˙O2− of AgI0.3BiOI/CFO. | |
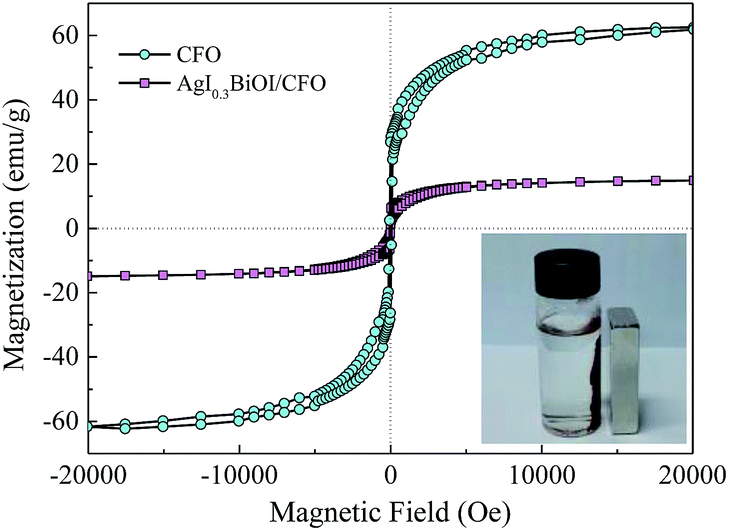
3.2.6. Magnetic performance. Fig. 10 shows the magnetic hysteresis loops of CFO and AgI0.3BiOI/CFO. CFO evidently displayed a strong magnetic response to the varied magnetic field. Since AgI0.3BiOI/CFO only contained 20 wt% of CFO and the components of AgI and BiOI have no magnetism, the magnetic saturation (Ms) value of AgI0.3BiOI/CFO (14.5 emu g−1) was smaller than that of CFO (61.9 emu g−1). However, from the inset of Fig. 10, it found that AgI0.3BiOI/CFO demonstrated a relatively strong enough magnetic separation performance and could be employed as a promising magnetic photocatalyst in the future.
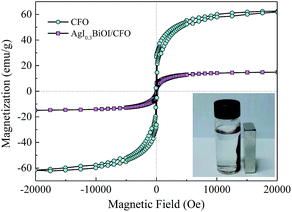 |
| | Fig. 10 Magnetization curves of CFO and AgI0.3BiOI/CFO. | |
3.3. Mechanism study
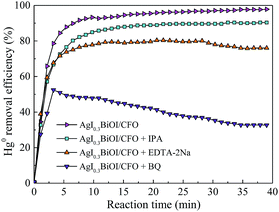
The trapping experiments for reactive radicals were performed to clarify the functions of photogenerated e−–h+ pairs during Hg0 removal process. It should be noticed that prior to turning on the light source, 15 mL of isopropyl alcohol (IPA), 0.8 g of ethylene diamine tetraacetic acid disodium salt (EDTA–2Na) and 0.8 g of benzoquinone (BQ) were added along with 0.2 g AgI0.3BiOI/CFO composite into the reaction liquid as the scavenger of ˙OH, h+ and ˙O2−, respectively.24 As shown in Fig. 11, the Hg0 removal efficiency of AgI0.3BiOI/CFO decreased by about 7% in the presence of IPA when compared to that of without any scavenger, indicating that ˙OH radicals had little impact on Hg0 removal. In contrast, an obvious reduction and a drastic inhibition were observed after the addition of EDTA–2Na and BQ, respectively, implying that h+ and ˙O2− radicals were the primary reactive substances for Hg0 removal.
 |
| | Fig. 11 Effect of different scavengers on Hg0 removal. | |
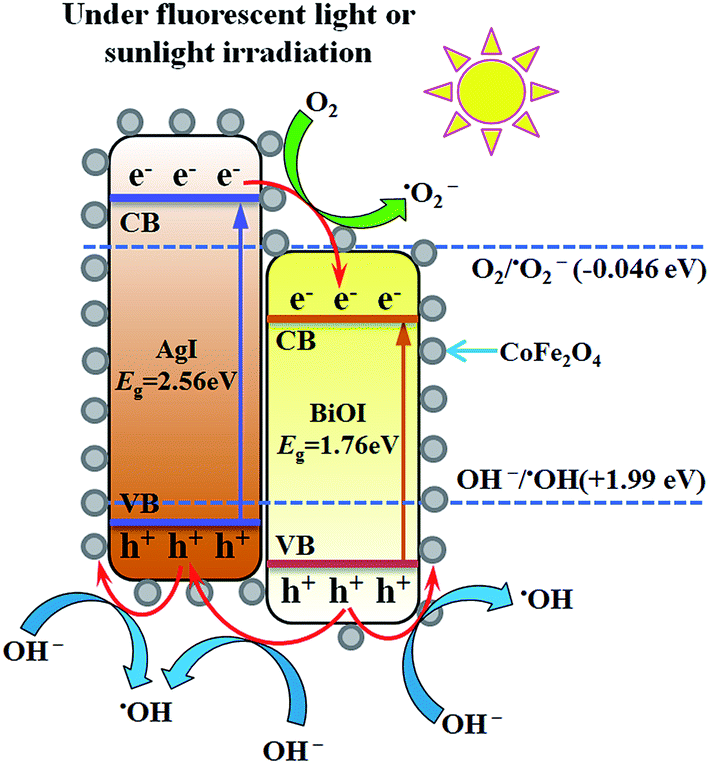
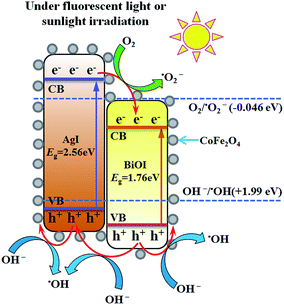
Based on above discussion, a schematic illustration of energy bands matching among AgI, BiOI and CoFe2O4 and possible ways of charges transfer were depicted in Fig. 12. Under visible light or FSL irradiation, the photoexcited e−–h+ pairs would be produced due to lower band gap energies of AgI (2.56 eV) and BiOI (1.76 eV). Since the CB edge of BiOI was more positive than that of AgI, the electrons on the CB of AgI would transfer to the CB of BiOI. Because the VB potential of AgI was more negative than that of BiOI correspondingly, the holes on the VB of BiOI would migrate to the VB of AgI.22,47 For BiOI and AgI, the electrons on the CB potential of AgI can be trapped by adsorbed O2 to generate ˙O2− since the CB potential of AgI was more negative than single electron reduction potential of O2 (−0.046 eV vs. NHE).47 The OH− or H2O can be oxidized by holes to yield ˙OH radicals since the VB potential of BiOI and AgI was more positive than ˙OH/OH− (1.99 eV vs. NHE).22,24 Moreover, since the CB potential (0.65 eV) and VB potential (1.98 eV) of CoFe2O4 was between −0.046 and 1.99 eV, the e−–h+ pairs did not have enough redox capacity to generate reactive species such as ˙OH and ˙O2− and were prone to recombine,38 thus only 6% of Hg0 removal efficiency was observed for pure CFO. However, due to lower VB potential of CFO than that of AgI and BiOI, the holes on the VBs of AgI and BiOI would transfer to the VB of CFO,33 thereby facilitating an effective separation of e−–h+ pairs and the presence of some amounts of reactive ˙OH. This might be the reason why binary BiOI/CFO and AgI/CFO composite achieved a higher Hg0 removal efficiency than single component. Taken together, the recombination of photogenerated e−–h+ pairs over AgI0.3BiOI/CFO could be greatly suppressed in such a way and much more electrons and holes could react with O2, OH− to generate ˙O2− and ˙OH radicals. In view of photo-electrochemistry, the products such as ˙OH, ˙O2− and h+ have enough oxidation capability to oxidize Hg0. Among them, the ˙O2− and h+ radicals were confirmed to play crucial roles in Hg0 removal.
 |
| | Fig. 12 Schematic mechanism of AgI0.3BiOI/CFO for enhanced Hg0 removal. | |
3.4. Stability of photocatalyst
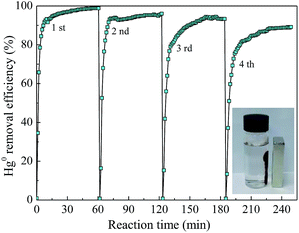
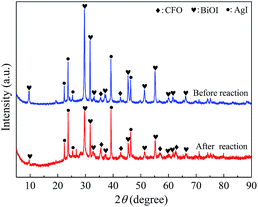
To understand the recyclability and durability of composite AgI0.3BiOI/CFO, four successive runs of photocatalytic experiments were performed under FSL irradiation. As shown in Fig. 13, AgI0.3BiOI/CFO exhibited a slight decline in Hg0 removal efficiency with the recycling tests increasing. About 87% of Hg0 removal efficiency was obtained after four successive runs, suggesting that AgI0.3BiOI/CFO was stable during the photocatalytic oxidation process. Furthermore, the inset of Fig. 13 clearly demonstrated that AgI0.3BiOI/CFO after four successive runs still had a good magnetic separation performance. To further investigate the structure change of as-prepared sample after Hg0 removal test, AgI0.3BiOI/CFO after four successive tests were also characterized by XRD technique. As revealed in Fig. 14, the crystal structure of used AgI0.3BiOI/CFO was almost identical to its fresh sample except two new peaks at 56.94° and 62.68° belonged to CoFe2O4,37 indicating a relatively stable crystal structure of the composite in the photocatalytic process.
 |
| | Fig. 13 Hg0 removal of consecutive experiments by AgI0.3BiOI/CFO. | |
 |
| | Fig. 14 XRD patterns of AgI0.3BiOI/CFO before and after four successive runs. | |
3.5. Simultaneous removal of SO2 and Hg0
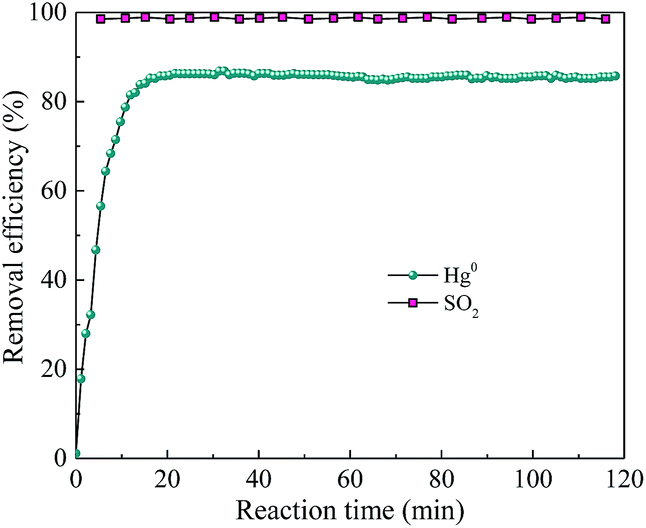
Considering that the current mature SCR technology is effective to remove NO and has been widely adopted by power plant, the simultaneous removal of SO2 and Hg0 over AgI–BiOI/CFO composite under alkaline condition was preliminary explored. As shown in Fig. 15, when 200 ppm of SO2 was introduced into the reaction system, Hg0 removal efficiency has little inhibition and still maintained at 85% with the solution pH adjusted to 11 by adding NaOH solution. Also, nearly 98% of SO2 removal efficiency was observed, suggesting that a certain degree of alkaline solution can not only restrain the inhibition of SO2 on Hg0 removal but also can effectively remove SO2 from flue gas.
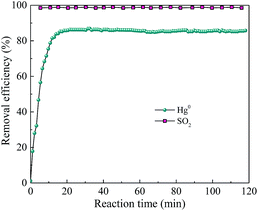 |
| | Fig. 15 Simultaneous removal of Hg0 and SO2 over AgI0.3BiOI/CFO. | |
4. Conclusions
A series of magnetically separable ternary AgI–BiOI/CoFe2O4 photocatalysts with different AgI contents were successfully prepared by a solvothermal and sequent coprecipitation method. The as-prepared ternary AgI–BiOI/CoFe2O4 composites showed greatly enhanced Hg0 removal activity under FSL irradiation, especially AgI0.3BiOI/CFO. AgI0.3BiOI/CFO demonstrated much stable Hg0 oxidation ability and enough magnetic property to be reused in the photocatalytic process. The DRS and photocurrent analyses indicated that the enhanced Hg0 removal efficiency over AgI0.3BiOI/CFO could be ascribed to the synergistic effect between AgI, BiOI and CoFe2O4 under FSL irradiation, which could lead to an efficient e−–h+ pairs separation and much more photogenerated reactive species. The trapping experiment result indicated the ˙O2− and h+ radicals were the primary reactive substances contributing to the oxidation of Hg0. This work can improve the insight into the wet photocatalytic oxidation process of Hg0 using magnetic photocatalyst under FSL irradiation and serve as a guide in using magnetic photocatalyst in the field of flue gas treatment.
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (No. 51676064, 51306046, and 51576086), the Fundamental Research Funds for the Universities of Henan Province (No. NSFRF140204) and the Young Core Instructor Project in the Higher Education Institutions of Henan Province (No. 2016GGJS-038).
References
- A. P. Dastoor and Y. Larocque, Atmos. Environ., 2004, 38, 147–161 CrossRef CAS
 .
. - E. G. Pacyna, J. M. Pacyna, K. Sundseth, J. Munthe, K. Kindbom, S. Wilson, F. Steenhuisen and P. Maxson, Atmos. Environ., 2010, 44, 2487–2499 CrossRef CAS .
- X. Zhang, B. X. Shen, S. W. Zhu, H. Xu and L. H. Tian, J. Hazard. Mater., 2016, 320, 556–563 CrossRef CAS PubMed .
- S. J. Zhao, Z. Qu, N. Q. Yan, Z. Li, W. F. Zhu, J. pan, J. F. Xu and M. D. Li, RSC Adv., 2015, 5, 30841–30850 RSC .
- State Environmental Protection Administration of China (SEPA), Emission Standard of Air Pollution for Thermal Power Plants, GB 13223-2011, Beijing, 2011 Search PubMed .
- C. F. You and X. C. Xu, Energy, 2010, 35, 4467–4472 CrossRef CAS .
- K. C. Galbreath and C. J. Zygarlicke, Fuel Process. Technol., 2000, 65, 289–310 CrossRef .
- R. K. Srivastava, N. Hutson, B. Martin, F. Princiotta and J. Staudt, Environ. Sci. Technol., 2006, 40, 1385–1393 CrossRef CAS PubMed .
- F. M. Zhan, C. T. Li, G. M. Zeng, S. S. Tao, Y. J. Xiao, X. Zhang, L. K. Zhao, J. Zhang and J. F. Ma, Chem. Eng. J., 2013, 232, 81–88 CrossRef CAS .
- Y. X. Liu, J. Zhang and J. F. Pan, Energy Fuels, 2014, 28, 2135–2143 CrossRef CAS .
- Y. Zhao, R. L. Hao, P. Zhang and S. H. Zhou, Fuel, 2014, 136, 113–121 CrossRef CAS .
- N. D. Hutson, R. Krzyzynska and R. K. Srivastava, Ind. Eng. Chem. Res., 2008, 47, 5825–5831 CrossRef CAS .
- H. Q. Wang, S. Y. Zhou, L. Xiao, Y. J. Wang, Y. Liu and Z. B. Wu, Catal. Today, 2011, 175, 202–208 CrossRef CAS .
- Y. Li and C.-Y. Wu, Environ. Sci. Technol., 2006, 40, 6444–6448 CrossRef CAS PubMed .
- J. Wu, C. E. Li, X. Y. Zhao, Q. Wu, X. M. Qi, X. T. Chen, T. Hu and Y. Cao, Appl. Catal., B, 2015, 176–177, 559–569 CrossRef CAS .
- Y. Yuan, J. Y. Zhang, H. L. Li, Y. Li, Y. C. Zhao and C. G. Zheng, Chem. Eng. J., 2012, 192, 21–28 CrossRef CAS .
- X. M. Qi, M. L. Gu, X. Y. Zhu, J. Wu, H. M. Long, K. He and Q. Wu, Chem. Eng. J., 2016, 285, 11–19 CrossRef CAS .
- G. P. Dai, J. G. Yu and G. Liu, J. Phys. Chem. C, 2012, 116, 15519–15524 CAS .
- Y. P. Bi, S. X. Ouyang, N. Umezawa, J. Y. Cao and J. H. Ye, J. Am. Chem. Soc., 2011, 133, 6490–6492 CrossRef CAS PubMed .
- C. Hu, T. W. Peng, X. X. Hu, Y. L. Nie, X. F. Zhou, J. H. Qu and H. He, J. Am. Chem. Soc., 2010, 132, 857–862 CrossRef CAS PubMed .
- X. Zhang, Z. H. Ai, F. L. Jia and L. Z. Zhang, J. Phys. Chem. C, 2008, 112, 747–753 CAS .
- H. F. Cheng, B. B. Huang, Y. Dai, X. Y. Qin and X. Y. Zhang, Langmuir, 2010, 26, 6618–6624 CrossRef CAS PubMed .
- M. Cui, J. X. Yu, H. J. Lin, Y. Wu, L. H. Zhao and Y. M. He, Appl. Surf. Sci., 2016, 387, 912–920 CrossRef CAS .
- A. C. Zhang, L. X. Zhang, X. Z. Chen, Q. F. Zhu, Z. C. Liu and J. Xiang, Appl. Surf. Sci., 2017, 392, 1107–1116 CrossRef CAS .
- G. C. Xi, B. Yue, J. Y. Cao and J. H. Ye, Chem.–Eur. J., 2011, 17, 5145–5154 CrossRef CAS PubMed .
- W. Wu, C. Z. Jiang and V. A. L. Roy, Nanoscale, 2015, 7, 38–58 CAS .
- T. W. Ng, L. S. Zhang, J. S. Liu, G. H. Huang, W. Wang and P. K. Wong, Water Res., 2016, 90, 111–118 CrossRef CAS PubMed .
- Y. G. Xu, T. Zhou, S. Q. Huang, M. Xie, H. P. Li, H. Xu, J. X. Xia and H. M. Li, RSC Adv., 2015, 5, 41475–41483 RSC .
- W. Q. Zhang, M. Wang, W. J. Zhao and B. Q. Wang, Dalton Trans., 2013, 42, 15464–15474 RSC .
- A. Ren, C. B. Liu, Y. Z. Hong, W. D. Shi, S. Lin and P. Li, Chem. Eng. J., 2014, 258, 301–308 CrossRef CAS .
- T. P. Xie, L. J. Xu, C. L. Liu, J. Yang and M. Wang, Dalton Trans., 2014, 43, 2211–2220 RSC .
- Y. S. Fu, H. Q. Chen, X. Q. Sun and X. Wang, Appl. Catal., B, 2012, 111–112, 280–287 CrossRef CAS .
- R. Jiang, H.-Y. Zhu, J.-B. Li, F.-Q. Fu, J. Yao, S.-T. Jiang and G.-M. Zeng, Appl. Surf. Sci., 2016, 364, 604–612 CrossRef CAS .
- S. Duangjam, K. Wetchakun, S. Phanichphant and N. Wetchakun, Mater. Lett., 2016, 181, 86–91 CrossRef CAS .
- P. Sathishkumar, N. Pugazhenthiran, R. V. Mangalaraja, A. M. Asiri and S. Anandan, J. Hazard. Mater., 2013, 252–253, 171–179 CrossRef CAS PubMed .
- S. Singh and N. Khare, Mater. Lett., 2015, 161, 64–67 CrossRef CAS .
- L. Q. Jing, Y. G. Xu, S. Q. Huang, M. Xie, M. Q. He, H. Xu, H. M. Li and Q. Zhang, Appl. Catal., B, 2016, 199, 11–22 CrossRef CAS .
- L. Kong, Z. Jiang, T. C. Xiao, L. F. Lu, M. O. Jones and P. P. Edwards, Chem. Commun., 2011, 47, 5512–5514 RSC .
- A. R. Upreti, Y. Li, N. Khadgi, S. Naraginti and C. Zhang, RSC Adv., 2016, 6, 32761–32769 RSC .
- H. G. Kim, P. H. Borse, J. S. Jang, E. D. Jeong, O. S. Jung, Y. J. Suhd and J. S. Lee, Chem. Commun., 2009, 39, 5889–5891 RSC .
- J. C. Colmenares and R. Luque, Chem. Soc. Rev., 2014, 43, 765–778 RSC .
- D. H. Xia, L. L. Hu, C. He, W. Q. Pan, T. S. Yang, Y. C. Yang and D. Shu, Chem. Eng. J., 2015, 279, 929–938 CrossRef CAS .
- B. Cai, M. G. Zhao, Y. Ma, Z. Z. Ye and J. Y. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 1327–1333 CAS .
- D. A. Reddy, J. Choi, S. Lee, R. Ma and T. K. Kim, RSC Adv., 2015, 5, 67394–67404 RSC .
- J. L. Liang, C. Shan, X. Zhang and M. P. Tong, Chem. Eng. J., 2015, 279, 277–285 CrossRef CAS .
- H. Liu, W. R. Cao, Y. Su, Y. Wang and X. H. Wang, Appl. Catal., B, 2012, 111–112, 271–279 CrossRef CAS .
- J. Cao, Y. J. Zhao, H. L. Lin, B. Y. Xu and S. F. Chen, J. Solid State Chem., 2013, 206, 38–44 CrossRef CAS .
- J.-C. Wang, H.-C. Yao, Z.-Y. Fan, L. Zhang, J.-S. Wang, S.-Q. Zang and Z.-J. Li, ACS Appl. Mater. Interfaces, 2016, 8, 3765–3775 CAS .
- T. Yan, H. Y. Liu, P. C. Gao, M. Sun, Q. Wei, W. G. Xu, X. D. Wang and B. Du, New J. Chem., 2015, 39, 3964–3972 RSC .
- H. L. Wang, L. S. Zhang, Z. G. Chen, J. Q. Hu, S. J. Li, Z. H. Wang, J. S. Liu and X. C. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC .
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS .
- R. F. Dong, B. Z. Tian, C. Y. Zeng, T. Y. Li, T. T. Wang and J. L. Zhang, J. Phys. Chem. C, 2013, 117, 213–220 CAS .
- F. Dong, Y. J. Sun, M. Fu, Z. B. Wu and S. C. Lee, J. Hazard. Mater., 2012, 219–220, 26–34 CrossRef CAS PubMed .
- Z. H. Ai, W. K. Ho, S. C. Lee and L. Z. Zhang, Environ. Sci. Technol., 2009, 43, 4143–4150 CrossRef CAS PubMed .
- M. Sun, D. Z. Li, Y. B. Chen, W. Chen, W. J. Li, Y. H. He and X. Z. Fu, J. Phys. Chem. C, 2009, 113, 13825–13831 CAS .
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Hao Lua,
Zhijun Sunab,
Wei Shenga,
Lushi Sunb and
Jun Xiang*b
*a,
Hao Lua,
Zhijun Sunab,
Wei Shenga,
Lushi Sunb and
Jun Xiang*b