
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural phase transition and photoluminescence properties of wurtzite CdS:Eu3+ nanoparticles under high pressure
Rui Zhaoab,
Tianye Yanga,
Yang Luoa,
Mingyan Chuaia,
Xiaoxin Wuc,
Yanyan Zhangc,
Yanzhang Macd and
Mingzhe Zhang *a
*a
aState Key Laboratory of Superhard Materials, Jilin University, Changchun 130012, People's Republic of China. E-mail: zhangmz@jlu.edu.cn
bCollege of Computer, Jilin Normal University, Siping 136000, People's Republic of China
cCenter for Hight Pressure Science & Technology Advanced Research, Changchun 130012, China
dDepartment of Mechanical Engineering, Texas Technology University, Lubbock, TX 79409, USA
First published on 20th June 2017
Abstract
High-pressure behaviors of wurtzite CdS and CdS:Eu3+ nanoparticles (8–10 nm) were investigated by synchrotron radiation X-ray diffraction, Raman spectroscopy, and photoluminescence under high pressure at ambient temperature. The doping of Eu ions increases the phase transition pressure from wurtzite structure to rocksalt structure (CdS = 4.76 GPa and CdS:Eu = 5.22 GPa) and so does the bulk modulus (B0) of the initial and high pressure phases. This phenomenon can be attributed to the great impact on tensile strain along the c-axis of CdS nanoparticles, which is identified by the relationship of lattice contraction and the pressure obtained from Raman 1LO. The phase transitions of all samples are partly reversible. The Eu3+ ions luminescence from 5D0 → 7FJ (J = 1, 2) transition in CdS:Eu nanoparticles emerges obviously and changes during the phase transformation, which indicate the variation of the local symmetry of the Eu3+ ions. The new peak of 5D0 → 7F3 emerges at 7.26 GPa, persisting until the end of the whole experiment. The obtained CdS nanoparticles will hold promising potential in the fabrication of effective biological sensors and photodetectors for practical application under high pressure.
Introduction
Doping and high pressure provide unique insight into the electronic structure, localized states and crystal structure of nanomaterials. Thus, they have been widely used in nanomaterials science as effective technological process. Cadmium sulfide (CdS) is one of the most attractive wide bandgap semiconductors because of its superior optical properties, which have been widely applied in photocatalysis,1–3 solar cells,4,5 biological sensors,6,7 lasers,8,9 photodetectors,10,11 and so on. The dopants can strongly influence optical behaviours of nanomaterials, and doped nanomaterials are thus regarded as a new class of luminescent materials.12 To date, many efforts have been made to improve the optical properties of CdS through different synthesis methods and doping agents (transition-metal or lanthanide ions).13,14 Many interesting phenomena have been observed in doping samples, such as the shifting and broadening of spectra, high luminescent quantum efficiency, shorting of emission lifetime, upconversion emission, etc.Recently, a variety of lanthanide ions, including Nd3+, Sm3+, Eu3+, Tb3+, Gd3+, Er3+, and Yb3+, have been successfully doped into CdS QDs.15–20 Compared to the transition-metal ions, lanthanide ions are advantageous dopants because of their sharper emission signals with a unique spectroscopic signature for unambiguous spectral identification. Therein, the Eu3+ doping was demonstrated to be an effective way to enhance the performance of QDs in the photocatalysis and biomedical applications (immunoassay, detection of metal ions and imaging applications) as the edge over QDs, long fluorescence lifetime, sharp emission peaks with narrow band width, and lack of blinking and biocompatibility.21–23 Because of the non-degenerate emissive state (5D0), the Eu3+ ion acts as an excellent luminescent structural probe for the determination of the number of metal ion sites in a compound, their symmetry, and their respective population.24,25 For example, the fluorescence intensity of Eu3+ doped CdS NPs reduced obviously in the presence of the Hg2+ ions, which makes it as a fluorescence probe for the Hg2+ detection.26 However, the luminescent properties of europium are influenced by a number of factors such as the site symmetry, quantum dots, surfactants, morphology and crystal defect, phenomena like antenna effect and physical parameters like temperature.27 It is still debated whether or not lanthanide ions are really incorporated into the host lattice (ZnS or CdS) sites as the luminescence from lanthanide dopants in QDs also depends on their location in the host lattice.12
Recently, we have reported on the relation between magnetism and optical properties of Eu3+ doped CdS (CdS:Eu) Nanoparticles (NPs).28 The emission line width and relative intensities of the Eu3+ ions were found to depend on the value of the doping. Whereas, all of the studies mentioned above were conducted under ambient pressure.
Other studies reported that the luminescence intensity of Mn2+ of ZnS:Mn NPs with 1 and 3 nm size decreases dramatically and the bandwidth increases fast with increasing pressure.29 This indicated that the size of QDs and the pressure have a significant effect on the behavior of Mn2+ emission. Many studies indicated that high pressure can cause remarkable influence on anomalous luminescence. Grinberg et al. reported that the pressure reduced impurity-trapped exciton energy under lower pressure in Eu2+-doped fluorides. However, it was elevatory under higher pressures. In Pr3+-doped oxides, in all the considered cases, the pressure caused the impurity-trapped exciton energy to diminish.30 Kamińska et al. suggested that the probability of f–f radiative transitions (2F7/2 ↔ 2F5/2) of Yb3+ ions in gadolinium gallium garnetcrystals is sensitive to hydrostatic pressure application.31 This demonstration provides not only an efficient way to artificially tune the emission properties of phosphors by means of hydrostatic pressure, but also alternative candidates as potential pressure gauges for high pressure techniques. Obviously, the luminescence behaviours of nanomaterials could be greatly affected by many factors, such as doping, size, and high pressure. Therefore, it is of great interest to explore the structural stability and luminescence properties of nanosized CdS:Eu under high pressure.
In this paper, we have carried out an investigation of phase transition (PT) and optical properties of pure CdS and CdS:Eu NPs by synchrotron XRD pattern, photoluminescence, Raman scattering spectrum under high pressure.
Experimental
The pure CdS and CdS doped with 0.08 at% of Eu3+ was grown using the gas–liquid phase reaction.28 There were two reactive steps in the experiment process: firstly, HCl reacted with Na2S to form H2S gas according to the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; subsequently, H2S gas and the mixed reactive solution (Cd(COOCH3)2, Eu(COOCH3)3, the surface-active agent polyvinylpyrrolidone (PVP), and deionized water) reacted on the hemispherical crown's polished surface in the chamber. The structure and morphology of the pure CdS and CdS:Eu NPs were characterized by XRD, SAED, and HRTEM.
:1; subsequently, H2S gas and the mixed reactive solution (Cd(COOCH3)2, Eu(COOCH3)3, the surface-active agent polyvinylpyrrolidone (PVP), and deionized water) reacted on the hemispherical crown's polished surface in the chamber. The structure and morphology of the pure CdS and CdS:Eu NPs were characterized by XRD, SAED, and HRTEM.
In situ angle-dispersive synchrotron XRD measurements under high pressure was generated by a diamond anvil cell (DAC) with 16:3:1 methanol–ethanol–water mixture as the pressure medium. Pressure was calibrated by the energy shift of the R1 luminescence line of a ruby crystal. The high-pressure ADXRD experiments of CdS and CdS:Eu NPs were carried out at the beamline B1 of Cornell High Energy Synchrotron Source (CHESS) in Cornell University and the beamline X17C of National Synchrotron Light Source (NSLS) in Brookhaven National Laboratory. A diamond anvil cell was used to generate high pressure. A hole with a diameter of 150 μm was drilled in a T301 stainless steel gasket and used as the sample chamber. The pressure was determined from the frequency shift of ruby R1 fluorescence line. The FIT2D software was used to transfer the XRD images into intensity versus diffraction angle (2θ) pattern.
The photoluminescence (PL) and Raman measurements were performed in a gasketed diamond-anvil cell (DAC) at room temperature under hydrostatic pressure. Some powder samples, together with a piece of ruby chip, were placed in a stainless steel gasket with a hole 150 μm in diameter. A small ruby chip is used for in situ pressure calibration, utilizing the R1 ruby fluorescence method. Silicone oil was used as a pressure transmitting medium. For the measurement of the Raman and PL emission spectra, a Renishaw in Via Raman system with a laser was used as an excitation source. The wavelengths were collected with 488 nm and 532 nm, respectively.
Results and discussion
The morphology, phase structure, and crystal size of the samples are observed and measured by means of X-ray diffraction (XRD), transmission electron microscope (TEM), high-resolution transmission electron microscopy (HRTEM), selected-area electron diffraction (SAED), Raman, energy dispersive X-ray spectroscopy (EDS) and chemical elements mapping. XRD characterizations (Fig. 1a) of pure CdS and CdS:Eu NPs are carried out to identify the phase structure. It can be seen from the XRD pattern that all of the diffraction peaks are indexed to the standard wurtzite (WZ) structured CdS (JCPDS no. 75-1545, space group: P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) 3mc (no. 186)). It is obvious that all peak positions of the CdS:Eu NPs slightly shift to higher angles compared to those of the pure CdS. A lattice compression phenomenon is indicated as a result of the substitution of smaller ion radius Eu3+ (0.0947 nm) for the Cd2+ (0.097 nm). The lattice parameters are a = 4.128 Å, c = 6.755 Å, and V0 = 99.68 Å3 for CdS:Eu NPs, which are smaller than those of undoped CdS NPs (a = 4.147 Å, c = 6.796 Å, V0 = 101.22 Å3).
3mc (no. 186)). It is obvious that all peak positions of the CdS:Eu NPs slightly shift to higher angles compared to those of the pure CdS. A lattice compression phenomenon is indicated as a result of the substitution of smaller ion radius Eu3+ (0.0947 nm) for the Cd2+ (0.097 nm). The lattice parameters are a = 4.128 Å, c = 6.755 Å, and V0 = 99.68 Å3 for CdS:Eu NPs, which are smaller than those of undoped CdS NPs (a = 4.147 Å, c = 6.796 Å, V0 = 101.22 Å3).
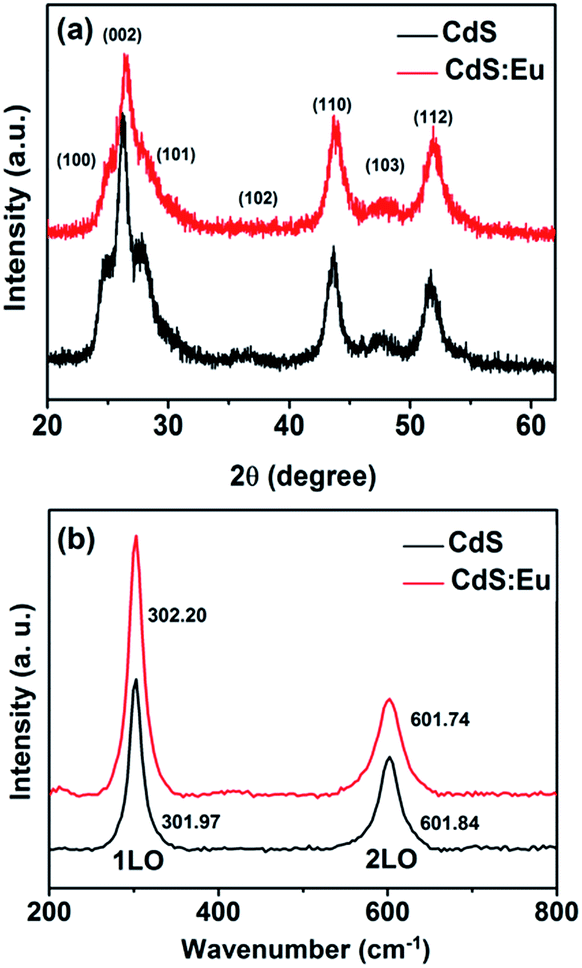 | ||
| Fig. 1 XRD patterns (a) and Raman spectra (b) of pure CdS and CdS:Eu NPs with dopant content 0.08 at%. | ||
The structural information of pure CdS and CdS:Eu NPs were further investigated by Raman spectra at ambient conditions, which are shown in Fig. 1b. These Raman spectra are dominated by the progression in the longitudinal optical (LO) phonon mode. Each spectrum has two peaks. The first stronger peak is the A1 longitudinal optical (1LO) mode. The weaker other is the overtone of the LO mode (2LO). Two typical vibrational peaks at 301.97 and 601.84 cm−1 are observed for the CdS sample, respectively. For the CdS:Eu sample, two peaks are located at 302.2 and 601.74 cm−1. They are in good agreement with the reported measurement.32,33 Obviously, the Raman spectrum of CdS:Eu is essentially independent with the doping.
A representative HRTEM image of the as-prepared CdS and CdS:Eu NPs are shown in Fig. 2a–d. These NPs have average (with standard deviations) length and diameter of 8–10 nm. The SAED pattern indicates that the diffraction rings correspond to the XRD pattern without any impurities, revealing only a single phase (hexagonal wurtzite) in CdS and CdS:Eu NPs.
 | ||
| Fig. 2 HRTEM images (a) and SAED pattern (b) of pure CdS NPs. HRTEM images (c) and SAED pattern (d) of CdS:Eu NPs. | ||
The TEM image of CdS:Eu NPs characterizes the morphologies of the samples in the insets of Fig. 3a. The energy-dispersive X-ray spectrum (EDS, Fig. 3a) confirms the presence of S, Cd, and Eu elements in CdS:Eu NPs, and the relative atom ratios are 48.83%, 51.08%, and 0.08%, respectively. The chemical element mapping analysis (Fig. 3b–d) shows that the elements including sulfur, cadmium and europium are homogenously distributed throughout the whole sample, which suggests a high purity of the final products.
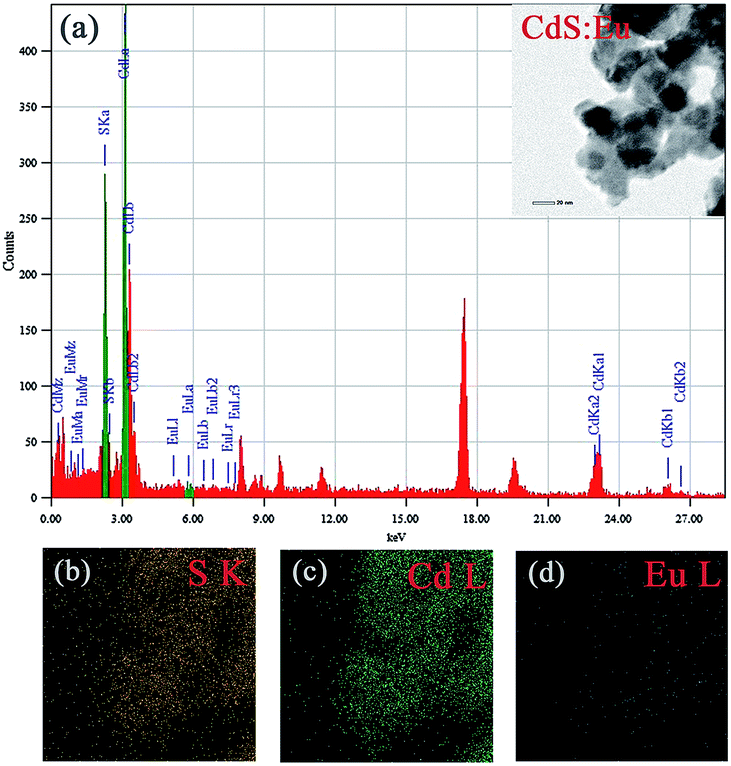 | ||
| Fig. 3 (a) EDS of CdS:Eu NPs, the insets is the TEM images of CdS:Eu NPs. (b–d) Chemical element mapping data corresponding to the TEM images. | ||
Synchrotron XRD patterns of CdS and CdS:Eu NPs samples collected at different pressures during compression and decompression are shown in Fig. 4. It can be observed that low pressure of the two samples stabilize the wurtzite structure. With increased pressure, all diffraction peaks shift to lower d values and the peak widths broaden gradually. Fig. 4a suggests that the phase transition starts at 4.97 GPa with two new diffraction peaks (*) which are not belonging to wurtzite structure appearance. The new phase completely appears at 5.90 GPa. The diffraction peaks of new high-pressure phase can be indexed to the cubic rock salt (RS) phase (JCPDS no. 21-829). All peaks of the original phase completely vanish at 13.60 GPa, which reveals the two-phase coexistence region in range of 4.97 and 13.60 GPa. The high pressure phase is stable up to the highest pressure of 18.31 GPa for pure CdS NPs. The new phase of CdS:Eu NPs is determined to initiate at 5.22 GPa and end at 5.96 GPa as shown in Fig. 4b. The new peak of CdS:Eu NPs at 5.22 GPa is marked with *. The enlarged view is displayed in the inset of Fig. 4b. The original phase of CdS:Eu NPs is not present after 14.27 GPa. The pressure range of phase coexistence of WZ and RS is from 5.22 GPa to 14.27 GPa due to the Eu doping of the sample. It is clearly that PT of CdS:Eu NPs is slightly higher than PT of CdS NPs.
 | ||
| Fig. 4 The compression and decompression XRD patterns of the CdS and the CdS:Eu NPs under high pressure. | ||
It can be detected that part of the peaks of WZ-CdS NPs and WZ-CdS:Eu NPs reappear when the pressure drops to 0 GPa as seen in Fig. 4a and b. It indicates that the phase transition process is partly reversible.
The bulk modulus reflects the hardness of a material and is important in many different industries. The volume dependence on pressure is shown in Fig. 5a and b. It can be observed that the volume collapse of the two samples was 13.3% and 17.4%, respectively, which is not sensitive to the Eu doping in CdS NPs. Bulk modulus for both phases are determined using third-order Murnaghan's equation of state.
| P = (3/2)B0[(V0/V)7/3 − (V0/V)5/3] × {1 + (3/4)(B′0 − 4) × [(V0/V)2/3 − 1]} | (1) |
 | ||
| Fig. 5 The volume dependence on pressure of CdS and CdS:Eu NPs. | ||
| WZ | RS | ||||||
|---|---|---|---|---|---|---|---|
| Sample | a (Å) | c (Å) | V0 (Å3) | B0 (GPa) | a (Å) | V0 (Å3) | B0 (GPa) |
| CdS | 4.147 | 6.796 | 101.22 | 39.88 ± 2.27 | 5.420 | 159.19 | 110.17 ± 4.30 |
| CdS:Eu | 4.128 | 6.755 | 99.68 | 52.51 ± 1.71 | 5.413 | 158.59 | 123.73 ± 3.01 |
High pressure Raman spectroscopy studies of CdS and CdS:Eu NPs were also carried out. From Fig. 6a and b, one can easily determine that the Raman peaks shift to higher wavenumbers linearly and the Raman peak intensities diminish rapidly with increasing pressure. Obviously, the pressure induced a decrease in bond length. The Raman shifts of CdS and CdS:Eu NPs as a function of pressure are shown in Fig. 7. Raman 1LO mode of CdS and CdS:Eu NPs disappeared at 6.38 and 8 GPa, respectively. The disappearance of the 1LO mode is also attributed to the wurtzite to rocksalt phase transition as Raman scattering of the rocksalt phase is inactive.33 However, this two pressures are not the beginning of phase transition of two samples. Fig. 7a illustrates that the Raman peaks of 1LO and 2LO phonon mode have blueshifts linearly before 4.76 GPa for CdS NPs. The inset of Fig. 7a provides a more evident identification. The nonlinear change of the Raman shifts indicates the occurrence of the structural phase transition from a wurtzite to a rocksalt phase.36 Thus, PT of CdS NPs should be 4.76 GPa, which amends accurately the previous XRD patterns under high pressure. Similarly, PT of CdS:Eu NPs is determined to 5.22 GPa combining the XRD data in Fig. 4b and Raman data in Fig. 7b. The A1 (LO) phonon mode corresponds to atomic oscillations along the c-axis. The peak value of this mode is sensitive to lattice strain along the c-axis.37 The slopes (dωi/dp) of 1LO wavenumber shift of CdS and CdS:Eu NPs before PT are 4.54 cm−1 GPa−1 and 4.48 cm−1 GPa−1, respectively, suggesting that the phonon frequency blueshift of 1LO phonon mode of CdS NPs is faster than CdS:Eu NPs. In other words, the pressure has more influences on the lattice tensile strain along the c-axis for CdS NPs.
 | ||
| Fig. 6 The Raman spectra under several applied pressures at room temperature. The excitation wavelength is 488 nm. (a) CdS; (b)CdS:Eu. | ||
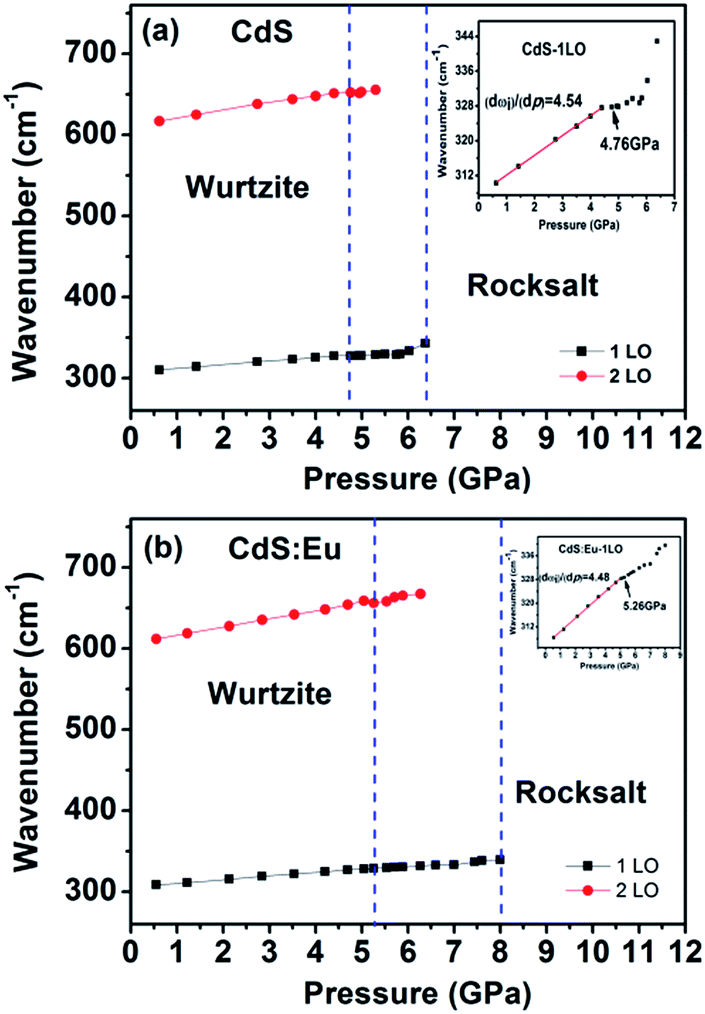 | ||
| Fig. 7 Raman shifts of CdS (a) and CdS:Eu (b) as a function of pressure. The insets are corresponding to the wavenumber shifts of 1LO as a function of pressure. | ||
The phonon frequency shift due to the lattice contraction is given by,
| Δω/ω0 = (1 + 3Δc/c)−γ − 1 | (2) |
where Δω is the 1LO phonon energy shift from its bulk value ω0 (305 cm−1) and γ is the Grüneisen parameter (1.1 for CdS). Δc/c is the lattice contraction of c-axis since CdS LO phonons are of vibrations along the c-axis.37–39 The relationship between lattice contraction and the pressure is shown in Fig. 8. It is clearly that the pressure effects the lattice contractions of CdS NPs more strongly than CdS:Eu NPs, which is in agreement with the results of phase transition. The phase transition can be attributed to an obvious structural transition due to the breakdown of the crystal symmetry. The greater impact on tensile strain along c-axis of CdS NPs result in the early phase transition compared to CdS:Eu NPs.
 | ||
| Fig. 8 Lattice contraction (Δc/c) as a function of pressure for CdS and CdS:Eu NPs. | ||
The photoluminescence (PL) spectra of CdS and CdS:Eu NPs obtained using an excitation wavelength of 532 nm are presented in Fig. 9 at ambient. Three optical vibrational Raman active modes are observed at approximately 540.69 nm, 549.64 nm and 558.85 nm in Fig. 9a, which are assigned to fundamental optical phonon mode (LO), the first over tone mode (2LO), and the second overtone (3LO) of CdS, respectively. The similar condition that the Raman peaks are detected in PL spectra was reported by Cheng.40 These Raman peaks of CdS are in agreement with the previous reported values.41–43 At lower doping concentration, there are no significant changes for three Raman peaks of CdS:Eu NPs in Fig. 9b. Their positions are at approximately 540.71 nm, 549.64 nm and 558.93 nm. However, it is reported that upon excitation of the CdS host, the energy from non-radiative recombination of electron–hole pairs can be transferred to the high lying energy levels of the Eu3+ ions; these excited levels decay non-radiatively to the long-lived 5D0 level and characteristic Eu3+5 D0 → 7FJ (J = 0–4) emission takes place.23 Obviously, efficient energy transfer from CdS NPs host to the Eu3+ ions centres have occurred in Fig. 9b, suggesting that the Eu3+ ions are indeed incorporated in the CdS NPs host.
 | ||
| Fig. 9 Photoluminescence spectra of CdS and CdS:Eu NPs at ambient. | ||
Three emission band of 5D0 → 7FJ (J = 0–4) transition are shown in Fig. 9b. The first emission band close to 579 nm may be ascribed to 5D0 → 7F0. The second emission band (located at 587.7, 592, 595.7 nm) can be fully resolved; these peaks correspond to the Stark components of the 5D0 → 7F1 transition of the Eu3+ ions, which is dominating in the inversion symmetry. The third emission band (located at 612.5, 615.8, 619. 5, 623.6 nm) are corresponding to 5D0 → 7F2 transition.36,44 These emission bands are similar to other Eu3+ doped systems because the 4f energy levels of the Eu3+ ions are hardly affected by the crystal field due to the shielding effect of the 5s2 5p6 electrons. The intensity of the emission peak at 5D0 → 7F2 (electric dipole transition), which is very sensitive to the local environment of the Eu3+ ions, is stronger than that of the 5D0 → 7F1 (magnetic dipole transition). This indicates the low site symmetry of the Eu3+ ions in CdS:Eu NPs. The 5D0 → 7F2 transition can be observed only when the lattice environment is distorted and contains noninversion symmetry.44
Fig. 10 shows the PL spectra dependence of the pressure for CdS NPs. The peak of 3LO at 558.93 nm is neglected due to its so weak under high pressure. The other two peaks positions are slightly redshift with increasing pressure, meanwhile, the intensities of the two peaks rapidly decrease. A redshift is observed due to the pressure induced redshift of bandgap energy. Above the pressure of 6.50 GPa, the peaks of 1LO and 2LO disappeared just like the Raman data, which indicating that CdS NPs undergo a phase transition from a direct bandgap WZ structure to an indirect bandgap RS structure, which will cause the quenching of emission.45,46 The peak positions of 1LO and 2LO as a function of pressure are shown in the inset of Fig. 10. It is obviously that the peak positions are linear redshift with increasing pressure up to 4.00 GPa. The nonlinear changes of the peak positions indicate that the phase transition have started at 4.88 GPa. The peak of 1LO can be detected up to 10.46 GPa, which is consistent with XRD and Raman results.
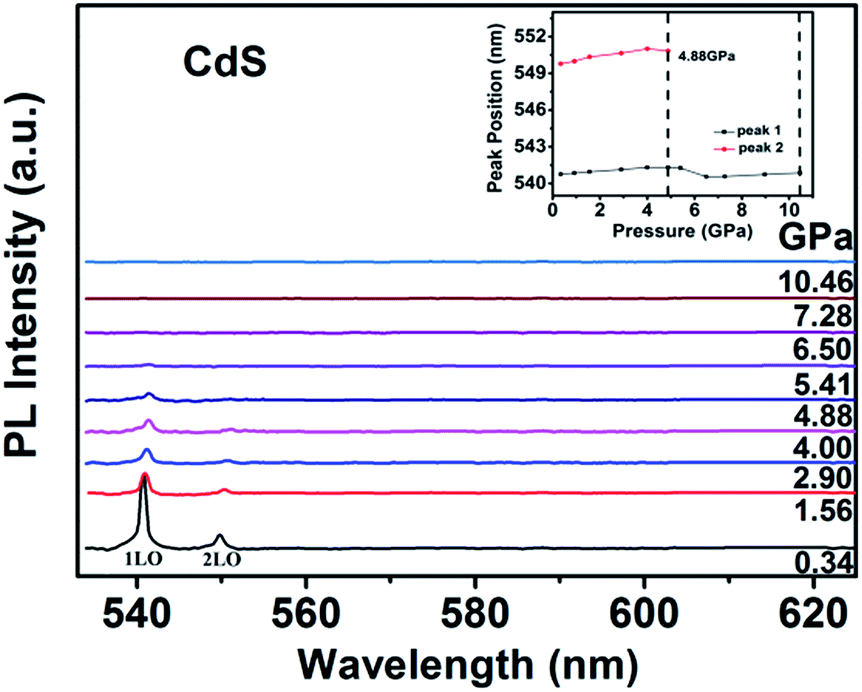 | ||
| Fig. 10 Photoluminescence spectra of CdS NPs as a function of pressure. | ||
It is well known that CdS nanomaterials play important roles in biological applications such as cellular labelling, selective ion probes, and deep-tissue imaging. Thus, it is expected to be potential biological materials under high pressure. So, it is necessary to probe slight changes of the local micro-structure of a variety of CdS crystal structure under high pressure. The monitoring of the doped Eu3+ ions luminescence can be used as an effective probe to reveal slight changes in structure and the local crystalline environment of CdS NPs under high pressure.
The evolution of the luminescence spectra of CdS:Eu NPs at room temperature are shown in Fig. 11. It is differ from the luminescence spectra of CdS NPs that the peaks of diamond and Si oil are not eliminated. The peak of 5D0 → 7F0 is not displayed as the influence of the diamond peak. The peaks of 1LO and 2LO of CdS:Eu NPs is slight redshift with the increasing pressure and distinct up to 6.18 GPa. The peaks of 5D0 → 7FJ (J = 1, 2) are displayed in the whole course of compression up to 20.69 GPa. There are several obvious changes in the luminescence spectra during loading pressure. As an illustration, the change of 5D0 → 7F2 transition is shown in the inset of Fig. 11a up to 6.18 GPa. The intensity of the peak of 5D0 → 7FJ (J = 1, 2) gets stronger with increasing pressure up to PT (5.20 GPa) and then weaker, which can be attributed to the new site symmetry of the Eu3+ ions as the phase transition from hexagonal WZ phase to cubic RS phase and corresponds with the X-ray diffraction and Raman results.
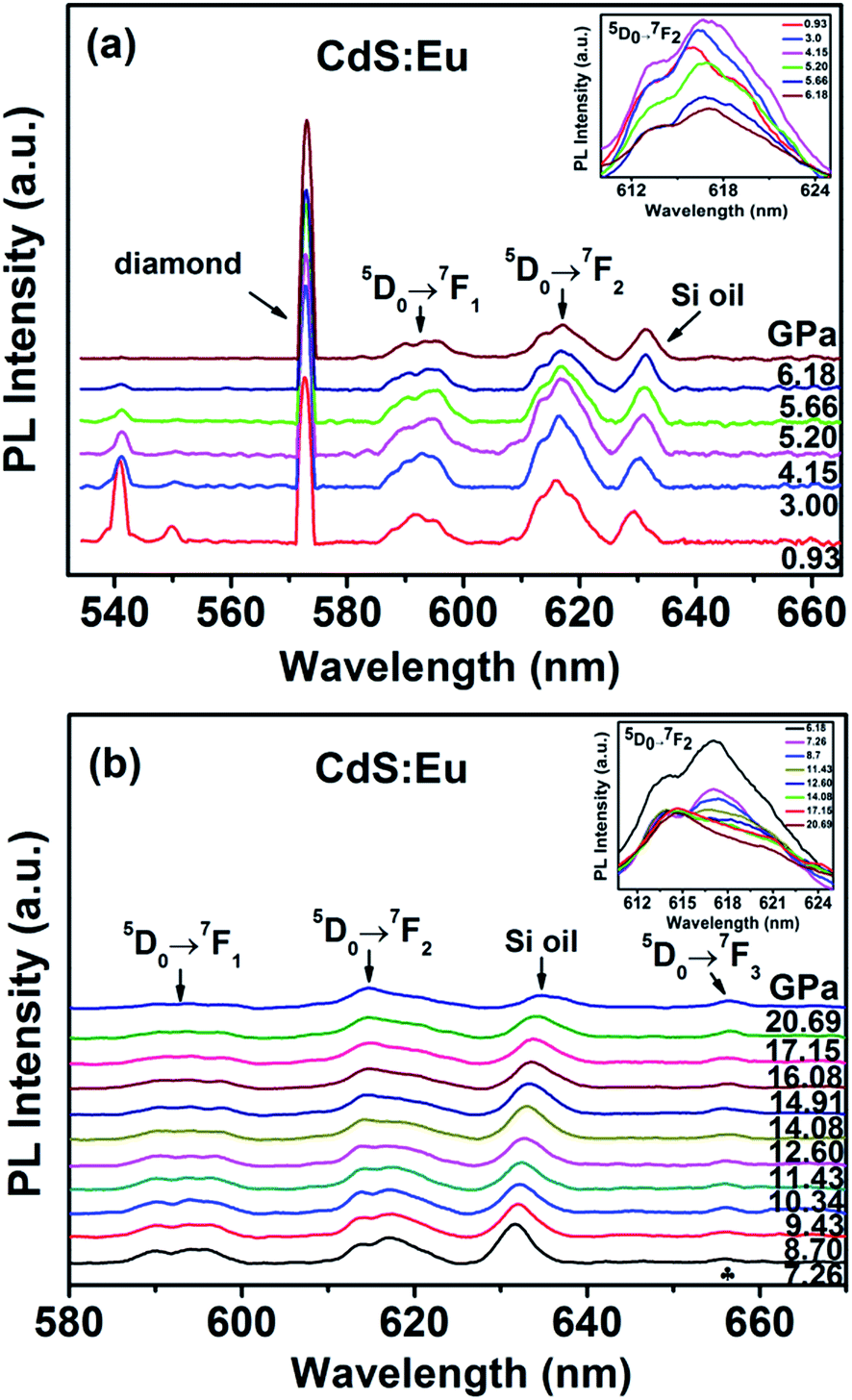 | ||
| Fig. 11 Photoluminescence spectra of CdS:Eu NPs as a function of pressure. | ||
When the increased pressure exceeds 12.60 GPa to 20.69 GPa, the intensities of 5D0 → 7F1 transitions gradually lose their intensity and are barely detectable. Similar to the change of 5D0 → 7F1 transition, the number of the peaks from the 5D0 → 7F2 transition starts to decrease at 14.08 GPa in the inset of Fig. 11b. Therefore, it is clearly that the coexistence of the two phases range of 5.20 GPa to 14.08 GPa. We assume that the coexistence of the two phases causes deviation from symmetry, thus, the PL spectra of 5D0 → 7FJ (J = 1, 2) in range of 5.20 GPa to 14.08 GPa is differ from the peaks of WZ CdS:Eu NPs and RS CdS:Eu NPs.
Meanwhile, at 7.26 GPa, a new peak due to 5D0 → 7F3 appears at 655.90 nm and becomes stronger with increasing pressure. It indicates that the point symmetry of the site of the Eu3+ ions in CdS:Eu NPs is shifted toward the noncentrosymmetric point group.36 In conclusion, a series of PL spectra of CdS and CdS:Eu NPs under high pressure indicate the changes of the local symmetry of the Eu3+ ions and the variation of the crystal field experienced by Eu3+ in the host material with increasing pressure.
Conclusions
The high-pressure behaviors of hexagonal wurtzite CdS and CdS:Eu NPs (8–10 nm) were investigated using angle-dispersive synchrotron radiation X-ray diffraction, Raman, and photoluminescence measurements. According to the results of Synchrotron XRD and high pressure Raman spectroscopy, the phase transition of CdS NPs starts at 4.76 GPa and ends at 13.60 GPa. The phase transition of CdS:Eu NPs starts at 5.22 GPa and ends at 14.27 GPa. PT of CdS:Eu NPs is slightly higher than CdS NPs. It can be attributed to the greater impact on tensile strain along c-axis of CdS NPs, which identified by the relationship between lattice contraction and the pressure obtained from Raman 1LO. The volume collapse of CdS and CdS:Eu NPs are 13.3% and 17.4%, respectively. The bulk modulus of hexagonal WZ and cubic RS structure of CdS NPs are estimated at 41.08 ± 3.52 and 100.58 ± 12.14 GPa. The bulk modulus of CdS:Eu NPs are 51.59 ± 5.71 and 88.43 ± 9.30 GPa for two structures, respectively. The reversibility of the phase transition was also found by ADXRD. The changes on Eu3+ luminescence peak intensity from the 5D0 → 7FJ (J = 1, 2) transition in CdS:Eu are observed at 5.20 GPa, which corresponds to the pressure-induced phase transition of CdS:Eu NPs from WZ to RS structure. The new peak of 5D0 → 7F3 is emerged at 7.26 GPa until the end of whole experiment. It is of great value for the application of CdS nanoparticles in biological sensors and photodetectors under high pressure.Acknowledgements
We would like to thank Zhongwu Wang for his technical support with the synchrotron X-ray diffraction measurements at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation and the National Institute of Health/National Institute of General Medical Science under NSF award DMR-0936384. This work was funded by the National Science Foundation of China, no. 11174103 and 11474124, and Specialized Research Fund for the Doctoral Program of Higher Education of China, no. 20130061110012.References
- Z. Zhou, F. Han, L. Guo and O. V. Prezhdo, Phys. Chem. Chem. Phys., 2016, 18, 16862–16869 RSC.
- K. Wu, Y. Du, H. Tang, Z. Chen and T. Lian, J. Am. Chem. Soc., 2015, 137, 10224–10230 CrossRef CAS PubMed.
- S. Saha, G. Das, J. Thote and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 14845–14851 CrossRef CAS PubMed.
- Y. Wang, H. Fu, Y. Wang, L. Tan, L. Chen and Y. Chen, Phys. Chem. Chem. Phys., 2016, 18, 12175–12182 RSC.
- Q. Zhang, X. Guo, X. Huang, S. Huang, D. Li, Y. Luo, Q. Shen, T. Toyoda and Q. Meng, Phys. Chem. Chem. Phys., 2011, 13, 4659–4667 RSC.
- R. N. Mitra, M. Doshi, X. Zhang, J. C. Tyus, N. Bengtsson, S. Fletcher, B. D. G. Page, J. Turkson, A. J. Gesquiere, P. T. Gunning, G. A. Walter and S. Santra, Biomaterials, 2012, 33, 1500–1508 CrossRef CAS PubMed.
- M. Gaceur, M. Giraud, M. Hemadi, S. Nowak, N. Menguy, J. P. Quisefit, K. David, T. Jahanbin, S. Benderbous, M. Boissière and S. Ammar, J. Nanopart. Res., 2012, 14, 1–15 CrossRef.
- J. Zhang, D. Li, R. Chen and Q. Xiong, Nature, 2013, 493, 504–508 CrossRef CAS PubMed.
- Y. Ma, X. Li, Z. Yang, H. Yu, P. Wang and L. Tong, Appl. Phys. Lett., 2010, 97, 153122 CrossRef.
- Y. Xi, C. Hu, C. Zheng, H. Zhang, R. Yang and Y. Tian, Mater. Res. Bull., 2010, 45, 1476–1480 CrossRef CAS.
- Q. Li and R. M. Penner, Nano Lett., 2005, 5, 1720–1725 CrossRef CAS PubMed.
- P. Wu and X.-P. Yan, Chem. Soc. Rev., 2013, 42, 5489–5521 RSC.
- A. Rmili, F. Ouachtari, A. Bouaoud, A. Louardi, T. Chtouki, B. Elidrissi and H. Erguig, J. Alloys Compd., 2013, 557, 53–59 CrossRef CAS.
- D. Wu, Y. Jiang, Y. Zhang, Y. Yu, Z. Zhu, X. Lan, F. Li, C. Wu, L. Wang and L. Luo, J. Mater. Chem., 2012, 22, 23272–23276 RSC.
- K. D. Nisha, M. Navaneethan, Y. Hayakawa, S. Ponnusamy and C. Muthamizhchelvan, J. Alloys Compd., 2011, 509, 5816–5821 CrossRef CAS.
- X. Wang, D. Li, Y. Guo, X. Wang, Y. Du and R. Sun, Opt. Mater., 2012, 34, 646–651 CrossRef CAS.
- R. Martin-Rodriguez, R. Geitenbeek and A. Meijerink, J. Am. Chem. Soc., 2013, 135, 13668–13671 CrossRef CAS PubMed.
- S. Khajuria, S. Sanotra, J. Ladol and H. N. Sheikh, J. Mater. Sci.: Mater. Electron., 2015, 26, 7073–7080 CrossRef CAS.
- L. Saravanan, R. Jayavel, A. Pandurangan, J. Liu and H. Miao, Mater. Res. Bull., 2014, 52, 128–133 CrossRef CAS.
- H. Sekhar, G. Trivikrama Rao, P. Harshavardhan Reddy and D. Narayana Rao, J. Alloys Compd., 2013, 562, 38–42 CrossRef CAS.
- P. Mukherjee, C. M. Shade, A. M. Yingling, D. N. Lamont, D. H. Waldeck and S. Petoud, J. Phys. Chem. A, 2011, 115, 4031–4041 CrossRef CAS PubMed.
- J. Planelles-Aragó, E. Cordoncillo, R. A. S. Ferreira, L. D. Carlos and P. Escribano, J. Mater. Chem., 2011, 21, 1162–1170 RSC.
- S. Kumar, Z. Jindal, N. Kumari and N. K. Verma, J. Nanopart. Res., 2011, 13, 5465–5471 CrossRef CAS.
- S. V. Eliseeva and J.-C. G. Bunzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- Z. P. Li, J. H. Wang, Y. Y. Hou, X. Bai, H. W. Song, Q. J. Zhou, T. Wei, Y. Lia and B. B. Liu, RSC Adv., 2015, 5, 3130–3134 RSC.
- H.-Q. Chen, J. Fu, L. Wang, B. Ling, B.-b. Qian, J.-g. Chen and C.-l. Zhou, Talanta, 2010, 83, 139–144 CrossRef CAS PubMed.
- S. S. Syamchand and G. Sony, J. Lumin., 2015, 165, 190–215 CrossRef CAS.
- R. Zhao, P. Wang, B.-b. Yao, T.-t. Hu, T.-y. Yang, B.-x. Xiao, S.-m. Wang, C.-h. Xiao and M.-z. Zhang, RSC Adv., 2015, 5, 17582–17587 RSC.
- F. H. Su, Z. L. Fang, B. S. Ma, K. Ding, G. H. Li and W. Chen, J. Phys. Chem. B, 2003, 107, 6991–6996 CrossRef CAS.
- M. Grinberg, J. Electrochem. Soc., 2010, 157, G100 CrossRef CAS.
- A. Kamińska, S. Biernacki, S. Kobyakov, A. Suchocki, G. Boulon, M. O. Ramirez and L. Bausa, Phys. Rev. B, 2007, 75, 174111 CrossRef.
- L. Zeiri, I. Patla, S. Acharya, Y. Golan and S. Efrima, J. Phys. Chem. C, 2007, 111, 11843–11848 CAS.
- H. M. Fan, Z. H. Ni, Y. P. Feng, X. F. Fan, Z. X. Shen and B. S. Zou, J. Raman Spectrosc., 2007, 38, 1112–1116 CrossRef CAS.
- G. Yin, H. Yin, X. Wang, M. Sun, L. Zhong, R. Cong, H. Zhu, W. Gao and Q. Cui, J. Alloys Compd., 2014, 611, 24–29 CrossRef CAS.
- R. Zhao, P. Wang, T. Yang, Z. Li, B. Xiao and M. Zhang, J. Phys. Chem. C, 2015, 119, 28679–28684 CAS.
- Q. G. Zeng, Z. J. Ding, Z. M. Zhang and Y. Q. Sheng, J. Phys. Chem. C, 2010, 114, 4895–4900 CAS.
- C. Hu, X. Zeng, J. Cui, H. Chen and J. Lu, J. Phys. Chem. C, 2013, 117, 20998–21005 CAS.
- J.-Y. Zhang, X.-Y. Wang, M. Xiao, L. Qu and X. Peng, Appl. Phys. Lett., 2002, 81, 2076–2078 CrossRef CAS.
- K.-Y. Lee, J.-R. Lim, H. Rho, Y.-J. Choi, K. J. Choi and J.-G. Park, Appl. Phys. Lett., 2007, 91, 201901 CrossRef.
- C. W. Cheng, E. J. Sie, B. Liu, C. H. A. Huan, T. C. Sum, H. D. Sun and H. J. Fan, Appl. Phys. Lett., 2010, 96, 071107 CrossRef.
- P. Elavarthi, A. A. Kumar, G. Murali, D. A. Reddy and K. R. Gunasekhar, J. Alloys Compd., 2016, 656, 510–517 CrossRef CAS.
- M. Thambidurai, N. Muthukumarasamy, S. Agilan, N. Sabari Arul, N. Murugan and R. Balasundaraprabhu, J. Mater. Sci., 2011, 46, 3200–3206 CrossRef CAS.
- M. Thambidurai, N. Muthukumarasamy, D. Velauthapillai and C. Lee, . Mater. Sci.: Mater. Electron., 2013, 24, 4535–4541 CrossRef CAS.
- C. Gong, Q. Li, R. Liu, Y. Hou, J. Wang, X. Dong, B. Liu, X. Tan, J. Liu, K. Yang, B. Zou, T. Cui and B. Liu, J. Phys. Chem. C, 2014, 118, 22739–22745 CAS.
- S. H. Tolbert, A. B. Herhold, C. S. Johnson and A. P. Alivisatos, Phys. Rev. Lett., 1994, 73, 3266–3269 CrossRef CAS PubMed.
- K. Jacobs, D. Zaziski, E. C. Scher, A. B. Herhold and A. Paul Alivisatos, Science, 2001, 293, 1803 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |