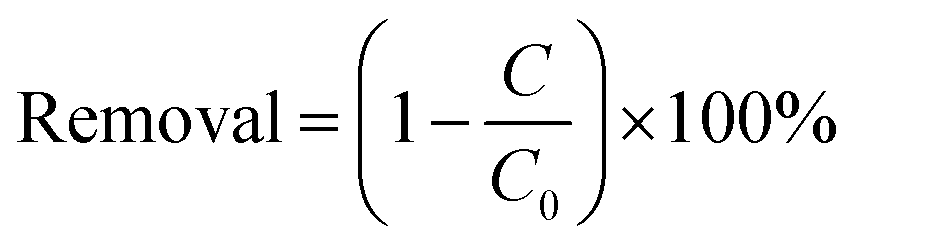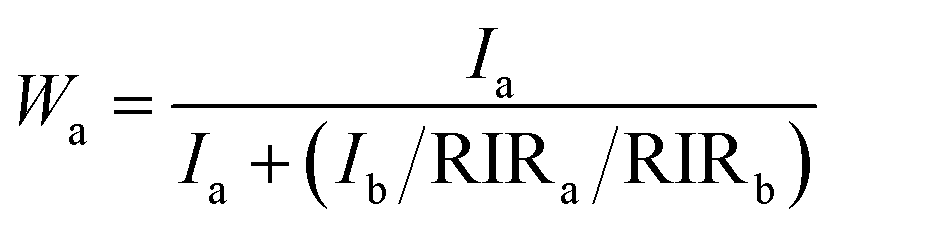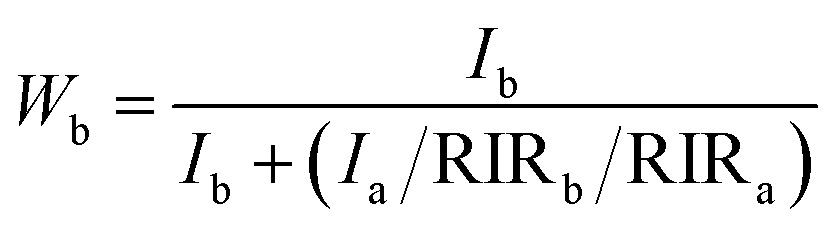DOI:
10.1039/C7RA05213H
(Paper)
RSC Adv., 2017,
7, 36269-36278
Additive dependent synthesis of bismuth oxybromide composites for photocatalytic removal of the antibacterial agent ciprofloxacin and mechanism insight
Received
9th May 2017
, Accepted 7th July 2017
First published on 21st July 2017
Abstract
Bismuth oxybromide composites were synthesized by solvothermal synthesis, applying triethanolamine (TEOA), sodium hydroxide (NaOH) and ammonium hydroxide (NH3 H2O) as the additives, which were denoted as S-TEOA, S-NaOH and S-NH3, respectively. The obtained samples were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), electron spin resonance spectrometry (ESR), Brunauer–Emmett–Teller (BET) measurement, and UV-Vis absorption spectrometry. The results showed that the samples consisted of the same phases containing BiOBr and Bi24O31Br10, and possess different structures, morphology and optical absorption properties depending on the additive. Their photoactivities were evaluated by degradation of ciprofloxacin (CIP) under visible light (λ ≥ 420 nm) irradiation. The apparent rate constant value of the sample S-TEOA is 2.62 and 4.17 times higher than that of the samples S-NaOH and S-NH3. The possible formation mechanism of the sample S-TEOA and adsorption mode of CIP species on the prepared samples are discussed on the basis of the experimental results.
1. Introduction
Ciprofloxacin (CIP) as a broad-spectrum antibacterial agent has been widely used for treating bacterial infections. The widespread use and difficult degradation of CIP1,2 may pose serious threats to the ecosystem and human health by inducing proliferation of bacterial drug resistance.3 Therefore, the employment of an appropriate approach to degrade CIP is of great importance. The photocatalytic degradation has been acknowledged as a promising technology for removal and degradation of CIP.4–6 The catalyst plays a key role in the photocatalytic degradation process. As a new class of promising layered materials for photocatalytic energy conversion and environmental remediation, Bismuth oxyhalides BiOX (X = Cl, Br, or I) have been intensively investigated.7–11 Among these BiOX photocatalysts, BiOBr possessing a layered structure composed of an alternating arrangement of (Bi2O2)2+ slabs and double slabs of Br−, has attracted great interest owing to its relatively superior catalytic activity and stability under visible light irradiation.12–16 As we all know, the photocatalytic properties of catalysts are closely related to their microstructure, grain size and morphology,17 and the morphology and microstructure of catalysts depend on the preparation method.18 Li et al. synthesized BiOCl nanosheets with tunable lamella thickness and dominantly exposed (001) facets via hydrothermal method using P123 and mannitol as surfactants.19 Xiong et al. synthesized a series of BiOBr nanosheets with tunable exposing proportion of (010) facets using different n-alcohols as solvent, the exposure of (010) facets, size and thickness of these BiOBr nanosheets could be well controlled by tuning the n-alcohols.20 Liu et al. prepared BiOBr/Bi24O31Br10 heterojunction photocatalysts applying ethylene glycol for solvent and NaOH as pH regulator,21 Li et al. reported the synthesis of BiOBr/Bi24O31Br10 heterojunction which was constructed by a route of one-step self-combustion of ionic liquids,22 however, the evolution process of the product, the degradation mechanism and adsorption mode between catalysts and pollutants were not further explored in these researches.
In recent years, some literatures reported triethanolamine (TEOA) as a regulator was employed to preparing complexes, which will be potential application in catalysis, magnetism and medicine.23–25 In these researches, TEOA plays important roles in the synthesis and crystallization process, as it has excellent ability in adjusting the pH value and the usability as counterion. Moreover, TEOA readily coordinates to metal ions to form complexes and stabilizes the anions by hydrogen bonding interactions because of its properties as both a tertiary amine and a primary alcohol.26
In this paper, we prepared bismuth oxybromide composites by one pot solvothermal synthesis method using TEOA, NaOH and NH3 H2O as the additive, respectively. And compared with the obtained samples, the sample obtained using TEOA as additive held the best photodegradation activity of CIP under visible light irradiation. We explored the formation mechanism of the sample S-TEOA, and found the additive TEOA may act as the complex agent except for the pH regulator. Meanwhile the adsorption modes of CIP species on the products were discussed according to the experimental results.
2. Experimental details
2.1. Materials
Bismuth nitrate pentahydrate (Bi(NO3)3·5H2O, AR), cetyltrimethyl ammonium bromide (CTAB, AR), ciprofloxacin (CIP), ethylene glycol ((HOCH2)2, EG, AR), triethanolamine (TEOA), NaOH, NH3 H2O, all reagents were used directly for the experiment without any further purification. Distilled water was used throughout this study.
2.2. Preparation of bismuth oxybromide composites
Firstly, a total of 8 mmol Bi(NO3)3·5H2O was dissolved into 30 mL of ethylene glycol (EG) stirring until a transparent solution was obtained. Subsequently, pH value of the solution was adjusted to 8 using TEOA, 1 mol L−1 NaOH aqueous solution and NH3 H2O (30.0%), respectively. Then another 30 mL of EG solution containing 8 mmol of cetyltrimethylammonium bromide (CTAB) was dropped into the above solution. The resultant precursor solution was poured into a 100 mL Teflon-lined autoclave after stirring for 30 min. Finally, the autoclave was kept at 160 °C for 16 h and allowed to cool down to room temperature naturally. The precipitate was washed with absolute ethanol and distilled water for several times, and dried at 60 °C in air. The samples obtained with adding TEOA, NaOH aqueous solution and NH3 H2O were denoted as S-TEOA, S-NaOH and S-NH3, respectively.
2.3. Characterization
XRD patterns were acquired with a Bede D1 System multifunction X-ray diffractometer employing Cu Kα (λ = 1.5418 Å) radiation. The voltage and current were 40 kV and 40 mA, respectively. Scanning electron microscopy (SEM) images were taken with a JSM 6701F field emission scanning electron microscope. Brunauer–Emmett–Teller (BET) surface areas were determined by nitrogen adsorption–desorption using a JW-BK132F analyzer. X-ray photoelectron spectroscopy (XPS) measurements were conducted on ThermoFisher Scientific Spectrometer using Al Kα radiation as the excitation source under vacuum at 2 × 10−6 Pa. JEOL JES-FA200 electron spin resonance (ESR) spectrometer (300 K, 9.063 GHz, X-band) was used for ESR analysis. The g factor was obtained by taking the signal of manganese as standard. UV-vis diffuse reflectance spectra were obtained using a TU-1900 spectrophotometer using BaSO4 as a reference and were converted from reflection to absorbance by the Kubelka–Munk method. UV-vis absorption spectra were obtained using a Shimadzu UV-2501PC spectrophotometer. Fourier transform infrared spectra were recorded at room temperature with a KBr pellet on Nicolet 5700 spectrometer.
2.4. Photocatalytic and active species experiments
The photocatalytic activity of the samples was evaluated by degrading of colourless CIP aqueous solution. A 300 W Xe lamp with 420 nm cut off filter was used as the light source. In order to maintain constant room temperature during visible light irradiation process, a water bath was used. In each experiment, 0.15 g of the as prepared photocatalyst was added to 250 mL the aqueous solution of CIP (20 mg L−1). Before irradiation, the suspension was treated by ultrasonication for 10 min, and then magnetically stirred in dark for 40 min to ensure the establishment of adsorption–desorption equilibrium of the CIP on the catalyst surface. Subsequently, at intervals of every 20 min, about 5 mL of suspension was sampled and separated by the filter membrane of a syringe to remove the catalyst particles. The absorbance of the solution was analyzed by using a UV-vis spectrophotometer. The removal efficiency of target pollutant (CIP) was calculated by the following equation:| |
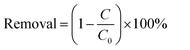 | (1) |
where C0 is the concentration of CIP after adsorption equilibrium, C is the residual concentration of CIP at different illumination intervals.
The photodegradation of CIP follows pseudo first-order kinetics, which can be expressed as:
where
k is the apparent reaction rate constant (min
−1).
For detecting the active species during photocatalytic reactivity, hydroxyl radicals (˙OH), superoxide radical (O2˙−) and holes (h+) were investigated by adding 1.0 m M isopropanol (IPA) (a quencher of ˙OH), 1 m M TEOA (a quencher of h+), respectively. The method was similar to the former photocatalytic activity test. Nitroblue tetrazolium (NBT, 2.5 × 10−5 M, exhibiting an absorption maximum at 259 nm) was used to determine the amount of O2˙− generating from photocatalyst. The production of O2˙− was quantitatively analyzed by detecting the concentration of NBT with Shimadzu UV-2501PC spectrophotometer. The method was similar to the former photocatalytic activity test with NBT replacing the pollutant CIP.
3. Result and discussion
3.1. XRD analysis
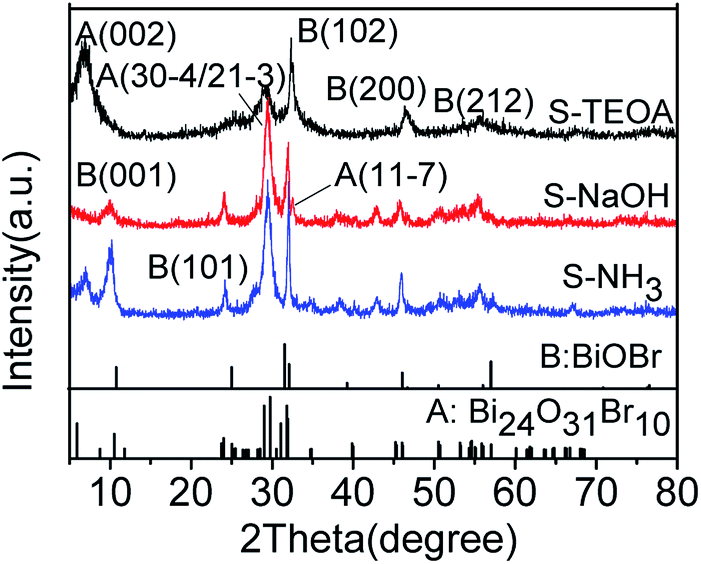
The XRD patterns of the samples S-TEOA, S-NaOH and S-NH3 are exhibited in Fig. 1. It can be seen that the three samples show a coexistence of BiOBr (JCPDS#09-0393 a = 3.926 Å, b = 3.926 Å, c = 8.103 Å) and Bi24O31Br10 (PDF#75-0888, a = 10.13 Å, b = 4.008 Å, c = 29.97 Å). Compared with the corresponding diffraction pattern of the sample S-TEOA, the (002) facet diffraction peak of Bi24O31Br10 disappears and the (001) facet diffraction peak of BiOBr appears in the sample S-NaOH. While the (002) facet diffraction peak of Bi24O31Br10 and the (001) facet diffraction peak of BiOBr appears in the sample S-NH3, but the peak intensity of (002) facet is weaker than that in the sample S-TEOA. The experimental results illustrate although the three samples contain the same phases, the two phases have different growth orientation. The sample S-TEOA obviously grows up along c-axis. The RIR method is employed to calculate the phase composition of the hybrids quantitatively, according to the following equations:27| |
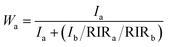 | (3) |
| |
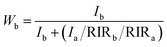 | (4) |
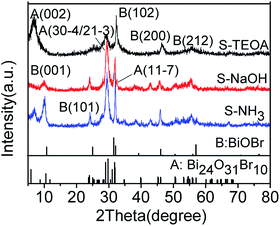 |
| | Fig. 1 The XRD patterns of the sample S-TEOA, S-NaOH and S-NH3. | |
The RIR values of BiOBr and Bi24O31Br10 phase read from the PDF database are 14.5 and 8.56, respectively. In calculation process, the “WPF Refinement” function of the “MDI JADE 5.0” software was employed. The weight percentages of BiOBr and Bi24O31Br10 in the samples are summarized in Table 1. According to the Table 1, the three samples contain much more Bi24O31Br10 than BiOBr.
Table 1 The weight percentages of BiOBr and Bi24O31Br10 in the obtained samples and the pseudo-first order rate constants K for CIP degradation under visible light irradiation over the samples
| Sample |
Phase composition (wt%) |
K (min−1) |
Correlation coefficient (R) |
| BiOBr |
Bi24O31Br10 |
| S-TEOA |
36.7 |
63.3 |
0.01644 |
0.9906 |
| S-NaOH |
26.7 |
73.3 |
0.00628 |
0.9957 |
| S-NH3 |
35.8 |
64.2 |
0.00394 |
0.9980 |
3.2. Crystal morphology and microstructure analysis
Fig. 2 shows that the SEM images of the obtained samples, the sample S-TEOA is composed of microspheres with a diameter of ca. 2–4 μm assembled by nanosheets and few of lamellates stacking on the surface of microspheres (Fig. 2(a)). Magnifying a microsphere, the microspheres exhibit marigold-like superstructure (Fig. 2(b)). The sample S-NaOH and S-NH3 are both composed of irregular thin sheets. By comparison, the thin sheets in the sample S-NH3 stack more closely and are much bigger than the sample S-NaOH (Fig. 2(e) and (f)). This indicates TEOA can improve the information of marigold-like role on the information and growth of thin sheets. It implies TEOA might act as complex agent and pH regulator, but NaOH and NH3 H2O work as pH regulators.
 |
| | Fig. 2 SEM images of the obtained samples. (a, b) S-TEOA, (c, d) S-NaOH, (e, f) S-NH3. | |
The image (Fig. 3(a)) shows the sample is consisted of nanosheets, which is consistent with the SEM images. From the Fig. 3(b), the HRTEM image shows the (102) lattice fringes of BiOBr with d value of 0.282 nm and (304) lattice fringes of Bi24O31Br10 with d values of 0.308 nm.
 |
| | Fig. 3 TEM images (a, b) of the sample S-TEOA. | |
3.3. The XPS spectra
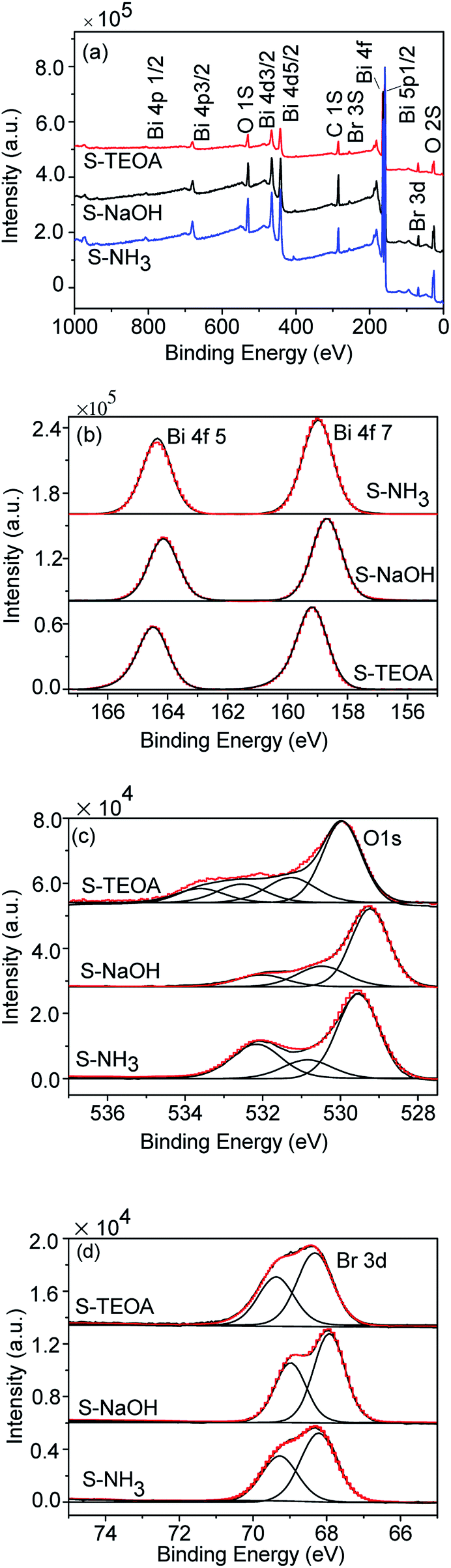
Fig. 4(a) shows the survey spectra of the samples S-TEOA, S-NaOH and S-NH3. This reveals the prepared samples are constituted of Bi, O, and Br elements. The high resolution XPS spectra Bi 4f, O 1s and Br 3d of the samples are shown in Fig. 4(b–d). From Fig. 4(b–d), the peak positions of Bi 4f, O 1s and Br 3d of the three samples are different, which imply the elements Bi, O and Br are in the distinct chemical environment. From Fig. 4(b), the Bi 4f spectrums are the symmetric Gauss two peaks structure, which are attributed to Bi3+.28,29 As shown in Fig. 4(c), the O 1s region are fitted into several peaks, we can classify these peaks into crystal lattice oxygen (Olatt) located at low binding energy (up to 530 eV) and the adsorbed oxygen (Oads) located at high binding energy (beyond 530 eV). From the Fig. 4(c), the sample S-TEOA has more Oads species than that of S-NaOH and S-NH3. Fig. 4(d) shows high-resolution XPS spectra for the Br 3d region. The binding energy could be assigned to Br at the monovalent oxidation state. Although the binding energies of Br 3d in the three samples shift, the energy differences of the overlapping spin–orbit components (Δ = 1.1 eV) keep constant. The XPS analysis results further illustrate the three samples have different surface structures, which agree with the XRD results.
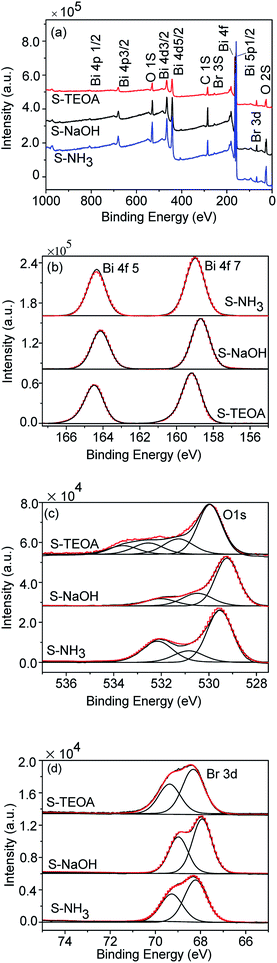 |
| | Fig. 4 XPS spectra of the sample S-TEOA, S-NaOH and S-NH3 (a) survey, (b) Bi 4f, (c) O 1s, (d) Br 3d. | |
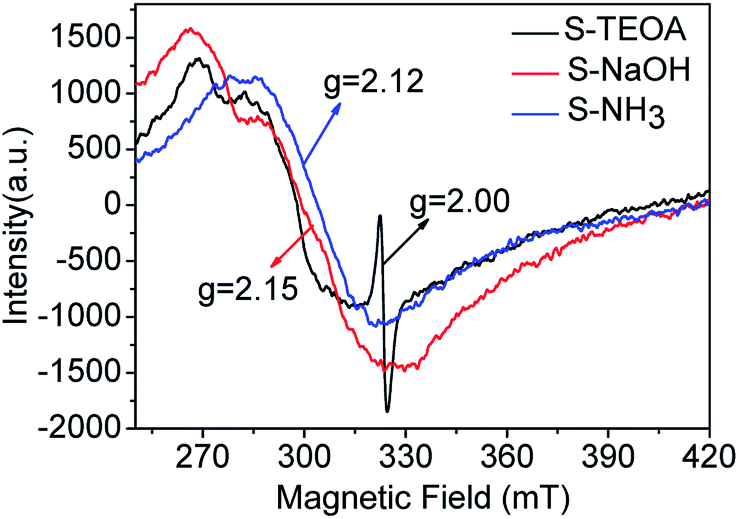
In order to verify the oxygen vacancy on the surface of the samples, the ESR technique was carried out. As can be seen from the Fig. 5, a strong ESR signal of the sample S-TEOA at g = 2.000 is ascribed to the surface oxygen vacancy feature.22 While for the sample S-NaOH and S-NH3, no ESR signal at g = 2.000 is found. Therefore, it can be concluded that there are a good number of oxygen vacancies on the surface of the sample S-TEOA, but it is absent on the surface of the sample S-NaOH and S-NH3.
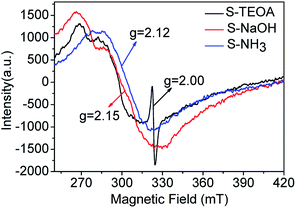 |
| | Fig. 5 ESR signals of the sample S-TEOA, S-NaOH and S-NH3. | |
3.4. Optical absorption properties
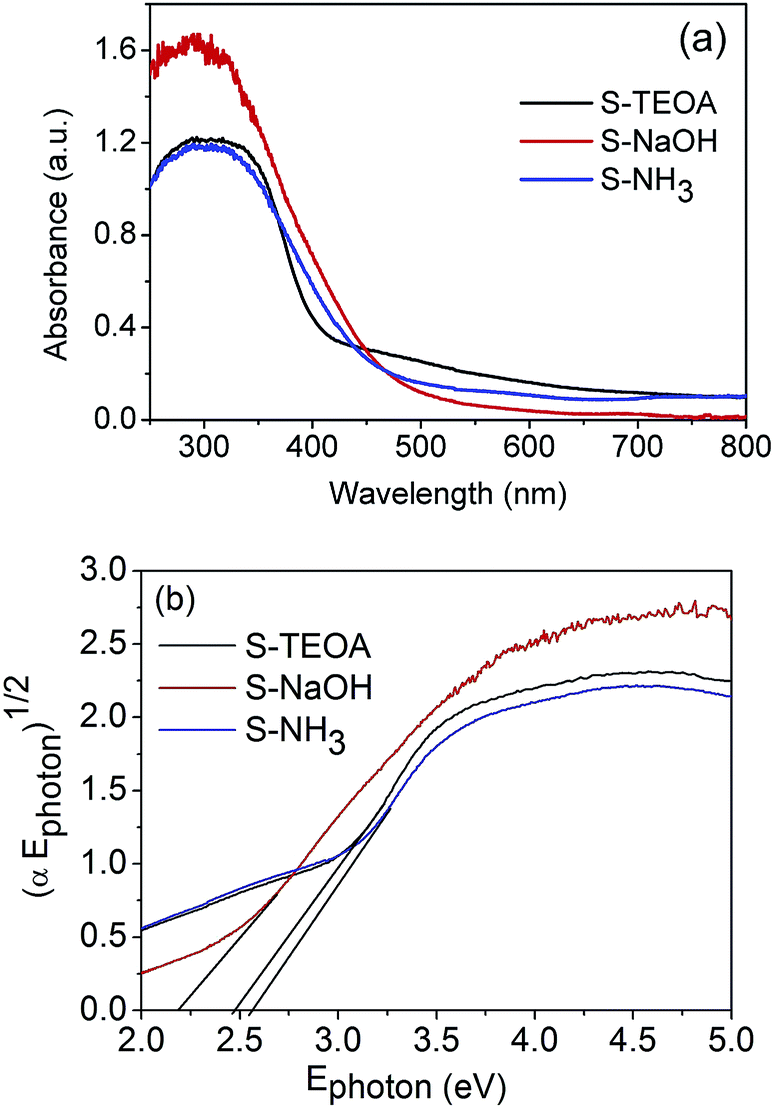
The optical properties of the as-prepared samples were investigated through the diffuse reflectance spectra (DRS) analysis. According to the Fig. 6(a), the three samples can absorb the visible light with the wavelength of above 420 nm. It was reported that the intensity of visible light with a wavelength range of 500 to 600 nm for a Xe lamp is larger than that of radiation wavelength below 500 nm.17 Therefore the sample S-TEOA can absorb more adequate the light source of Xe lamp than the sample S-NaOH and S-NH3. The band gap of semiconductor can be calculated according to the formula:| | |
α(Ephoton) = A(Ephoton − Eg)n/2,
| (5) |
where α, Ephoton, A and n are the absorption coefficient, the photo energy, constant and an integer, respectively. The n is equal to 1 for an indirect band gap, and it is 4 for a direct band gap.30 Then the band gap of the sample S-TEOA, S-NaOH and S-NH3 is estimated to be 2.48 eV, 2.19 eV and 2.52 eV, respectively. It is further instructive of their property of visible light absorption.
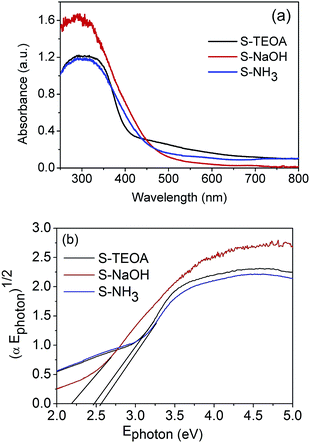 |
| | Fig. 6 (a) UV-vis diffuse reflectance spectra and (b) plots of (αEphoton)1/2 versus Ephoton curves of the sample S-TEOA, S-NaOH and S-NH3. | |
3.5. BET surface area and pore size distribution
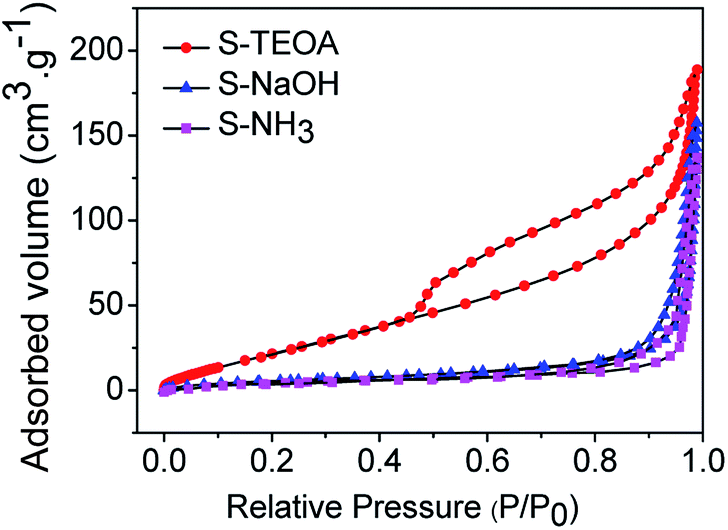
Fig. 7 shows the nitrogen adsorption–desorption isotherms of the as prepared samples. They all show hysteresis loops at P/P0 > 0.45. According to the IUPAC classification,31 the three samples can be clearly classified as typical type IV adsorption–desorption isotherms, which correspond to mesoporous solids. Furthermore, the hysteresis loop of S-TEOA is type H3, indicating the presence of slit-shaped pores with non-uniform size and shape.32 The hysteresis loop of S-NaOH and S-NH3 are type H4, which is often associated with narrow slit-like pores.33 The BET surface areas were estimated to be 28.20, 23.24 and 22.82 m2 g−1 for samples S-TEOA, S-NaOH and S-NH3, respectively. A larger BET surface area provides more active sites for the photochemical reaction, leading to an enhancement of the photocatalytic performance.34,35
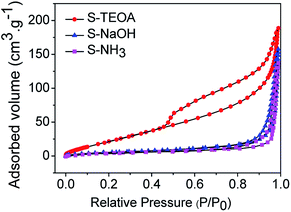 |
| | Fig. 7 Nitrogen adsorption–desorption isotherms of the sample S-TEOA, S-NaOH and S-NH3 (inset is pore size distribution curves of the samples calculated on the basis of the BJH equation). | |
3.6. Fluorescence emission spectra
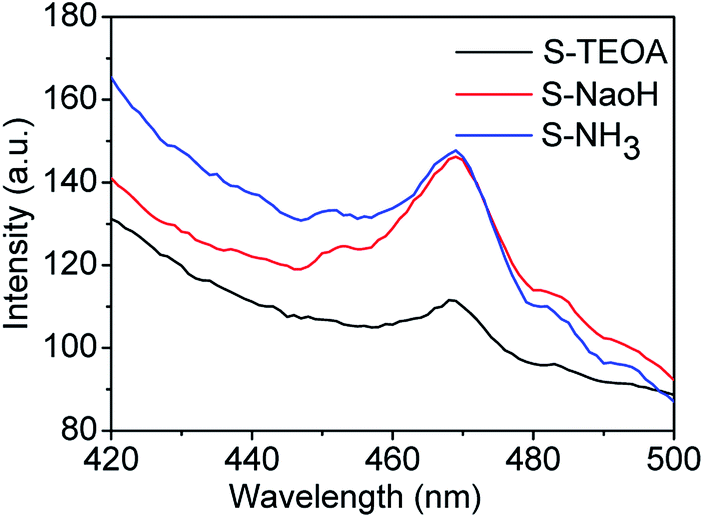
Fluorescence emission spectra have been used to reveal the migration, transfer and recombination of photo-generated electrons and holes, the relative lower PL emission intensity represents means the lower recombination rate of the photo-generated carriers and higher photocatalytic activity.36,37 The fluorescence emission spectra of the as prepared samples using an excitation wavelength of 360 nm are shown in Fig. 8. It can be seen the sample S-TEOA displays the lowest PL intensity than the other two samples, corresponding to the highest efficient separation of photo-generated electrons and holes under light irradiation.
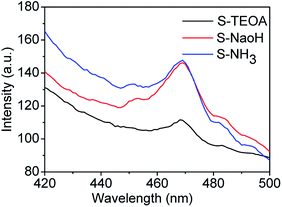 |
| | Fig. 8 PL spectra of the prepared samples. | |
3.7. Photocatalytic activity
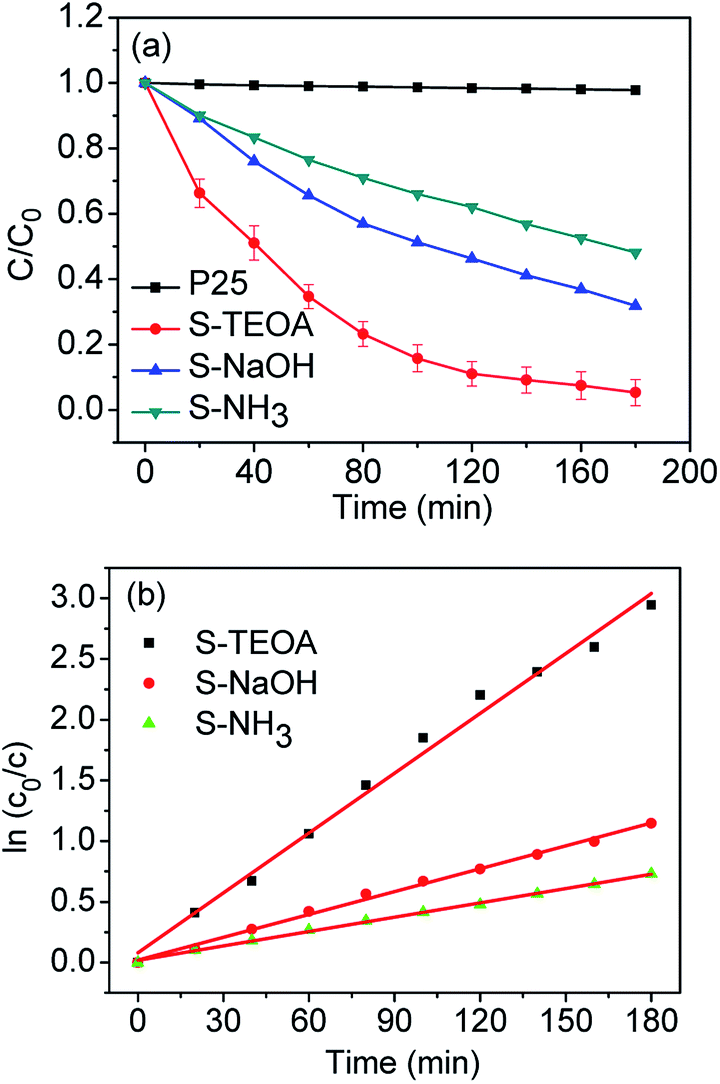
In order to reach the adsorption–desorption equilibrium between CIP and the catalyst, CIP aqueous containing the catalyst was ultrasounded for 10 min and stirred for 40 min in dank. After the adsorption–desorption equilibrium, adsorption rates of the obtained samples were 45.07% for S-TEOA, 31.10% for S-NaOH and 30.55% for S-NH3, which indicate the sample S-TEOA shows higher adsorption capability than the sample S-NaOH and S-NH3.
Fig. 9(a) presents the variation of CIP concentration (C/C0) as the function of reaction time in the presence of the obtained samples under visible light. For comparison, P25 TiO2 was used as a reference. As it can be seen from Fig. 9(a), P25 nearly has no photocatalytic ability under visible light irradiation, because it is a kind of ultraviolet-light responsive photocatalyst. With the increase of the irradiation time, the concentration of CIP solution decrease step by step. After irradiation for 180 min, 94.8% CIP in suspension can be photodegraded in the presence of the sample S-TEOA, however, 68.0% and 51.9% of CIP for the sample S-NaOH and S-NH3, respectively. The photocatalytic degradation kinetic of CIP in the presence of the three samples was investigated (Fig. 9(b)). The linear relationship between ln(C0/C) and irradiation time follows pseudo first-order kinetics. The calculated reaction rate constants and relative coefficients are listed in Table 1. The apparent rate constant value of the sample S-TEOA is 2.62 and 4.17 times higher than that of the sample S-NaOH and S-NH3, respectively.
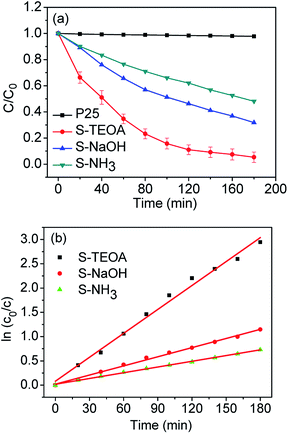 |
| | Fig. 9 (a) Photocatalytic performance for CIP solution under visible light irradiation of CIP solution containing 150 mg of P25 and the samples S-TEOA, S-NaOH, S-NH3. (b) Plots of ln(C0/C) as a function of visible light irradiation time for photodegradation of CIP solution containing 150 mg of the samples S-TEOA, S-NaOH, S-NH3. | |
According to the above characterization of the prepared samples, we infer that excellent photoactivity of the sample S-TEOA could be attributed the following factors. Firstly, the hierarchical flower-like morphology assembled with nanosheets in S-TEOA could obtain stronger visible-light absorbance via improving light multi reflections, and the flower-like morphology could provide more efficient transportation of reactants.38 Secondly, the sample S-TEOA with larger SBET and larger pore volume enables adequate photocatalyst-pollutants contact and adsorbs more active species, which are benefit to photocatalytic activity.39 Thirdly, more exposure percentage of (001) facet of the sample S-TEOA and the abundant surface oxygen vacancies favor to enhance photoactivity.40
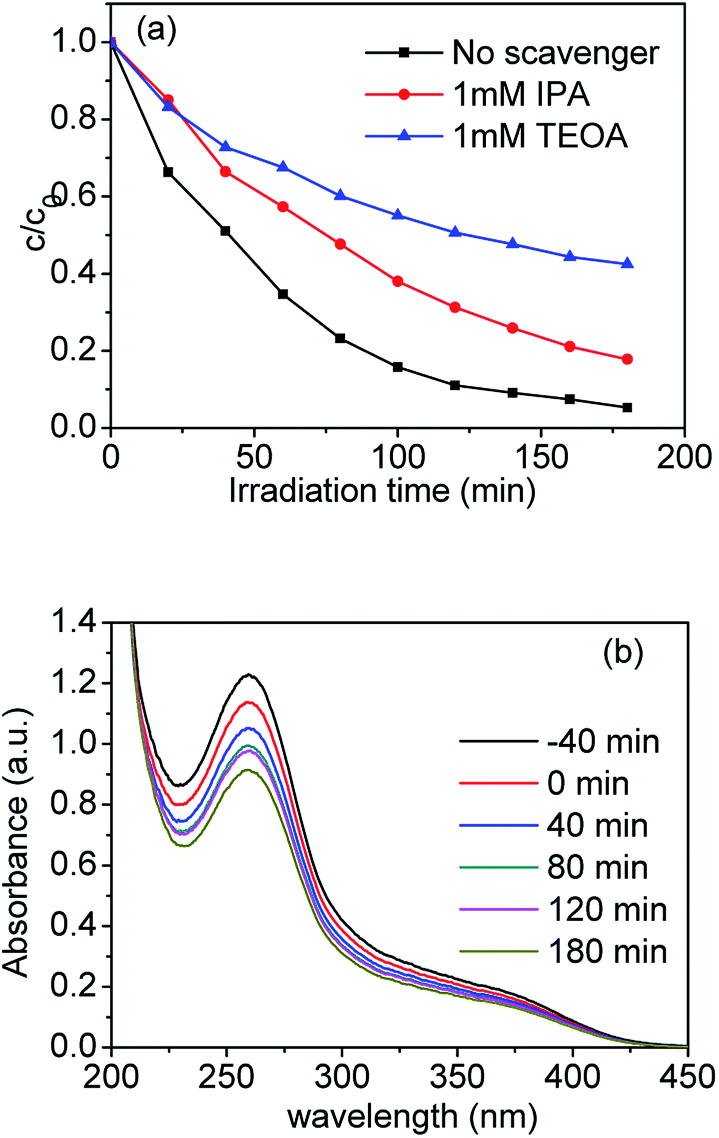
As well know, various primary reactive species, such as the hydroxyl radical ˙OH, photogenerated hole h+ and superoxide radical O2˙− can be formed during the photocatalytic degradation process in the UV-vis/semiconductor system.41–44 In order to confirm the main reactive species in the photocatalytic system, we carried out radical trapping experiments. We used triethanolamine (TEOA) as a quencher of holes (h+) and isopropanol (IPA) as a quencher of ˙OH. According to Fig. 10(a), after adding 1 mmol L−1 IPA into the suspension containing the sample S-TEOA, its photodegradation efficiency was reduced by ca. 13% in comparison with no IPA. It indicates OH˙ can be produced and plays some roles in photodegradation process. When adding 1 mmol L−1 TEOA into the suspension containing the sample S-TEOA, the degradation efficiency for CIP was reduced by ca. 40% compared with no scavenger. It indicates holes play an important role in photocatalytic process.
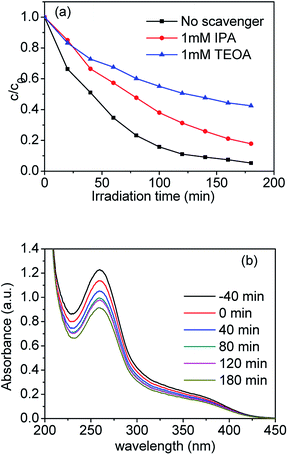 |
| | Fig. 10 (a) Time course of the photodegradation for CIP over the S-TEOA photocatalyst in the presence of various radicals scavengers and (b) UV-vis absorption spectra of NBT in the S-TEOA suspension under the visible light irradiation (λ ≥ 420 nm). | |
In this work, nitroblue tetrazolium (NBT) was used to determine the amount of O2˙− generated from the photocatalytic system. Fig. 10(b) shows the UV-vis absorption spectra of NBT in the suspensions of S-TEOA under visible light irradiation. It is obvious that the maximum absorbance declines with irradiation time increasing. It indicates that photo-generated electrons can react with O2 to produce O2˙− in the suspension of S-TEOA under visible light irradiation (eqn (6)).
According to above experimental results, holes and ˙OH and O2˙− are reaction active species during the photocatalytic process.
Since the valence band potential of Bi24O31Br10 (2.79 eV) is more positive than the E of (˙OH/OH−) (2.38 eV vs. NHE), E of (˙OH/H2O) (2.27 eV vs. NHE) and the oxidation potential of water (1.23 eV vs. NHE), hvb+ in the valence band of Bi24O31Br10 can oxidize water molecules to form ˙OH (eqn (7) and (8)). The conduction band potential of BiOBr (0.48 eV) is more negative than the electrode potential of O2 capturing photogenerated electrons to form hydrogen peroxide (O2˙−). Therefore ˙OH can be produced by reduction of O2 by photogenerated electrons (eqn (9)–(12)).45 In the end, the adsorbed CIP are oxidized by photogenerated holes (h+) and hydroxyl radicals (˙OH) and superoxide radical (O2˙−) to the final products (eqn (13)).
| | |
H2O + h+ → ˙OH + H+ + 2.27 eV
| (7) |
| | |
OH− + h+ → ˙OH + 2.38 eV
| (8) |
| | |
2H+ + O2 + 2e → H2O2 + 0.69 eV
| (9) |
| | |
2H+ + O2 + e → H2O2 + 1.71 eV
| (10) |
| | |
H+ + HO2 + e → H2O2 + 1.42 eV
| (11) |
| | |
H2O2 + hν → ˙OH + ˙OH
| (12) |
| | |
CIP + h+ + ˙OH + O2˙− → CO2 + H2O + small moleculars
| (13) |
3.8. Plausible formation mechanism of Bi24O31Br10/BiOBr
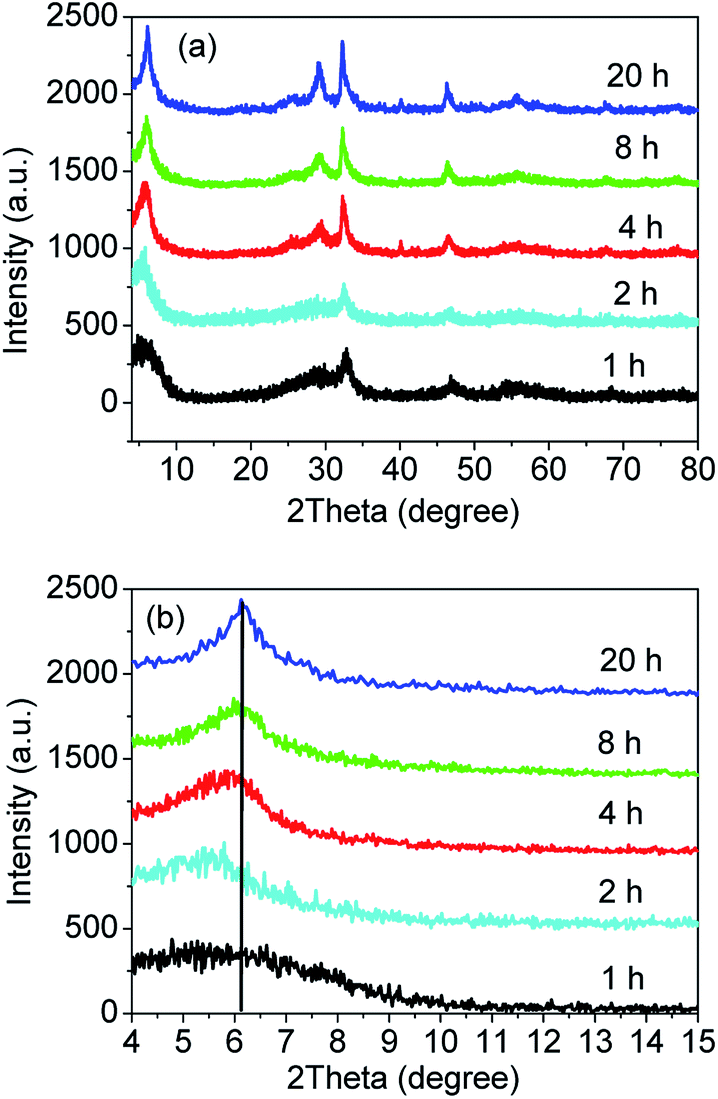
In order to explore the formation process of the sample S-TEOA during the solvothermal treatment, the samples obtained at different solvothermal times were characterized by XRD and SEM. Fig. 11(a) shows the XRD patterns of the sample S-TEOA under different thermal treatment time (t denoted as reaction time). When treatment for one hour, the intensities of diffraction peaks of the sample is very low, which indicates the poor crystallinity. But we can still index the diffraction peaks to Bi24O31Br10 and BiOBr. During the experimental process, we found after the EG suspension containing CTAB was added into the EG solution containing Bi(NO3)3 and TEOA, the mixed suspension was clear after stirring for 30 min. It is possibly due to the formation of one bismuth complex ion (Bi(TEOA)3+) using TEOA as the ligand (eqn (14)). The literature46 reported that the TEOA complex ion of Bi3+ could transform to more stable Bi(TEOA)(OH)2+ complex ion when pH value of the solution is up to 7 (eqn (15)). After adding CTAB into the Bi(TEOA)(OH)2+ solution, the CTAB species can be resolved into the solution which illustrates a new species arise, in other words, the Br ions maybe react with complex ion Bi(TEOA)(OH)2+ into the Bi(TEOA)(OH)Br2 species (eqn (16)).46| |
Bi3+ + TEOA ⇔ Bi(TEOA)3+, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = 9.2 K = 9.2
| (14) |
| | |
Bi(TEOA)3+ + OH− ⇔ Bi(TEOA)(OH)2+, logK = 10.3
| (15) |
| | |
Bi(TEOA)(OH)2+ + CTAB ⇔ Bi(TEOA)(OH)Br2
| (16) |
| | |
Bi(TEOA)(OH)Br2 → Bi24O31Br10 + BiOBr + TEOA
| (17) |
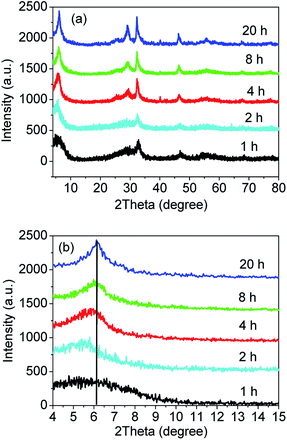 |
| | Fig. 11 XRD patterns (a) and magnified patterns (b) of the sample S-TEOA-t (t = 1, 2, 4, 8, 20 h). | |
While TEOA coordinating with Bi3+, the bismuth ion chelates by polydentate coordination with three hydroxyls of TEOA,47 then the rich oxygen Bi24O31Br10 and BiOBr will be formed gradually in the process of heating. From Fig. 11(a), with reaction time increasing, the diffraction peaks become stronger which indicates the crystal grains grow bigger. Magnified the (002) facet diffraction peak (Fig. 11(b)), with the reaction time prolong, the (002) diffraction peaks of Bi24O31Br10 are stronger and the diffraction peaks move to the higher angle gradually. According to the Bragg's equation: 2d(hkl)sinθ = nλ, where d(hkl) is interplanar spacing of (hkl) facet, θ is the Bragg angle, λ is the wavelength of incident X-ray, the d(002) of Bi24O31Br10 decreases gradually. It is possibly because that complexes of TEOA with Bi insetting into the interlayer of Bi24O31Br10 at the preliminary reaction lead to (002) crystal plane expansion. With the thermal treatment time increasing and the crystal grain ripening, more and more Bi(TEOA)(OH)Br2 complexes transform into Bi24O31Br10 and BiOBr. Meanwhile the TEOA releases from interlayer of Bi24O31Br10.
According to the eqn (3) and (4), the weight percentages of BiOBr and Bi24O31Br10 in the sample S-TEOA-t were calculated and listed in Table 2. It is can be seen from Table 2 that weight percentage of Bi24O31Br10 lifted up and then declined slowly with the increase of reaction time. It is maybe because that at reaction initial stage (up to 2 h), the complex Bi(TEOA)(OH)Br2 is increasingly decomposed into Bi24O31Br10 and BiOBr phase. Beyond 2 h, the reaction process is an Ostwald-ripening one and during the process BiOBr phase grows up at the cost of slight dissolution of Bi24O31Br10.
Table 2 The weight percentage of BiOBr and Bi24O31Br10 in the obtained product S-TEOA-t
| Time (t) |
1 h |
2 h |
4 h |
8 h |
16 h |
20 h |
| Bi24O31Br10 (wt%) |
69.9 |
76.0 |
70.5 |
68.3 |
63.3 |
65.1 |
| BiOBr (wt%) |
30.1 |
24.0 |
29.5 |
31.7 |
36.7 |
34.9 |
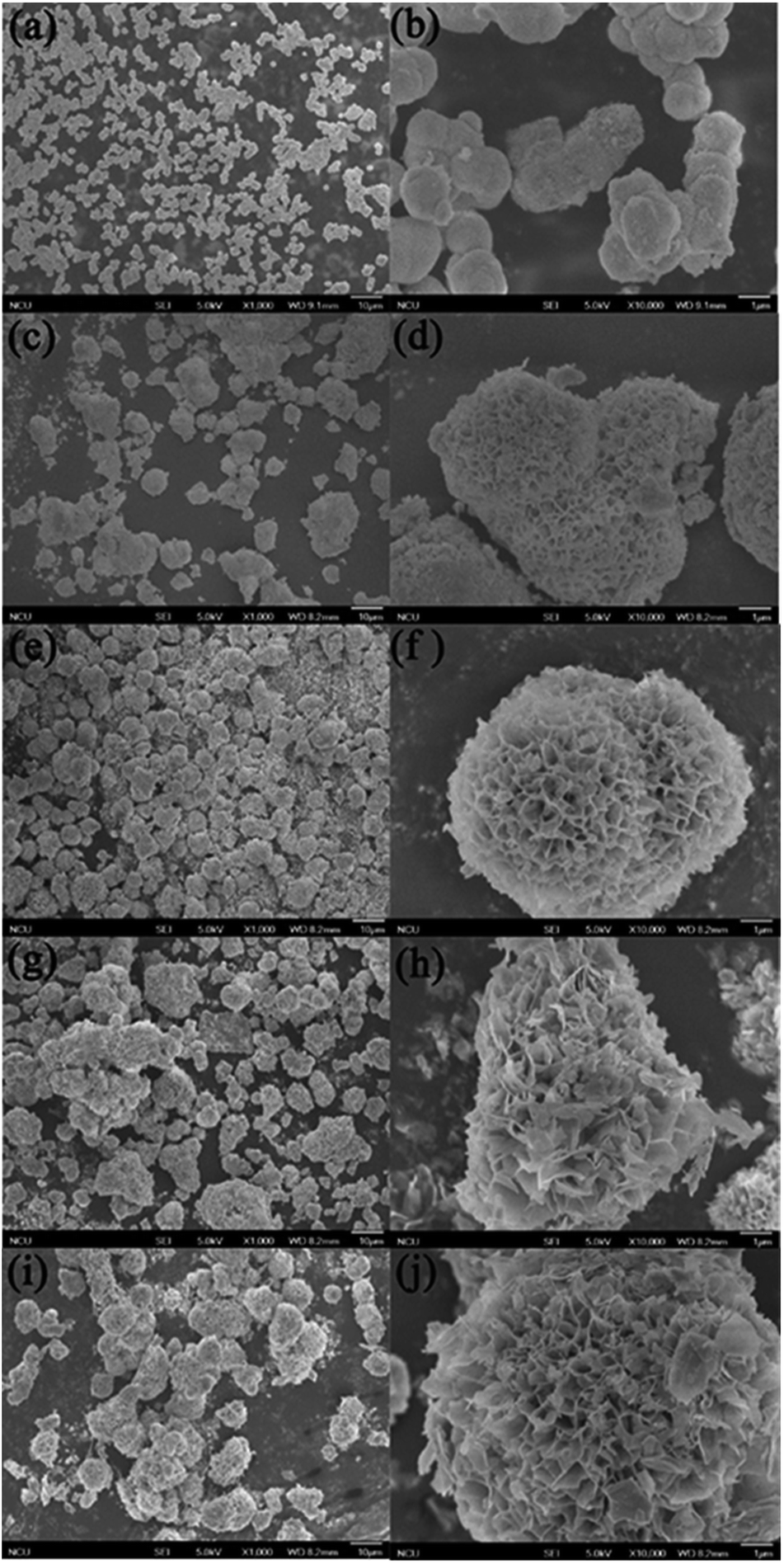
The SEM images of the sample S-TEOA-t (t = 1 h, 2 h, 4 h, 8 h, 20 h) are shown in Fig. 12 According the Fig. 12(a, c, e, i and j), with the increase of reaction time, the marigold-like microspheres grow up gradually. Magnify the single microsphere, we can obviously find out the petal-like superstructure of microsphere increase, which lead to the interspaces of petal enlarge (Fig. 12(b, d, f, h and j)). It agrees with the XRD analysis result.
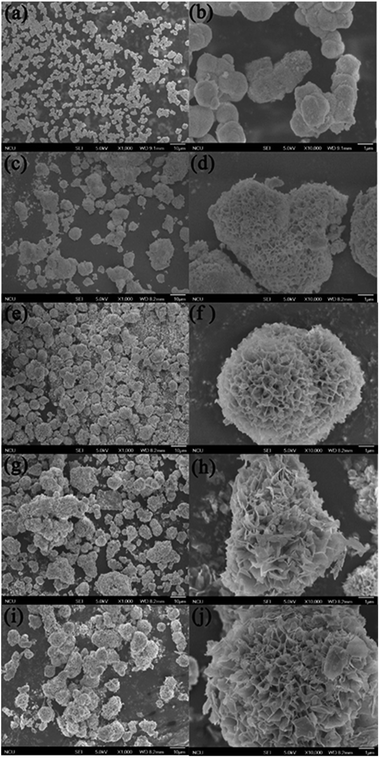 |
| | Fig. 12 SEM images of samples obtained during different solvothermal times: (a, b) 1 h, (c, d) 2 h, (e, f) 4 h, (g, h) 8 h, (i, j) 20 h. | |
3.9. Chemical interaction between CIP and the interface of photocatalysts
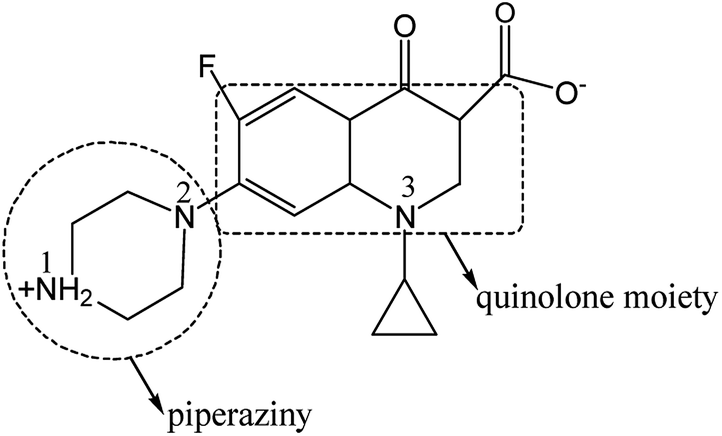
The photocatalytic efficiency is influenced by the adsorption site and mode of CIP on the photocatalyst. So it is necessary to further study on the interaction between CIP and catalyst. CIP possesses ionogenic functional groups (carboxylic acid group, N1 and N2 amino group on piperazine band and N3 amino group on band).48 According to the literature, charged CIP molecular varies with pH value as its pKa1 = 6.0 and pKa2 = 8.8, respectively.49 The pH value of the photocatalytic reaction system was determined to be ca. 6.4, close to the neutral. So, most of CIP species are amphoteric rather than nonionic. Carboxylic acid group is negatively charged by losing one hydrogen and N1 group is positively charged by obtaining one hydrogen (Scheme 1). Functional groups of the CIP species possibly interact with the interface of photocatalyst via electrostatic attraction, coordination with metal ions and H-bonding. Infrared vibrational spectroscopy usually provides a powerful tool to distinguish these binding modes. Fig. 13 shows the FTIR spectra of the sample S-TEOA, S-NaOH and S-NH3 after adsorbing CIP using the CIP solid powder as a reference 0.15 g of the samples S-TEOA, S-NaOH and S-NH3 was added into the 250 mL of CIP solution with the concentration of 20 mg L−1, respectively. And the suspension was ultrasounded 10 min and stirred for 40 min in dark in order to reach the adsorption equilibrium, then filtered and dried at 60 °C in air.
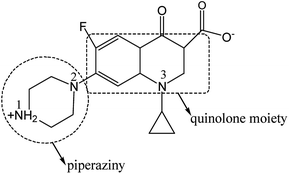 |
| | Scheme 1 CIP molecular structure in pure water. | |
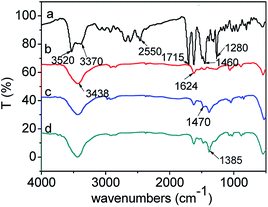 |
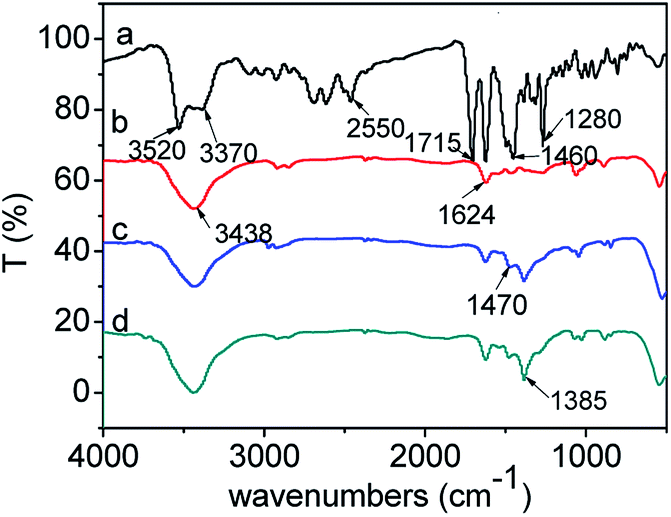
| | Fig. 13 FTIR spectra of CIP powder and CIP species adsorbed onto the samples: (a) CIP powder, (b) CIP/S-TEOA, (c) CIP/S-NaOH, (d) CIP/S-NH3. | |
For the pure CIP powder, the stretching vibration of the carboxyl group is located at 1715 cm−1. The OH stretching vibration of the adsorbed water is at ca. 3520 cm−1, and the C–N stretching vibration peak is at ca. 1280 cm−1. The 1630 and 1460 cm−1 can be attributed to the amide doublet stretching vibration and the N–H bending vibration peak,50,51 respectively (Fig. 13(a)). After adsorption of CIP on the obtained samples, the stretching vibrations of the carboxyl group at 1715 cm−1 over the three samples disappear. Meanwhile, the two new peaks appear at ca. 1624 and 1385 cm−1, which can be attributed to antisymmetric (νas) and symmetric (νa) stretching vibration of the carboxyl group.51 This splitting vibration was also observed in the RhB coordination with TiO2.52,53 Generally, the frequency difference (Δ) between the antisymmetric and symmetric stretching vibration of carboxyl group in various chemical environment is in the order of –(monodentate) > –(ionic) ∼ (bridging) > –(bidentate)–.54–57 In our work, the Δ value (239 cm−1) of carboxyl group indicates that carboxyl group links with the Bi3+ by monodentate coordination. Additionally, we find that the intensities of the splitting vibration are distinct among the three samples. As for the sample CIP/S-TEOA, the transmittance of vibration at 1385 cm−1 is the weakest. In reverse, it indicates that the interaction between the carboxyl group and Bi ions makes the vibration absorption peak of carboxyl group being stronger. Oxygen vacancy on the surface of the sample S-TEOA should be responsible for stronger vibration absorption of carboxyl group. Since the oxygen atoms escape from the sample surface, the positively charged oxygen vacancies decrease the constraint of adjacent Bi ions. It is likely that these Bi ions coordinating with carboxyl groups lead to increase vibration dipole moments of carboxyl groups, corresponding to the vibration absorption peak intensity being stronger.
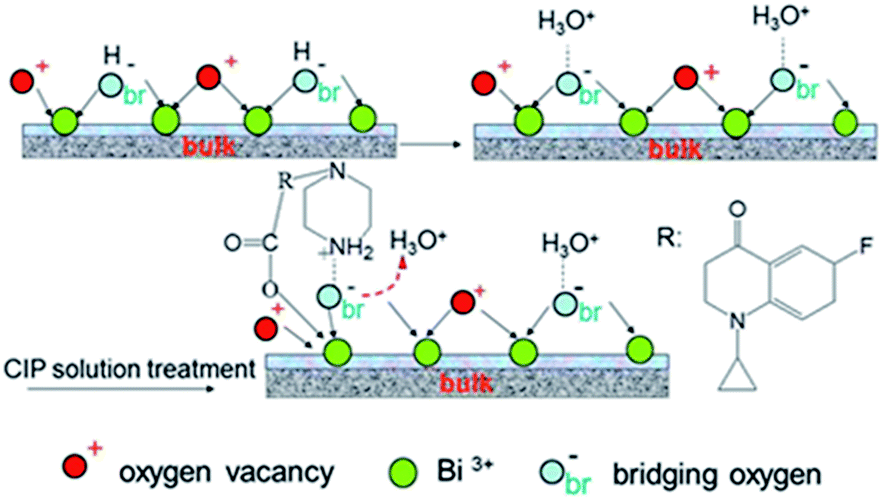
From the Fig. 13, we can find the N–H bending vibration peak at ca. 1460 cm−1 of the pure CIP powder shifts to the ca. 1470 cm−1. It is believed that a new linkage between positively charged NH2 and the surface of the samples can be formed. But due to the stronger interaction between the positively charged NH2 and the sample S-TEOA, the peak at ca. 1470 cm−1 almost disappears. It is well known that the water molecules can bond with surface bridging hydroxyls (HObr) of materials through interaction between H2O and HObr. During the adsorption process of CIP species, due to water molecules bonding interaction, HObr donates its proton to H2O, forming H3O+–Obr− structures. Simultaneously, the positively charged NH2 groups of CIP species displace the H3O+ and interact with Obr− by the electrostatic adsorption. According to the above experimental results and analysis, the possible adsorption modes of CIP species on the sample S-TEOA can be illustrated in Scheme 2. The closely interaction between CIP species and the surface of the sample S-TEOA via electrostatic attraction, coordination with metal ions benefit to the transference of electrons and holes on the interface. The proper adsorption site and mode of CIP play an important role on the improvement of photocatalytic activity of the sample S-TEOA.
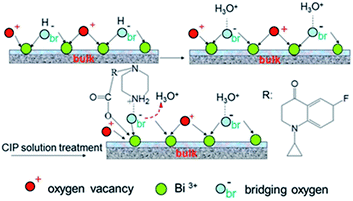 |
| | Scheme 2 Diagrammatic adsorption modes of CIP on the surface of the sample S-TEOA. | |
4. Conclusions
In this paper, Bismuth oxybromide composites have been synthesized through a facile solvothermal process. The obtained samples are consisted of the same phases containing BiOBr and Bi24O31Br10. The microstructure, morphology and optical property of the samples are distinct depending on the additives. The sample S-TEOA obtained using TEOA as coordination agent and pH regulator has high percentage of (001) facet of Bi24O31Br10, surface oxygen vacancies, excellent optical absorption and adsorption properties. Therefore, the sample S-TEOA has the highest photocatalytic activity for degradation of CIP under visible light irradiation. Additionally, the adsorption modes between the CIP species and surface of the sample S-TEOA are monodentate coordination and electrostatic interaction. This work is important to understanding the application of TEOA for preparing high effective photocatalysts.
Acknowledgements
The financial support of this study from the National Natural Science Foundation of China (21563020, 21465017), Jiangxi Province Science and Technology University Ground Plan Project (KJLD 14007) are gratefully acknowledged.
Notes and references
- Y. Pico, V. Andreu and B. Anal, Anal. Bioanal. Chem., 2007, 387, 1287–1299 CrossRef CAS PubMed.
- I. Tantis, L. Bousiakou, G. Karikas and P. Lianos, Photochem. Photobiol., 2015, 14, 603–607 RSC.
- T. Paul, P. L. Miller and T. J. Strathmann, Environ. Sci. Technol., 2007, 41, 4720–4727 CrossRef CAS PubMed.
- K. Zheng, X. Y. Zheng, F. Yu and J. Ma, RSC Adv., 2016, 6, 3625–3631 RSC.
- Y. Yan, S. F. Sun, Y. Song, X. Yan, W. S. Guan, X. L. Liu and W. D. Shi, J. Hazard. Mater., 2013, 250, 106–114 CrossRef PubMed.
- J. X. Xia, Y. P. Ge, D. X. Zhao, J. Di, M. X. Ji, S. Yin, H. M. Li and R. Chen, CrystEngComm, 2015, 17, 3645–3651 RSC.
- Q. Z. Wang, D. H. Jiao, J. H. Lian, Q. Ma, J. Yu, H. H. Huang, J. B. Zhong and J. Z. Li, J. Alloys Compd., 2015, 649, 474–482 CrossRef CAS.
- D. Wu, B. Wang, W. Wang, T. C. An, G. Y. Li, T. W. Ng, H. Y. Yip, C. M. Xiong, H. K. Lee and P. K. Wong, J. Mater. Chem. A, 2015, 3, 15148–15155 CAS.
- W. W. Lee, C. S. Lu, C. W. Chuang, Y. J. Chen, J. Y. Fu, C. W. Siao and C. C. Chen, RSC Adv., 2015, 5, 23450–23463 RSC.
- S. Y. Chou, W. H. Chung, L. W. Chen, Y. M. Dai, W. Y. Lin, J. H. Lin and C. C. Chen, RSC Adv., 2016, 6, 82743–82758 RSC.
- Y. R. Jiang, H. P. Lin, W. H. Chung, Y. M. Dai, W. Y. Lin and C. C. Chen, J. Hazard. Mater., 2015, 283, 787–805 CrossRef CAS PubMed.
- K. L. Li, W. W. Lee, C. S. Lu, Y. M. Dai, S. Y. Chou, H. L. Chen, H. P. Lin and C. C. Chen, J. Taiwan Inst. Chem. Eng., 2014, 45, 2688–2697 CrossRef CAS.
- H. L. Chen, W. W. Lee, W. H. Chung, H. P. Lin, Y. J. Chen, Y. R. Jiang, W. Y. Lin and C. C. Chen, J. Taiwan Inst. Chem. Eng., 2014, 45, 1892–1909 CrossRef CAS.
- H. P. Lin, W. W. Lee, S. T. Huang, L. W. Chen, T. W. Yeh, J. Y. Fu and C. C. Chen, J. Mol. Catal. A: Chem., 2016, 417, 168–183 CrossRef CAS.
- Y. R. Jiang, S. Y. Chou, J. L. Chang, S. T. Huang, H. P. Lin and C. C. Chen, RSC Adv., 2015, 5, 30851–30860 RSC.
- S. T. Huang, Y. R. Jiang, S. Y. Chou, Y. M. Dai and C. C. Chen, J. Mol. Catal. A: Chem., 2014, 391, 105–120 CrossRef CAS.
- Y. Gu, Z. D. Xu, L. Guo and Y. Q. Wan, CrystEngComm, 2014, 16, 10997–11006 RSC.
- Y. Q. Wan, X. F. Wang, Y. Gu, L. Guo and Z. D. Xu, Appl. Surf. Sci., 2016, 366, 59–66 CrossRef CAS.
- Y. J. Li, Q. Wang, B. C. Liu and J. Zhang, Appl. Surf. Sci., 2015, 349, 957–969 CrossRef CAS.
- X. Y. Xiong, L. Y. Ding, Q. Q. Wang, Y. X. Li, Q. Q. Jiang and J. C. Hu, Appl. Catal., B, 2016, 188, 283–291 CrossRef CAS.
- Z. S. Liu, H. S. Ran, J. N. Niu, P. Z. Feng and Y. B. Zhu, J. Colloid Interface Sci., 2014, 431, 187–193 CrossRef CAS PubMed.
- F. T. Li, Q. Wang, J. R. Ran, Y. J. Hao, X. J. Wang, D. S. Zhao and S. Z. Qiao, Nanoscale, 2015, 7, 1116–1126 RSC.
- R. D. Hancock, I. Cukrowski, J. Baloyi and J. Mashishi, J. Chem. Soc., Dalton Trans., 1993, 19, 2895–2899 RSC.
- L. Zhang, Y. Zhang, Z. M. Hao and F. Luo, Z. Anorg. Allg. Chem., 2010, 636, 1991–1997 CrossRef CAS.
- R. E. Bachman, K. H. Whitmire, J. H. Thurston, A. Gulea, O. Stavila and V. Stavila, Inorg. Chim. Acta, 2003, 346, 250–255 CrossRef.
- M. H. Chisholm, A. M. Macintosh, J. C. Huffman, D. D. Wu, E. R. Davidson, R. J. H. Clark and S. Firth, Inorg. Chem., 2000, 39, 3544–3550 CrossRef CAS PubMed.
- J. Cao, B. D. Luo, H. L. Lin, B. Y. Xu and S. F. Chen, Appl. Catal., B, 2012, 111, 288–296 CrossRef.
- Z. H. Ai, W. K. Ho, S. C. Lee and L. Z. Zhang, Environ. Sci. Technol., 2009, 43, 4143–4150 CrossRef CAS PubMed.
- X. W. Liu, H. Q. Cao and J. F. Yin, Nano Res., 2011, 4, 470–482 CrossRef CAS.
- J. Xu, W. Meng, Y. Zhang, L. Li and C. S. Guo, Appl. Catal., B, 2011, 107, 355–362 CrossRef CAS.
- D. H. E. K. S. W. Sing, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef.
- J. S. Valente, F. Tzompantzi, J. Prince, J. G. H. Cortez and R. Gomez, Appl. Catal., B, 2009, 90, 330–338 CrossRef CAS.
- H. Adelkhani, M. Ghaemi and M. Ruzbehani, Int. J. Electrochem. Sci., 2011, 6, 123–135 CAS.
- J. Di, J. X. Xia, Y. P. Ge, H. P. Li, H. Y. Ji, H. Xu, Q. Zhang, H. M. Li and M. N. Li, Appl. Catal., B, 2015, 168, 51–61 CrossRef.
- J. Xu, W. Meng, Y. Zhang, L. Li and C. S. Guo, Appl. Catal., B, 2011, 107, 355–362 CrossRef CAS.
- K. Nagaveni, M. S. Hegde and G. Madras, J. Phys. Chem. B, 2004, 108, 20204–20212 CrossRef CAS.
- H. M. Fan, T. F. Jiang, H. Y. Li, D. J. Wang, L. L. Wang, J. L. Zhai, D. Q. He, P. Wang and T. F. Xie, J. Phys. Chem. C, 2012, 116, 2425–2430 CAS.
- L. Z. Zhang and J. C. Yu, Chem. Commun., 2003, 16, 2078–2079 RSC.
- J. Di, J. X. Xia, Y. P. Ge, H. P. Li, H. Y. Ji, H. Xu, Q. Zhang, H. M. Li and M. N. Li, Appl. Catal., B, 2015, 168, 51–61 CrossRef.
- J. Li, L. Z. Zhang, Y. J. Li and Y. Yu, Nanoscale, 2014, 6, 167–171 RSC.
- S. K. Sana, U. Vladimir, F. Sveta, P. Inna and S. Yoel, Appl. Catal., B, 2012, 117, 148–155 Search PubMed.
- H. P. Lin, C. C. Chen, W. W. Lee, Y. Y. Lai, J. Y. Chen, Y. Q. Chen and J. Y. Fu, RSC Adv., 2016, 6, 2323–2336 RSC.
- C. T. Yang, W. W. Lee, H. P. Lin, Y. M. Dai, H. T. Chia and C. C. Chen, RSC Adv., 2016, 6, 40664–40675 RSC.
- S. Y. Chou, C. C. Chen, Y. M. Dai, J. H. Lin and W. W. Lee, RSC Adv., 2016, 6, 33478–33491 RSC.
- J. J. Guo, S. X. Ouyang, P. Li, Y. J. Zhang, T. Kako and J. H. Ye, Appl. Catal., B, 2013, 134, 286–292 CrossRef.
- R. D. Hancock, I. Cukrowski, J. Baloyi and J. Mashishi, J. Chem. Soc., Dalton Trans., 1993, 19, 2895–2899 RSC.
- W. T. Miller, J. Am. Chem. Soc., 1940, 62, 2707–2709 CrossRef CAS.
- A. Salma, S. Thoroe-Boveleth, T. C. Schmidt and J. Tuerk, J. Hazard. Mater., 2016, 313, 49–59 CrossRef CAS PubMed.
- J. L. Vazquez, M. Berlanga, S. Merino, O. Domènech, M. Vinas, M. T. Montero and J. Hernandez-Borrell, Photochem. Photobiol., 2001, 73, 14–19 CrossRef CAS PubMed.
- M. Das, D. Mishra, P. Dhak, S. Gupta, T. K. Maity, A. Basak and P. Pramanik, Small, 2009, 24, 2883–2893 CrossRef PubMed.
- P. Patra, S. Mitra, N. Debnath, P. Pramanik and A. Goswami, Bull. Mater. Sci., 2014, 37, 199–206 CrossRef CAS.
- L. Pan, J. J. Zou, X. W. Zhang and L. Wang, J. Am. Chem. Soc., 2011, 133, 10000–10002 CrossRef CAS PubMed.
- A. Couzis and E. Gulari, Langmuir, 1993, 9, 3414–3421 CrossRef CAS.
- J. B. Priebe, M. Karnahl, H. Junge, M. Beller, D. Hollmann and A. Bruckner, Angew. Chem., Int. Ed., 2013, 52, 11420–11424 CrossRef CAS PubMed.
- Y. X. Weng, L. Li, Y. Liu, L. Wang and G. Z. Yang, J. Phys. Chem. B, 2003, 107, 4356–4363 CrossRef CAS.
- S. W. Boettcher, M. H. Bartl, J. G. Hu and G. D. Stucky, J. Am. Chem. Soc., 2005, 127, 9721–9730 CrossRef CAS PubMed.
- B. S. Manhas and A. K. Trikha, J. Indian Chem. Soc., 1982, 59, 315–319 CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b