
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled nanoparticles based on a carboxymethylcellulose–ursolic acid conjugate for anticancer combination therapy
Yan-xue Liua,
Ke-feng Liua,
Chun-xiao Lia,
Lu-ying Wanga,
Jing Liua,
Jing Hea,
Jiandu Lei *ab and
Xingyong Liu*b
*ab and
Xingyong Liu*b
aBeijing Key Laboratory of Lignocellulosic Chemistry, Beijing Forestry University, Beijing 100083, PR China. E-mail: ljd2012@bjfu.edu.cn; Fax: +86-10-62337251; Tel: +86-10-62337251
bCollege of Chemistry and Environmental Engineering, Sichuan University of Science & Engineering, Zigong 643000, PR China
First published on 21st July 2017
Abstract
A new self-assembled nanoparticle platform based on a carboxymethylcellulose (CMC)–ursolic acid (UA) conjugate is presented for the first time. The CMC–UA conjugate was synthesized by introducing the hydrophobic drug UA into the hydrophilic polymer molecule CMC, and then another anticancer drug, hydroxycamptothecin (HCPT), was encapsulated into the self-assembled nanoparticles (CMC–UA/HCPT NPs) of the conjugate formed by the nanoprecipitation method. The obtained nanoparticles possessed appropriate size (∼40 nm), high encapsulation efficiency (∼17.53 wt% HCPT) and drug-loading efficiency (∼29.62 wt% UA). Cell experiments indicated that CMC–UA/HCPT NPs exhibited higher cytotoxicity than free UA and free HCPT, owing to the longer blood retention time than free drug (7.3-fold UA, 2.5-fold HCPT) and effective cellular uptake. Anti-tumor capacity, mice survival rate and tumor growth inhibition of UA and HCPT were also significant. Moreover, the evaluation of the side effects clearly and elaborately certified that NPs could reduce the risk of hypersensitivity reactions substantially. Therefore, CMC–UA/HCPT NPs is a further prospective anticancer drug delivery system.
1. Introduction
Cancer is a group of diseases involving abnormal cell growth with the potential to invade or spread to other parts of the body. Growing cancer incidence rate and increasing mortality trends in recently published global cancer statistics worldwide implied that more efforts should be made and challenges overcome to treat cancer.1–3 In all strategies for fighting cancer, chemotherapy plays a crucial role in clinical treatment.As adjuvant therapy, chemotherapeutic agents have been used independently or integrated with other treatment.4 Ursolic acid (UA), a pentacyclic triterpenoid compound that is isolated from various medicinal plants, seems to show a promising inhibitory effect in different tumor cell lines. UA has recently attracted great attention for its potential as a chemopreventive and chemotherapeutic agent.5,6 UA as a single chemotherapy agent kills the rapidly growing and dividing cancerous cells, but it also destroys the growing normal cells with resulting adverse reactions, such as congestive heart failure, in clinical treatment.7 In addition, UA is also limited in the treatment of cancer because of its insolubility, relatively short half-life, and low bioavailability.8,9 Therefore, a convenient and safe delivery system urgently needs to be established to maximize the therapeutic efficacy at tumor sites while minimizing the side effects.10
Application of nanotechnologies in oncology has greatly impacted cancer diagnosis and therapy in the past decade.11–14 Nanoparticle drugs have been exploited to circumvent the low bioavailability, rapid degradation and inactivation of drugs, and can rapidly enhance the permeability and retention (EPR) effect, and their ability to reduce the off-target toxicities of chemotherapeutics in tumors has been explored, leading to safe delivery systems and stable efficacy.15–18 Recently, the use of UA-loaded poly-D,L-lactic-co-glycolic acid nanoparticles (UA-NPs) as drug carrier was evaluated for differential tumor targeting effects in B16F10 melanoma cells. Smooth spherical nanoparticles of small size with relatively narrow size distribution (∼154 nm) were produced through a single emulsification technique, and about 4% drug-loading efficiency and high encapsulation efficiency (∼40%) were obtained. The technetium-99m radiolabelled UA-NPs exhibited slower blood clearance and comparatively high uptake in the tumor region.19 In addition, a nanoparticle-based drug carrier composed of chitosan, UA and folate (FA–CS–UA-NPs) was reported for MCF-7 cells. An average particle size of ∼160 nm and encapsulation efficiency of 50% were obtained. FA–CS–UA-NPs could effectively diminish off-target effects and increase local drug concentrations of UA, and were easily internalized by cancer cells through a folate receptor-mediated endocytic pathway. In vivo experiments revealed that nanoparticles could significantly decrease the breast cancer burden in the MCF-7 xenograft mouse model.20,21 Although delivery systems of UA-loaded nanoparticles have been reported,8,19–21 in great part methods and strategy need to be established for synthesis of UA-loaded nanoparticles in order to improve the drug loading and efficiency of drug release, and especially to reduce particle size in order to improve carrier tissue penetration.
Carboxymethylcellulose (CMC) is a polysaccharide that is plentifully available worldwide. CMC, a highly biocompatible and biodegradable material, is ideal for drug delivery systems as it possesses many favorable pharmaceutical properties such as non-toxicity, high stability and drug-binding capacity, and long half-life in vivo. Ernsting et al. used CMC as a polymer backbone for the delivery of docetaxel. In vitro, the cytotoxicity of the optimal conjugate formulation was improved by ∼2–40-fold compared with free docetaxel.22 Dai et al. designed a delivery system for the insoluble anticancer drug betulinic acid (BA) using CMC nanoparticles, as well as developing a cell-specific targeting F–PEG–CMC–BA nanocarrier to deliver another anticancer drug (hydroxycamptothecin). The obtained F–PEG–CMC–BA/HCPT nanoparticles (FPCB/HCPT NPs) had 6.4-fold and 6.0-fold the blood circulation half-life of free BA and free HCPT, respectively.23
In the present study, we present a new drug delivery nanoparticle system for an insoluble anticancer drug using the CMC–UA conjugate. The CMC–UA conjugate was first synthesized by introducing the hydrophobic drug UA into the hydrophilic polymer molecule CMC, and then another anticancer drug, hydroxycamptothecin, was encapsulated in the self-assembled nanoparticles of the conjugate by a nanoprecipitation method. In addition, the in vitro and in vivo antitumor activities of CMC–UA/HCPT NPs nanoparticles were investigated.
2. Experimental
2.1. Materials
Carboxymethylcellulose (CMC) sodium salt 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 P with an average molecular weight of 270 kDa and degree of substitution (DS) of 0.82 was purchased from CP Kelco (Atlanta, GA, USA), and is an FDA and EU food-grade material. Ursolic acid (UA) was purchased from Chengdu Preferred Biotechnology Co., Ltd (Chengdu, Sichuan, PR China). Acetic anhydride, dimethylsulfoxide (DMSO), sulfuric acid, glacial acetic acid, diethyl ether, methanol, potassium bromide (KBr), 10-hydroxycamptothecin, acetonitrile, pyridine (Py), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide HCl (EDC·HCl), and 4-dimethylaminopyridine (DMAP) were purchased from Sigma-Aldrich (Oakville, ON, CA). All other reagents were also purchased from Sigma-Aldrich.
000 P with an average molecular weight of 270 kDa and degree of substitution (DS) of 0.82 was purchased from CP Kelco (Atlanta, GA, USA), and is an FDA and EU food-grade material. Ursolic acid (UA) was purchased from Chengdu Preferred Biotechnology Co., Ltd (Chengdu, Sichuan, PR China). Acetic anhydride, dimethylsulfoxide (DMSO), sulfuric acid, glacial acetic acid, diethyl ether, methanol, potassium bromide (KBr), 10-hydroxycamptothecin, acetonitrile, pyridine (Py), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide HCl (EDC·HCl), and 4-dimethylaminopyridine (DMAP) were purchased from Sigma-Aldrich (Oakville, ON, CA). All other reagents were also purchased from Sigma-Aldrich.
2.2. Cell culture
Penicillin and streptomycin, Gibco Dulbecco's phosphate-buffered saline (DPBS), and Gibco Dulbecco's modified Eagle's medium (DMEM) were all bought from Invitrogen. Fetal bovine serum (FBS) was from HyClone. Cell-Counting Kit-8 (CCK-8) was supplied by Dojindo Laboratories. The mouse breast cancer cell line 4T1, obtained from the 410.4 tumor strain without mutagenesis screen 6-thioguanlinol resistance cell line, was purchased from the Peking University Health Science Center (Beijing, PR China) and maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum at 37 °C and 5% CO2 for a maximum of 10 passages.2.3. Animals and ethics
Female BALB/c mice, 4–6 weeks of age and weight 18–20 g, were purchased from Beijing Hfk Bioscience Co., Ltd. All the animal experiments were consistent with the guidelines set by the National Institutes of Health (NIH Publication no. 85-23, revised 1985) and were approved by the Experimental Animal Ethics Committee, Beijing.2.4. Preparation of the acetylated CMC
Carboxymethylcellulose (CMC) sodium salt was first converted to a water-insoluble form by acetylation reaction in organic solvent. It was suspended in 20% sulfuric acid with vigorous stirring at room temperature for 2 h. The CMC–COOH precipitate was obtained by centrifuging at 10000 rpm for 30 min, and washed with water using ultrasonically assisted homogeneous stirring until the water tested neutral, and then washed with glacial acetic acid three times afterwards. CMC–COOH was resuspended in glacial acetic acid (0.2 g mL−1) at −5 °C for 30 min, and acetic anhydride (0.35 g mL−1) and sulfuric acid (8.5 g mL−1) were added to the chilled CMC–COOH at 50 °C and vigorously stirred for 3 h till the solution clarified. The precipitate was obtained from the mixed solution by adding deionized water, and then washed till neutral by centrifuging at 10000 rpm for 15 min. The acetylated carboxymethylcellulose (CMC-Ac) was frozen at −20 °C and dried by vacuum freeze dryer at −56 °C for 48 h.
2.5. Synthesis of CMC–UA
The preparation of acetylated carboxymethylcellulose (CMC-Ac, Mw = 515.68 g mol−1, 100 mg) was dissolved in dry pyridine (3 mL) in a 25 mL glass vial at 35 °C for 2 h. EDC·HCl (230 mg, 1.20 mmol) was dissolved in 1 mL dry pyridine, DMAP (10 mg, 0.08 mmol) was dissolved in dry pyridine (0.1 mL), and UA (200 mg, 0.5 mmol) was dissolved in 1 mL dry pyridine. Dissolved EDC·HCl and DMAP reagents were added to the CMC-Ac solution at 35 °C for 1.5 h, followed by addition of the UA. The solution was stirred at 35 °C for 48 h with protection from light, and passed through nitrogen to exclude water vapor. The solvent was removed by rotary evaporation (45 °C, 5 mbar), and the compound was precipitated in diethyl ether (1:3, v/v), and centrifuged at 4000 rpm for 30 min. The supernatant was removed, and the obtained precipitate was washed three times with diethyl ether (3 × 10 mL) by centrifuging at 4000 rpm for 10 min. The resulting precipitate was dissolved in deionized water and then dialyzed in PBS solution using a dialysis membrane with MWCO 3.5 kDa for 12 h with three changes of liquid. The liquid in the dialysis membrane was transferred to a centrifuge tube, and then frozen and dried by a vacuum freeze dryer at −56 °C for 48 h.
2.6. 1H-NMR measurements and FTIR measurements
A structural analysis of the CMC–UA derivative was carried out by 1H-NMR. The 1H-NMR experiment was carried out using a Bruker Avance 300 spectrometer operating at a frequency of 400.12 MHz equipped with a 5 mm probe. Dry CMC (5 mg) was dissolved in D2O (Sigma), and UA and CMC–UA derivative were dissolved in CDCl3 (Sigma) at 10 mg mL−1 concentration and the NMR spectra were recorded at 80 °C. The spectra were accumulated with a 301 pulse, an acquisition time of 3.98 s, a recycle time (relaxation delay) of 8 s, 128 scans and a sweep width of 8278.15 Hz, resulting in 32 K complex data points.To characterize the CMC, UA and the as-synthesized CMC–UA derivatives, FTIR spectroscopy was performed using a Bruker Tensor 27 series FTIR spectrometer in the region of 400–4000 cm−1 with 2 cm−1 resolution and 16 scans. The test samples were prepared by mixing 0.2 g of CMC, UA or CMC–UA powder together with 1 g of KBr to make a pellet.
2.7. Preparation of CMC–UA/HCPT nanoparticles
The preparation of CMC–UA/HCPT nanoparticles was adapted from the self-assembly method24,25 described previously. Simply, CMC–UA (5 mg) and HCPT (2 mg) were dissolved in 1 mL DMSO, and added dropwise to a vortexing solution of 5 mL PBS in a 15 mL round-bottomed flask for about 15 min. The resulting CMC–UA/HCPT NPs solutions were transferred to an 8000 MWCO cartridge, and dialyzed against 100 mL PBS solution for 3 h with three exchanges of dialysate. The particles were filtered through a 25 mm Millipore PVDF filter (0.2 μm), and transferred to a Vivaspin centrifugal filter unit (10000 MWCO), and then spun at 4000 rpm to concentrate the particles. The size of the particles was determined by dynamic light scattering using a particle analyzer (Zetasizer Nano-ZS, Malvern Instruments Ltd, Malvern, UK). CMC–UA NPs was prepared similarly to CMC–UA/HCPT NPs.
2.8. TEM analysis
CMC–UA/HCPT NPs and CMC–UA/HCPT NPs were diluted 100× in deionized water, and 2 μL aliquots of solution were pipetted onto the surface of Formvar-coated copper TEM grids (TedPella, Redding, CA) and allowed to air-dry. Analysis was performed on a JEM-100CXa transmission electron microscope at an acceleration voltage of 100 kV.2.9. Hemolysis assay
The hemolytic activity of the conjugate solutions was investigated as reported earlier.26,27 Briefly, fresh blood samples were collected through cardiac puncture from healthy rats. EDTA-Na2 was immediately mixed into about 10 mL of blood to prevent coagulation. Precipitated red blood cells (RBCs) were collected by means of centrifuging at 1500 rpm at ∼0–4 °C for 15 min. The erythrocytes precipitation was washed till clear using ice-cold DPBS, and were diluted in ice-cold DPBS at a final concentration of 5 × 108 cells mL−1. 1 mL CMC–UA NPs solution and 1 mL CMC–UA/HCPT NPs solution with a concentration range from 1 mg mL−1 to 0.1 mg mL−1 were separately mixed with 1 mL erythrocyte suspension, then incubated for 1 h at 37 °C under constant shaking. Centrifugation took place at 1500 rpm at 4 °C for 15 min; the supernatant was analyzed for hemoglobin release at 541 nm using an Infinite M200 microplate spectrophotometer (Tecan, Switzerland). DPBS and 1% Triton X-100 in DPBS were used as negative control (0% lysis) and positive control (100% lysis), respectively. Hemoglobin release was calculated as:| (ODsample − ODnegative control)/(ODpositive control − ODnegative control) × 100%. |
Hemolysis was determined though three independent experiments.
2.10. Determination of drug loading and in vitro drug release
CMC–UA/HCPT NPs samples were assayed for UA and HCPT content by UV assay. Briefly, the pure drugs UA and HCPT were dissolved in methanol and acetonitrile at five different concentrations in order to obtain standard curves. CMC–UA/HCPT NPs was hydrolysed in dilute hydrochloric acid (10%, v/v, HCl) to obtain mixed precipitates of the free UA and HCPT, and the precipitates were sedimented by centrifuging at 4000 rpm. The obtained precipitates were dissolved in 88% (v/v) methanol–water solution to measure the content of UA and HCPT in CMC–UA/HCPT NPs by UV absorbance at 210 nm and 254 nm.The release of UA and HCPT from CMC–UA/HCPT NPs was analyzed by dialysis. First, CMC–UA/HCPT NPs PBS solution (0.5 mg mL−1, pH = 7.4) was dialysed (dialysis bag: MWCO 3 kDa) by immersion in PBS buffer (pH = 7.4, 37 °C) with gentle agitation. PBS medium (100 μL) was withdrawn at timed intervals and the UA and HCPT concentration in the CMC–UA/HCPT NPs PBS solution was determined by HPLC [UA: 210 nm, 88:12 mixtures (v/v) of methanol–water as mobile phase, flow rate of 0.6 mL min−1; HCPT: 254 nm, 30:70 mixture (v/v) of acetonitrile–water as mobile phase, flow rate of 0.6 mL min−1] using a C18 reverse phase column. Each stability profile represents the average of three independent runs with the same sampling schedules. Drug-loading efficiency (DLE) of UA and encapsulation efficiency (EE) of HCPT were calculated as follows:
| DLE (%) = (weight of UA in nanoparticles/weight of the nanoparticles) × 100% |
| EE (%) = (weight of HCPT in nanoparticles/initial amount of drug) × 100% |
Simultaneously, esterase (30 units) was added into the dialysis bag, the previous procedure repeated, and the UA and HCPT release in the presence of esterase was studied.
2.11. Toxicity analysis in vitro
CCK-8 assay was used for cell viability evaluation of different samples. Briefly, 4T1 breast cancer cells were seeded at a density of 5 × 103 cells per well in 200 μL culture medium in a 96-well plate (Corning, USA) and incubated for 24 h. Then, the cells were treated with UA, HCPT, CMC–UA NPs and CMC–UA/HCPT NPs at 37 °C in a humidified incubator with 5% CO2 for 72 h, the samples of UA having been dissolved in DMSO (Merck, Darmstadt, Germany) and diluted into tissue culture medium before assay. The UA dose ranged from 0.1 to 100 μg mL−1, and CMC–UA NPs and CMC–UA/HCPT NPs doses were the same as UA. The HCPT dose was equal to the HCPT content in CMC–UA/HCPT NPs. CCK-8 solution (20 μL) was added to each well of the plate and incubated for 2 h at 37 °C. The absorbance at 450 nm was measured using an Infinite M200 microplate spectrophotometer. Percentage viability was normalized to cell viability in the absence of the samples. IC50 was calculated as the polymer concentration that inhibited the growth of 50% of cells relative to non-treated cells according to Unger et al.28 IC50 was calculated using the Boltzmann sigmoidal function from Origin 8.6 (OriginLab, Northampton, USA). Data are representative of three independent experiments. We evaluated the synergistic effects between UA and HCPT in the CMC–UA/HCPT NPs by using the combination index: combination index (CI) = UA1/UA0 + HCPT1/HCPT0, where UA1 and HCPT1 represent the IC50 values of UA and HCPT in CMC–UA/HCPT NPs, and UA0 and HCPT0 represent the IC50 values of UA and HCPT. CI < 1 denotes drug synergism, while CI > 1 shows an antagonistic effect.2.12. Cellular uptake study
Cellular uptake and distribution of HCPT from the developed nanoparticles were observed by confocal laser scanning microscopy (CLSM, TCS SP5, Leica). 4T1 cells were trypsinized and seeded onto culture slides (BD Falcon, Bedford, MA) at a density of 1.0 × 105 cells per mL. One milliliter of cell suspension was added to 4 cm2 confocal Petri dishes at 37 °C overnight. After 24 h of incubation, free HCPT (IC50) and CMC–UA/HCPT NPs (IC50) were added along the inner wall, and incubated at 37 °C for 4 h. The drug solution was removed, and the cells were washed three times, and then fixed with 4% formaldehyde solution for 15 min before the formaldehyde was aspirated. One milliliter of 0.5 μg mL−1 DAPI solution was added. After 5 min, the DAPI solution was aspirated, and the cells were rinsed three times with PBS. Nanoparticles were detected by the absorbance of HCPT at 488 nm.2.13. Pharmacokinetic (PK) study
Twenty-four tumor-free healthy BALB/c female mice were randomly divided into four groups. Groups 1 and 2 were treated with free UA and free HCPT, respectively, and groups 3 and 4 with CMC–UA NPs and CMC–UA/HCPT NPs, respectively, injected via the tail vein. After intravenous administration, blood samples were collected at 0.125, 0.25, 0.5, 1, 3, 5, 10, 24, 48, and 72 h from the orbital plexus and centrifuged immediately at 3000 rpm for 10 min at 4 °C. Plasma (100 μL) was mixed with 50 μL of 0.1 N NaOH for 15 min in a water bath at 37 °C to determine the level of total UA or HCPT in each plasma sample. HCl (0.1 N, 50 mL) was added, followed by 100 μL methanol for 2 min, sonication for 5 min and centrifugation at 4000 rpm for 5 min. The clear supernatant was dissolved in 100 μL of methanol for HPLC (C18, 5 mm, 4.6 × 250 mm) analysis. A gradient of 60% acetonitrile in 0.05% trifluoroacetate was used at a flow rate of 1 mL min−1. Blood UA and HCPT levels, with the unit of percentage of injected dose per gram (% ID per g), were plotted against time after injection.2.14. Subcutaneous tumor efficacy models
Subcutaneous tumor xenograft models were established in the right auxiliary flank region of BALB/c female mice (6–7 weeks) by injecting 5 × 105 4T1 cells in 200 μL DMEM medium per mouse. Treatments were initiated when tumor volume reached 50–100 mm3. Mice were randomly divided into six groups (n = 6) and intravenously injected with PBS (control), free UA (10 mg kg−1), free HCPT (10 mg kg−1), or CMC–UA NPs or CMC–UA/HCPT NPs equal to the free UA concentration, on days 0, 2, 4, 6, and 8. In the cure phase, mice were monitored for tumor sizes and body weights at daily intervals. The relative tumor volume (RTV) was calculated at each measurement time point. For efficacy studies, the percentage of tumor growth inhibition (% TGI) was calculated using the following formula: % TGI = [(C − T)/C] × 100%, where C is the mean tumor volume of the control group and T is the mean tumor volume of the treatment group.2.15. Detection of allergic reaction
Detection of allergic reaction is very important in order to protect against toxic side effects of chemotherapeutical drugs. Five groups of tumor-bearing mice (n = 6) were used in allergy testing studies of five samples (control, UA, HCPT, CMC–UA NPs and CMC–UA/HCPT NPs). The five samples were administered via tail intravenous injection every two days (UA: 10 mg kg−1; HCPT: 5 mg kg−1; CMC–UA NPs and CMC–UA/HCPT NPs: equal to the free UA concentration). After 10 days, blood from mice in the different groups was collected and centrifuged, and serum samples were analyzed by mouse IgE ELISA.2.16. Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA). All graphical data are reported as mean ± SD. Significance levels were set at *p < 0.05.3. Results and discussion
In cancer treatment, chemotherapy has many drawbacks such as limited efficacy, severe toxic side effects, and the tendency to induce drug resistance. To overcome such long-standing challenges, the combination of various therapeutic strategies to treat cancer is an important and promising strategy to improve therapeutic efficiency and overcome drug resistance.29 UA and HCPT mentioned in this paper could achieve a synergistic effect of two kinds of drugs in breast cancer treatment.Recently, the self-assembly of colloidal nanoparticles (NPs) has emerged as a powerful concept for devising novel nanomaterials, becoming a natural starting point for the bottom-up fabrication of devices in nanotechnology. Nanoparticles are explosively increasing in importance as vehicles for chemotherapy agents because of their ability to enhance drug delivery efficacy and reduce drug side effects.30,31
Therefore, a CMC drug-loading and self-assembly system was established to load two different anticancer drugs (UA and HCPT), prolonging the delivery half-life based on the self-assembly design of colloidal nanoparticles, and achieving a biocompatible and biodegradable drug-delivery system that is a simple and reliable approach for combination therapy for cancer. The designed method for CMC–UA/HCPT NPs self-assembly is shown in Fig. 1 and 2.
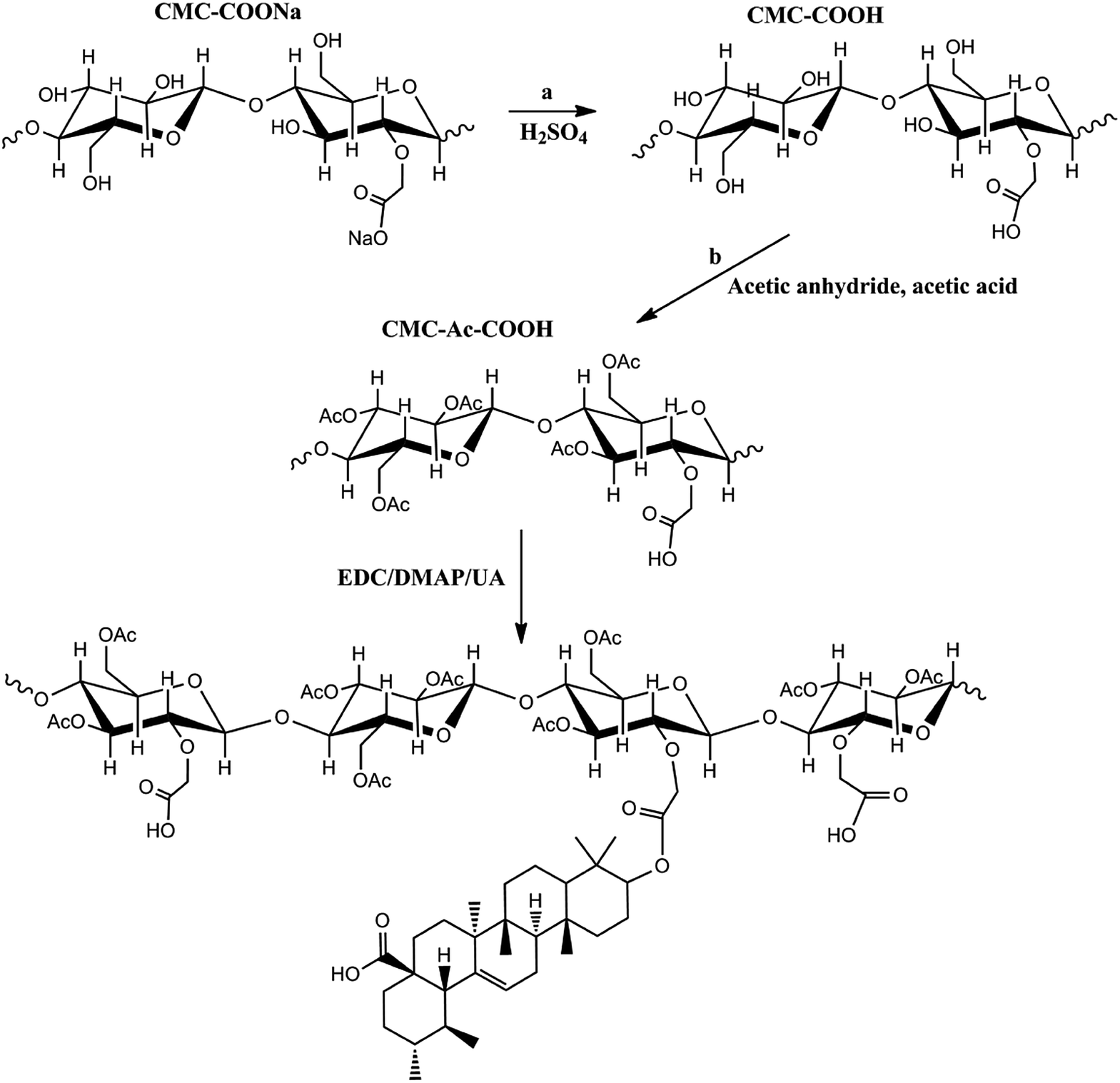 | ||
| Fig. 1 Scheme showing the synthesis of the carboxymethylcellulose–ursolic acid conjugate (CMC–UA). Note that the distribution of UA substitutions is random. | ||
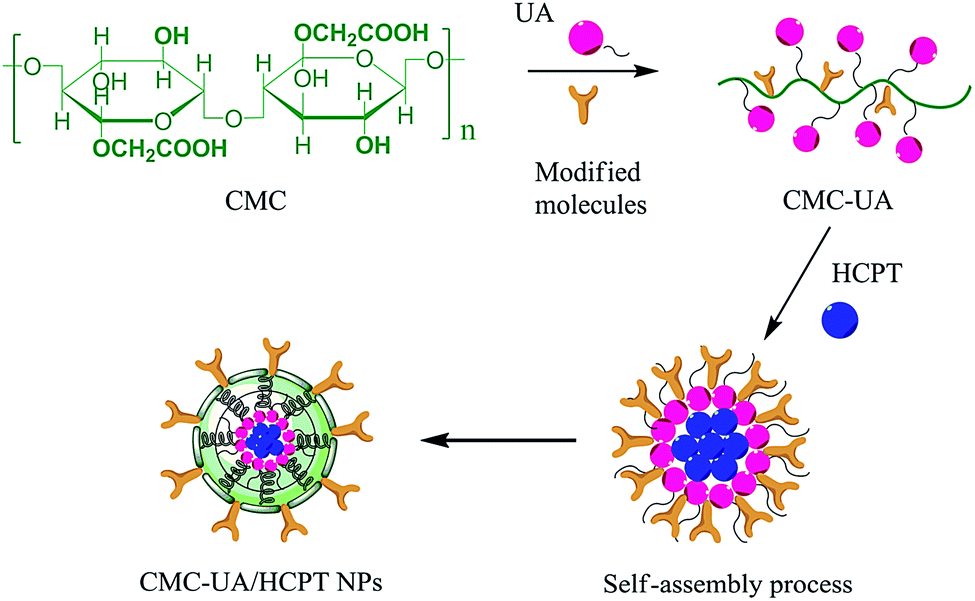 | ||
| Fig. 2 Schematic of the conjugate synthesis and CMC–UA/HCPT NPs preparation. | ||
3.1. Preparation of CMC–UA and drug loading
CMC–UA was synthesized successfully by catalytic esterification. The structures of UA, CMC and CMC–UA were characterized by 1H-NMR (Fig. 3). The partial structure of UA can be identified from peaks between 0.8 and 2.2 ppm (CDCl3), and peaks at 3.0–4.8 are characteristic of CMC (D2O). The 4-methylene proton peaks at 3.25 of UA moved to 4.20, and vinyl proton peaks at 5.28 of UA moved to 5.30, which confirmed the formation of ester bonds between CMC and UA. By 1H-NMR analysis, the peak assignments were identical in all polymer products, varying only in the integration of peaks assigned to UA.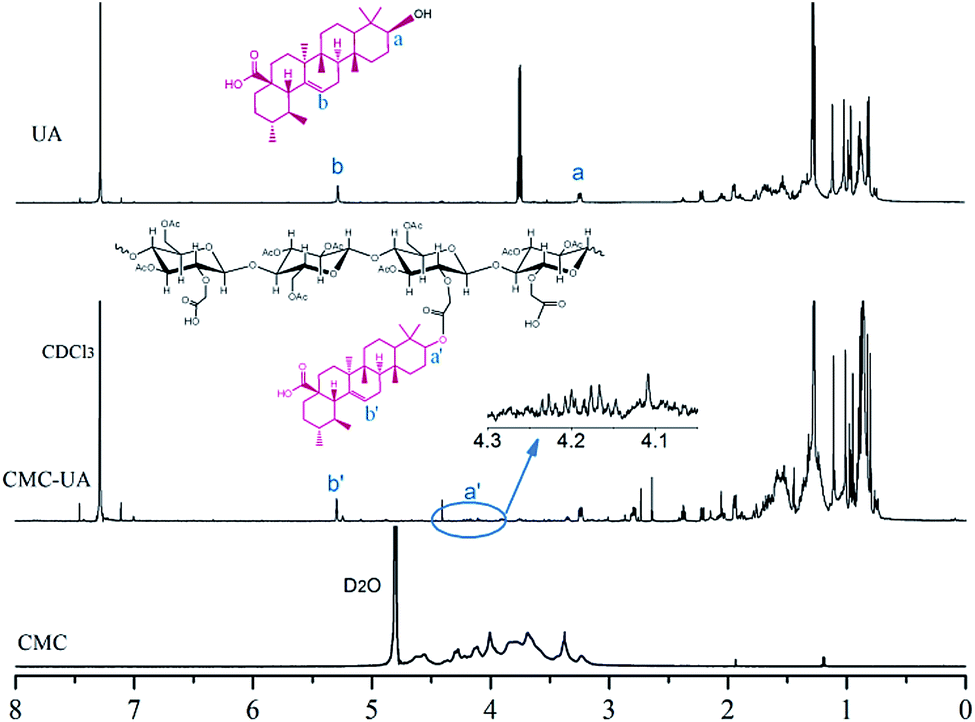 | ||
| Fig. 3 1H-NMR spectrum of CMC–UA in CDCl3. | ||
In the FTIR spectrum of CMC–UA a characteristic absorption peak is observed at 1600 cm−1 for carboxylate (–COO–) asymmetric stretching vibration.32–34 The bands at 1110 cm−1 and 1064 cm−1 are assigned to the ether bonds stretching vibration. Compared with the FTIR spectrum of CMC, CMC–UA (Fig. 4) has a more visible characteristic band at 1732 cm−1 corresponding to carbonyl stretching vibration in the ester group, which means ursolic acid is successfully attached to carboxymethylcellulose.
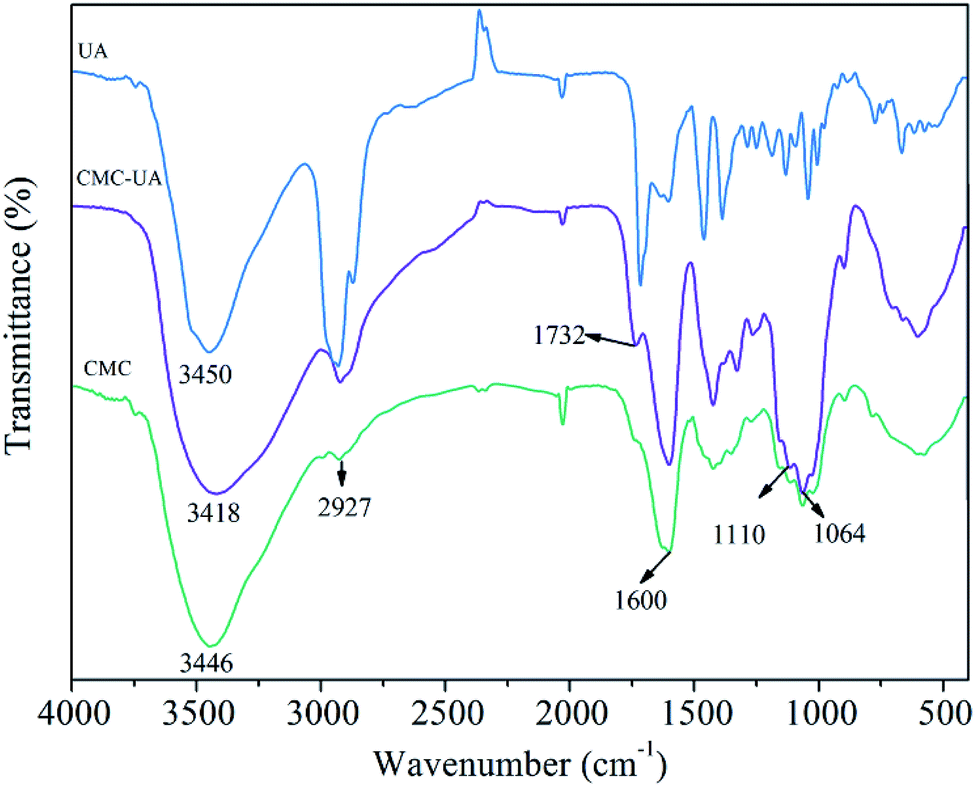 | ||
| Fig. 4 FTIR spectra of CMC, UA, CMC–UA. | ||
Polymer conjugates were prepared with a range of UA wt% feeds (10, 20, 30, 40, 50, 80 and 90%). As shown in Fig. 5a, the UA wt% in CMC–UA at first increased with increasing feed UA% concentrations, but then did not change much when feed UA% concentrations were more than 50%. For the preferred composition of 50 wt% UA feed, the CMC–UA contained 29.62 ± 2.49 wt% UA. The highest drug-loading rate reached was 32.11%. Conjugates prepared across the 10–90 wt% feed range of composition were tested for the size of particles formed: as shown in Fig. 5b, conjugates prepared with 15–60 wt% UA feed yielded particles ranging from 60 to 25 nm. A recent report indicated that the particle size of the drug-loaded nanoparticles was more conducive to transmission in the human body when it was below 100 nm, which not only improved drug-loading, but also increased the specific surface area for drug delivery.35 It was reported that drug delivery systems with a particle size between 30 and 200 nm would be suitable for intravenous drug delivery, and lead to preferred accumulation of the drug delivery systems at the tumor site by enhanced permeability and retention (EPR).31,36 Although many studies have reported that the cellulose nanoparticles delivery system could be accumulated at the solid tumor site in a passive targeting manner by an EPR effect,37–40 particles of 20–60 nm have not previously been reported.
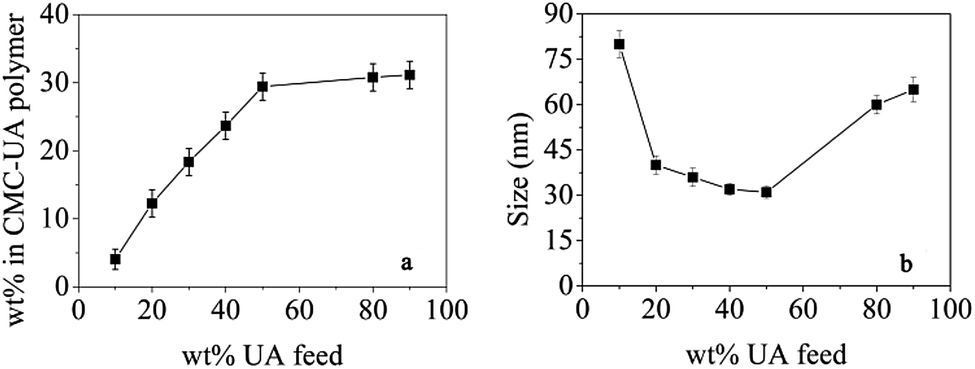 | ||
| Fig. 5 Optimization of CMC–UA/HCPT NPs drug (UA)-loading rate and particle size. (a) UA was fed into the reactions with CMC at different ratios. The mass fractions of UA in the resulting conjugates were calculated. (b) CMC–UA conjugates formed nanoparticles with a defined UA content. | ||
3.2. Self-assembled CMC–UA/HCPT nanoparticles and drug encapsulation
Camptothecin (CPT) is a pentacyclic indole alkaloid that was isolated from a native Chinese tree, Camptotheca accuminata, in 1966. 10-Hydroxycamptothecin (HCPT) has shown relatively low toxicity and displayed significant broad-spectrum antitumor activity against various types of cancers, such as breast cancer, lung cancer, hepatoma, leukemia, colorectal cancer and gastric carcinoma.41–43 However, HCPT is a hydrophobic drug and also sensitive to pH changes, both of which properties make it difficult to prepare the desired nanoparticles.44 In our study, HCPT as another model chemotherapy drug was encapsulated into the CMC–UA NPs by self-assembly for combination therapy (Fig. 6). | ||
| Fig. 6 Scanning TEM images of CMC–UA/HCPT NPs showing the particle size. | ||
Simply, CMC–UA and CMC–UA/HCPT were dissolved in DMSO (5–50 mg mL−1) for 10 min, and then slowly added into phosphate buffer solution (0.05–1 mg mL−1) at a stirring speed of 500 rpm. From Table 1, it can be observed that the smallest sizes of CMC–UA and CMC–UA/HCPT nanoparticles were approximately 32 nm and 40 nm, respectively. Such a small nanoparticle system suggested its potential for effective tumor targeting in vivo. Increase in particle size after being loaded with HCPT might be due to the insertion of the hydrophobic drug into the nanoparticles. The particle size distributions of CMC–UA (Fig. 7a) and CMC–UA/HCPT (Fig. 7b) nanoparticles were relatively concentrated, and better dispersion was obtained in the investigation. The variation in size of the nanoparticles was observed over an interval of 28 days (Fig. 7c). The nanoparticles were discovered to be surprisingly stable, with better stability for CMC–UA NPs and CMC–UA/HCPT NPs.
| Compound | DLE of UA (wt%) | DLE of HCPT (wt%) | Size (nm) | Zeta potential (mV) |
|---|---|---|---|---|
| CMC–UA NPs | 29.62 ± 1.19 | — | 32.17 ± 2.25 | 6.73 ± 0.51 |
| CMC–UA/HCPT NPs | 22.07 ± 1.21 | 17.53 ± 1.79 | 40.33 ± 3.28 | 8.14 ± 0.66 |
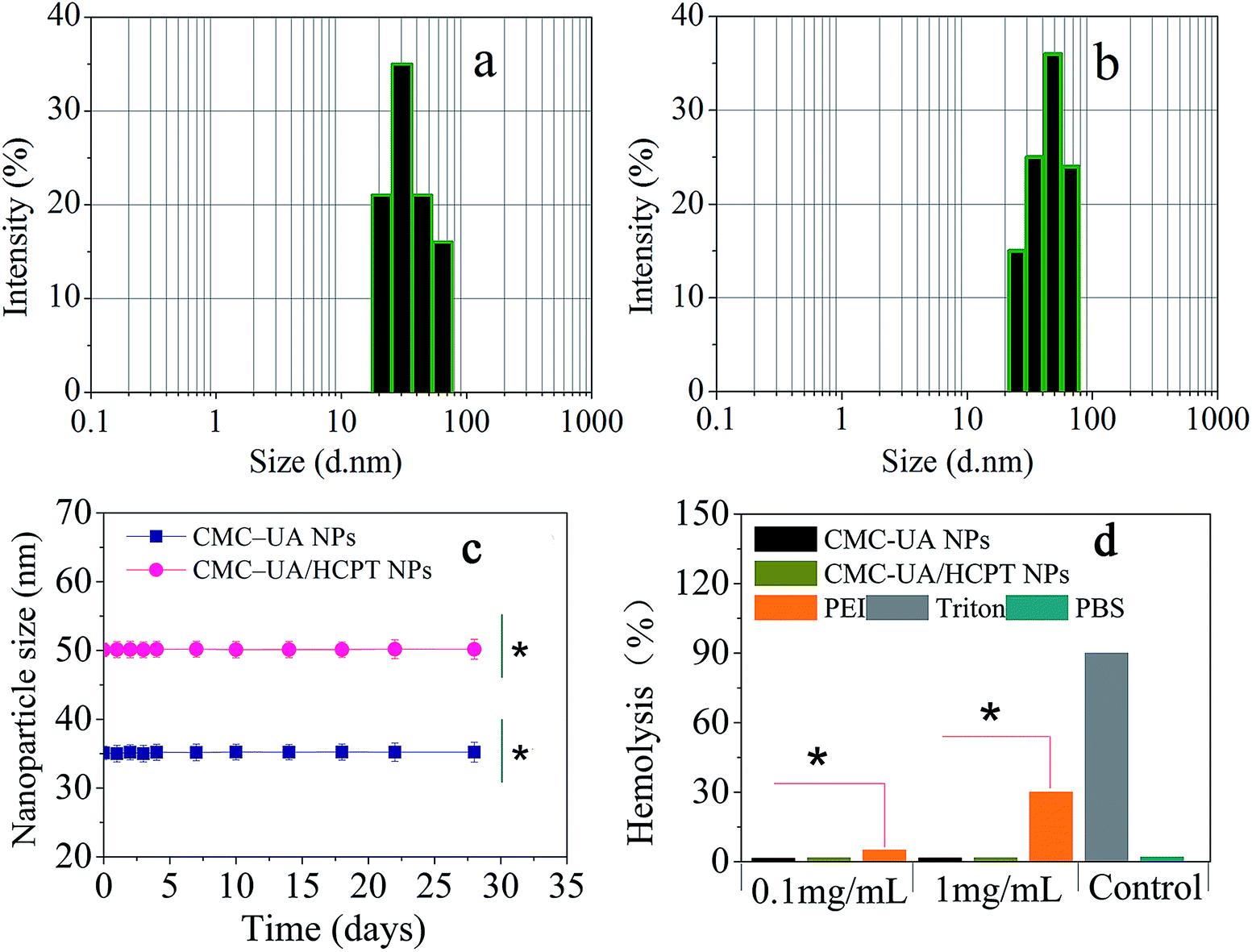 | ||
| Fig. 7 Characterization of CMC–UA/HCPT nanoparticles. Dynamic light scattering (DLS) of CMC–UA (a) and CMC–UA/HCPT (b) in PBS solution. (c) The particle size distribution of the nanoparticles during 28 days of storage at 4 °C (*p < 0.05). (d) In vitro hemolysis assay of CMC–UA NPs and CMC–UA/HCPT NPs compared with PEI25K and Triton X-100 measured at 541 nm. Values are reported as the mean ± SD for triplicate samples. | ||
The zeta potential is a key factor to be considered when evaluating the stability of a colloidal dispersion. Most charged functional groups are responsible for active nanoparticle interaction with cells. As positively charged particles have the greatest efficiency in cell-membrane penetration and cellular internalization, they form the primary platform as carriers for drug delivery. In Table 1 CMC–UA NPs and CMC–UA/HCPT NPs both have a positive surface charge (ζ = 6.73 mV and 8.14 mV). Interparticle interactions may be partly responsible for the ability of the nanoparticles to easily disperse, enabling CMC–UA NPs and CMC–UA/HCPT NPs to be easily resuspended after sedimentation.
3.3. Hemolysis study
In order to research the suitability of CMC–UA NPs and CMC–UA/HCPT NPs for drug delivery in the blood circulation and avoid allergic reactions due to injection, the effect of the nanoparticles on blood cells has to be considered and so was determined. Erythrocytes were incubated with two concentrations of nanoparticles (CMC–UA NPs and CMC–UA/HCPT NPs), 1 mg mL−1 and 0.1 mg mL−1, for 1 h at 37 °C. Hemolysis was determined by measuring the amount of hemoglobin released in the supernatant at 541 nm. Triton X-100 was chosen as a positive control, which could induce full hemoglobin release. As seen from Fig. 7d, the samples at concentrations of 1 mg mL−1 and 0.1 mg mL−1 showed hemoglobin release similar to blank values (<5%), obviously lower than with similar concentrations of PEI25K, a significant cationic polymer.3.4. In vitro drug release
Owing to the pH-dependent decomposition of the ester bond, we would expect pH-responsive UA and HCPT release behavior. The release kinetics of UA and HCPT from CMC–UA/HCPT NPs in solutions at varying pH values (5.0, 7.4) that simulate biological fluids were measured by HPLC analysis in vitro. As expected, CMC–UA/HCPT NPs was very slowly hydrolyzed and released UA at pH = 5.0 or 7.4 without the burst release phenomenon commonly found in drug-loaded nanoparticles (Fig. 8). However, CMC–UA/HCPT NPs was quickly hydrolyzed and released UA in the presence of esterase, which is abundant in the cytoplasm. The release of UA and that of HCPT were obviously pH-dependent, presenting a similar increment tendency with release time. The CMC–UA/HCPT NPs could therefore act as a prodrug for simultaneous release of UA and HCPT. The resulting release data are shown in Fig. 8a. Adding esterase promoted HCPT release due to the hydrolysis of CMC–UA/HCPT NPs (Fig. 8b).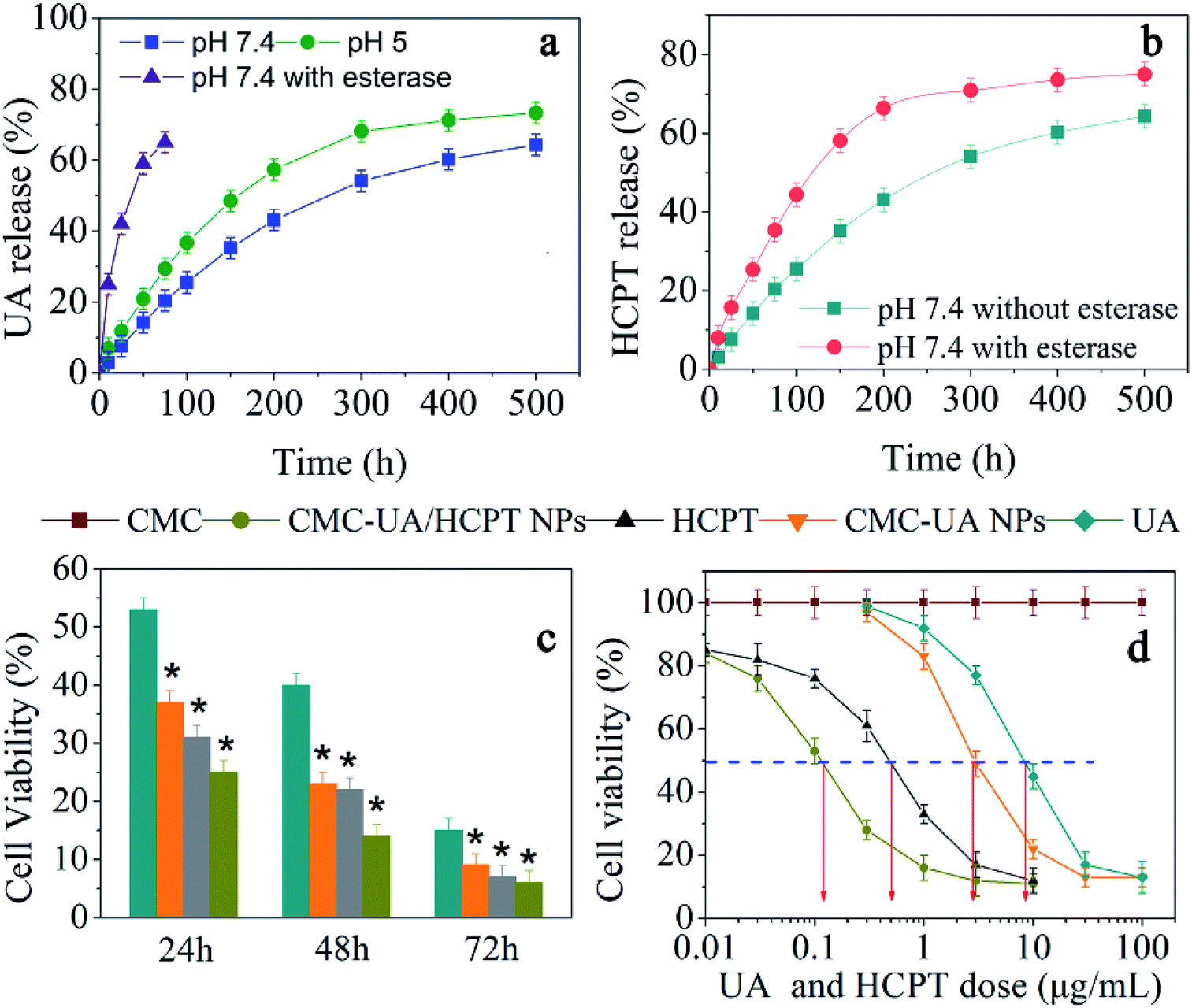 | ||
| Fig. 8 UA release kinetics (a) at different pH, and HCPT release kinetics with esterase and without esterase (b) in PBS at pH 7.4 and 37 °C from the CMC–UA/HCPT NPs. Error bars are based on at least triplicate measurements. (c) Cellular cytotoxicity of UA, HCPT, CMC–UA NPs and CMC–UA/HCPT NPs in 4T1 cells. Cell viability of 4T1 cells treated with 8 μg mL−1 of UA, HCPT, and nanoparticles (equivalent to native UA) was measured by CCK-8 assay (n = 3, error bars represent standard deviation). (d) CCK-8 assay of UA, HCPT and nanoparticles at different concentrations in the 4T1 cell line (n = 3, error bars represent standard deviation). | ||
3.5. Cytotoxicity evaluation studies of CMC–UA/HCPT NPs in vitro
In vitro cytotoxicity should be considered to ensure the safety of the nanoparticles before they can be tested in the human system.45 To examine the cytotoxicity of UA, HCPT, and the nanoparticles, a CCK-8 assay was conducted after incubating 4T1 cells with the different drugs. The response of the cells was tested in vitro by seeding the cells with various concentrations of CMC–UA NPs, CMC–UA/HCPT NPs, free UA, and free HCPT for 24, 48 or 72 h. As shown in Fig. 8c, 9 μg mL−1 UA and 11 μg mL−1 HCPT could result in cell death, the extent of which was dependent upon the length of incubation. The time-dependent cytotoxic effect of the CMC–UA/HCPT NPs was evident: 25.1%, 14.0% and 6.3% of 4T1 cells survived after 24 h, 48 h, 72 h at 10 μg mL−1. To compare potential drug efficacy, IC50 was estimated from the survival curves in Fig. 8d. The results showed that the IC50 of the samples is in the order UA > CMC–UA NPs > HCPT > CMC–UA/HCPT NPs (Table 2). With the CMC–UA/HCPT NPs (22.07 wt% UA and 17.53 wt% HCPT), IC50 of 4T1 cells was 0.12 μg mL−1, and the calculated combination index (CI) of UA and HCPT in the CMC–UA/HCPT NPs was 0.05. This suggested that CMC–UA/HCPT NPs achieved a significant synergistic effect by co-delivery of the two different anticancer drugs UA and HCPT.| Compound | IC50 | Concentration (μg mL−1) |
|---|---|---|
| UA | 7.44 | 0.52018 |
| HCPT | 0.41 | 0.03479 |
| CMC–UA NPs | 1.88 | 0.14235 |
| CMC–UA/HCPT NPs | 0.12 | 0.00250 |
3.6. Cellular uptake of CMC–UA/HCPT NPs
It is well known that the unfavorable pharmaceutical properties of traditional chemotherapeutics, including poor water solubility and short circulation time, limit their clinical application. Nanotechnology-based drug delivery systems (e.g., dendrimer, liposome, polymeric micelle, nanohydrogel, etc.) have shown significant promise in overcoming the aforementioned limitations and have demonstrated encouraging results in breast cancer cell therapy in recent years.46 Moreover, nanoparticle-mediated combined delivery of two drugs has shown synergistic effects in multiple cancers. To provide evidence of cell compatibility and evaluate the drug delivery efficiency of nanoparticles, we incubated 4T1 cells with drug-loaded nanoparticles for 4 h. In order to detect the cellular uptake, the concentrations of free HCPT and CMC–UA/HCPT NPs in medium were the same as their respective IC50 values. The fluorescence of HCPT (green) and DAPI (blue) was visualized. CMC–UA/HCPT NPs (green fluorescence) was more effectively delivered than free HCPT which had almost no attachment to the surface of 4T1 cells (Fig. 9). HCPT-loaded nanoparticles, such as liposomes, micelles, and polymer nanoparticles, have been shown to be taken up by cells through an endocytic pathway, thereby allowing them to escape from the effect of P-glycoprotein. HCPT in nanoparticles could maintain a high intracellular HCPT concentration in the cytoplasm. The results demonstrated that increased green fluorescence due to CMC–UA/HCPT NPs is densely located in the cytoplasm region near the cell membrane and primarily located on the cell surface.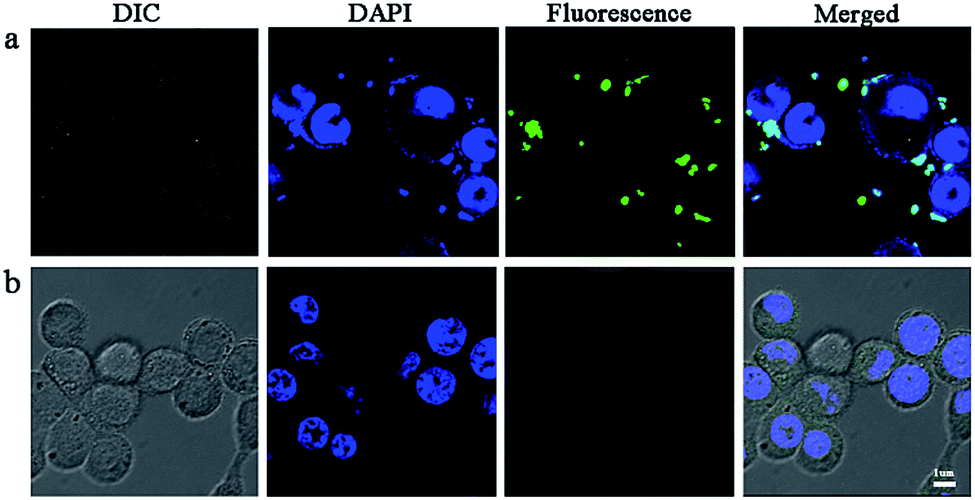 | ||
| Fig. 9 Confocal microscopic pictures of 4T1 cells incubated with (a) free HCPT and (b) CMC–UA/HCPT NPs at an equivalent HCPT concentration of 0.4 μg mL−1 (IC50) for 4 h at 37 °C. | ||
3.7. Pharmacokinetics experiment
An optimal distribution of anticancer agents in vivo is closely related to the enhancement of therapeutic efficacy.47 To confirm the relation between efficacy and drug biodistribution, a pharmacokinetics study was undertaken by intravenous injection of HCPT, UA, CMC–UA NPs and CMC–UA/HCPT NPs into 4T1-bearing mice. The results, displayed in Fig. 10, showed that HCPT and UA concentrations in plasma gradually decreased with time for CMC–UA NPs and CMC–UA/HCPT NPs administered by intravenous injection. Obviously, the HCPT and UA of CMC–UA/HCPT NPs and CMC–UA NPs are retained at higher concentration in the plasma up to 55 h after injection, whereas the concentrations of HCPT and UA were almost undetectable after 4 h and 5.5 h, respectively. Disappearance of free UA and free HCPT from the blood circulation after intravenous injection was very rapid, with the plasma concentration below 10%. However, CMC–UA NPs and CMC–UA/HCPT NPs showed remarkably prolonged clearance, with UA levels of 18.9% and 23.7% ID per g at 24 h after administration. For HCPT levels, CMC–UA/HCPT NPs exhibited 15.1% ID per g at 25 h after administration.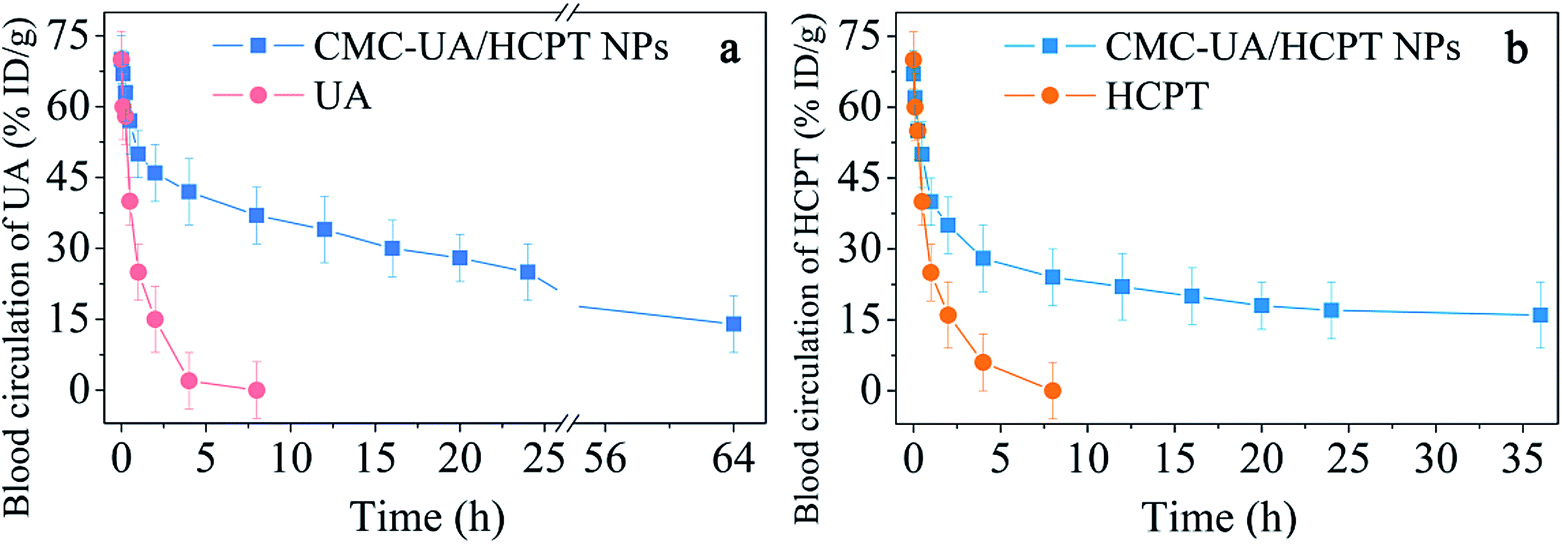 | ||
| Fig. 10 Blood circulation level and half-life of CMC–UA/HCPT NPs compared with free UA (a), and CMC–UA/HCPT NPs compared with free HCPT (b). Error bars are based on six mice per group at each time point. | ||
CMC–UA NPs and CMC–UA/HCPT NPs could extend the blood circulation half-life of UA from 1 h to 4.5 h and 7.3 h, respectively, which were far longer (4.5- and 7.3-fold) than the value for free UA. CMC–UA/HCPT NPs could extend the blood circulation half-life of HCPT from 1.2 h to 3.0 h, which was 2.5-fold that of free HCPT.
3.8. In vivo anticancer activity studies of CMC–UA/HCPT NPs
For humane reasons, animals were killed after the tumor volume reached 5000 mm3 or at the end of the experiment (>6 weeks). The drug delivery efficacies of nanoparticles were considered for equivalent doses of 10 mg kg−1 UA and 10 mg kg−1 HCPT, respectively (Fig. 11a). The two most important goals in cancer treatment are prolonged survival without reduction in the quality of life.23 4T1 tumor-bearing mice were used to test the in vivo anticancer activity of CMC–UA/HCPT NPs, and the results are shown in Fig. 11.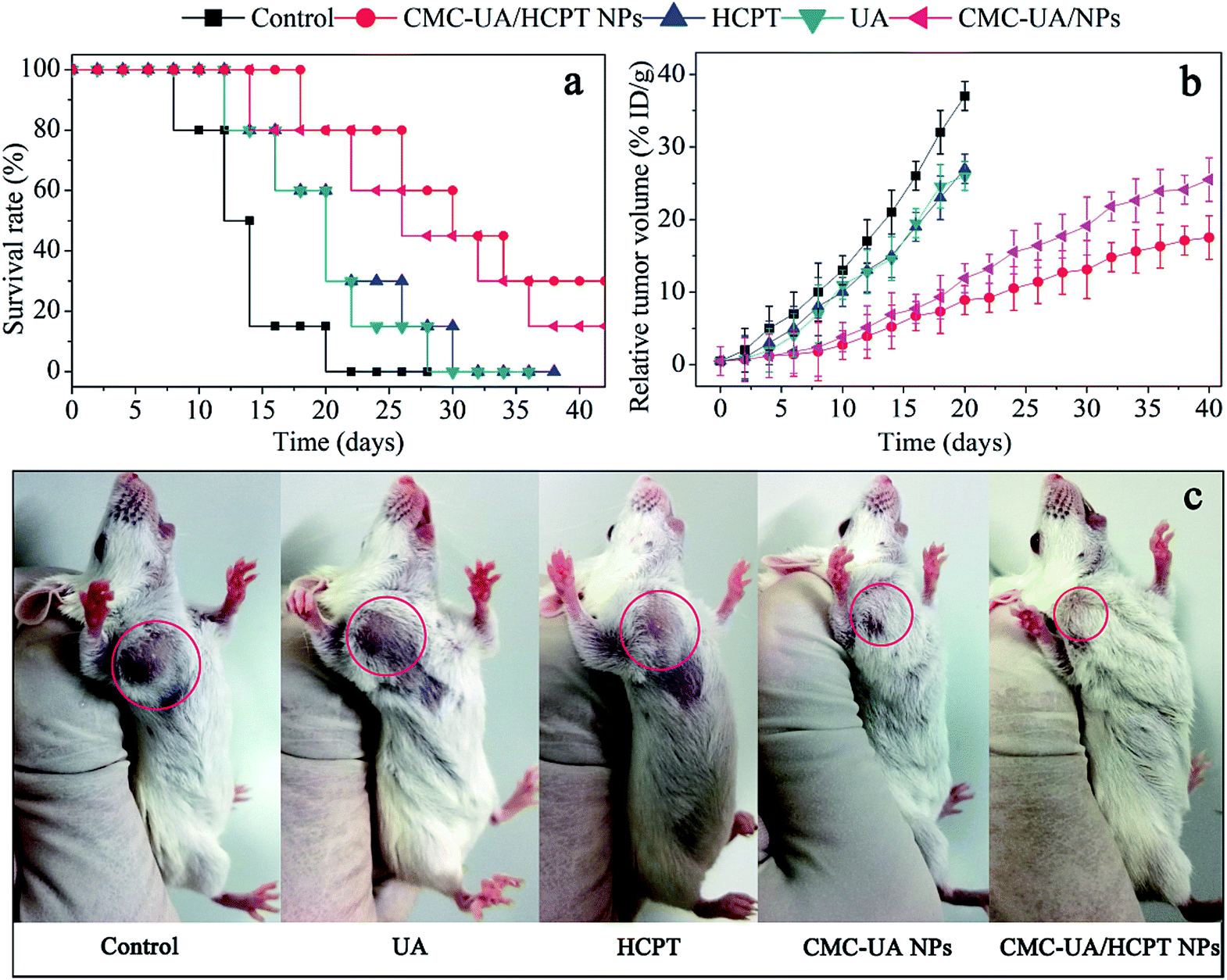 | ||
| Fig. 11 In vivo antitumor activity of free UA, free HCPT, and nanoparticles in the subcutaneous mouse model of 4T1. (a) Survival of mice in different treatments. (b) Tumor volumes of mice during treatment with different groups. (c) Tumor photographs from each treatment group on day 28. | ||
After 40 days' treatment, a significant difference in the tumor volumes in the mice of the experimental groups can be seen in Fig. 11b. 4T1 tumor-bearing mice treated with the nanoparticles showed a considerable survival advantage in comparison with the free UA and free HCPT. Anti-tumor capacity of UA, HCPT, CMC–UA NPs and CMC–UA/HCPT NPs was in the order CMC–UA/HCPT NPs > CMC–UA NPs > UA and free HCPT. Survival rate and TGI with CMC–UA/HCPT NPs were 88.6% (30 days) and 93.5% (25 days), while those with UA were 13.3% (30 days) and 42.9% (25 days), and those with HCPT were 12.8% (30 days) and 35.2% (25 days) (Fig. 11c, Table 3). These results indicate that the tumor volumes in the CMC–UA/HCPT NPs-treated group were much smaller than those in the groups treated with UA or HCPT injection. Throughout the experiment, no obvious changes of the average body weights were observed in all treated mice (Fig. 12a), which suggested the drugs were safe at such a dose. These findings coincide with the foregoing results of in vitro evaluations.
| Compound | Mean TV ± SDa (mm3) | RTVa | TGIa (%) | Curesb (%) |
|---|---|---|---|---|
| a Mean tumor volume (TV), relative tumour volume (RTV), and percentage tumour growth inhibition (TGI) data were recorded at day 25.b Percentage cures were recorded at day 30. | ||||
| Control | 5059 ± 1526 | 39.0 ± 17.3 | 0 | 0 |
| UA | 2834 ± 1074 | 23.6 ± 13.8 | 42.9 | 13.3 |
| HCPT | 3247 ± 1189 | 25.9 ± 15.9 | 35.2 | 12.8 |
| CMC–UA NPs | 538 ± 167 | 4.5 ± 1.4 | 88.9 | 65.4 |
| CMC–UA/HCPT NPs | 362 ± 129 | 3.0 ± 1.0 | 93.5 | 88.6 |
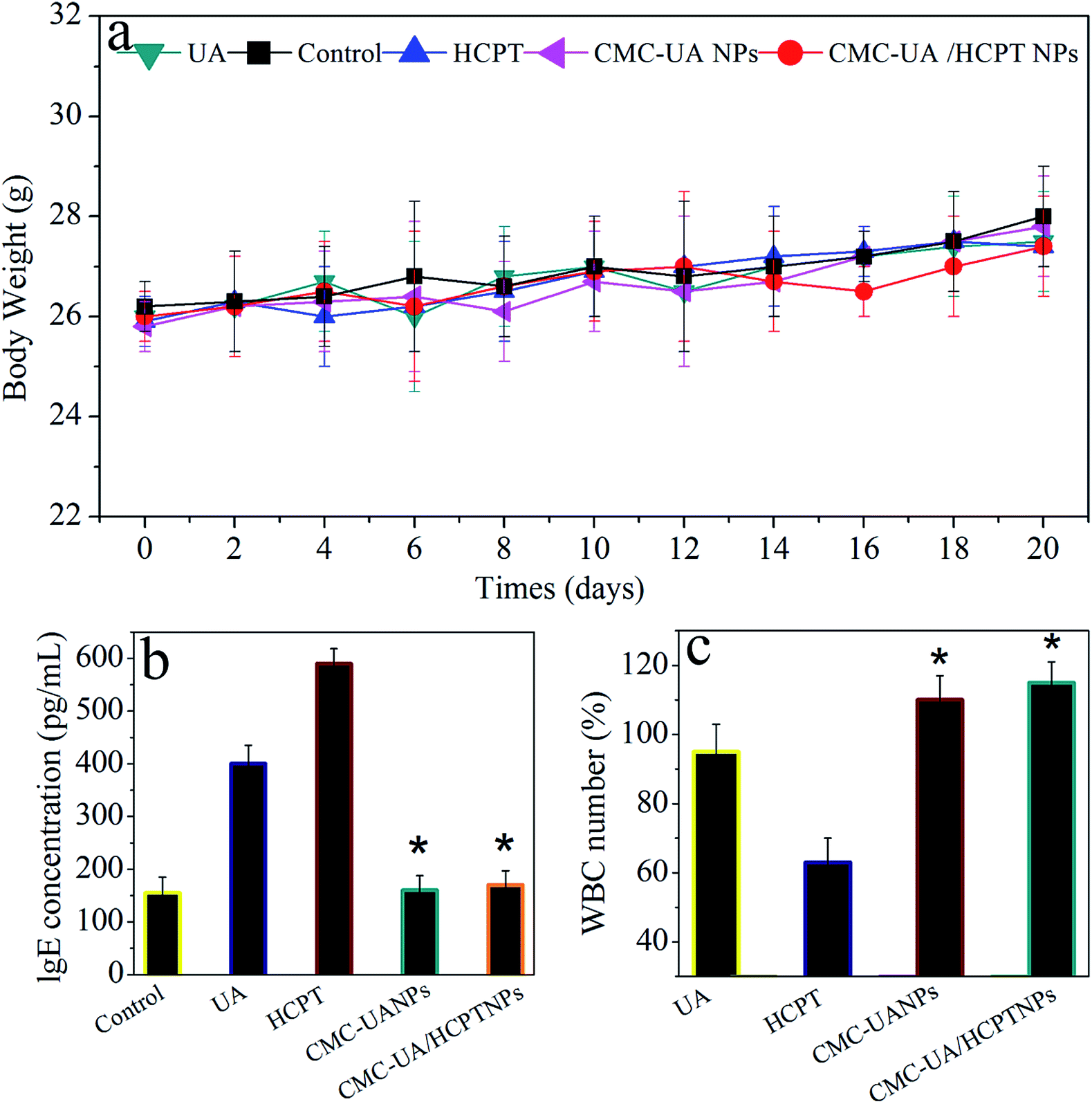 | ||
| Fig. 12 (a) The animal weights were recorded once per week and expressed over the 28 day observation. (b) IgE levels of mice treated with free UA, free HCPT, and nanoparticles for 30 min. (c) White blood cell (WBC) changes during four administrations in normal mice with free UA, free HCPT, and nanoparticles. The blood sample was collected from mice on day 2 after the last dosage treatment. Data are means ± SD; n = 6. | ||
3.9. Evaluation of the side effects
Parameter IgE levels (UA, HCPT, CMC–UA NPs and CMC–UA/HCPT NPs) were selected for rapid evaluation of type I hypersensitivity reactions. From Fig. 12b and c, we can see that 4T1 tumor-bearing mice treated with UA and HCPT displayed a higher IgE level than the control group, and no significant change in the CMC–UA NPs and CMC–UA/HCPT NPs groups. This may be attributed to the bad water solubility of UA and HCPT, meanwhile, it is clearly an elaborately certification that nanoparticles could reduce the risk of hypersensitivity reactions substantially. The blood of mice after treatment with different formulations was also collected to test the WBC count, which is often used as an indicator of hematologic toxicity. No discernible decreases in the WBC number of the mice treated with the CMC–UA NPs and CMC–UA/HCPT NPs groups were observed, indicating that the nanoparticles designed in this study could avoid severe hematologic toxicity.4. Conclusions
This study strongly suggests that combined therapy for eliminating bulk tumor cells could achieve a synergistic anti-tumor effect, and thus may be an attractive strategy for cancer treatment. Biodegradable polymer nanoparticles showed a higher drug-loading efficiency (29.62%) and encapsulation efficiency (17.53%) than the literature had reported. Especially, the appropriate nanoparticle size (∼20 nm–60 nm) was more conducive to phagocytosis of cells than the size of >100 nm. In vitro assays demonstrated that the functionalized nanoparticles exhibited enhanced cellular uptake, cell apoptosis induction and cell-viability inhibition ability in 4T1 cells. The cellular uptake and apoptosis indicated that nanoparticles were able to capture and kill tumor cells. Confocal microscopy analysis showed that nanoparticles can actively capture 4T1 cells. In addition, drug delivery systems of CMC–UA/HCPT NPs remarkably suppressed tumor growth by eliminating bulk tumor cells in a 4T1 orthotopic tumor murine model and realized a satisfactory effect.Acknowledgements
This study was supported by the Special Fund for Beijing Common Construction Project, and National Natural Science Foundation of China (No. 21406013 and No. 21576029), State Forestry Administration 948 Project of China (No. 2014-4-35) and the Sichuan Science and Technology Department (No. 2017JY0139).Notes and references
- W. Q. Chen, R. S. Zheng, P. D. Baade, S. Zhang, H. M. Zeng, F. Bray, A. Jema, X. Q. Yu and J. He, Ca-Cancer J. Clin., 2016, 66(2), 115–132 CrossRef PubMed.
- L. A. Torre, R. L. Siegel, E. M. Ward and A. Jemal, Cancer Epidemiol., Biomarkers Prev., 2016, 25(1), 16–27 CrossRef PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, CA: Cancer J. Clin., 2016, 66(1), 7–30 CrossRef PubMed.
- H. Banu, D. K. Sethi, A. Edgar, A. Sheriff, N. Rayees, N. Renuka, S. M. Faheem, K. Premkumar and G. Vasanthakumar, J. Photochem. Photobiol., B, 2015, 149, 116–128 CrossRef CAS PubMed.
- G. Juncu, A. Stoica-Guzun, M. Stroescu, G. Isopencu and S. I. Jinga, Int. J. Pharm., 2015, 510(2), 485–492 CrossRef PubMed.
- M. K. Shanmugam, X. Dai, A. P. Kumar, B. K. H. Tan, G. Sethi and A. Bishayee, Biochem. Pharmacol., 2013, 85(11), 1579–1587 CrossRef CAS PubMed.
- D. D. V. Hoff, M. W. Layard, P. Basa, H. L. Davis, A. L. V. Hoff, M. Rozencweig and F. M. Muggia, Ann. Intern. Med., 1979, 91(5), 710–717 CrossRef.
- H. Zhang, X. Li, J. Ding, H. Xu, X. Dai, Z. Hou, K. Zhang, K. Sun and W. Sun, Int. J. Pharm., 2013, 441(1–2), 261–268 CrossRef CAS PubMed.
- Y. Wang, J. Song, S. F. Chow, A. H. Chow and Y. Zheng, Int. J. Pharm., 2015, 494(1), 479–489 CrossRef CAS PubMed.
- V. H. Shargh, H. Hondermarck and M. Liang, Int. J. Pharm., 2016, 515(1–2), 527–534 CrossRef CAS PubMed.
- J. R. Heath and M. E. Davis, Annu. Rev. Med., 2008, 59(2)(9), 251–265 CrossRef PubMed.
- A. Z. Wang, R. Langer and O. C. Farokhzad, Medicine, 2012, 63(63), 185–198 CrossRef CAS PubMed.
- A. Sarkar, S. Ghosh, S. Chowdhury, B. Pandey and P. C. Sil, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860(10), 2065–2075 CrossRef CAS PubMed.
- S. Wang, J. Zhang, Y. Wang and M. Chen, Nanomedicine, 2015, 12(2), 411–420 CrossRef PubMed.
- S. Chakraborti, S. Chakraborty, S. Saha, A. Manna, S. Banerjee, A. Adhikary, S. Sarwara, K. T. Hazrac, T. Dasb and P. Chakrabarti, Free Radical Biol. Med., 2017, 103, 35–47 CrossRef CAS PubMed.
- Z. Cai, N. Chattopadhyay, K. Yang, Y. L. Kwon, S. Yook, J. P. Pignol and R. M. Reilly, Nucl. Med. Biol., 2016, 43(12), 818–826 CrossRef CAS PubMed.
- P. Xu, Q. Meng, H. Sun, Q. Yin, H. Yu, Z. Zhang, M. Cao, Y. Zhang and Y. Li, Biomaterials, 2015, 64, 10–20 CrossRef CAS PubMed.
- J. Yao, J. Feng, X. Gao, D. Wei, T. Kang, Q. Zhu, T. Jiang, X. Wei and J. Chen, Biomaterials, 2016, 113, 1–17 CrossRef PubMed.
- R. Baishya, D. K. Nayak, D. Kumar, S. Sinha, A. Gupta, S. Ganguly and M. C. Debnath, Pharm. Res., 2016, 33(11), 1–13 Search PubMed.
- H. Jin, J. Pi, F. Yang, J. H. Jiang, X. P. Wang, H. H. Bai, M. T. Shao, L. Huang, H. Y. Zhu, P. H. Yang, L. H. Li, T. Li, J. Y. Cai and Z. W. Chen, Sci. Rep., 2016, 6, 30782 CrossRef CAS PubMed.
- H. Jin, J. Pi, F. Yang, C. Wu, X. Chen, H. H. Bai, D. Huang, J. H. Jiang, J. Y. Cai and Z. W. Chen, Appl. Microbiol. Biotechnol., 2016, 100(15), 6643–6652 CrossRef CAS PubMed.
- M. J. Ernsting, W. L. Tang, N. Maccallum and S. D. Li, Bioconjugate Chem., 2011, 22(12), 2474–2486 CrossRef CAS PubMed.
- L. Dai, K. Liu, C. Si, J. He, J. Lei and L. Guo, J. Mater. Chem. B, 2015, 3(32), 6605–6617 RSC.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4(7), 3591–3605 CrossRef CAS PubMed.
- T. D. Nguyen, E. Jankowski and S. C. Glotzer, ACS Nano, 2011, 5(11), 8892–8903 CrossRef CAS PubMed.
- J. Nguyen, T. W. J. Steele, O. Merkel, R. Reul and T. Kissel, J. Controlled Release, 2008, 132(3), 243–251 CrossRef CAS PubMed.
- R. Reul, J. Nguyen and T. Kissel, Biomaterials, 2009, 30(29), 5815–5824 CrossRef CAS PubMed.
- F. Unger, M. Wittmar and T. Kissel, Biomaterials, 2007, 28(9), 1610–1619 CrossRef CAS PubMed.
- Q. Chen, X. Wang, C. Wang, L. Feng, Y. Li and Z. Liu, ACS Nano, 2015, 9(5), 5223–5233 CrossRef CAS PubMed.
- J. P. Coelho, G. Tardajos, V. Stepanenko, A. Rödle, G. Fernández and A. Guerreromartínez, ACS Nano, 2015, 9(11), 11241–11248 CrossRef CAS PubMed.
- Y. Li, J. Lin, X. Yang, Y. Li, S. Wu, Y. Huang, S. Ye, L. Xie, L. Dai and Z. Hou, ACS Appl. Mater. Interfaces, 2015, 7(32), 17573–17581 CAS.
- J. L. Xiao, Z. X. Lu and Y. Q. Li, Ind. Eng. Chem. Res., 2015, 54(3), 790–797 CrossRef CAS.
- T. Plyduang, L. Lomlim, S. Yuenyongsawad and R. Wiwattanapatapee, Eur. J. Pharm. Biopharm., 2014, 88(2), 351–360 CrossRef CAS PubMed.
- S. Y. Lü, M. Z. Liu and B. L. Ni, Chem. Eng. J., 2010, 160(2), 779–787 CrossRef.
- J. D. Obayemi, Y. Danyuo, S. Dozie-Nwachukwu, O. S. Odusanya, N. Anuku, K. Malatesta, W. Yu, K. E. Uhrich and W. O. Soboyejo, Mater. Sci. Eng., C, 2016, 66, 51–65 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303(5665), 1818–1822 CrossRef CAS PubMed.
- M. A. Bruckman, A. E. Czapar, A. Vanmeter, L. N. Randolph and N. F. Steinmetz, J. Controlled Release, 2016, 231, 103–113 CrossRef CAS PubMed.
- J. Varshosaz, F. Hassanzadeh, H. S. Aliabadi, F. R. Khoraskani, M. Mirian and B. Behdadfar, Int. J. Biol. Macromol., 2016, 93, 1192–1205 CrossRef CAS PubMed.
- M. J. Ernsting, M. Murakami, E. Undzys, A. Aman, B. Press and S. D. Li, J. Controlled Release, 2012, 162(3), 575–581 CrossRef CAS PubMed.
- M. J. Ernsting, B. Hoang, I. Lohse, E. Undzys, P. Cao, T. Do, B. Gill, M. Pintilie, D. Hedley and S. D. Li, J. Controlled Release, 2015, 206, 122–130 CrossRef CAS PubMed.
- G. Liu, S. Li, Y. Huang, H. Wang and Y. Jiang, Chem. Eng. Sci., 2016, 155, 405–414 CrossRef CAS.
- Z. Liu, Q. Zheng, W. Chen, M. Wu, G. Pan, K. Yang, X. Li, S. Man, Y. Teng, P. Yu and W. Gao, Eur. J. Med. Chem., 2016, 125, 760–769 CrossRef PubMed.
- M. Han, X. Liu, Y. Guo, Y. Wang and X. Wang, Int. J. Pharm., 2013, 455(1–2), 85–92 CrossRef CAS PubMed.
- Z. Yang, W. Gong, Z. Wang, B. Li, M. Li, X. Xie, H. Zhang, Y. Yang, Z. Li, Y. Li, F. Yu and X. Mei, Int. J. Pharm., 2015, 490(1–2), 412–428 CrossRef CAS PubMed.
- W. Wu, R. Li, X. Bian, Z. Zhu, D. Ding, X. Li, Z. Jia, X. Jiang and Y. Hu, ACS Nano, 2009, 3(9), 2740 CrossRef CAS PubMed.
- Z. Dong, L. Feng, W. Zhu, X. Sun, M. Gao, H. Zhao, Y. Chao and Z. Liu, Biomaterials, 2016, 110, 60–70 CrossRef CAS PubMed.
- H. Wang, J. Feng, G. Liu, B. Chen, Y. Jiang and Q. Xie, Nanomedicine, 2016, 12(4), 881–891 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |