
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA fluorescent photochromic diarylethene based on naphthalic anhydride with strong solvatochromism†
Shangjun Chen *a,
Xin Lib and
Liwen Songc
*a,
Xin Lib and
Liwen Songc
aKey Laboratory of Resource Chemistry of Ministry of Education, Shanghai Key Laboratory of Rare Earth Functional Materials, Department of Chemistry, Shanghai Normal University, Shanghai, 200234, P. R. China. E-mail: jshchen@shnu.edu.cn
bDivision of Theoretical Chemistry and Biology, School of Biotechnology, KTH Royal Institute of Technology, SE-10691 Stockholm, Sweden
cShanghai Key Laboratory of Functional Materials Chemistry, East China University of Science and Technology, Shanghai 200237, P. R. China
First published on 8th June 2017
Abstract
A novel “push–pull” diarylethene molecule consisting of an electron withdrawing ethene bridge (1,8-naphthalic anhydride) and two moderate electron donating side chains (2,5-dimethylthiophene) has been designed and synthesized. The photochromism study, together with density functional theory calculations, revealed that the molecule exhibits reversible fluorescence switching capacity upon photo-isomerization and remarkable solvatochromism with red shift of the fluorescence maximum by more than 150 nm owing to intramolecular charge transfer.
Introduction
Research on bistable diarylethenes has received much attention on account of their photo-isomerization states with distinct absorption spectra, promising fatigue resistance, and excellent thermal stability.1–6 In particular, diarylethene compounds exhibiting fluorescence modulation properties, in which the fluorescence could be switched on and off reversibly via the photochromic isomerization between the open- and closed-ring isomers, are desirable for their potential applications in logic gates, information storage and biological sensing by taking the advantages of fluorescence such as sensitivity, convenience, and relative cheapness.7–13Rational molecular structure designs, whether on the side chains or the ethene bridge of the diarylethenes, aiming at regulating the fluorescence have been well reported.14–22 One of the efficient approaches is to replace the traditional ethene bridges (usually perfluorocyclopentene or cyclopentene) with new aromatic fluorophore building blocks.23–26 Owing to the diversity of the fluorophores as well as the remarkable structure changes of the aromatic fluorophore groups during photochromic processes, the fluorescence of the diarylethenes can be regulated efficiently and versatility. For example, an interesting fluorescence turn-on photochromic system in which the closed ring-isomer shows unusual intense fluorescence has been reported, with help of a modulated thiophene ring as ethene bridge.27,28 Employing coumarin chromophore as the central ethene bridge, Yokoyama's group have achieved an amazing photochromic fluorescence photoswitching controlled by both photochromic reactions and pH.29 Recently, our group has also advanced multiple responsive photochromic fluorescence systems, which can be trigged by stimuli such as solvent, ions, electrochemical oxidation and reduction, by introducing six-membered ring based ethene bridges such as naphthalimide or benzothiadiazoles into molecular framework of diarylethenes.30–32
Solvatochromism, which usually describes the distinct change of the absorption and emission spectra that accompanies a change in solvent polarity, is commonly used in chemical and biological research areas to monitor microenvironmental polarity or to study the conformation of proteins.33,34 A compound exhibiting evident solvatochromism typically possesses an electron donor and an acceptor, which makes the intramolecular charge-transfer (ICT) extremely sensitive to even small changes in local microenvironment that originates from the variation of solvent polarity. Recently, by introducing different electron-accepting moieties onto the molecule skeleton of tetraphenylethene, Tang et al. developed a series of aggregation-induced emission molecules with remarkable solvatochromism for the construction of light-emitting diodes or fluorescent visualizers for intracellular imaging.35,36 More recently, in order to fluorescently distinguish two types of amyloids in tissue, an aminonaphthalene 2-cyanoacrylate (ANCA) derivative with 20 nm maximal emission wavelength shift when bound to amyloid deposits has been reported.37 However, diarylethene molecules with strong solvatochromism and photoswitching properties are relatively rare.
Naphthalic anhydrides are one kinds of widely used fluorescent chromophores with high fluorescence efficiency and chemical stability. Moreover, it is expected that connection of electron donating groups at the C-4 or C-5 positions of the naphthalic ring would generate a “push–pull” molecule with an ICT excited state and considerable dipole character.38,39 Aim at constructing new diarylethene molecule with novel photo-switchable fluorescence properties and distinguished solvatochromism, a new diarylethene compound, BTE-O, was designed by directly incorporating a fluorescent moiety of 1,8-naphthalic anhydrides as a six-membered aryl unit for the center ethene bridge and further introducing two moderate electron donating units 2,5-dimethylthiophene as the side chains. The easily obtained new compound displayed reversible fluorescence switching capacity upon photo-isomerization and exhibits strong ICT character between the naphthalic anhydride based ethene bridge and two substituted thiophene groups, leading to strong solvatochromism with a red shift of the fluorescence maximum greater than 150 nm.
Experimental
Materials and methods
NMR spectra were recorded on a Bruker AM-400 spectrometer using tetramethylsilane (TMS) as the internal reference. CDCl3 was used as the solvent. HRMS spectra were measured on a Waters LCT Premier XE spectrometer using methanol or acetonitrile as solvents. UV-vis absorption spectra and fluorescence spectra were recorded on a Varian Cary 100 spectrophotometer and a Varian Cary Eclipse fluorescence spectrophotometer, respectively. Fluorescence quantum yields of BTE-O in different solvents were determined relative to standard quinine sulphate (Φf = 0.55) within an error of ±5%. Fluorescence lifetimes were obtained on an Edinburgh Lifespec-Ps spectrofluorometer (FL920). The photochromic reaction was conducted in situ by continuous irradiation using an Hg/Xe lamp (Hamamatsu, LC8 Lightningcure, 200 W) equipped with a narrow-band interference filter (Shenyang HB optical Technology) for λirr = 365 nm and a broad-band interference filter (Shenyang HB optical Technology) for λirr > 500 nm. Absorption changes were monitored using a CCD camera mounted on a Princeton spectrometer instrument. The photochromic reaction quantum yields were evaluated by the standard procedures using 3,3′-(perfluorocyclopent-1-ene-1,2-diyl) bis(2-methylbenzo[b]thiophene (BTF6) as the reference for photocyclization. The rates of isomerization in the initial stage of the reaction (0–5%) were compared to a reference whose Φo–c (35%) in hexane was known, which resulted in 5% uncertainties in the calculations of the quantum yields.40Synthesis
All the chemicals were of analytical grade and commercially available unless otherwise stated and used as received without further purification. All organic solvents were purified by standard methods prior to use. The intermediate BTE-N (Scheme 1) was prepared according to our established methods previously reported.30 BTF6 was obtained by procedures similar to those in ref. 41.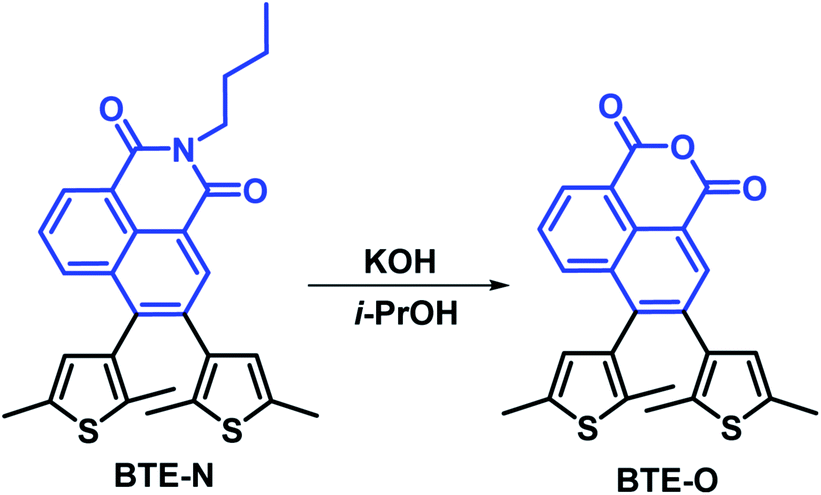 | ||
| Scheme 1 Synthetic rout of BTE-O. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to yield a yellow solid (57%). 1H NMR (400 MHz, CDCl3, ppm): δ = 8.62 (d, J = 7.3 Hz, 1H, naphthalene-H), 8.59 (s, 1H, naphthalene-H), 8.17 (d, J = 8.5 Hz, 1H, naphthalene-H), 7.74 (t, J = 7.4 Hz, 1H, naphthalene-H), 6.43 (s, 1H, thienyl-H), 6.20 (s, 1H, thienyl-H), 2.44 (s, 1H, –CH3), 2.34 (s, 1H, –CH3), 2.20 (s, 1H, –CH3), 1.90 (s, 1H, –CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 160.87, 160.59, 142.06, 136.71, 136.45, 135.97, 135.69, 135.51, 134.71, 133.91, 132.87, 132.68, 132.01, 129.57, 127.64, 127.43, 126.84, 118.78, 117.42, 15.20, 15.06, 13.85. Mass spectrometry (the ESI positive ion mode for [M + H]+): calculated C24H19O3S2, 419.0776, found: 419.0779.
:1, v/v) to yield a yellow solid (57%). 1H NMR (400 MHz, CDCl3, ppm): δ = 8.62 (d, J = 7.3 Hz, 1H, naphthalene-H), 8.59 (s, 1H, naphthalene-H), 8.17 (d, J = 8.5 Hz, 1H, naphthalene-H), 7.74 (t, J = 7.4 Hz, 1H, naphthalene-H), 6.43 (s, 1H, thienyl-H), 6.20 (s, 1H, thienyl-H), 2.44 (s, 1H, –CH3), 2.34 (s, 1H, –CH3), 2.20 (s, 1H, –CH3), 1.90 (s, 1H, –CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 160.87, 160.59, 142.06, 136.71, 136.45, 135.97, 135.69, 135.51, 134.71, 133.91, 132.87, 132.68, 132.01, 129.57, 127.64, 127.43, 126.84, 118.78, 117.42, 15.20, 15.06, 13.85. Mass spectrometry (the ESI positive ion mode for [M + H]+): calculated C24H19O3S2, 419.0776, found: 419.0779.Computational details
Density functional theory (DFT) calculations were employed to optimize the ground state geometries of BTE-O and the ring-closed isomer of BTE-O (c-BTE-O), using the B3LYP functional42 and the 6-31G(d) basis set.43 The polarizable continuum model44 was used to model the solvent effects of cyclohexane. At the optimized geometries, the excitation energies and oscillator strengths were calculated by time-dependent DFT (TDDFT) calculations using the CAM-B3LYP functional45 and the 6-311+G(d,p) basis set,46 and the absorption spectra were obtained by Gaussian broadening of the stick spectra with a full-width at half-maximum of 0.175 eV. All calculations were carried out using the Gaussian09 program package.47Results and discussion
Molecule design and synthesis
As shown in Scheme 1, BTE-O was conventionally obtained by alkali treatment of BTE-N with potassium hydroxide. The chemical structure of BTE-O was well confirmed by 1H NMR, 13C NMR and HRMS (see the electronic ESI† for details).Photochromism of BTE-O in solvents
BTE-O was found to dissolve easily in both cyclohexane and acetonitrile to give similar colorless solutions characterised by an intense absorption band in the range of 230–250 nm and a moderately intense band at 270–350 nm. Irradiating the solutions with 365 nm light induced obvious changes in the colour of the solutions from colorless to light green in cyclohexane and deep green in acetonitrile, respectively (Fig. 1), thereby resulting in broad absorption bands centered at 412 and 645 nm (cyclohexane), as well as 414 and 665 nm (acetonitrile) in the visible range. The colour changes of the solutions were due to the formation of c-BTE-O, a ring-closed isomer of BTE-O with an extended conjugation system, as shown in Scheme 2.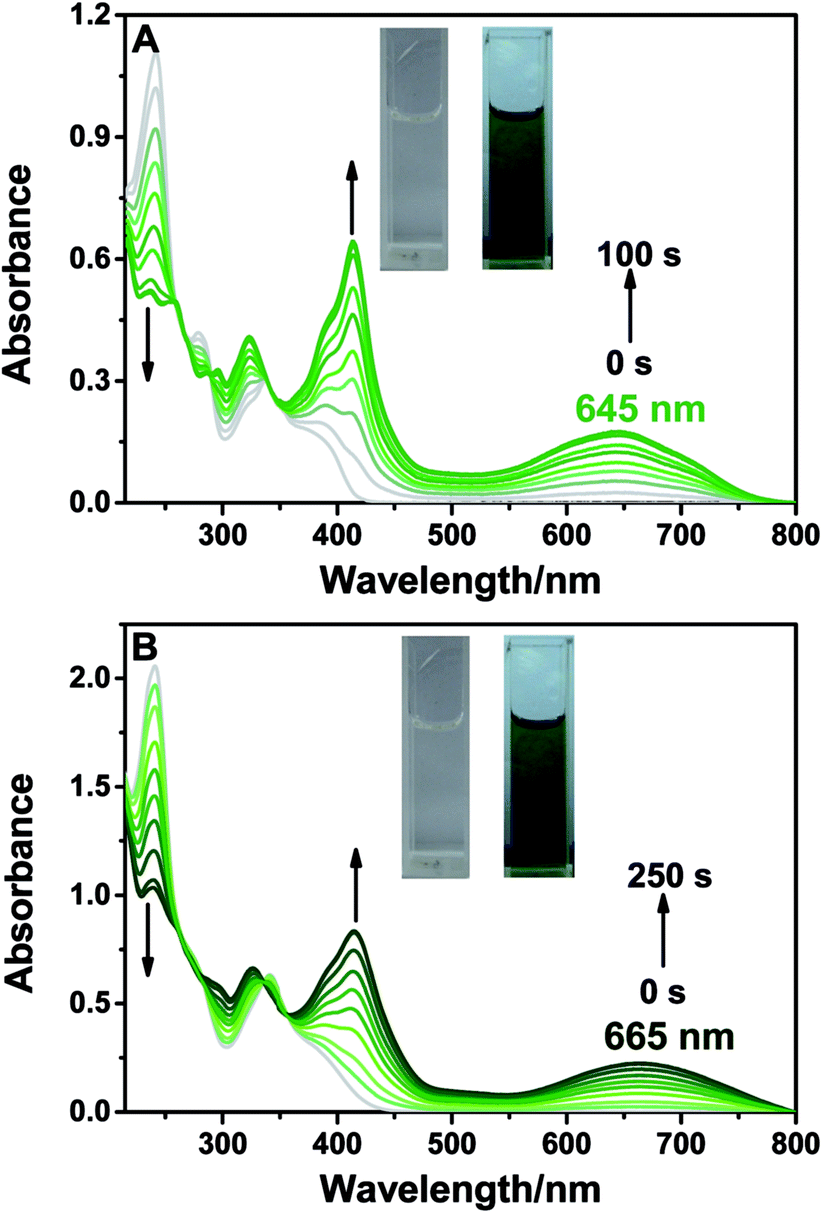 | ||
| Fig. 1 Absorption spectra changes of BTE-O upon irradiation at 365 nm in different solutions (5.0 × 10−5 M) (A) cyclohexane and (B) acetonitrile. Insets: photographic images of BTE-O in (A) cyclohexane and (B) acetonitrile before (left) and after (right) photochromism (5.0 × 10−4 M). | ||
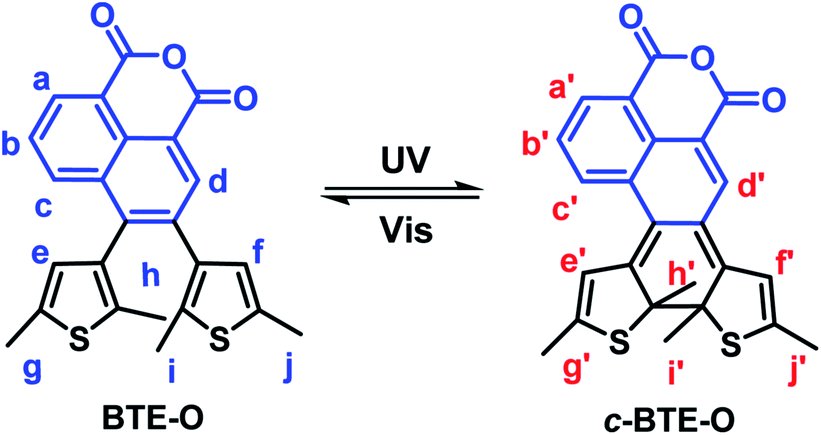 | ||
| Scheme 2 Chemical structure and photochromism of BTE-O. | ||
In fact, due to the large chemical environment changes between the ring-open and -closed forms, distinct 1H NMR spectroscopic changes were also observed for BTE-O before and after 365 nm photo-irradiation in C6D6, which further confirm the typical photocyclization process. The proton signals of the methylene moieties on thiophene rings located at δ = 1.72, 1.96 (Hh, Hi), and δ = 2.06, 2.08 ppm (Hj, Hk) for BTE-O in C6D6, respectively (Fig. S1†). The different chemical shifts of the two groups of methyl protons are on account of the asymmetric organic framework of the central ethene bridge 1,8-naphthalic anhydride. When irradiated at 365 nm, BTE-O was converted to c-BTE-O, and new signals of H′h, H′i, H′j, and H′k located at δ = 1.59, 1.70, 2.16, and 2.24 ppm after photocyclization with respect to that of the open form were observed. More obvious signal changes were found for the hydrogens attached to on the thiophene rings and the ethene bridge due to π delocalization of 1,8-naphthalic anhydride. It is generally accepted that the proton resonances on the six-membered heterocycles ethene bridges shift upfield after photocyclization reaction of diarylethene due to the aromaticity loss of heterocycles.48,49 As expected, the two hydrogen protons located on thiophene (He, Hf) in BTE-O shifted from δ = 6.29 and 6.18 ppm to δ = 5.88 and 5.36 ppm, respectively. In addition, the NMR signals of Ha, Hb, Hc and Hd on 1,8-naphthalic anhydride also shifts upfield after photocyclization.
Moreover, a high-performance liquid chromatography (HPLC) analysis was also used to monitor the photocyclization reaction. The conversion ratio from BTE-O to c-BTE-O in the photostationary state (365 nm) in cyclohexane was 75%, obtained from the corresponding integrated areas of HPLC peaks detected at the isobestic wavelength of 348 nm. The cyclization quantum yield was estimated to be 21.5% according to that of BTF6.
Fluorescence switching and solvatochromism
BTE-O exhibited intense fluorescence in both polar and nonpolar solution due to the existence of the fluorophore unit 1,8-naphthalic anhydride (Fig. 2). Moreover, the fluorescence can be reversibly switched on and off via the photochromic reactions between the ring-open and closed isomers of BTE-O. Upon irradiation at 365 nm, fluorescence of BTE-O is quenched in both nonpolar cyclohexane and polar acetonitrile, as reflected by the fluorescence intensity drop by more than 95% at the photostationary state. The disappearance of fluorescence could be ascribed to the possible quenching channel of Förster resonance energy transfer (FRET) between the ring-open and closed isomers due to the spectra overlap between the absorption and fluorescence spectra.48 Upon further irradiation with visible light (>500 nm), which transfer c-BTE-O to the original open isomer, the fluorescence could be recovered. The fluorescence quantum yield of BTE-O was found to be 0.011 in cyclohexane. A decrease in fluorescence quantum yield was observed as the solvent polarity increases. The fluorescence quantum yields of BTE-O were measured to be 0.009, 0.007, and 0.004 in tetrahydrofuran, dichloromethane, and acetonitrile, respectively.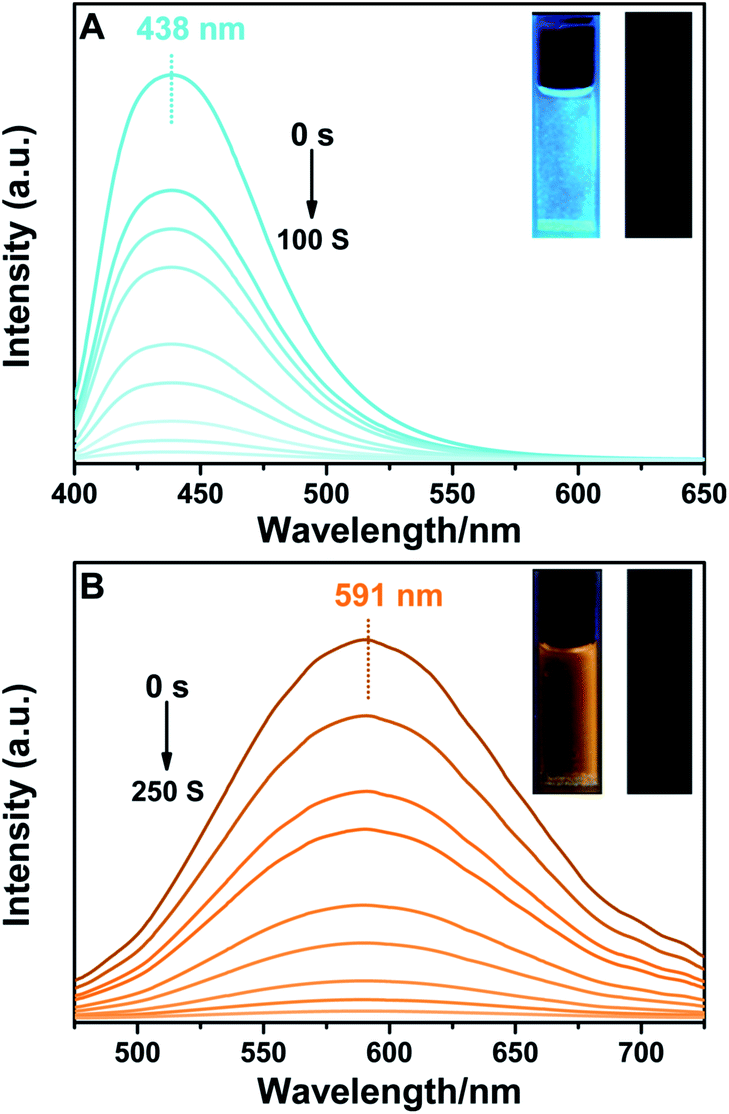 | ||
| Fig. 2 Fluorescence spectra changes of BTE-O upon irradiation at 365 nm in (A) cyclohexane and (B) acetonitrile. Inset: fluorescent photographic images in (A) cyclohexane and (B) acetonitrile before (left) and after (right) photoirradiation (excitation wavelength: 380 nm, 5.0 × 10−5 M). | ||
More interestingly, a remarkable change of emission characteristics both in terms of the position of spectroscopic emission peak as well as fluorescence lifetime induced by polarity of the solvents, which is commonly reckoned as a special type of solvatochromism, was observed for BTE-O.31 As shown in Fig. 3A, the fluorescence maximum of BTE-O gradually red shifted from 438 nm in nonpolar cyclohexane to 511 nm in chloroform, 520 nm in tetrahydrofuran, 533 nm in dichloromethane, 577 nm in acetone, and 591 nm in polar acetonitrile. The solvent polarity trigged a significant bathochromic shift (153 nm) of the luminescent wavelength of BTE-O with increasing solvent polarity from cyclohexane to acetonitrile.
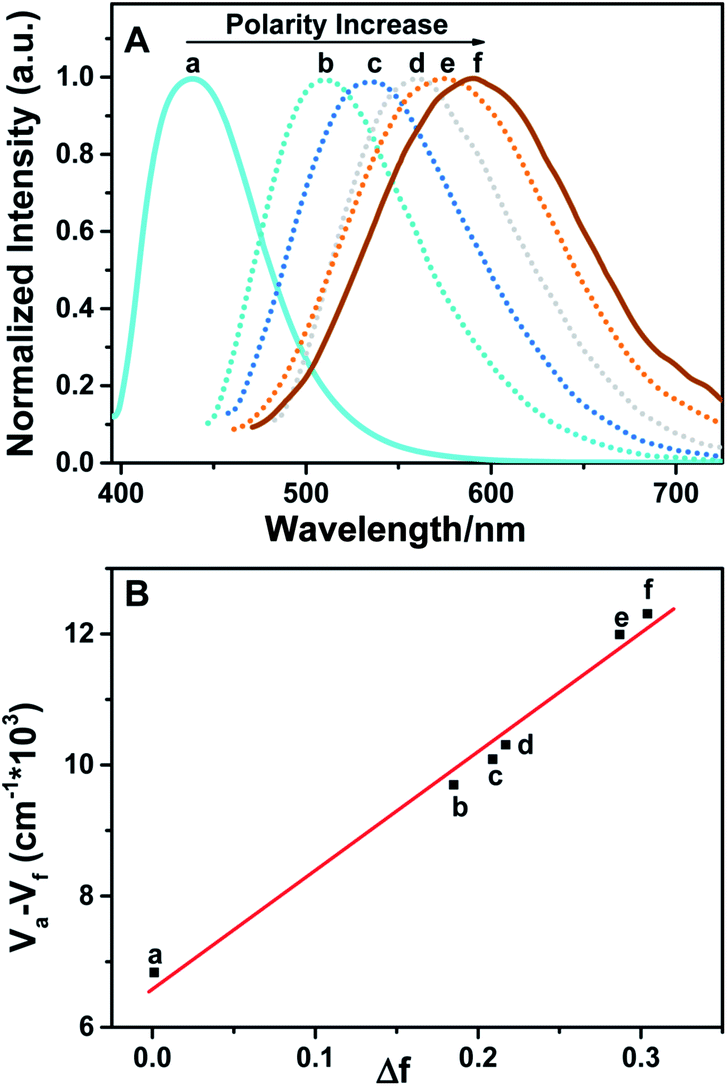 | ||
| Fig. 3 Normalized fluorescence spectra (A) and Lippert–Mataga plot (B) of BTE-O in solvents with different polarity (excitation wavelength: 380 nm, 5.0 × 10−5 M): (a) cyclohexane λmax = 438 nm), (b) chloroform λmax = 511 nm), (c) tetrahydrofuran λmax = 520 nm), (d) dichloromethane λmax = 533 nm), (e) acetone λmax = 577 nm), and (f) acetonitrile λmax = 591 nm). | ||
Meanwhile, the fluorescence lifetime was measure to be 0.35 ns (34.56%) and 5.27 ns (65.44%) fitted with double exponential decaying with χ2 of 1.199 in cyclohexane, while in acetone, the value was prolonged to 0.98 ns (77.07%) and 6.34 ns (22.93%) with χ2 of 1.308. Based on our previous investigation, the distinct solvatochromic shifts as well as the increase on the fluorescence lifetime of BTE-O in different solvents could be attributed to the ICT effect.50,51 In general, the ICT excited state is stabilized by the presence of polar solvent molecules, leading to lowered excitation energy and hence remarkable red-shift of the maximal emission peak. The difference in dipole moments between the excited and the ground states was estimated by the Lippert–Mataga equation, which shows the solvent dependence of the Stokes' shift for a compound. The difference in the maximum absorption and emission wavelengths, is fitted to the following equations:
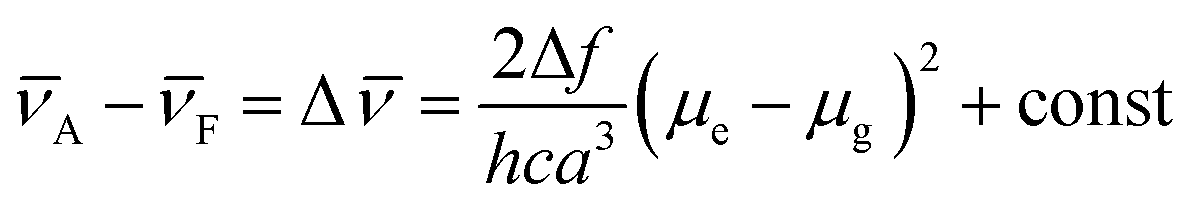 | (1) |
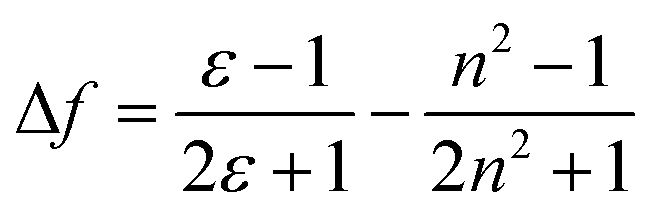 | (2) |
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) A and F are the wavenumbers (cm−1) of the absorbance and fluorescence emission, h is Plank's constant, c is the velocity of light, ε and n are the solvent dielectric constant and refraction index. Δf is known as orientation polarizability. μe and μg are the dipole moments of the excited and the ground states.52,53 As illustrated in Fig. 3B, the Lippert–Mataga plot of the Stokes shift (Δ) as a function of orientation polarizability (Δf) shows a good linear relationship, which strongly suggests that the spectral behaviour of BTE-O emission is indeed related to ICT characteristic.54
A and F are the wavenumbers (cm−1) of the absorbance and fluorescence emission, h is Plank's constant, c is the velocity of light, ε and n are the solvent dielectric constant and refraction index. Δf is known as orientation polarizability. μe and μg are the dipole moments of the excited and the ground states.52,53 As illustrated in Fig. 3B, the Lippert–Mataga plot of the Stokes shift (Δ) as a function of orientation polarizability (Δf) shows a good linear relationship, which strongly suggests that the spectral behaviour of BTE-O emission is indeed related to ICT characteristic.54
Density functional theory (DFT) calculations
To further understand the impressive solvatochromism of BTE-O, density functional theory (DFT) calculations and time-dependent DFT (TDDFT) were performed using the Gaussian09 program package. The simulated absorption spectra of BTE-O and c-BTE-O in cyclohexane are shown in Fig. S2.† Compared with experimental measurement, the excitation energies are slightly overestimated by TDDFT calculations using the CAM-B3LYP functional. As shown in Fig. S3,† the HOMO and LUMO of BTE-O are located on the thiophene and the 1,8-naphthalic anhydride units, respectively, forming an efficient charge-transfer excitation. Based on the TDDFT results, the absorption bands in 300–400 nm region for BTE-O are attributed to electronic excitations from the occupied orbitals (mainly HOMO-2 and HOMO) to LUMO (Fig. S3 and Table S1†). For BTE-O, HOMO−2 → LUMO is a mixture between local excitation and charge-transfer excitation, whereas HOMO → LUMO is pure charge-transfer excitation. After photoisomerization, the new absorption band at around 645 nm is attributed to HOMO → LUMO of c-BTE-O, which is a local excitation (Fig. S4 and Table S1†). The new bands at around 400 nm are contributed by HOMO → LUMO+1 and HOMO−1 → LUMO excitations of c-BTE-O. Although the absorption spectra of BTE-O show only minor solvent polarity dependence, the experimentally measured emission of BTE-O shows significant red-shift as the polarity of solvent increases (Fig. 3a). The linear relationship between the emission wavenumber and the relationship between the emission wavenumber and the solvent polarity is plotted in Fig. 4. According to methods reported by Pasman et al., the excited state dipole moment of BTE-O is estimated as 26.8 Debye, assuming a solvent radius shell of 5.84 Å as determined by DFT calculations.55 This is in fair agreement with the excited state dipole moment calculated by TDDFT in cyclohexane (20.5 Debye) and in acetonitrile (24.0 Debye). When the excited state has a large dipole, the excited molecule is notably stabilized by polar solvent. Consequently, the excitation energy decreases and the fluorescence maximum wavelength increases.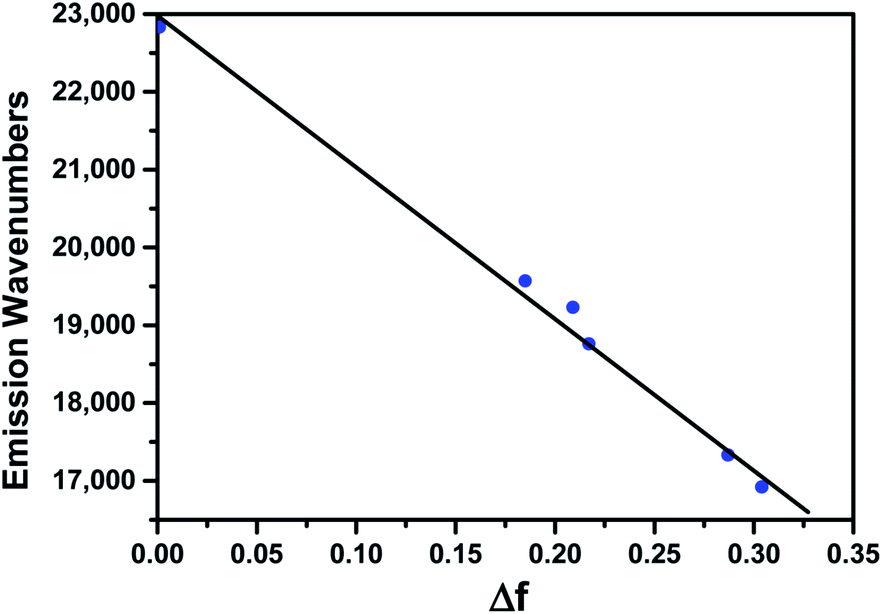 | ||
| Fig. 4 Fitted linear relationship between emission wavenumber (in cm−1) and Δf. | ||
Conclusions
In summary, a novel “push–pull” diarylethene molecule BTE-O containing an 1,8-naphthalic anhydride ethene bridge and two substituted 2,5-dimethylthiophene groups is reported. BTE-O exhibits intense fluorescence owing to the existence of aromatic naphthalic ring and the fluorescence can be switched on and off reversibly upon photo-isomerization between the ring-open and -closed isomers of BTE-O. In addition, as demonstrated by experimental data and theoretical calculations, the fluorescence is highly solvent dependent due to intermolecular charge transfer between the electron donating side chains and the electron withdrawing ethene bridge. This provides solvatochromic fluorescence emission with very large shift exceeding 150 nm, which is of interest for potential applications such as monitoring microenvironmental polarity in chemical and biological systems.Acknowledgements
This work was supported by NSFC/China (21503136), Ministry of education of China (PCSIRT_IRT_16R49), Shanghai Sci. & Tech. and Edu. Committee (14YF1409200) and Shanghai Gaofeng & Gaoyuan Project for University Academic Program Development. We also thank the Swedish National Infrastructure for Computing (SNIC) for providing computational resources for project SNIC 2015-16/10.Notes and references
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS PubMed.
- C. Jia, A. Migliore, N. Xin, S. Huang, J. Wang, Q. Yang, S. Wang, H. Chen, D. Wang, B. Feng, Z. Liu, G. Zhang, D.-H. Qu, H. Tian, M. A. Ratner, H. Q. Xu, A. Nitzan and X. Guo, Science, 2016, 352, 1443–1445 CrossRef CAS PubMed.
- T. Tsuruoka, R. Hayakawa, K. Kobashi, K. Higashiguchi, K. Matsuda and Y. Wakayama, Nano Lett., 2016, 16, 7474–7480 CrossRef CAS PubMed.
- S. Z. Pu, Q. Sun, C. B. Fan, R. J. Wang and G. Liu, J. Mater. Chem. C, 2016, 4, 3075–3093 RSC.
- S. Fredrich, R. Göstl, M. Herder, L. Grubert and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1208–1212 CrossRef CAS PubMed.
- H. Tian and Y. L. Feng, J. Mater. Chem., 2008, 18, 1617–1622 RSC.
- C. Yun, J. You, J. Kim, J. Huh and E. Kim, J. Photochem. Photobiol., C, 2009, 10, 111–129 CrossRef CAS.
- J. Andreasson and U. Pischel, Chem. Soc. Rev., 2015, 44, 1053–1069 RSC.
- F. M. Raymo and M. Tomasulo, J. Phys. Chem. A, 2005, 109, 7343–7352 CrossRef CAS PubMed.
- G. Li, L. Ma, G. Liu, C. Fan and S. Pu, RSC Adv., 2017, 7, 20591–20596 RSC.
- S. Chen, W. Li, X. Li and W.-H. Zhu, RSC Adv., 2015, 5, 87626–87634 RSC.
- C. G. P. Taylor, J. R. Piper and M. D. Ward, Chem. Commun., 2016, 52, 6225–6228 RSC.
- T. Fukaminato, T. Sasaki, T. Kawai, N. Tamai and M. Irie, J. Am. Chem. Soc., 2004, 126, 14843–14849 CrossRef CAS PubMed.
- S.-J. Lim, B.-K. An, S. D. Jung, M.-A. Chung and S. Y. Park, Angew. Chem., Int. Ed., 2004, 43, 6346–6350 CrossRef CAS PubMed.
- M. Bossi, V. Belov, S. Polyakova and S. W. Hell, Angew. Chem., Int. Ed., 2006, 45, 7462–7465 CrossRef CAS PubMed.
- M. Balter, S. Li, M. Morimoto, S. Tang, J. Hernando, G. Guirado, M. Irie, F. M. Raymo and J. Andreasson, Chem. Sci., 2016, 7, 5867–5871 RSC.
- F. Hu, C. Jiang, W. Liu, J. Wang, J. Yin and S. H. Liu, Dyes Pigm., 2017, 136, 161–167 CrossRef CAS.
- Y. Cai, Z. Guo, J. Chen, W. Li, L. Zhong, Y. Gao, L. Jiang, L. Chi, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2016, 138, 2219–2224 CrossRef CAS PubMed.
- X. X. Zhang, R. J. Wang, C. B. Fan, G. Liu and S. Z. Pu, Dyes Pigm., 2017, 139, 208–217 CrossRef CAS.
- H. Dong, M. Luo, S. Wang and X. Ma, Dyes Pigm., 2017, 139, 118–128 CrossRef CAS.
- M. Berberich, A.-M. Krause, M. Orlandi, F. Scandola and F. Würthner, Angew. Chem., Int. Ed., 2008, 47, 6616–6619 CrossRef CAS PubMed.
- W. Li, C. Jiao, X. Li, Y. Xie, K. Nakatani, H. Tian and W. Zhu, Angew. Chem., Int. Ed., 2014, 53, 4603–4607 CrossRef CAS PubMed.
- S. Chen, L.-J. Chen, H.-B. Yang, H. Tian and W. Zhu, J. Am. Chem. Soc., 2012, 134, 13596–13599 CrossRef CAS PubMed.
- H.-H. Liu and Y. Chen, J. Mater. Chem., 2011, 21, 1246–1249 RSC.
- J. Guérin, A. Léaustic, J. Berthet, R. Métivier, R. Guillot, S. Delbaere, K. Nakatani and P. Yu, Chem.–Asian J., 2017, 12, 853–859 CrossRef PubMed.
- S.-C. Pang, H. Hyun, S. Lee, D. Jang, M. J. Lee, S. H. Kang and K.-H. Ahn, Chem. Commun., 2012, 48, 3745–3747 RSC.
- Q. Ai, S. Pang and K.-H. Ahn, Chem.–Eur. J., 2016, 22, 656–662 CrossRef CAS PubMed.
- K. Suzuki, T. Ubukata and Y. Yokoyama, Chem. Commun., 2012, 48, 765–767 RSC.
- X. Meng, W. Zhu, Q. Zhang, Y. Feng, W. Tan and H. Tian, J. Phys. Chem. B, 2008, 112, 15636–15645 CrossRef CAS PubMed.
- W. Zhu, X. Meng, Y. Yang, Q. Zhang, Y. Xie and H. Tian, Chem.–Eur. J., 2010, 16, 899–906 CrossRef CAS PubMed.
- W. Zhu, L. Song, Y. Yang and H. Tian, Chem.–Eur. J., 2012, 18, 13388–13394 CrossRef CAS PubMed.
- Z. Yang, J. Cao, Y. He, J. H. Yang, T. Kim, X. Peng and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4563–4601 RSC.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- X. Y. Shen, Y. J. Wang, E. Zhao, W. Z. Yuan, Y. Liu, P. Lu, A. Qin, Y. Ma, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2013, 117, 7334–7347 CAS.
- R. Hu, C. F. A. Gómez-Durán, J. W. Y. Lam, J. L. Belmonte-Vázquez, C. Deng, S. Chen, R. Ye, E. Pena-Cabrera, Y. Zhong, K. S. Wong and B. Z. Tang, Chem. Commun., 2012, 48, 10099–10101 RSC.
- K. Cao, M. Farahi, M. Dakanali, W. M. Chang, C. J. Sigurdson, E. A. Theodorakis and J. Yang, J. Am. Chem. Soc., 2012, 134, 17338–17341 CrossRef CAS PubMed.
- X. Qian, Z. Zhu and K. Chen, Dyes Pigm., 1989, 11, 13–20 CrossRef CAS.
- I. Grabtchev and T. Philipova, Dyes Pigm., 1995, 27, 321–325 CrossRef CAS.
- S. Fukumoto, T. Nakashima and T. Kawai, Angew. Chem., Int. Ed., 2011, 50, 1565–1568 CrossRef CAS PubMed.
- K. Uchida, E. Tsuchida, Y. Aoi, S. Nakamura and M. Irie, Chem. Lett., 1999, 28, 63–64 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, A. Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. A. damo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Yang, Y. Xie, Q. Zhang, K. Nakatani, H. Tian and W. Zhu, Chem.–Eur. J., 2012, 18, 11685–11694 CrossRef CAS PubMed.
- S. H. Kawai, S. L. Gilat, R. Ponsinet and J.-M. Lehn, Chem.–Eur. J., 1995, 1, 285–293 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- B. Valeur, Molecular fluorescence: principles and applications, Wiley-VCH Verlag GmbH, New York, 2001 Search PubMed.
- A. Arjona-Esteban, M. Stolte and F. Würthner, Angew. Chem., Int. Ed., 2016, 55, 2470–2473 CrossRef CAS PubMed.
- M. Chen, H. Nie, B. Song, L. Li, J. Z. Sun, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 2901–2908 RSC.
- A. Marini, A. Muñoz-Losa, A. Biancardi and B. Mennucci, J. Phys. Chem. B, 2010, 114, 17128–17135 CrossRef CAS PubMed.
- P. Pasman, F. Rob and J. W. Verhoeven, J. Am. Chem. Soc., 1982, 104, 5127–5133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic routes, and NMR and high resolution mass spectra of BTE-O. See DOI: 10.1039/c7ra05157c |
| This journal is © The Royal Society of Chemistry 2017 |