
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand-free Pd(0)/SiO2-catalyzed aminocarbonylation of aryl iodides to amides under atmospheric CO pressure†
Qinhua Hu,
Lele Wang,
Chen Wang,
Yubin Wu,
Zhengxin Ding and
Rusheng Yuan *
*
State Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou 350116, P. R. China. E-mail: yuanrs@fzu.edu.cn
First published on 27th July 2017
Abstract
An efficient and facile route for CO-based carbonylation of aryl iodides with amines to synthesize amides has been established by using SiO2 supported Pd(0) as the catalyst in a mild basic environment (K2CO3). This ligand-free heterogeneous reaction model can afford amide products in good to excellent yields (up to 99%) under atmospheric CO pressure and moderate temperature. The supported catalyst also displayed a broad substrate scope, good functional group tolerance and good recyclability. These features render the as-provided carbonylation approach sustainable and applicable in organic synthesis.
Introduction
Aromatic amides are fundamental structural motifs in numerous natural products, materials, agrochemicals, and pharmaceutical agents.1,2 An effective and straightforward method to synthesize amides is transition-metal-catalyzed carbonylation of aryl halides using CO gas as a carbonyl group source because CO is not only inexpensive and readily available, but also an excellent ligand for transition metals due to its ability to act as a σ-donor and a π-acceptor.3–5 The conversion of CO to a carbonyl group is consistent with the demands of green chemistry.6The homogeneous palladium-catalyzed carbonylation of aryl halide with CO is well-known as a direct and efficient protocol for the synthesis of aromatic amides.7–11 Although these homogeneous palladium catalysts exhibited excellent selectivity and yields, the practical applications still suffer from the problems of separation and reuse of expensive palladium catalysts. In addition, the palladium residues in the product could be a serious issue in pharmaceutical applications. This gives rise to the development of recyclable and efficient heterogeneous palladium catalysts in a variety of organic chemical fields. Various heterogeneous palladium catalysts have been reported for the synthesis of amides by aminocarbonylation of aryl halide with CO gas using a support such as silica, carbon, ZIF-8, MCM-41, ionic liquid phases and organic polymers.12–19 However, these carbonylation reactions over supported palladium catalysts proceeded at high CO pressure and high reaction temperature. Dang group have reported the atmospheric pressure aminocarbonylation of aryl iodides using palladium nanoparticles supported on MOF-5 at 120 °C.20 Adolfsson group have used a highly dispersed nanopalladium catalysts supported on mesocellular foam for the aminocarbonylation reaction of aryl iodides in the presence of 1 atm of carbon monoxide.21 Cai group have developed MCM-41-supported bidentate phosphine palladium(II) complex, silica-supported poly-γ-diphenylarsinopropylsiloxane palladium complex and SiO2-supported sulfur and phosphine mixed bidentate palladium complex as catalysts for the carbonylation of aryl halides under atmospheric pressure of carbon monoxide at 90–130 °C.22–24 It should be mentioned that the addition of ligands or cocatalysts was required in most supported palladium catalyzed carbonylation reactions at ambient CO pressure.
Herein, we report an efficient and ligand-free approach to synthesize aromatic amides by using Pd(0) supported on silica as a robust catalyst for the carbonylation of aryl iodides with amines at atmospheric CO pressure and moderate temperatures. This recycling catalytic system provides a convenient access to a series of amides from a wide range of aryl iodides and amines in good to excellent yields. When amine was replaced by methanol and styrene, the formation of esters and α,β-unsaturated ketones can also be realized through this carbonylative process.
Results and discussion
Initially we carried out the model reaction of iodobenzene (0.4 mmol) with aniline (2 mL) using Pd/SiO2 as catalyst and K2CO3 (2 equiv.) as base under 1 atm of CO and solvent-free conditions at 80 °C for 24 hours. To our delight, the 97% yield of desired product 2c was obtained (Table 1, entry 1). In the absence of catalyst, no product was obtained. In the case of only using Pd/SiO2 without K2CO3, a small amount of the desired product (10%) was obtained (Table 1, entry 2). This indicates that the catalyst and base are essential for the above carbonylation reaction. Various SiO2-supported transition-metal catalysts were screened. As shown in Table 1, different transition-metal catalysts had a great influence on the yields of 2c. Supported Ru, Co, and Ni catalysts only provided a trace amount of the desired product, and other transition-metal catalysts like Ir, Au, Cu, Pt and Ag gave no product (Table 1, entries 3–10). Other catalyst supports provided the desired product in moderate to good yields (from 42% to 84%) under similar reaction conditions (entries 11–18). Therefore, Pd/SiO2 catalyst was found to be the optimum catalyst for the carbonylation of iodobenzene.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Yield (%) | Entry | Catalyst | Yield (%) |
| a Reaction conditions: iodobenzene (0.4 mmol, 1 equiv.), aniline (2 mL), and CO (1 atm), 1 wt% Pd/SiO2 (0.5 mol%), K2CO3 (2 equiv.), no solvent.b Without K2CO3.c Active charcoal. Yields based on GC analysis. | |||||
| 1 | Pd/SiO2 | 97 | 10 | Ag/SiO2 | — |
| 2b | Pd/SiO2 | 10 | 11 | Pd/Al2O3 | 75 |
| 3 | Ni/SiO2 | Trace | 12c | Pd/C | 42 |
| 4 | Co/SiO2 | Trace | 13 | Pd/Bi2O3 | 84 |
| 5 | Ru/SiO2 | Trace | 14 | Pd/V2O5 | 64 |
| 6 | Ir/SiO2 | — | 15 | Pd/WO3 | 77 |
| 7 | Au/SiO2 | — | 16 | Pd/CeO2 | 76 |
| 8 | Cu/SiO2 | — | 17 | Pd/ZrO2 | 67 |
| 9 | Pt/SiO2 | — | 18 | Pd/TiO2 | 76 |
The different reaction parameters such as solvent, base, catalyst loading, temperature and time were tested to show their effect on this reaction. The presence of solvents like DMSO, acetonitrile and DMF provided the desired product in moderate yields, and only trace amounts of product were detected when using benzene and THF as solvents (Table 2, entries 1–5). From the yields of the model reaction with and without K2CO3, we found that the addition of base played a significant role in the formation of amide. Among the bases used, K2CO3 exhibited a best yield of 97% for the final product, while NaOH and CsCO3 were also effective with only a slight decrease in the yield (93% and 92%) (entries 7 and 9). When Na2CO3 and KOH were used, only trace amounts of product were detected (entries 8 and 10). Given that NaOH is more readily subject to deliquescence and CsCO3 is more expensive, K2CO3 is considered to be the optimal base. The catalyst loading from 1 wt% to 2 wt% had no obvious effect on the product yield. From the view of economy, 1 wt% catalyst loading was chosen. An obvious increase in reaction yields was observed from 50 °C to 80 °C, but a higher reaction temperature resulted in a slight decrease in the yields (Fig. 1a). The product yield increased gradually with reaction time, and reached the highest yield (97%) at 24 h (Fig. 1b). At this moment, iodobenzene has been exhausted almost completely. A longer reaction time than 24 h would result in the precipitation of N-phenylbenzamide 2C on the solid catalyst (Table S1†).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Catalyst loading (mol%) | Base | Yield (%) |
| a Reaction conditions: iodobenzene (0.4 mmol, 1 equiv.), aniline (2 mL), and CO (1 atm), reaction time: 24 h, reaction temperature: 80 °C, 1 wt% Pd/SiO2 (0.5 mol%), base (2 equiv.).b Solvent (2 mL), aniline (2 equiv.).c 1.5 wt% Pd/SiO2 (0.75 mol%).d 2 wt% Pd/SiO2 (1 mol%). Yields based on GC analysis. | ||||
| 1b | DMSO | 0.5 | K2CO3 | 74 |
| 2b | DMF | 0.5 | K2CO3 | 54 |
| 3b | Acetonitrile | 0.5 | K2CO3 | 78 |
| 4b | Benzene | 0.5 | K2CO3 | Trace |
| 5b | THF | 0.5 | K2CO3 | Trace |
| 6 | — | 0.5 | K2CO3 | 97 |
| 7 | — | 0.5 | NaOH | 93 |
| 8 | — | 0.5 | Na2CO3 | Trace |
| 9 | — | 0.5 | CsCO3 | 92 |
| 10 | — | 0.5 | KOH | Trace |
| 11c | — | 0.75 | K2CO3 | 96 |
| 12d | — | 1 | K2CO3 | 91 |
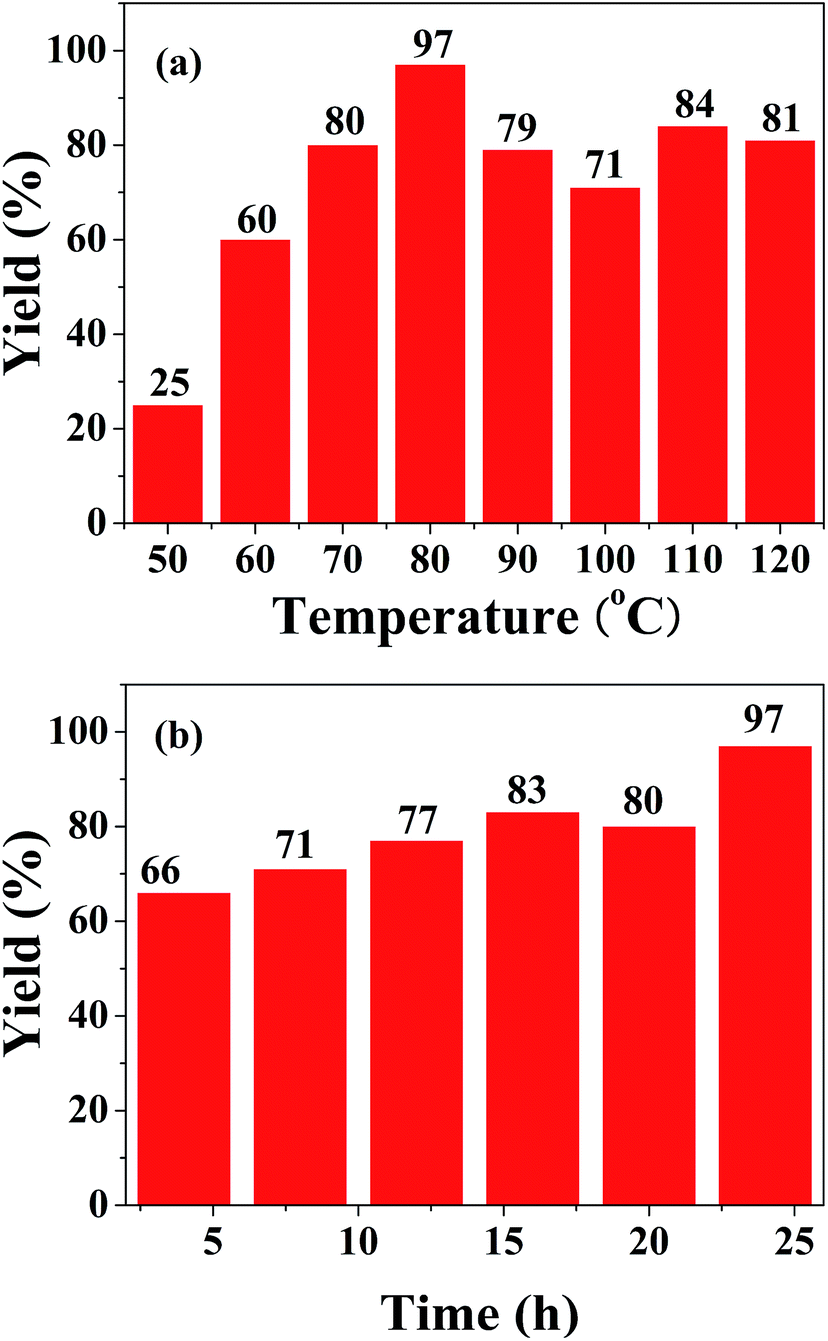 | ||
| Fig. 1 Effect of (a) temperature and (b) time for carbonylation reaction. Reaction conditions: iodobenzene (0.4 mmol, 1 equiv.), aniline (2 mL), and CO (1 atm), 1 wt% Pd/SiO2 (0.5 mol%), K2CO3 (2 equiv.). Yields based on GC analysis. | ||
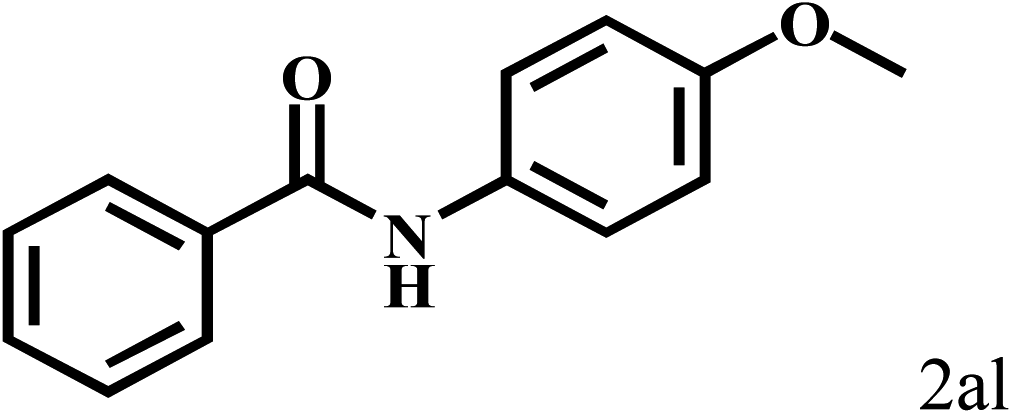
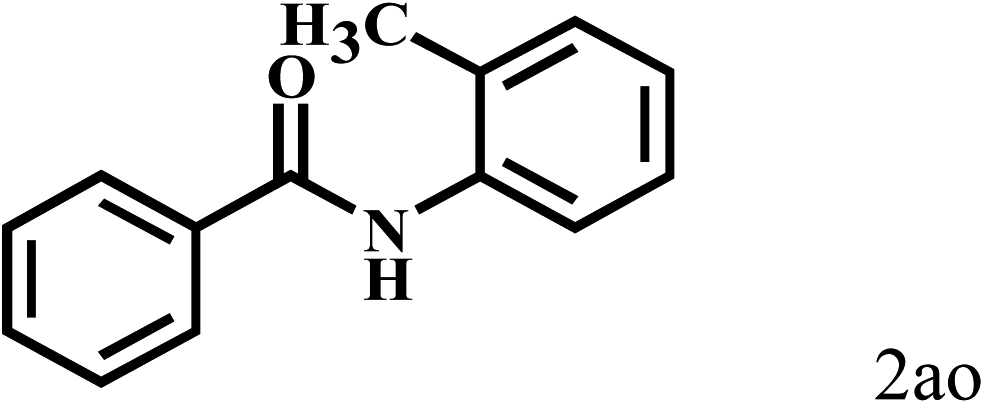
Having optimized the reaction conditions for the synthesis of amide, the substrate scope was further investigated. As shown in Table 3, aryl iodides bearing electron-donating and electron-withdrawing substituents on its phenyl ring were well tolerated, which gave the corresponding amides in moderate to high yields (56% to 99%) (entries 1–11). Sterically hindered o-iodophenol exhibited much lower reactivity (entry 8), as compared with that of p-iodophenol (98%). The o-, m- and p-substituted aryl iodides with –CF3 group also provided excellent yields (entries 4–6). Different substituted amines were also tested. Good yields of 84% to 99% were obtained for the amines with electron-donating groups (entries 12–16). It is noteworthy that p-substituted amines gave a slightly decreased yield compared with that of o- and m-substituted amines. The substituted amines with electron-withdrawing halogen groups were also tolerated, and gave 66% to 91% yields to the corresponding amides in the order I > Br > Cl (entries 18–21). When the amines that have a substituent attached to the amino group were used, the desired products were obtained in moderate yields (entries 22). In the case of using N-methyl-m-methylaniline, the yield of 2aq was reduced greatly possibly due to the steric hindrance effect (entry 17). Alkyl and cyclic amines were also reactive, but relatively low yields were obtained (entries 23–25) under the current conditions.
|
||||
|---|---|---|---|---|
| Entry | Aryl iodides | Amines | Products | Yield (%) |
| a Reaction conditions: aryl iodides (0.4 mmol, 1 equiv.), amines (2 mL), and CO (1 atm), 1 wt% Pd/SiO2 (0.5 mol%), K2CO3 (2 equiv.).b Amines (2 equiv.), acetonitrile (2 mL) as solvent.c Yields based on GC-MS analysis.d Amines (2 equiv.), acetonitrile (2 mL) as solvent, yields based on GC-MS analysis. Yields based on 1H NMR analysis. | ||||
| 1 | 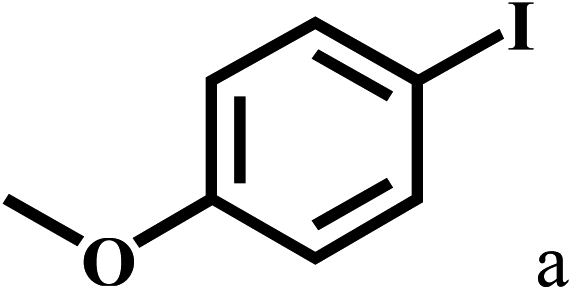 |
2b | 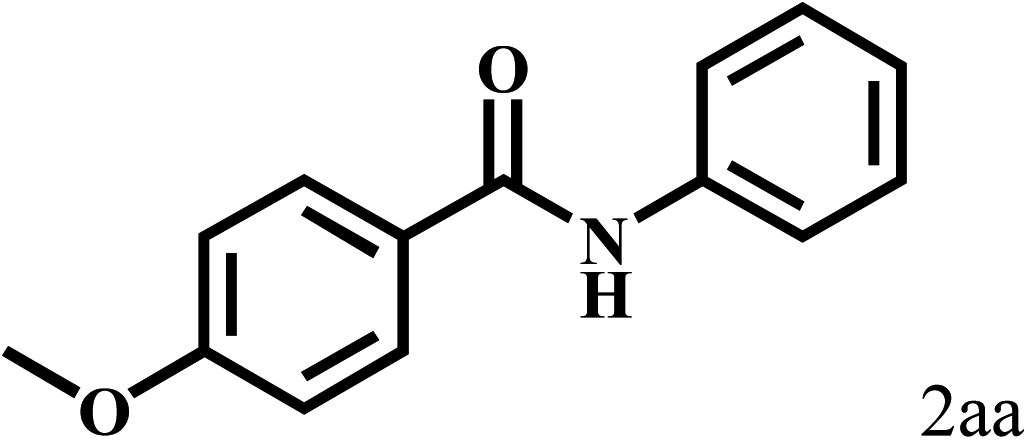 |
96 |
| 2 | 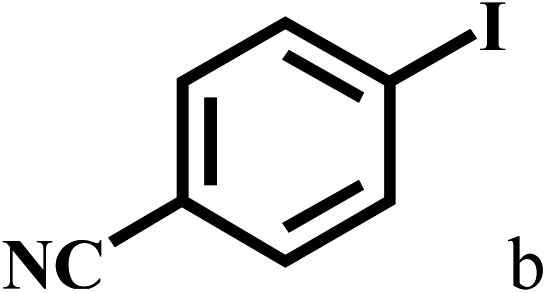 |
2b | 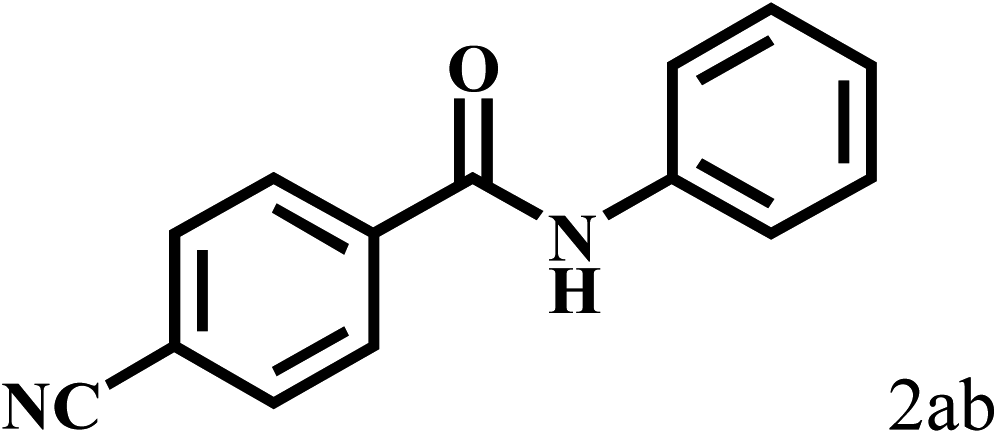 |
98 |
| 3 | 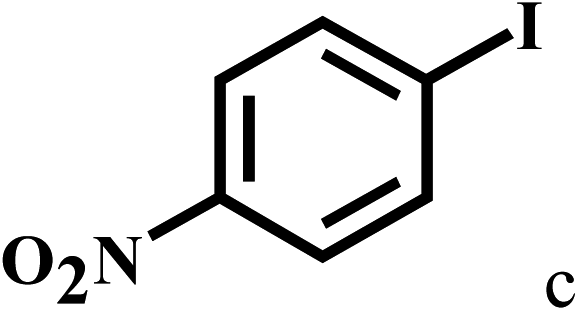 |
2b | 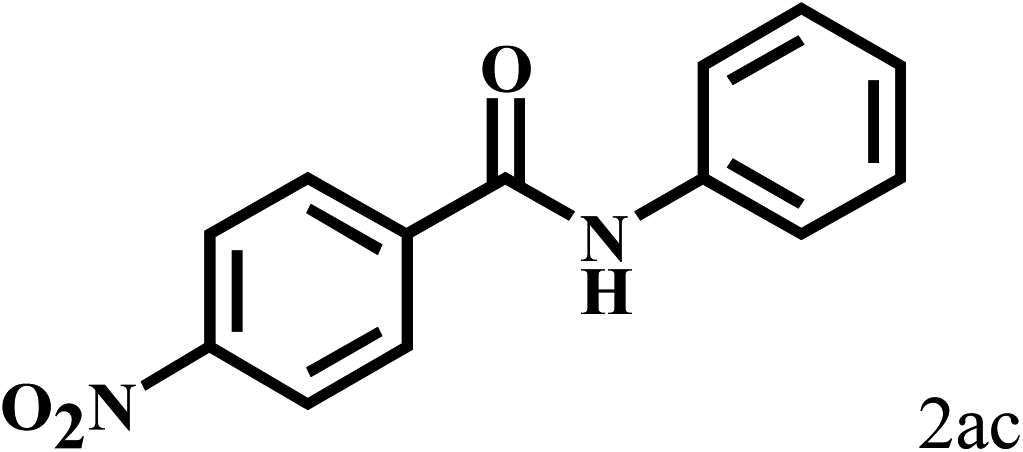 |
91 |
| 4 | 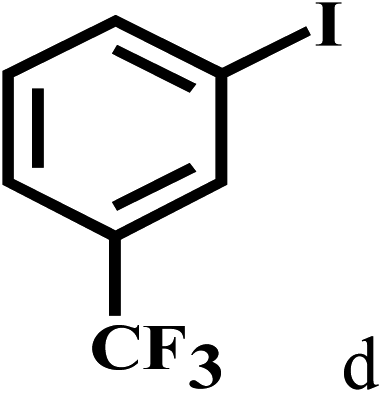 |
2b | 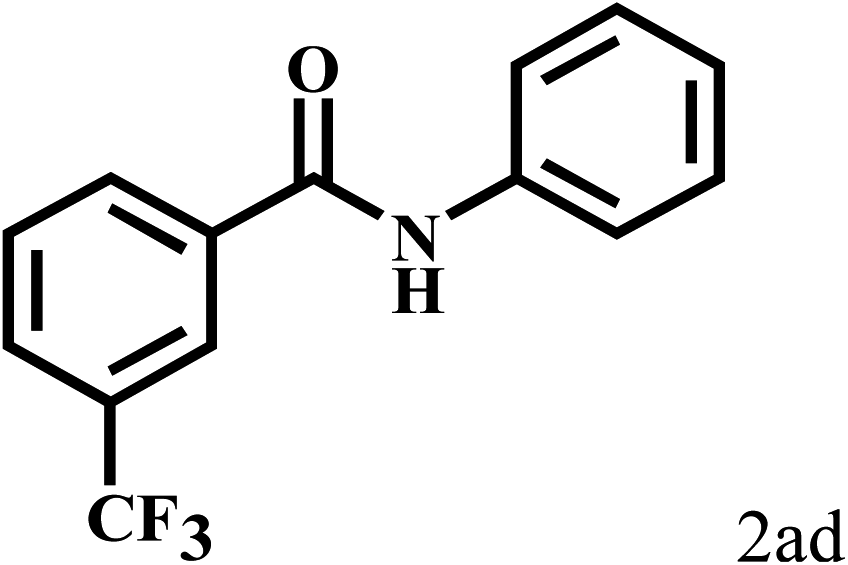 |
99 |
| 5 | 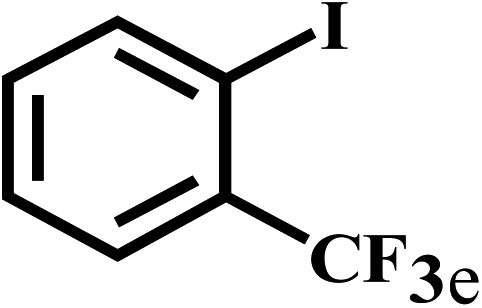 |
2b | 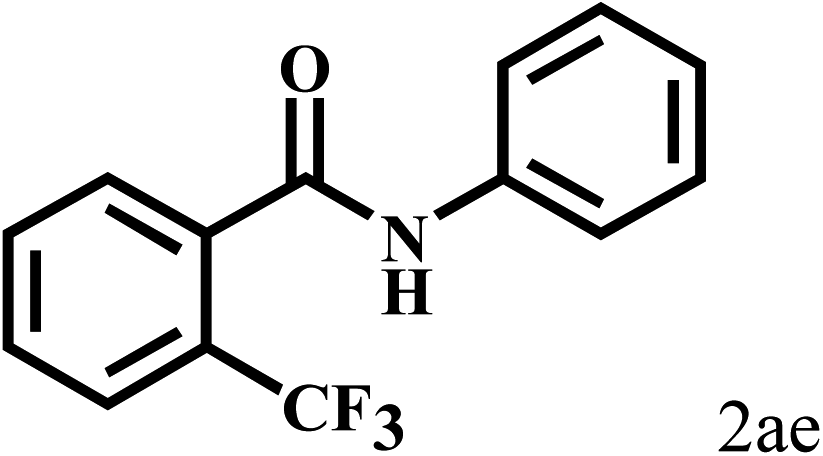 |
95 |
| 6 | 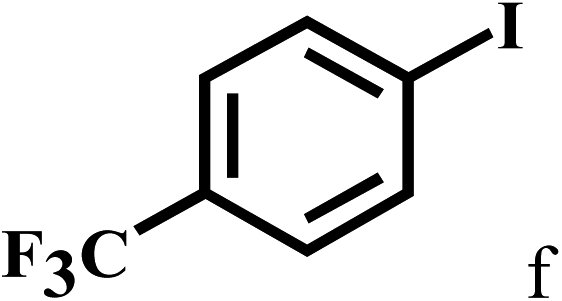 |
2b | 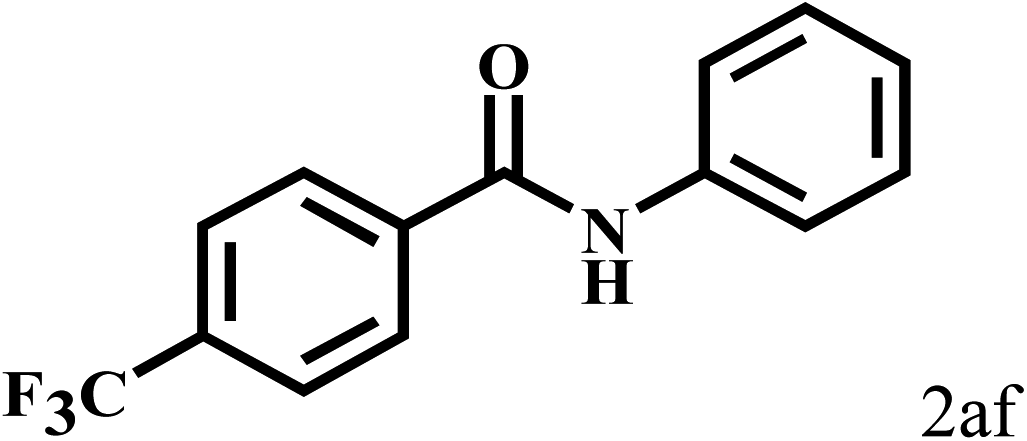 |
85 |
| 7 | 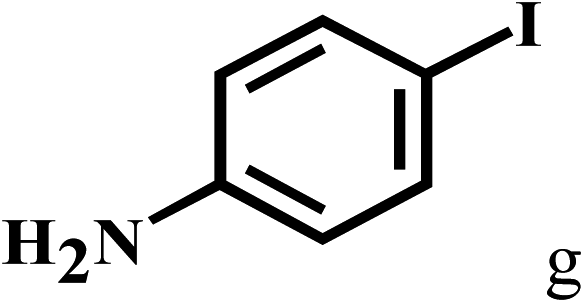 |
2b | 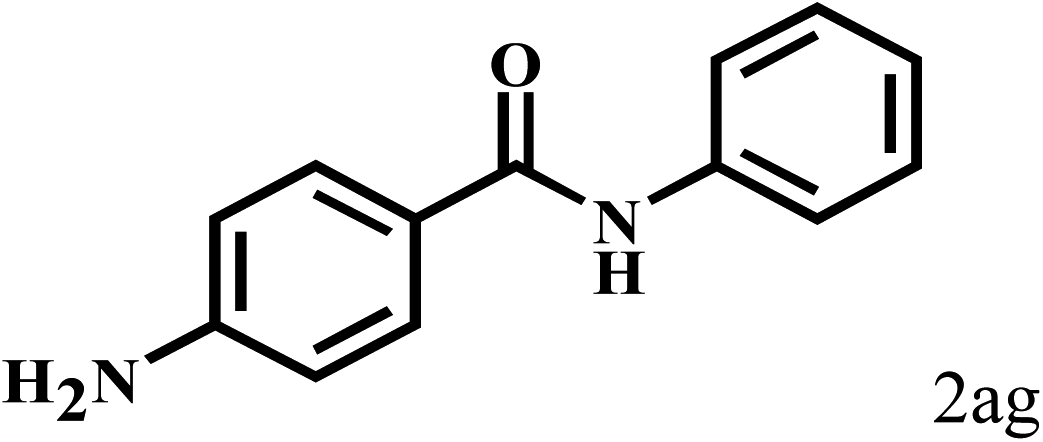 |
98 |
| 8c | 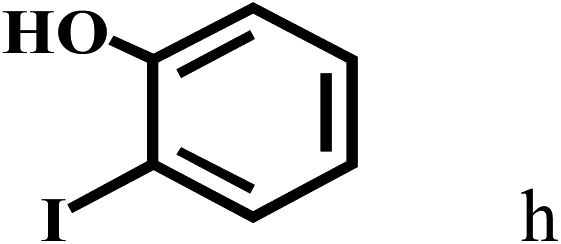 |
2b | 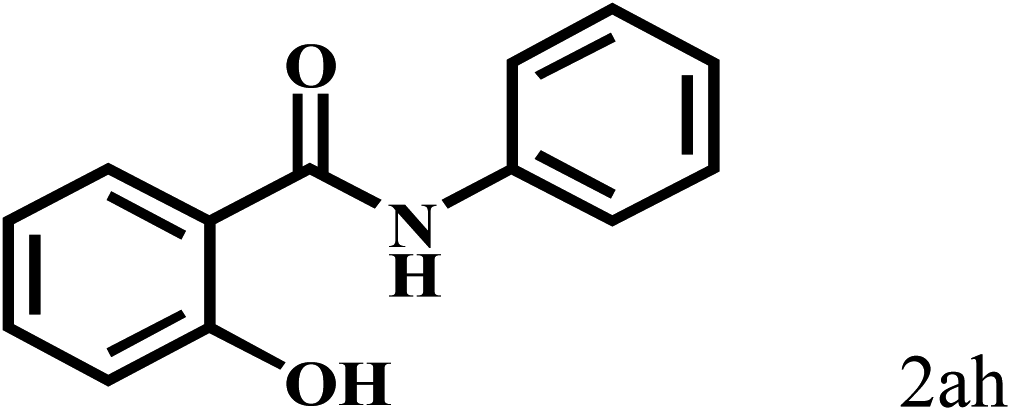 |
56 |
| 9 | 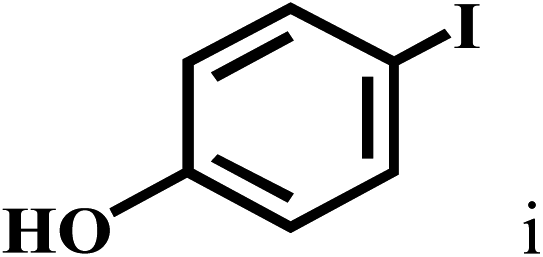 |
2b | 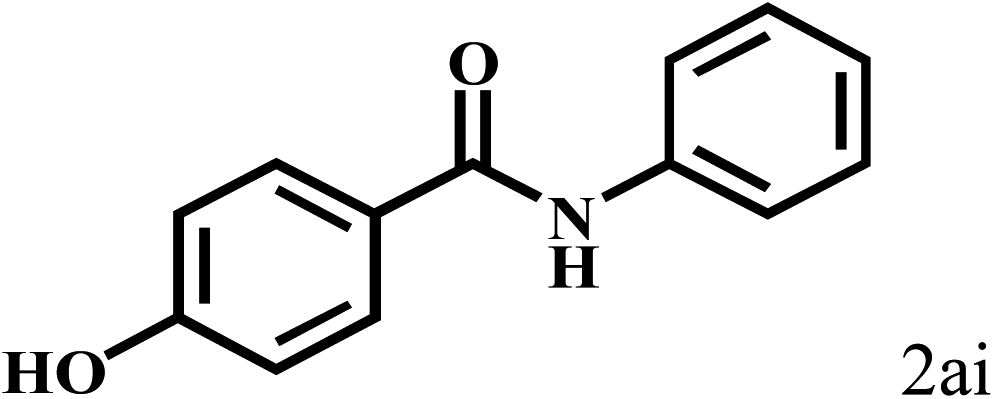 |
98 |
| 10 | 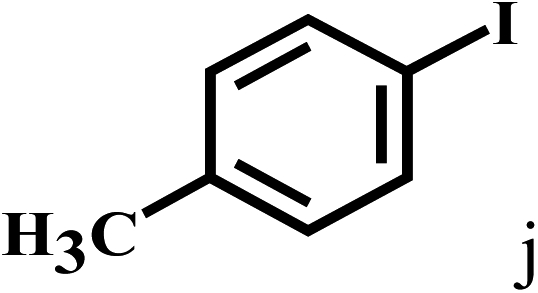 |
2b | 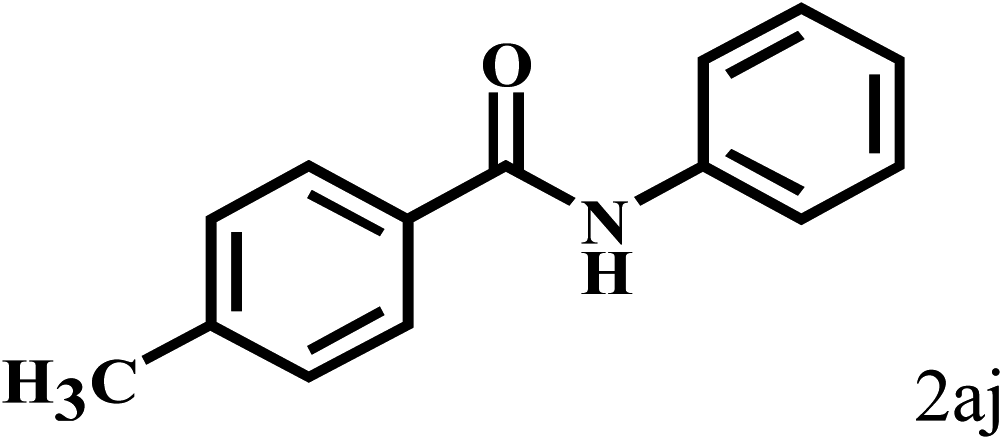 |
99 |
| 11 |  |
2b | 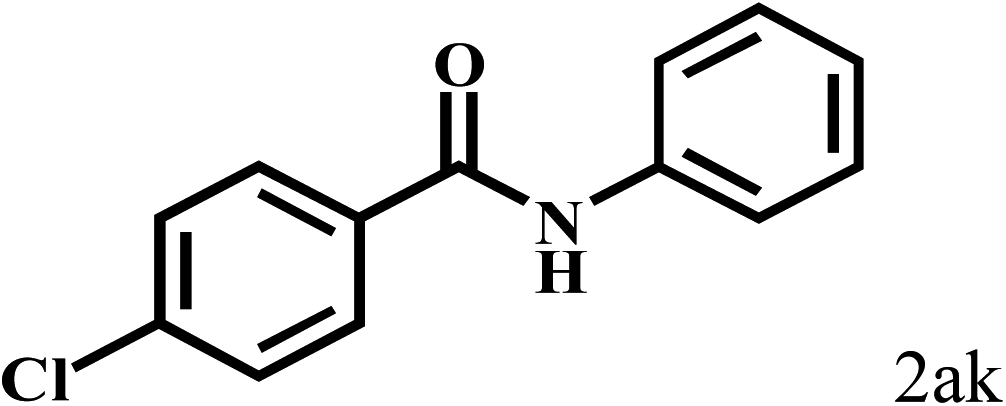 |
94 |
| 12b | 2a | 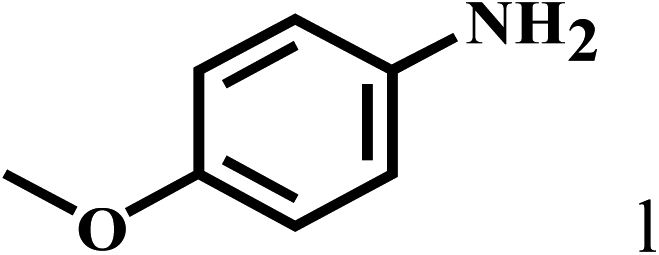 |
 |
89 |
| 13 | 2a | 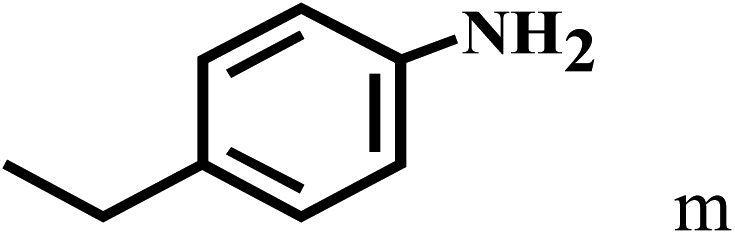 |
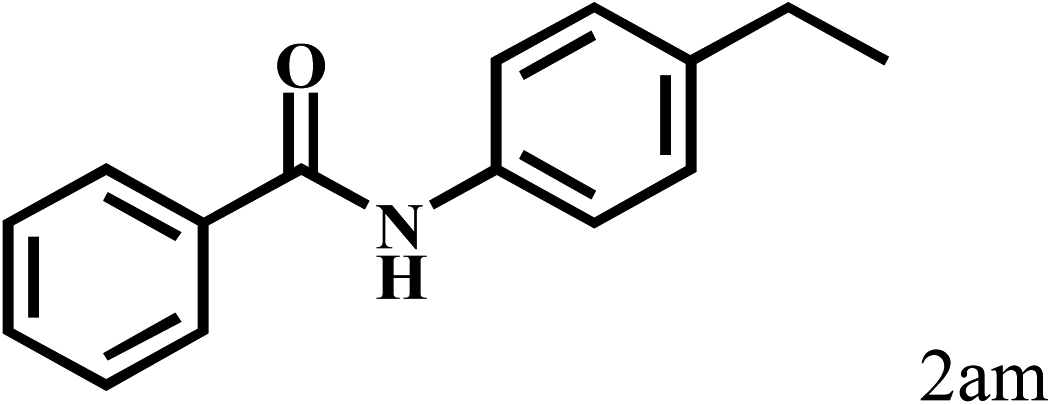 |
92 |
| 14b | 2a | 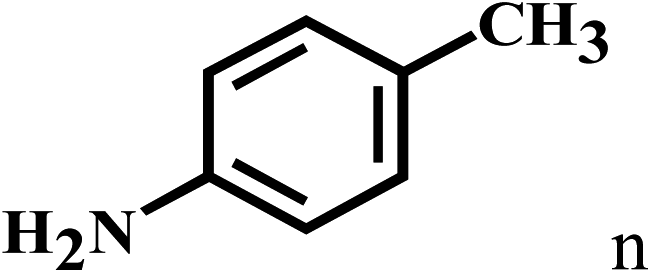 |
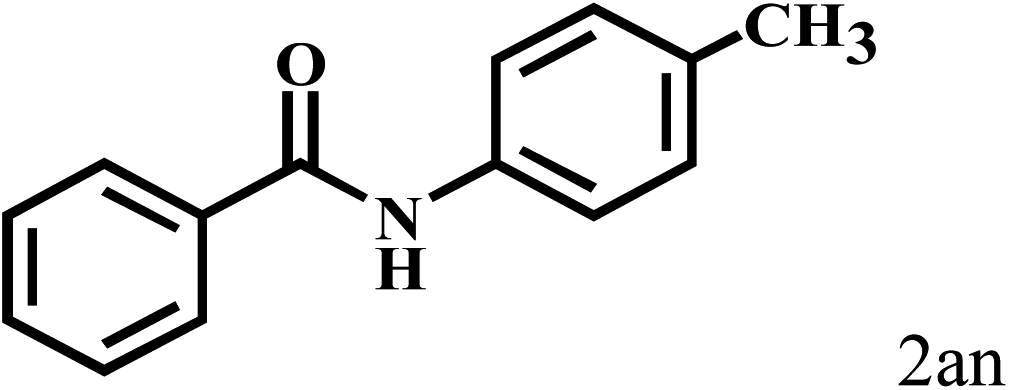 |
84 |
| 15 | 2a | 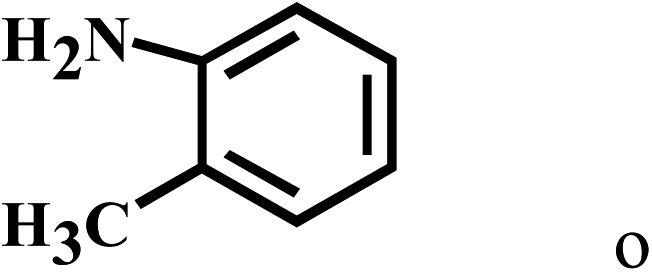 |
 |
99 |
| 16 | 2a | 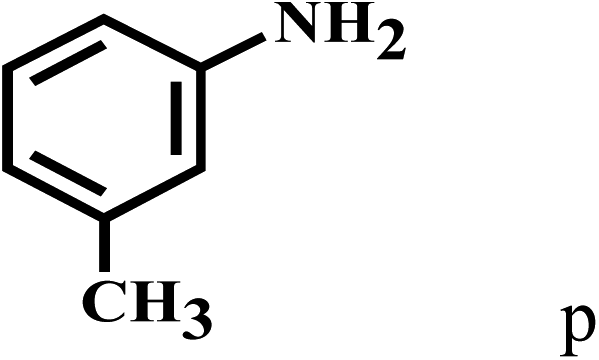 |
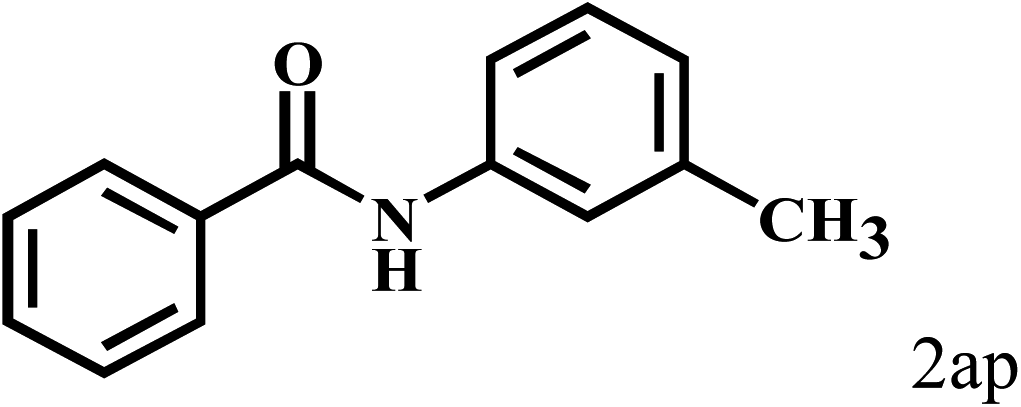 |
98 |
| 17c | 2a | 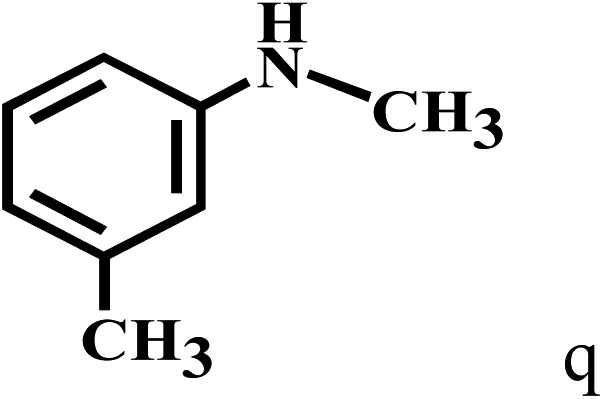 |
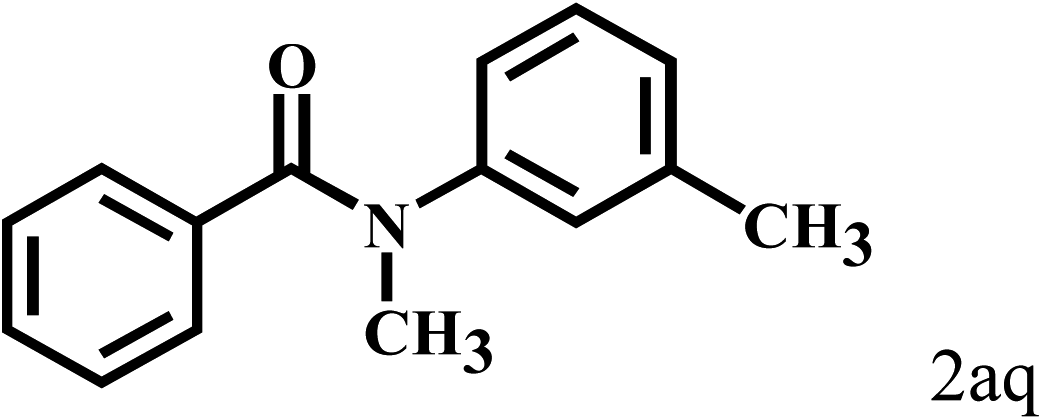 |
11 |
| 18b | 2a | 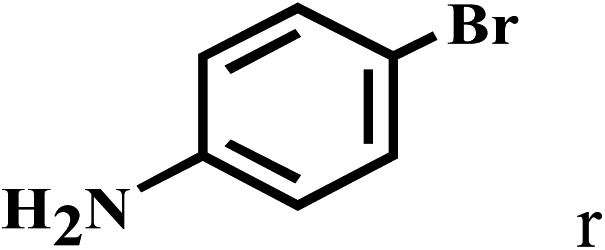 |
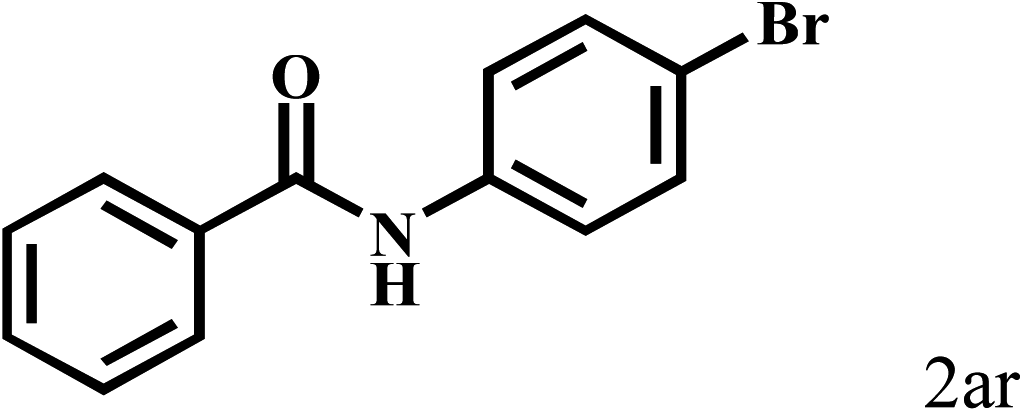 |
87 |
| 19d | 2a | 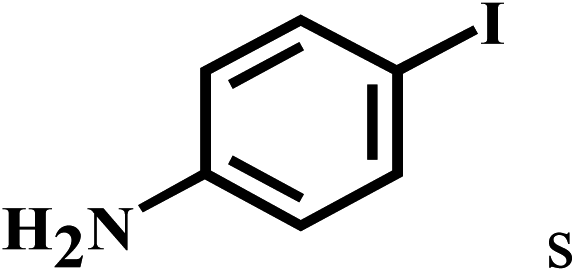 |
 |
91 |
| 20b | 2a | 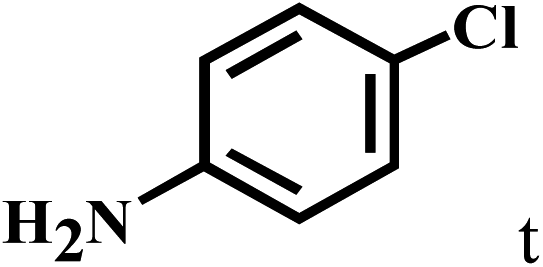 |
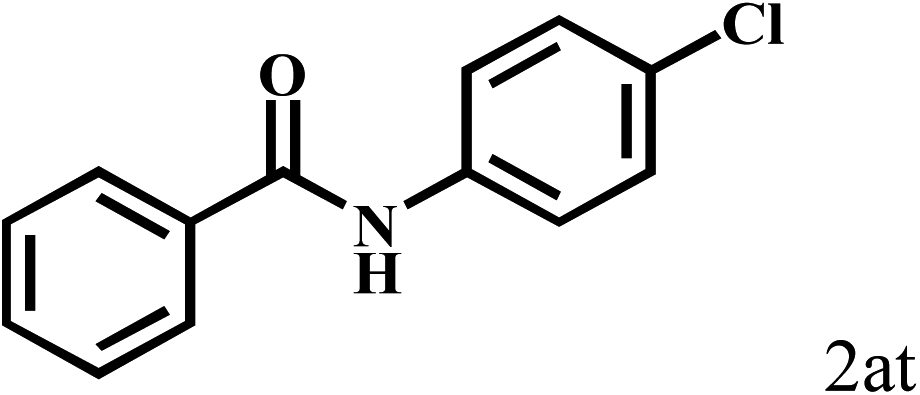 |
77 |
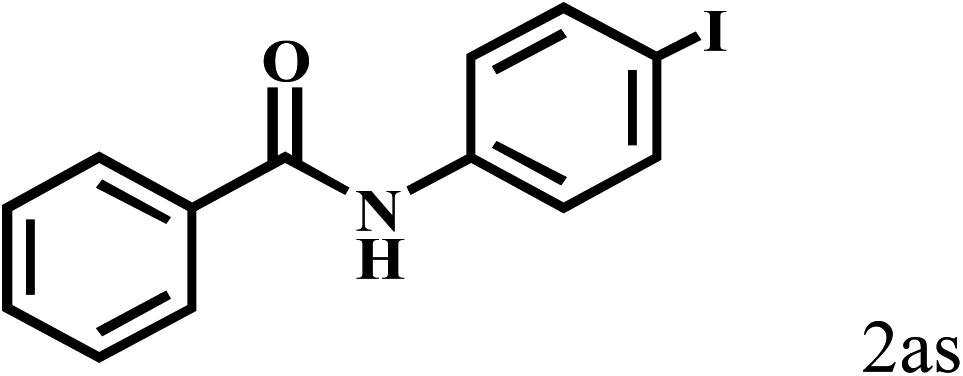
| 21c | 2a | 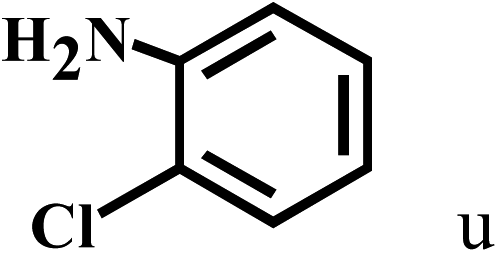 |
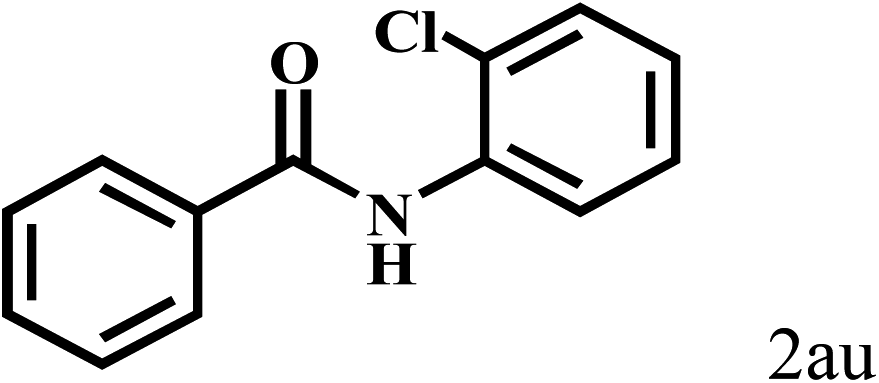 |
66 |
| 22c | 2a | 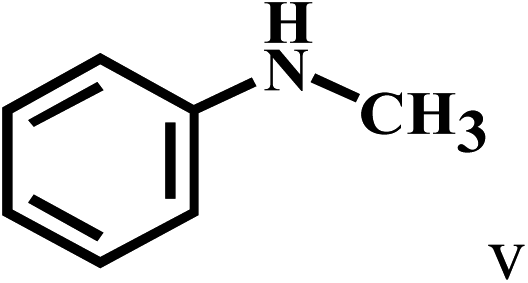 |
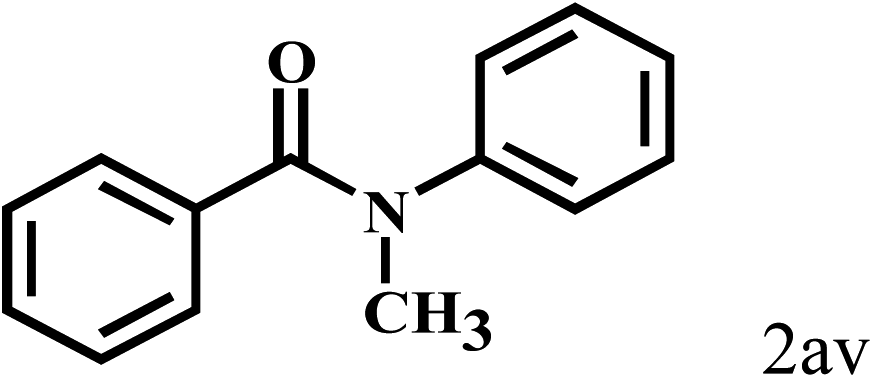 |
55 |
| 23 | 2a | 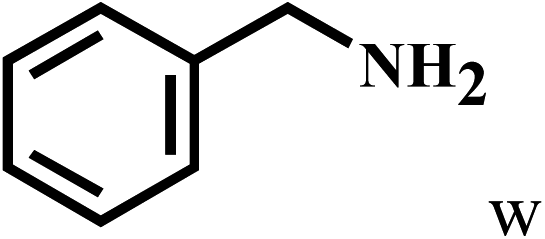 |
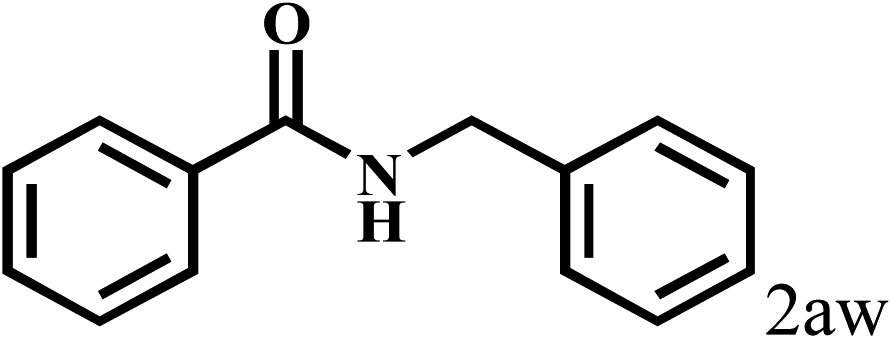 |
59 |
| 24 | 2a | 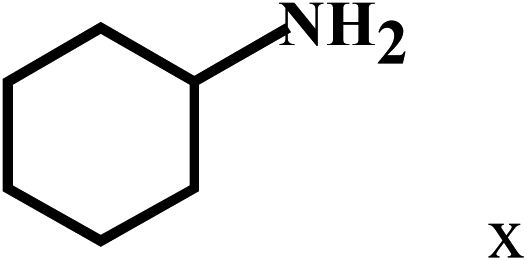 |
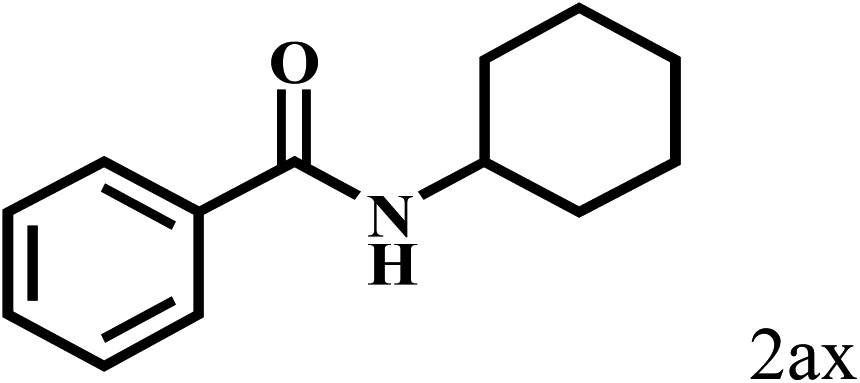 |
35 |
| 25c | 2a | 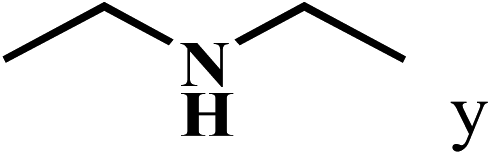 |
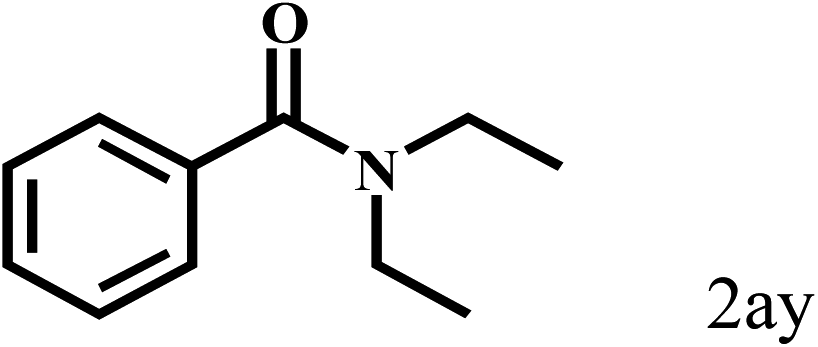 |
15 |
The recycling of Pd/SiO2 catalyst was evaluated for the aminocarbonylation of aryl iodides under the optimized reaction conditions. After five cycles, the results are shown in Fig. 2. There is little change in the yields of the desired product, which indicated that the recovered catalyst showed a good reaction activity. In order to check the leaching of palladium metal, the fresh catalyst, the third and fifth recycled catalyst were subjected to the inductively coupled plasma optical emission spectrometry (ICP-OES) technique. The palladium content of the above three samples were 1.06 wt%, 1.00 wt% and 0.94 wt%, respectively. That is to say, no obvious catalyst leaching was observed.
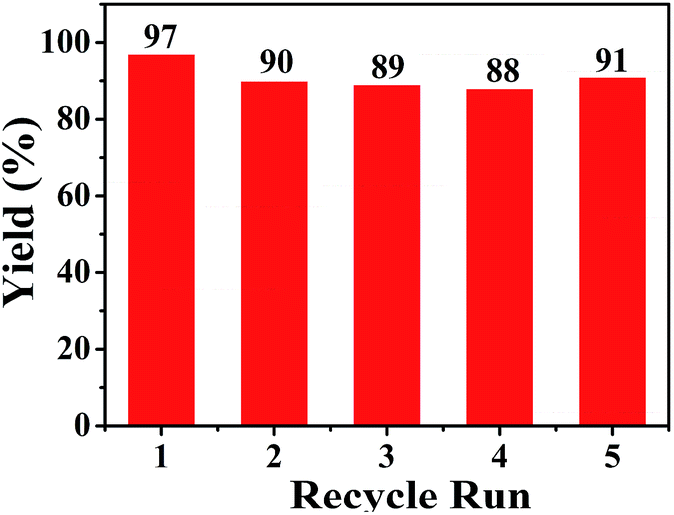 | ||
| Fig. 2 Recycle study of Pd/SiO2 for the synthesis of amide. Reaction conditions: iodobenzene (0.4 mmol, 1 equiv.), aniline (2 mL), and CO (1 atm), 1 wt% Pd/SiO2 (0.5 mol%), K2CO3 (2 equiv.). Yields based on GC analysis. | ||
The oxidation state of palladium in Pd/SiO2 catalyst before and after recycling was examined by XPS. As shown in Fig. 3, the peaks around 335.7 eV and 340.8 eV for the fresh Pd/SiO2 catalyst can be readily assigned to Pd(0) 3d5/2 and 3d3/2 bands, in agreement with the literature reports.25–27 After the Pd/SiO2 catalyst was recycled for five cycles, the two peaks were still located at the same binding energy. Further evidence for Pd was also provided by HRTEM images in Fig. 4. For both samples, well-resolved lattice fringes with an interplanar spacing of about 0.23 nm was observed, which corresponds to the (111) lattice plane of face-centered cubic Pd metal.28
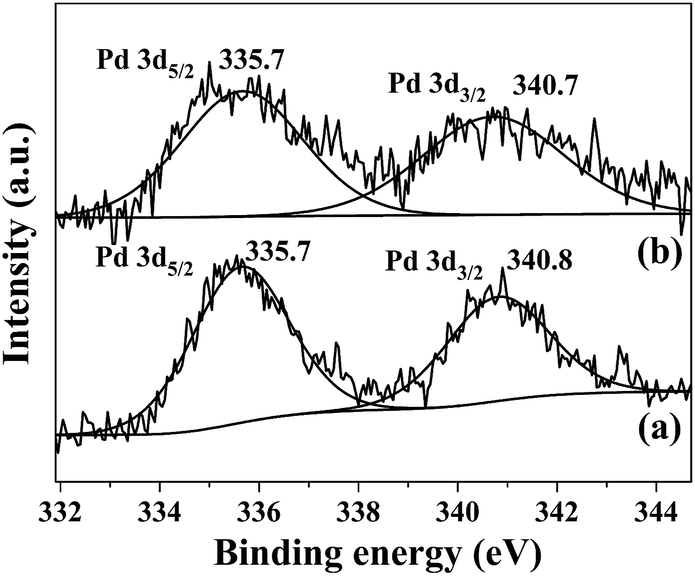 | ||
| Fig. 3 XPS spectra of (a) fresh Pd/SiO2 and (b) recycled Pd/SiO2 for five cycles. | ||
 | ||
| Fig. 4 HRTEM images of (a) fresh Pd/SiO2 and (b) recycled Pd/SiO2 for five cycles. | ||
In order to determine the reaction mechanism, we conducted a series of studies. The addition of 3 equiv. of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO, a typical radical scavenger) cannot inhibit the reaction, whereas only trace amounts of desired product was obtained with the addition of 3 equiv. of electron scavenger (1,4-benzoquinone, BQ) (Table 4). This indicated that the aminocarbonylation reaction proceeded via a single electron transfer process, rather than via a free radical pathway. When triethylamine was substituted for aniline in the model aminocarbonylation reaction, no amide product was found in the final reaction mixture. This is different from the case using primary or secondary amine as the substrate that has hydrogen atom bonded to N atom. Thus, we think that the base-promoted deprotonation of amine play an indispensable role in the occurrence of the aminocarbonylation reaction. After the reaction of iodobenzene with aniline, the pH of the final reaction mixture decreased. Meanwhile, a negligible amount of hydrogen was detected by gas chromatography. From these results, we can conclude that the hydrogen in the final reaction solution exists mainly in the form of hydrogen ion. Ion chromatography analysis detected a small amount of iodide ions, but potassium ions were not present. Thus, the base K2CO3 was not dissolved in solution, and the combination of iodide ion and hydrogen ion formed HI.
| Entry | Catalyst | Additive | Yield (%) |
|---|---|---|---|
| a Reaction conditions: iodobenzene (0.4 mmol, 1 equiv.), aniline (2 mL), and CO (1 atm), 1 wt% Pd/SiO2 (0.5 mol%), K2CO3 (2 equiv.). Yields based on GC analysis. | |||
| 1 | Pd/SiO2 | BQ | Trace |
| 2 | Pd/SiO2 | TEMPO | 88 |
| 3 | Pd/SiO2 | None | 97 |
On the basis of our results and previous reports.29,30 a plausible reaction mechanism of Pd/SiO2 catalyzed carbonylation process is proposed in Scheme 1. Initially, the complex C is formed by oxidative addition of aryl iodide B to Pd(0) species A under the reaction conditions. Next, the insertion of CO into the phenyl–palladium bond of complex C forms acylpalladium intermediate D. In the presence of a base, the nucleophilic amine coordinates with intermediate D to give complex E via the elimination of HI. Finally, reductive elimination from complex E affords amide product F and Pd(0) species A.
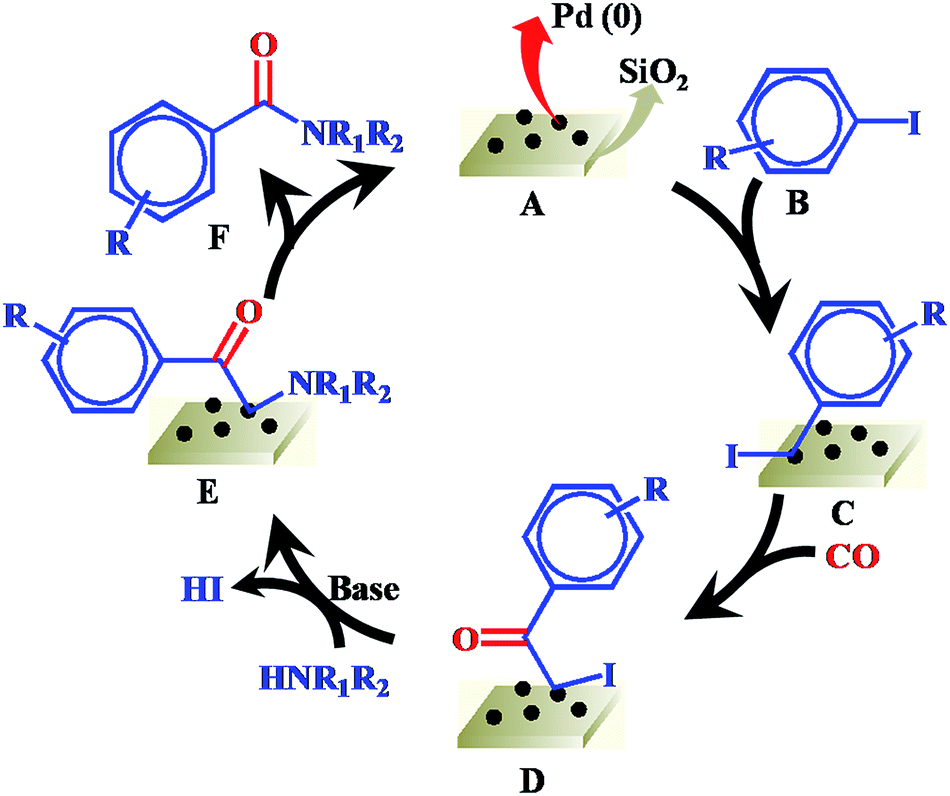 | ||
| Scheme 1 Plausible reaction mechanism. | ||
In addition, the proposed carbonylation method is also suitable for the formation of esters and α,β-unsaturated ketones when amine was replaced by methanol and styrene (Scheme 2). The reaction of iodobenzene with styrene did produce the phenyltrans–styrylketone product in acetonitrile as solvent at 80 °C for 40 h, albeit in low yield (<10%). When using methanol as staring material, methyl benzoate was also constructed effectively in 62% yield at 70 °C for 6 h. Although the above reaction conditions are not optimal, these results demonstrate the feasibility of our system for the synthesis of esters and α,β-unsaturated ketones.
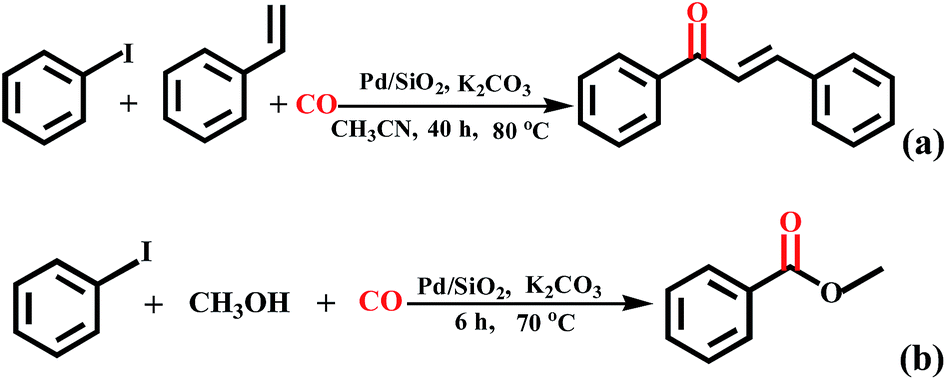 | ||
| Scheme 2 Extended reactions. | ||
Conclusions
An efficient, ligand-free Pd(0)/SiO2 catalyst has been applied to the carbonylation of aryl iodines with amines at atmospheric CO pressure and moderate temperature, which avoids the need for specialized pressure reactors and the dangers associated with handling presurized CO. The supported catalyst can be easily recycled and no obvious decrease in reaction activity was observed after five cycles. This prorocol offered a green and sustainable way to the construction of the amide products, and the reaction mode can also be applied to other synthetic reactions, such as the construction of esters and α,β-unsaturated ketones.Experimental section
General information
All reagents were purchased from commercial suppliers and used without further purification. All reactions were carried out in a dry 10 mL glass tube reactor, and the products were identified by GC, GC-MS and 400 MHz instrument. GC analysis was performed in a GC Agilent Technologies 7890A equipped with a flame ionization detector by using an HP-5 5% phenyl methyl siloxane column (30 m × 0.32 mm × 0.5 μm). GC-MS analysis was performed in a GC Agilent Technologies 7890B that was fitted with a mass detector Agilent Technologies 5977A MSD. 1H NMR spectra and 13C NMR spectra were recorded at 400 MHz and at 100 MHz in DMSO-d6 on a Bruker AVANCE III 400 MHz spectrometer. X-ray photoelectron spectroscopy (XPS) measurements of Pd/SiO2 were performed on a PHI Quantum 2000 XPS system, and the binding energies were calibrated to the C 1s peak at 284.8 eV of the surface adventitious carbon. Inductively coupled plasma optical emission spectrometry (ICP-OES) technique of Pd/SiO2 was performed on a OPTIMA 8000. High resolution transmission electron microscopy (HRTEM) imaging was obtained on a JEOL 2010F TEM at 200 kV accelerating voltage. The crude products were monitored by analytical thin layer chromatography using commercial thin layer chromatography, and purified by exquisite column chromatography silica gel (100–200 mesh).Preparation of palladium catalyst
We adopted precipitation deposition method to prepare palladium catalyst by using chloropalladic acid as the palladium source. First of all, 2 g milled commercial silica powders (300–400 mesh) was dissolved in 50 mL deionized water with mechanical agitation for 0.5 h. Then different amounts of chloropalladic acid solution was added into the as-obtained SiO2-water to make the load is 1 wt%, 1.5 wt%, 2 wt%, respectively. The pH of this solution was adjusted to 9 through slowly adding KOH solution and stirred for 7 hours at room temperature, followed by drying at 80 °C. After grinding, the dried solid was dissolved in deionized water and reduced by sodium borohydride solution under stirring at room temperature for 12 h. Finally, the Pd/SiO2 catalyst was washed with deionized water and centrifuged until the ionic degree less than 10, the obtained solid product was dried in a vacuum at 60 °C overnight. The preparation of other catalysts are similar to that of Pd/SiO2.General procedure of palladium catalyzed synthesis of amide
In a typical experiment, iodobenzene (0.4 mmol, 1 equiv.), aniline (2 mL), Pd/SiO2 (0.5 mol%), K2CO3 (2 equiv.) were added into a dry 10 mL glass tube reactor with a stirring bar. The tube reactor was sealed and evacuated by a vacuum pump. Then CO gas was introduced through a balloon being attached to the air inlet of the reactor. Finally, the reactor with carbon monoxide balloons was put into a stirred oil bath at 80 °C for 24 h (the subsequent reaction process is the same as that discussed above for the carbonylation reaction). In the case of amines on solid phase, acetonitrile was used as solvent. The amount of amine is 2 equiv., the reaction process remains unchanged. The reaction mixture was separated from the catalyst by simple centrifugation, and the liquid was analyzed by GC, GC–MS, and 1H NMR techniques.The recycling of palladium catalyst
The recyclability of Pd/SiO2 for the synthesis of amide was carried out under the optimized reaction conditions. After completing the reaction, the reactor was cooled to room temperature and the remaining CO gas was carefully vented out to a hood after 24 hours. The catalyst was separated from the mixture and washed with acetonitrile (2 mL) and deionized water (2 mL) to remove the residual organic material. After drying under vacuum, the obtained catalyst was used for a new reaction cycle.Acknowledgements
This work was financially supported by the National Nature Science Foundation of China 21643009, the National Basic Research Program of China 2014CB239303, the Natural Science Foundation of Fujian Province of China 2015J01046, and the Independent Research Project of State Key Laboratory of Photocatalysis on Energy and Environment 2014B01.Notes and references
- (a) G. Zhang, B. Gao and H. Huang, Angew. Chem., Int. Ed., 2015, 54, 7657 CrossRef CAS PubMed; (b) L. Zhang, S. Wang, S. Zhou, G. Yang and E. Sheng, J. Org. Chem., 2006, 71, 3149 CrossRef CAS PubMed; (c) Y. Li, F. Zhu, Z. Wang and X.-F. Wu, ACS Catal., 2016, 6, 5561 CrossRef CAS; (d) R. García-Álvarez, A. E. Díaz-Álvarez, J. Borge, P. Crochet and V. Cadierno, Organometallics, 2012, 31, 6482 CrossRef; (e) C. Bai, X. Yao and Y. Li, ACS Catal., 2015, 5, 884 CrossRef CAS.
- (a) J. W. Clader, J. Med. Chem., 2004, 47, 1 CrossRef CAS PubMed; (b) W. Wu, Z. Zhang and L. S. Liebeskind, J. Am. Chem. Soc., 2011, 133, 14256 CrossRef CAS PubMed; (c) S. Eichner, T. Eichner, H. G. Floss, J. Fohrer, E. Hofer, F. Sasse, C. Zeilinger and A. Kirschning, J. Am. Chem. Soc., 2012, 134, 1673 CrossRef CAS PubMed; (d) B. N. Naidu, M. E. Sorenson, T. P. Connolly and Y. Ueda, J. Org. Chem., 2003, 68, 10098 CrossRef CAS PubMed.
- (a) P. Nordeman, L. R. Odell and M. Larhed, J. Org. Chem., 2012, 77, 11393 CrossRef CAS PubMed; (b) S. Fukuoka, Ind. Eng. Chem. Res., 2016, 55, 4830 CrossRef CAS; (c) Y. Jo, J. Ju, J. Choe, K. H. Song and S. Lee, J. Org. Chem., 2009, 74, 6358 CrossRef CAS PubMed.
- E. C. Gaudino, D. Carnaroglio, K. Martina, G. Palmisano, A. Penoni and G. Cravotto, Org. Process Res. Dev., 2015, 19, 499 CrossRef.
- J. R. Martinelli, D. A. Watson, D. M. M. Freckmann, T. E. Barder and S. L. Buchwald, J. Org. Chem., 2008, 73, 7102 CrossRef CAS PubMed.
- X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1 CrossRef CAS PubMed.
- (a) H. Li, H. Neumann, M. Beller and X.-F. Wu, Angew. Chem., Int. Ed., 2014, 53, 3183 CrossRef CAS PubMed; (b) J. R. Martinelli, D. M. M. Freckmann and S. L. Buchwald, Org. Lett., 2006, 8, 4843 CrossRef CAS PubMed.
- (a) A. Schoenberg, I. Bartolet and R. F. Heck, J. Org. Chem., 1974, 39, 3318 CrossRef CAS; (b) A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327 CrossRef CAS; (c) A. Schoenberg and R. F. Heck, J. Am. Chem. Soc., 1974, 96, 7761 CrossRef CAS.
- (a) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef PubMed; (b) Q. Liu, G. Li, J. He, J. Liu, P. Li and A. Lei, Angew. Chem., Int. Ed., 2010, 49, 3371 CrossRef CAS PubMed; (c) J. R. Martinelli, T. P. Clark, D. A. Watson, R. H. Munday and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 8460 CrossRef CAS PubMed.
- (a) K. Okuro and H. Alper, J. Org. Chem., 1997, 62, 1566 CrossRef CAS; (b) S. Gao, M. Chen, M.-N. Zhao, W. Du, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, J. Org. Chem., 2014, 79, 4196 CrossRef CAS PubMed; (c) R. J. Perry' and B. D. Wilson, J. Org. Chem., 1993, 58, 7016 CrossRef.
- S. N. Gockel and K. L. Hull, Org. Lett., 2015, 17, 3236 CrossRef CAS PubMed.
- A. Satapathy, S. T. Gadge, T. Sasaki and B. M. Bhanage, RSC Adv., 2015, 5, 93773 RSC.
- J. Salvadori, E. Balducci, S. Zaza, E. Petricci and M. Taddei, J. Org. Chem., 2010, 75, 1841 CrossRef CAS PubMed.
- R. S. Mane and B. M. Bhanage, RSC Adv., 2015, 5, 76122 RSC.
- T. T. Dang, A. Chen and A. M. Seayad, RSC Adv., 2014, 4, 30019 RSC.
- Z. S. Qureshi, S. A. Revankar, M. V. Khedkar and B. M. Bhanage, Catal. Today, 2012, 198, 148 CrossRef CAS.
- B. Urbán, M. Papp, D. Srankó and R. Skoda-Földes, J. Mol. Catal. A: Chem., 2015, 397, 150 CrossRef.
- T. T. Dang, Y. Zhu, J. S. Y. Ngiam, S. C. Ghosh, A. Chen and A. M. Seayad, ACS Catal., 2013, 3, 1406 CrossRef CAS.
- M. Papp, P. Szabó, D. Srankó and R. Skoda-Földes, RSC Adv., 2016, 6, 45349 RSC.
- T. T. Dang, Y. Zhu, S. C. Ghosh, A. Chen, C. L. L. Chai and A. M. Seayad, Chem. Commun., 2012, 48, 1805 RSC.
- F. Tinnis, O. Verho, K. P. J. Gustafson, C.-W. Tai, J.-E. Bäckvalll and H. Adolfsson, Chem.–Eur. J., 2014, 20, 5885 CrossRef CAS PubMed.
- W. Hao, J. Sha, S. Sheng and M. Cai, Catal. Commun., 2008, 10, 257 CrossRef CAS.
- M. Cai, H. Zhao and Y. Huang, J. Mol. Catal. A: Chem., 2005, 238, 41 CrossRef CAS.
- M. Cai, Y. Huang, R. Hu and C. Song, J. Mol. Catal. A: Chem., 2004, 208, 17 CrossRef CAS.
- M. Esmaeilpour, A. Sardarian and J. Javidi, Catal. Sci. Technol., 2016, 6, 4005 CAS.
- Z. Li, J. Chen, W. Su and M. Hong, J. Mol. Catal. A: Chem., 2010, 328, 93 CrossRef CAS.
- M. L. Cubeiroa and J. L. G. Fierro, Appl. Catal., A, 1998, 168, 307 CrossRef.
- Q. Liu, K. K. Gath, J. C. Bauer, R. E. Schaak and J. H. Lunsford, Catal. Lett., 2009, 132, 342 CrossRef CAS.
- T. Xu and H. Alper, J. Am. Chem. Soc., 2014, 136, 16970 CrossRef CAS PubMed.
- S. Zheng, Y. Wang, C. Zhang, J. Liu and C. Xia, Appl. Organomet. Chem., 2014, 28, 48 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04985d |
| This journal is © The Royal Society of Chemistry 2017 |