
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSensitive electrochemical determination of rifampicin using gold nanoparticles/poly-melamine nanocomposite†
S. Amidia,
Y. Hosseinzadeh Ardakanib,
M. Amiri-Arefb,
E. Ranjbarib,
Z. Sepehric and
H. Bagheri *d
*d
aDepartment of Medicinal Chemistry, School of Pharmacy & Protein Technology Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran
bBiopharmaceutics and Pharmacokinetics Division, Department of Pharmaceutics, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran 14155-6451, Iran
cDepartment of Internal Medicine, Zabol University of Medical Sciences, Zabol, Iran
dChemical Injuries Research Center, Baqiyatallah University of Medical Sciences, Tehran, Iran. E-mail: h.bagheri@bmsu.ac.ir; h.bagheri82@gmail.com
First published on 16th August 2017
Abstract
In this work, a novel electrochemical sensing platform was successfully prepared by electrodeposition of gold nanoparticles on the electropolymerized layer of poly-melamine modified pre-anodized glassy carbon electrode. The surface morphology and electrochemical behaviour of the modified electrode (PMel-Aunano/GCE*) were characterized by scanning electron microscopy (SEM), electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV). The proposed sensor remarkably increased the oxidation responses of rifampicin (RIF) associated with a significant decrease in its peak over-potentials of about 0.10 V based on the integration of the beneficial effects of a conductive PMel (with excellent electrocatalytic activity) and gold nanoparticles (with prominent electrochemical properties and large surface area), which exhibits the properties of their individual components with synergistic effects. The results demonstrated that voltammetric determination of RIF achieved favourably and the modified electrode represents two oxidation peaks for rifampicin at potentials approximately 0.09 V (peak 1) and 0.70 V (peak 2) in phosphate buffer solution with a linear range of 0.08–15.00 μM and a detection limit of 0.03 μM. The developed sensor is stable, reproducible and feasible for long-term use in biological samples with satisfactory results for RIF detection.
Introduction
Rifampicin (RIF), 3-[(4-methyl-1-piperazinyl)imino] methyl rifamycin, also known as rifampin is a semi-synthetic macrocyclic antibiotic, which was introduced in 1967 in order to cope with tuberculosis.1 Tuberculosis (TB) is a contagious bacterial infection caused by Mycobacterium tuberculosis (MB-TB), which can affect many organs of body, especially the lungs. RIF is also used to fight with Neisseria meningitidis (a type of bacteria that can cause a serious infection called meningitis) infections in the noses or throats, as well as to treat Hansen's disease (HD) and other serious infections such as HIV, which inhibits bacterial DNA-dependent RNA polymerase.The concentration of anti-bacterial infection drugs is a key factor when encountering bacteria, and an insufficient concentration of drug leads to the development of drug resistance and the subsequent incomplete treatment of infectious disease.2 Incomplete treatment of bacterial infections is common, and the success in therapeutic action can be influenced by therapeutic regime, interrupted drug supply3–5 and physical or chemical interactions with other medications and nutrients, which may alter its bioavailability;6 on the other hand, an overdose of RIF can produce potential side-effects like hepatotoxicity, allergic rashes, appetite loss, nausea, or immunological disturbances.6 Therefore, sufficient RIF dosage should be based on an individualized clinical process using the best judgment of a physician trained to administer this antibiotic. In fact, due to the differences in the pharmacokinetics RIF and its bioavailability among different people, development of the therapeutic drug monitoring based on rapid and reliable analytical methods is particularly important. They should determine the RIF concentration to individualize doses for achieving efficient treatment.
So far, numerous analytical methods have been applied to quantify the RIF,3,7–14 that the most common described procedures are based on liquid chromatography (LC) including LC coupled with ultra-violet (UV)3 and tandem mass spectrometry (MS/MS)7 detections, high performance liquid chromatography (HPLC) coupled with UV,3,8 MS/MS9 and diode array10 detections and high performance thin layer chromatography (HPTLC).11 Despite the sensitivity of these methods, they often require time consuming complex pretreatment procedures and expensive apparatus for routine analyses. Electrochemical methods have the great advantages of high sensitivity, low cost and simple instrumentation without requiring tedious pretreatments. However, there are still a few electrochemical methods for RIF determination12–17 and common interest is dealing with developing novel electrochemical sensors for RIF detection.
Modification of the surface by electropolymerization is an effective way to enhance the catalytic activity of the electrochemical sensors in terms of enhancement in peak current and lowering of over-potential. Moreover, conductive polymer modified electrodes possess very much excellent characteristics in acting as electrochemical sensor, such as desirable stability, reproducibility, enhanced active sites, and homogeneity. Heteroatoms such as nitrogen, sulfur and amino groups present in the conducting polymers interact with metal nanoparticles to form a complex to stabilize the nanoparticles. Poly-melamine film is a new type of conductive polymer, which has attracted considerable attention for analytical purposes due to its high stability and abundant nitrogen functional groups.18,19 Recently, Su et al. developed the poly-melamine film modified screen-printed carbon electrode and illustrated its electrocatalytic activity by determination of gallic acid.18 Cotchim et al. reported a poly-melamine on the surface of graphene oxide-modified electrode for the determination of trinitrotoluene in soil samples via adsorptive cathodic stripping voltammetry.19
Noble metal nanoparticles have been widely utilized in recent years, due to their fabulous catalytic activities for electrochemical application.20,21 Due to the unique properties of gold nanoparticles, such as good conductivity, particular electrocatalytic ability and biocompatibility, several researches have been devoted to fabricate electrochemical sensors and biosensors based on gold nanoparticles.22–24 Electrochemical deposition could provide a rapid and easy alternative rather than the chemical synthesis method for the preparation of gold nanoparticles in a shorter time scales, which provides the particles with higher purity, more controllable and narrower particle size distribution.25–27 Conducting polymers can be ideal host matrices for gold nanoparticles because they have advantages of permitting a facile electronic charge flow through the polymer matrix in electrochemical processes.28,29 The presence of amine group on carbon materials is also helpful in preventing aggregation, to provide biocompatibility, and to self-assemble of metal particles. The presence of AuNPs on the poly-melamine could amplify the electrochemical and mechanical characteristics of the sensing platform.
Due to modification of conducting polymers with nanomaterials on carbon electrode surface is preferred than other metal electrode surfaces for electrocatalysis purposes, in the present work, a new, sensitive and green approach was developed to prepare a high performance electrochemical sensing platform based on the electroactive poly-melamine film and electrodeposited gold nanoparticles (PMel-Aunano) modified pre-anodized glassy carbon electrode (GCE*) for the quantitative determination of RIF. The fabricated sensor showed to be promising for RIF detection with many desirable features including very low detection limit, high sensitivity, decrease in over-voltage, reproducible responses, and acceptable anti-interferences ability.
Experimental
Reagents and instrumentation
RIF was purchased from Sigma-Aldrich. The reagents including hydrochloric acid (HCl, 37% purity), sulfuric acid (H2SO4, >95% purity), and sodium hydroxide pellets (NaOH, >97% purity) were procured from Sigma-Aldrich. Melamine (>99% purity) used for electropolymerization and Au salt (HAuCl4·4H2O) used in electro-deposition of gold nanoparticles were also purchased from Sigma-Aldrich. Phosphate buffer solutions (PBS) were prepared with Na2HPO4 and NaH2PO4 from Sigma-Aldrich. All of the chemicals were analytical grade and used without purification.Electrochemical experiments including CV, linear sweep voltammetry (LSV) and EIS techniques were carried out with an Ivium potentiostat/galvanostat (Vertex, Ivium Technologies, The Netherlands). The experimental conditions for measurements were controlled with Ivium software. The morphological characterization has been examined by means of scanning electron microscope (SEM, KYKY-EM 3200, China). The measurements were carried out by a conventional three-electrode cell consisting an Ag|AgCl|KCl (3 M) electrode as the reference, a Pt wire as the counter electrode, and a glassy carbon disk electrode (Ivium, The Netherlands), bare or modified, was used as working electrode.
Fabrication of PMel-Aunano/GCE*
First, GCE surface was polished mechanically, with 0.05 μm alumina slurry on a polishing cloth, rinsed thoroughly with Milli-Q water. Then, the electrode was sonicated for 5 min in mixture of ethanol and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) in ultrasonic cleaning bath. After the ultrasonication, it was rinsed with Milli-Q water again and dried with a nitrogen gas stream. The PMel-Aunano/GCE* was prepared according to the following procedure in three steps. First, the GCE was pre-anodized in 0.1 M PBS (pH 7.0) by applying potential of 2.0 V for 300 s and denoted as GCE*. Buffer solutions were purged with nitrogen gas before electrochemical pre-treatment. The activated carboxyl or hydroxyl groups on the pre-anodized GCE surface are prone to adsorb the melamine on the electrode surface in order to form PMel in the next step. Second, the PMel was electropolymerized on the GCE* by cyclic potential scanning from +0.1 to +1.8 V for 14 cycles at a scan rate 100 mV s−1 in a solution 0.1 M HCl and 2.0 mM melamine. In the third step, the gold nanoparticles were electrochemically deposited on the surface of prepared PMel/GCE* by cyclic potential scanning from −0.2 V to +1.2 V in 0.5 M H2SO4 solution containing 0.1 mM HAuCl4 (degassed with a N2 stream for at least 10 min and pH = 2.2–2.5) at a scan rate of 100 mV s−1 for 15 cycles. The as-fabricated PMel-Aunano/GCE* was used for the determination of RIF in PBS and real sample solution. Before voltammetric measurements, the modified electrode was cycled five times between 0.0 and +1.0 V (at scan rate of 100 mV s−1) in a PBS of pH 7.0 to obtain a reproducible response. After an accumulation time of 125 seconds on an open-circuit, the CVs and LSVs were recorded from −0.1 to +1.0 V at the scan rate of 100 mV s−1 for RIF determination.
:1, v/v) in ultrasonic cleaning bath. After the ultrasonication, it was rinsed with Milli-Q water again and dried with a nitrogen gas stream. The PMel-Aunano/GCE* was prepared according to the following procedure in three steps. First, the GCE was pre-anodized in 0.1 M PBS (pH 7.0) by applying potential of 2.0 V for 300 s and denoted as GCE*. Buffer solutions were purged with nitrogen gas before electrochemical pre-treatment. The activated carboxyl or hydroxyl groups on the pre-anodized GCE surface are prone to adsorb the melamine on the electrode surface in order to form PMel in the next step. Second, the PMel was electropolymerized on the GCE* by cyclic potential scanning from +0.1 to +1.8 V for 14 cycles at a scan rate 100 mV s−1 in a solution 0.1 M HCl and 2.0 mM melamine. In the third step, the gold nanoparticles were electrochemically deposited on the surface of prepared PMel/GCE* by cyclic potential scanning from −0.2 V to +1.2 V in 0.5 M H2SO4 solution containing 0.1 mM HAuCl4 (degassed with a N2 stream for at least 10 min and pH = 2.2–2.5) at a scan rate of 100 mV s−1 for 15 cycles. The as-fabricated PMel-Aunano/GCE* was used for the determination of RIF in PBS and real sample solution. Before voltammetric measurements, the modified electrode was cycled five times between 0.0 and +1.0 V (at scan rate of 100 mV s−1) in a PBS of pH 7.0 to obtain a reproducible response. After an accumulation time of 125 seconds on an open-circuit, the CVs and LSVs were recorded from −0.1 to +1.0 V at the scan rate of 100 mV s−1 for RIF determination.
Results and discussion
Electropolymerization of melamine on GCE*
After anodic treatment, background current increased. In addition, the pre-anodized PMel-Aunano/GCE* yielded larger peak currents than non pretreated PMel-Aunano/GCE. These observation showed that the anodization stage was done successfully. Due to the electropolymerization initiates only under proper conditions that can increase the radical cation concentration to a certain level, a positive enough potential associate with activated carbon platform was applied for the poly-melamine formation.30 The successive cyclic voltammograms in Fig. 1A are related to the electropolymerization process of 2.0 mM melamine in a 0.1 M HCl solution by 14 potential cycles from +0.1 to +1.8 V vs. Ag|AgCl|KCl (3 M) on the surface of GCE*. In the first cycle of potential, with the increase of potential to +1.8 V, the oxidation of amine starts to take place (Epa ∼ +1.6 V) and the radical cations are formed. In the second cycle, the newly formed radical cations undergo head-to-head coupling with NH–NH bonding of melamine to create dimer which are followed by anodic peak and formation of polymer during the next cycles.30 The typical redox behaviour of the polymer is observed at lower potentials with well-defined anodic and cathodic peaks at 0.69 V and 0.55 V, respectively. With increasing the number of potential cycles, the polymer peaks current continuously increased together with a decrease in the amount of monomers remained in solution, which is evidence of the gradually growth of conductive polymer on the surface of GCE* (Fig. 1Ab has higher magnification than Fig. 1Aa).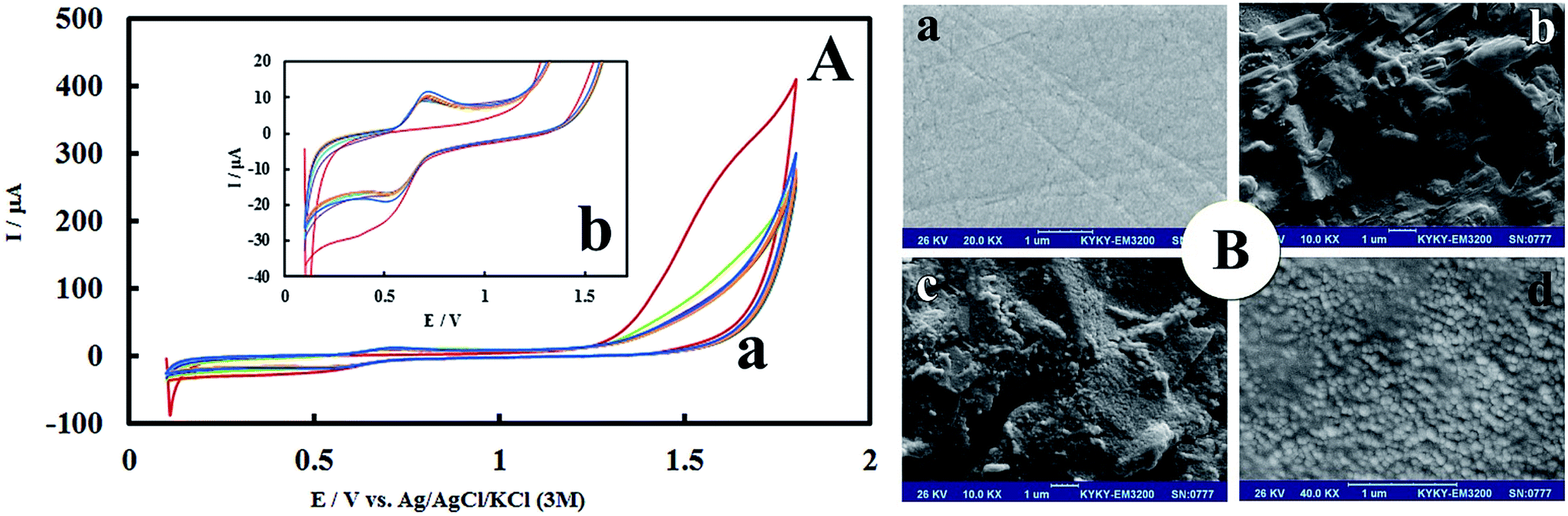 | ||
| Fig. 1 (A-a) Cyclic voltammograms of the growth of polymelamine film at the surface of pre-anodized GCE. Polymerization was carried out in 0.1 M HCl solution containing 2.0 mM melamine monomer at a scan ate 100 mV s−1 with the potential range of 0.1–1.8 V, 14 cycles from inner to outer. (Plot b is higher magnification of plot a). (B) SEM images of (a) bare GCE, (b) PMel/GCE*, (c) PMel-Aunano/GCE* and (d) PMel-Aunano/GCE* in higher magnification. | ||
Morphological and electrochemical characteristics of PMel-Aunano/GCE*
The morphology of PMel-Aunano nanocomposite on the surface of GCE* has been investigated by SEM images. The SEM images of the unmodified GCE and formed PMel film on the GCE* represents in Fig. 1B, respectively. After the electrodeposition of gold nanoparticles (PMel-Aunano/GCE*), it is obviously seen that many spherical gold nanoparticles were distributed and attached to the surface of the PMel (Fig. 1Bc and Bd). Furthermore, the electroactive feature of the PMel/GCE* and PMel-Aunano/GCE* modified layers were investigated by means of cyclic voltammetric study in PBS (pH 7.0). As shown in Fig. 2A (curve a), a couple of well-defined anodic and cathodic peaks were clearly observed attributing to the oxidation and reduction of the PMel at GCE*. The formal potential (E°′), which is estimated from the average of the redox peak potentials, was calculated as 0.259 V vs. Ag|AgCl|KCl (3 M). Peak-to-peak separation (ΔEp) for the PMel/GCE* was 0.182 V and the ratio of cathodic peak current over the anodic one is nearly unit. Thus, based on the obtained parameters, the electrochemical process of PMel is considered as a quasi-reversible redox process. Upon electrodeposition of gold nanoparticles on the PMel/GCE* by cyclic sweeping, the peak current for the PMel-Aunano/GCE* increased dramatically (Fig. 2A, curve b) with formal potential of 0.216 V and peak separation of 0.160 V. Indeed, the integration of PMel and gold nanoparticles in the modified film amplified the peak currents and reversibility of redox peaks, which can be related to the larger electroactive surface area of PMel-Aunano/GCE* and electrocatalytic behaviour of gold nanoparticles.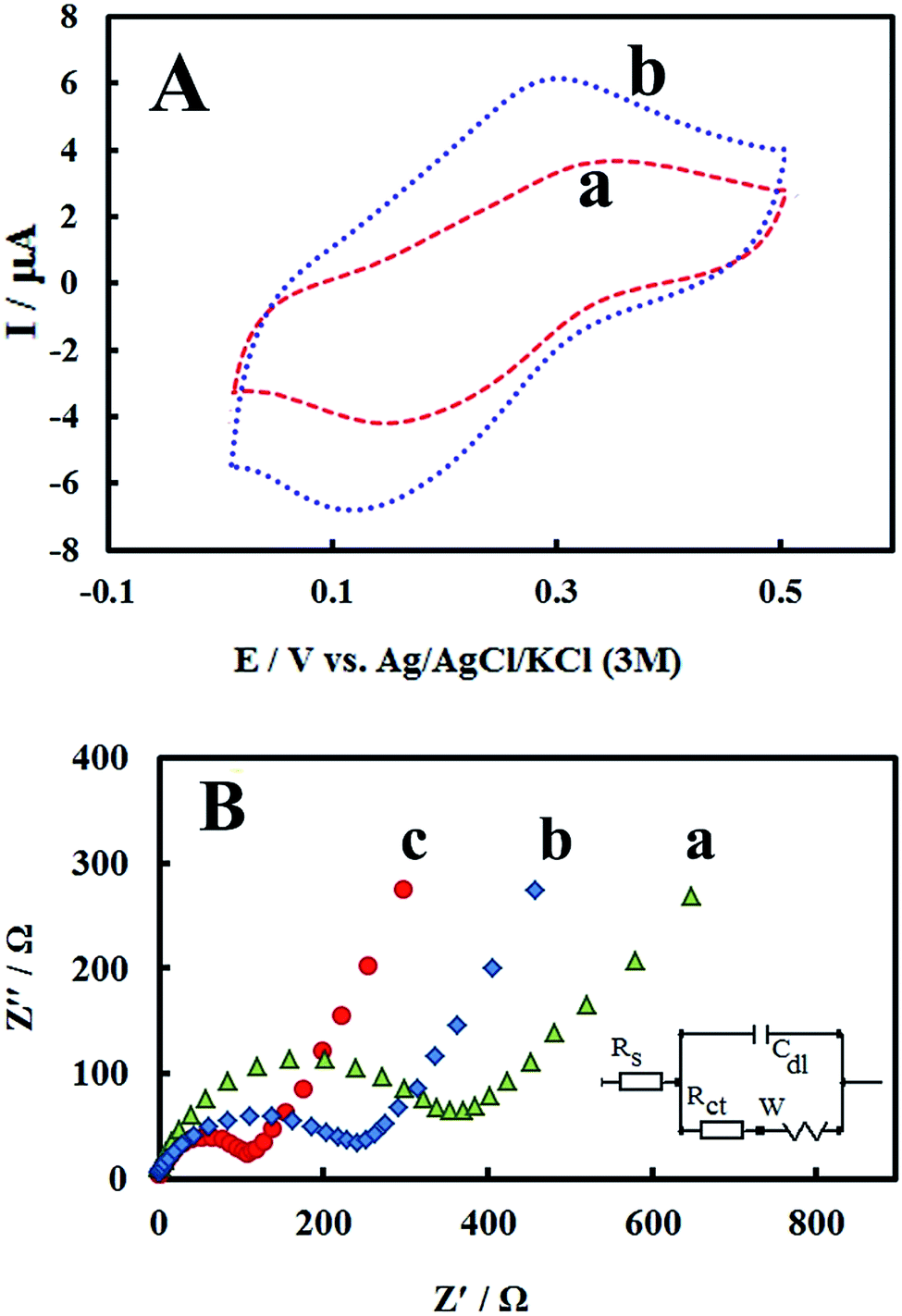 | ||
| Fig. 2 (A) Cyclic voltammograms of (a) PMel/GCE* and (b) PMel-Aunano/GCE* in a 0.1 M blank phosphate buffer solution (pH 7.0) at the scan rates of 100 mV s−1. (B) Nyquist plots for the faradic impedance measurements of a 5 mM solution of 1:1 K3[Fe(CN)6]/K4[Fe(CN)6] performed on (a) bare GCE, (b) PMel/GCE* and (c) PMel-Aunano/GCE*. Inset: the Randel's equivalent circuit model. | ||
The effective surface area of the different electrodes was estimated by recording cyclic voltammograms of 0.1 mM potassium ferrocyanide at different scan rates of 10–200 mV s−1.
From the slope of Ip vs. square root of scan rate (ν1/2) using Randles–Sevcik equation31 and considering the diffusion coefficient of ferrocyanide as 6.20 × 10−6 cm2 s−1,32,33 the effective surface areas for bare GCE, PMel/GCE* and PMel-Aunano/GCE* were calculated to be 0.058, 0.103 and 0.141 cm2, respectively. The results depicted the presence of PMel and gold nanoparticles at PMel-Aunano/GCE* clearly amplified the active surface area of the unmodified electrode.
EIS studies
As a powerful technique for probing the features of electrode surface, the EIS was applied to investigate the interfacial properties of electrode surface within the stepwise modification process.34–36 Fig. 2B shows the Nyquist plots recorded in 0.1 M KCl solution containing 5 mM of the redox probe K3[Fe(CN)6]/K4[Fe(CN)6]. As it can be seen, significant changes in the impedance data were obtained at bare GCE, PMel/GCE* and PMel-Aunano/GCE*. At bare GCE (Fig. 2B, curve a), a large semicircle domain with charge transfer resistance (Rct) equals to 405 Ω in the Fe[CN]63−/4− redox probe was observed. However, for the PMel/GCE* (Fig. 2B, curve b), the value of Rct was decreased obviously to about 276 Ω, implying that PMel acts as a suitable electron transfer platform for shuttling electrons between the electrochemical probe and the electrode, and exhibits a conducting behavior like wire. Upon electrodeposition of gold nanoparticles on the PMel/GCE* (Fig. 2B, curve c), the Rct was further decreased to the 128 Ω, indicating the synergistic electrocatalytic effect of gold nanoparticles and poly-melamine film towards the redox process of the probe.Electrooxidation of RIF at the PMel-Aunano/GCE*
The electrocatalytic performance of the modified electrode towards RIF oxidation was investigated by CV method. Fig. 3A shows the cyclic voltammograms of RIF (5.0 μM) at different electrodes including bare GCE, PMel/GCE* and PMel-Aunano/GCE* in 0.1 M PBS (pH 7.0). Electrochemical oxidation of RIF at the surface of bare GCE shows two very weak peaks, at potentials about 0.19 V (Epa1) and 0.81 V (Epa2) vs. Ag|AgCl|KCl (3 M), due to sluggish electron transfer (Fig. 3A, curve a).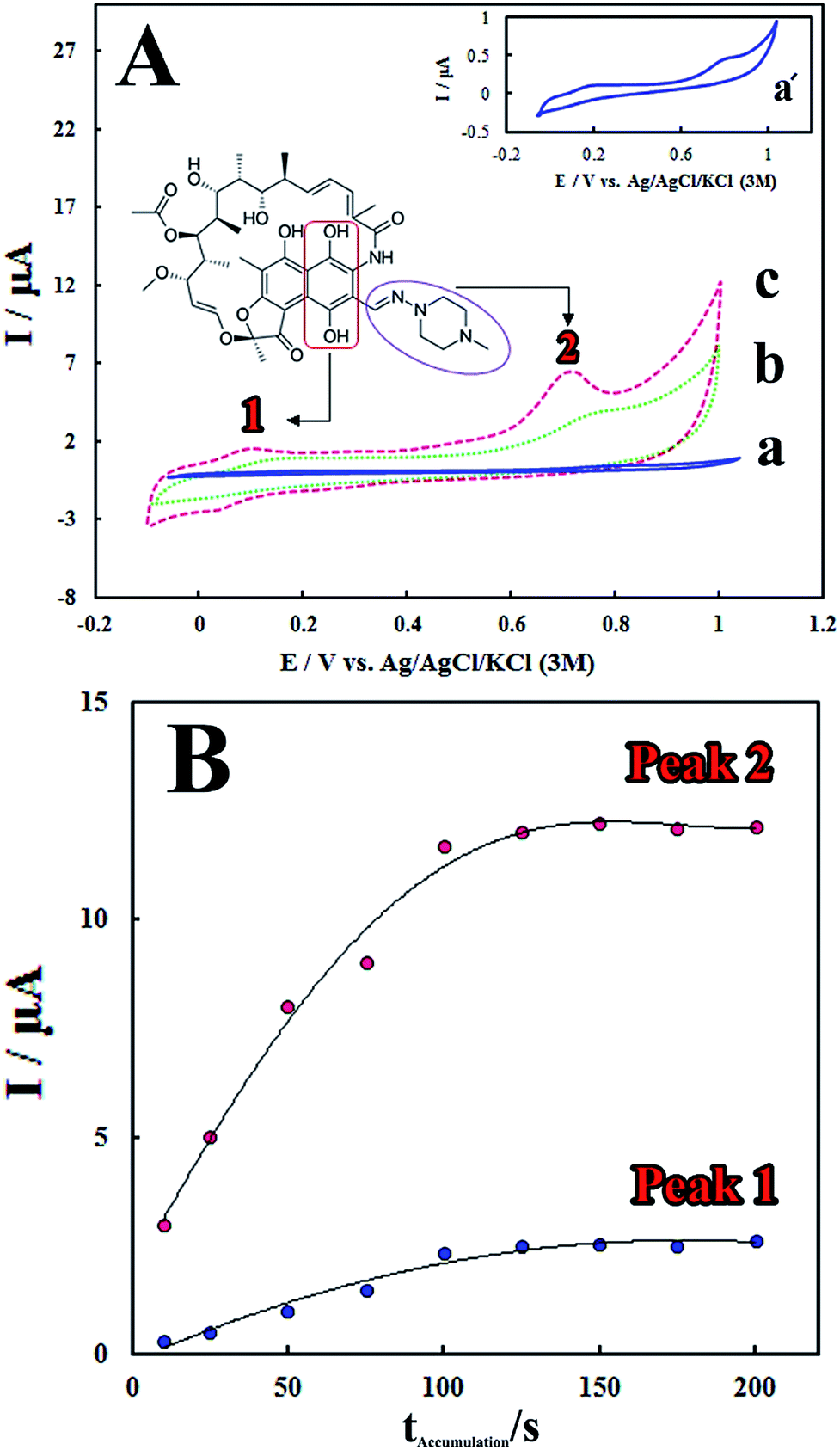 | ||
| Fig. 3 (A) Cyclic voltammograms after adding 5.0 μM RIF at the surface of (a) bare GCE, (b) PMel/GCE* and (c) PMel-Aunano/GCE* in a 0.1 M blank phosphate buffer solution (pH 7.0) at the scan rates of 100.0 mV s−1. Inset: RIF chemical structure and the corresponding moieties to the peaks I and II electron transfer process. (B) Effect of the accumulation time (tacc) on the peak (1) and (2) current responses from LSV for 10.0 μM RIF determination on the surface of the PMel-Aunano/GCE* in 0.1 M PBS (pH 7.0) at Eacc 0.0 V. | ||
The remarkable enhancement in the peak currents was observed at the PMel/GCE* (Fig. 3A, curve b), which is implied that the oxidation of RIF became facile in the presence of PMel film modified electrode. However, the values of RIF oxidation peaks currents obtained at the PMel-Aunano/GCE* (Epa1 = 0.09 V and Epa2 = 0.70 V) showed considerable negative shifts in the over-potentials of about 0.10 V (Fig. 3A, curve c) together with a significant increase in the peak currents compared to the unmodified electrode. It was concluded that the best results for electrocatalytic detection of RIF was obtained at the surface of PMel-Aunano/GCE* due the integration of the polymelamine (with excellent electrocatalytic activity) and gold nanoparticles (with prominent electrochemical properties and large surface area) which exhibits the properties of its individual components with synergistic effects.
Here, the electrochemical oxidation of RIF peak 1 at the PMel-Aunano/GCE* can be ascribed to the oxidation of 6,9-dihydroxynaphthalene moiety to the corresponding naphthoquinone, very close to that observed for the hydroquinone/quinone redox system and the electrochemical oxidation of RIF peak 2 may be assigned to the oxidation of piperazinyl-imino moiety of the RIF molecule (the corresponding moieties were shown in Fig. 3A).12,13
Accumulation time and potential can also significantly affect sensitivity of the determination method by altering the amount of the adsorbed RIF at the modified electrode surface. Fig. 3B shows that the peak currents of RIF increased gradually with increasing the accumulation time from 10 to 125 s. However, with further increasing the accumulation time, the peak currents tended to be almost stable. This observation implied that surface saturation happened at higher accumulation times (125–200 s). Therefore, the optimal accumulation time of 125 s was employed prior to RIF voltammetric determination. In addition, it was found that the accumulation potential has no influence on the RIF oxidation peak currents. Therefore, the accumulation step was carried out under the open-circuit condition for following experiments.
Effect of pH on the electrochemical oxidation of RIF
RIF shows two peaks onto the surface of PMel-Aunano/GCE*. Various parameters affect the peak potentials and peak currents. Optimal conditions for obtaining high peak currents and well defined peak shapes for the two are different. Since the potential of these peaks are far from each other. In some cases, one compound is interference for first oxidation peak, but second oxidation peak of analyte is interference free and vice versa. Therefore, according to the type of samples and interferences, one of them can be used for analysis. The electrochemical responses of RIF at the PMel-Aunano/GCE was carried out in PBS with different pHs ranging from 3.0 to 9.0. Typical LSV behaviours of 12.0 μM RIF at different PBS are shown in Fig. 4A. The peak currents for RIF gradually increased by raising the pH values from 3.0–7.0 and reaching to their maximum at pH = 7.0. Further increase of the pH to 9.0 caused the peak heights to decline. Hence, PBS of pH 7.0 was chosen as the optimum pH and used for all further experiments (Fig. 4B).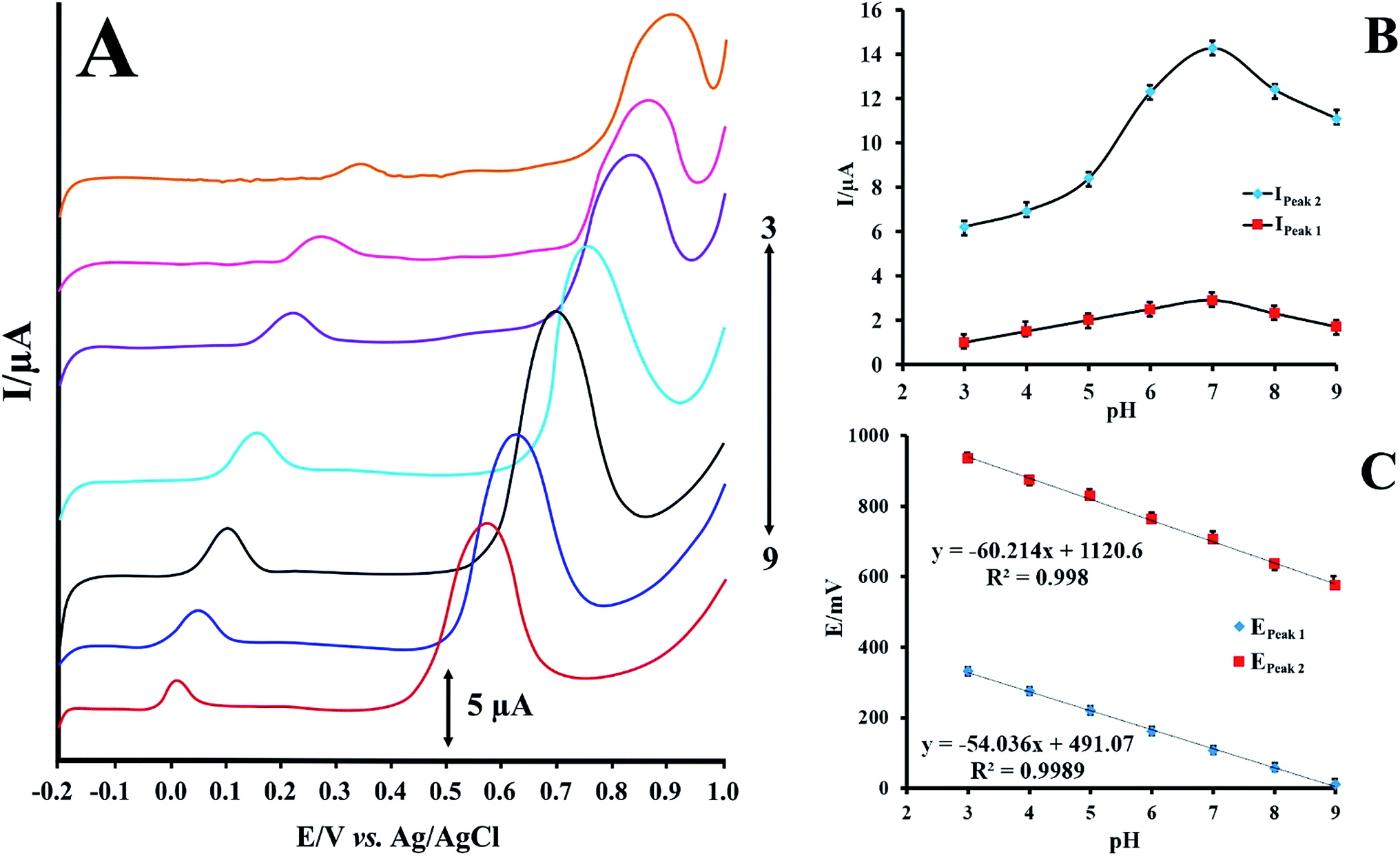 | ||
| Fig. 4 (A) LSVs for PMel-Aunano/GCE in 12.0 μmol L−1 RIF at various pHs (pH 3.0–9.0) (B) effect of pH on the peak currents of RIF (C) effect of pH on the peak potentials of RIF. | ||
As shown in Fig. 4C the oxidation peak potentials of peak 1 and peak 2 indicate the same trend and shifted to the negative direction with the increase of pH values, demonstrating that the protons directly involved in their electrode reaction processes.
The linear regression equations of peak 1 and peak 2 are expressed as follows respectively:
| E = −0.054 pH + 0.491, R2 = 0.998 | (1) |
| E = −0.060 pH + 1.120, R2 = 0.998 | (2) |
The resulted slopes of the plots in the pH range 3.0–9.0 also demonstrate that the number of electrons and protons are equal in the oxidation of RIF.
Analytical characteristics
The LSV technique was employed to obtain the linear ranges and detection limits for the determination of RIF at the surface of the proposed electrode. The anodic peak currents were proportional to RIF concentrations in the linear range of 0.08–15.00 μM (Fig. 5). The detection limit (3σ) for determination of RIF was estimated using the slope of the calibration plot of peak 2 (which has more sensitivity that the calibration plot of peak 1) and was found to be 0.03 μM. Table 1 displays a comparison between previously reported modified electrodes and the developed PMel-Aunano/GCE* for RIF determination, which implies that PMel-Aunano/GCE* exhibits comparable performance for RIF determination over other electrodes.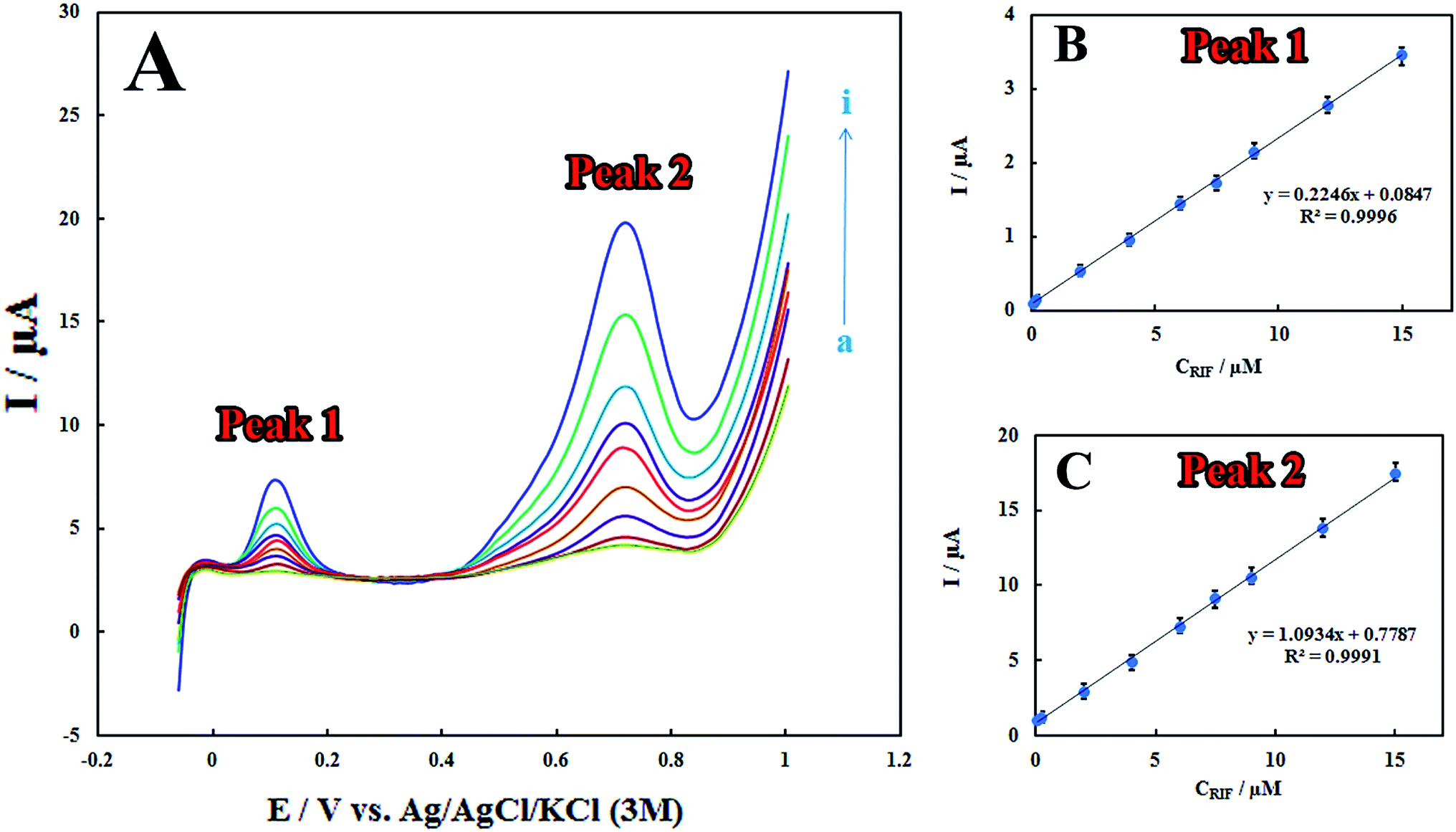 | ||
| Fig. 5 (A) LSVs for different concentrations of RIF (a) 0.08, (b) 0.2, (c) 2.0, (d) 4.0, (e) 6.0, (f) 7.5, (g) 9.0, (h) 12.0 and (i) 15.0 μM in 0.1 M PBS (pH 7.0) at the scan rates of 100.0 mV s−1 on the surface of PMel-Aunano/GCE*. (B and C) Linear calibration curves of peaks 1 and 2 currents vs. RIF concentrations. | ||
| Method | Modified electrode | pH | Decrease in overpotential of | Linear range (μM) | LOD (μM) | Rep.a (RSD) | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Epa1 (V) | Epa2 (V) | |||||||
| a Reproducibility.b Nickel hydroxide nanoparticles-reduced graphene oxide nanosheets.c Amperometry.d β-Cyclodextrin/polypyrrol platinum electrode.e Adsorptive square wave voltammetry.f Dysprosium hydroxide nanowires modified carbon paste electrode. | ||||||||
| LSV | Ni(OH)2/RGO/GCEb | 7.0 | 0.12 | 0.09 | 0.004–10 | 0.002 | 3.5 | 12 |
| DPV | Surfactant/CPE | 2.0 | 0.2 | 0.76 | 3.5 × 10−4–0.0054 | 0.0001 | 2.7 | 13 |
| Ampc | β-Cyclodextrin/Ppyr/Ptd | 7.0 | — | — | 2.61–25.23 | 1.69 | 3.51 | 14 |
| ASV | Lead film/GCE | 5.0 | 0.2 | — | 2.5 × 10−4–0.01 | 9 × 10−5 | — | 15 |
| AdSWVe | DyNW/CPEf | 5.0 | 0.03 | 0.03 | 1 × 10−5–0.0075 | 5 × 10−5 | — | 16 |
| AdSWV | PVP-capped CoFe2O4@CdSe/GCE | 2.0 | 0.219 | — | 10−10–10−1 | 4.5 × 10−11 | 2.62 | 17 |
| LSV | PMel-Aunano/GCE* | 7.0 | 0.10 | 0.11 | 0.08–15.00 | 0.03 | 2.8 | This work |
Furthermore, the stability of the modified electrode is investigated by examining its performance during one-month storage. A signal decrease of 3.3% and 5.7% was observed toward RIF oxidation peaks 1 and 2, respectively (Fig. 6). Also, in order to study the reproducibility of the electrode modification, five PMel-Aunano/GCE* electrodes were prepared independently. The relative standard deviations (RSDs) of measuring peak currents 1 and 2 for 5.0 μM RIF for five successive prepared electrodes were 2.0% and 2.8%, respectively, indicating a reasonable reproducibility of the modified electrode (Fig. S1 and S2†).
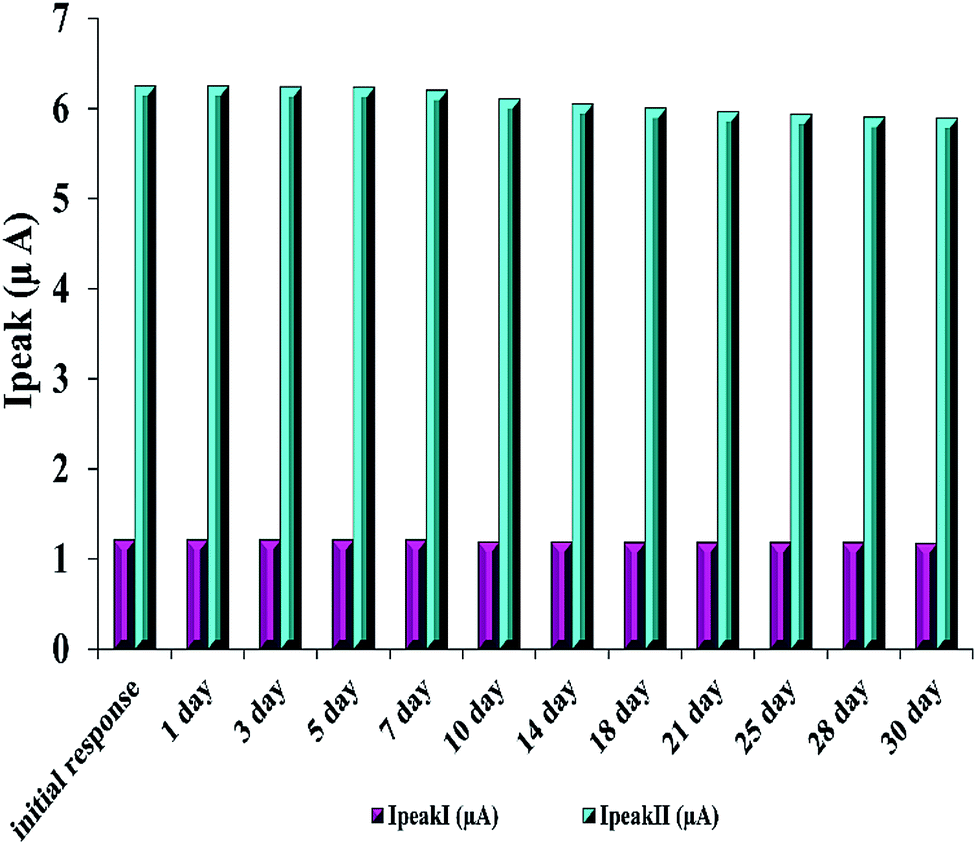 | ||
| Fig. 6 Stability of the modified electrode for 5.0 μM RIF. | ||
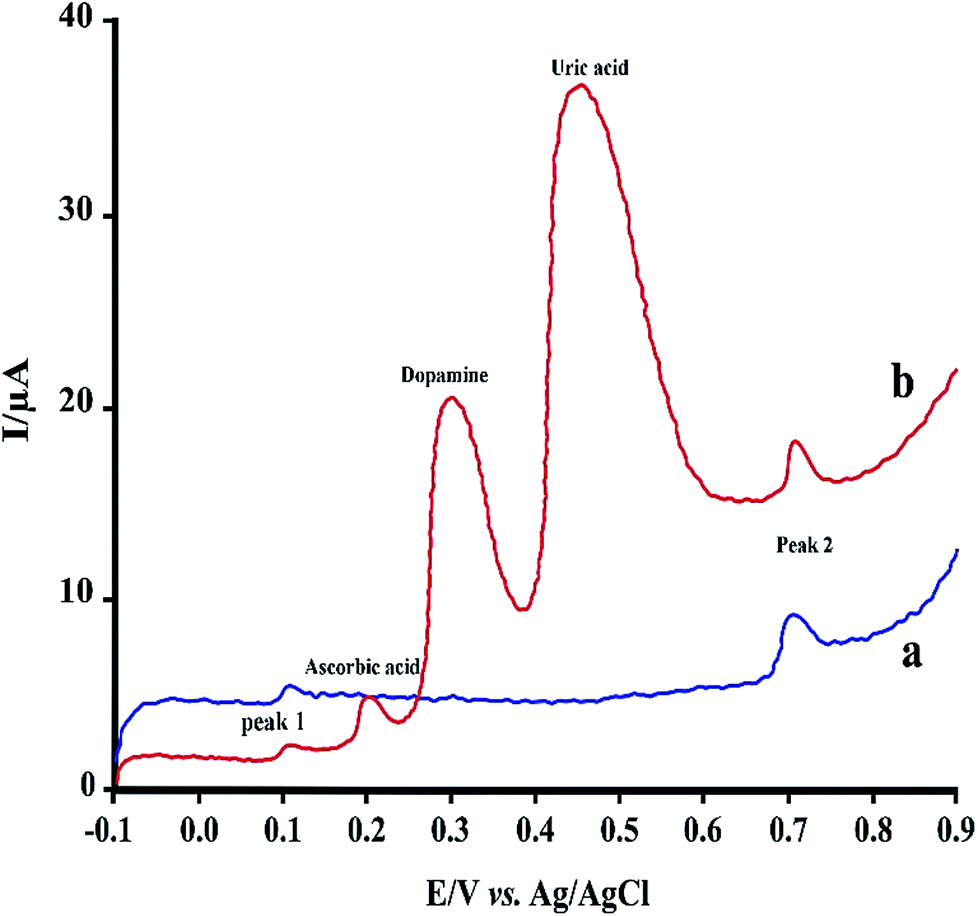 | ||
| Fig. 7 LS voltammograms of 2.0 μM RIF using PMel-Aunano/GCE (a) and after addition of some common interferences (b). | ||
Interference studies and real sample analysis
Under the optimized experimental conditions, the effect of some common interferences such as ascorbic acid, uric acid, dopamine and glucose on the voltammetric determination of RIF was examined. The results (Table 2) shows there are no significant change in the peak currents of RIF in the presence of these compounds (signal change less than 5%). Fig. 7 shows typical linear sweep voltammogram in the absence of interferences (a) and after addition of some mentioned species. Hence, the proposed sensor can be successfully applied for the determination of RIF in the presence of other interference compounds in the clinical preparations.| Interfering agent | Concentration (μM) | Signal change (%) | |
|---|---|---|---|
| Peak I | Peak II | ||
| Dopamine | 2.0 | +4.7 | +1.3 |
| Ascorbic acid | 2.0 | +3.5 | −1.0 |
| Glucose | 100 | −1.1 | +1.0 |
| Uric acid | 100 | +3.8 | −2.6 |
The feasibility and reliability of the proposed sensor in practical analysis was also tested in spiked human urine samples. Urine sample was originally obtained from a healthy volunteer (one of the authors). Sample collection was performed according to institutional ethic approval and informed consent was obtained from the volunteer. Urine sample was stored in a refrigerator immediately after its collection. For analysis, 5.0 mL of the sample was centrifuged for 15 min at 2000 rpm. The aliquot amounts of RIF were added to urine samples which did not contain the RIF. Each voltammetric measurements (LSV) was employed in triplicate by the standard addition method (n = 3) and the analysis data were summarized in Table 3. The PMel-Aunano/GCE* offers the satisfactory recoveries and RSD values for voltammetric determination of RIF in real samples analysis.
Conclusions
An extremely sensitive and selective electrochemical sensor was developed through modification of the pre-anodized GCE with poly-melamine film decorated with gold nanoparticles (PMel-Aunano/GCE*) for the determination of rifampicin. The proposed sensor remarkably increases the oxidation responses of rifampicin as well as shifts its oxidation potentials to the fewer ones. The developed sensor displayed desirable properties such as rapid preparation, wide linear concentration range, low detection limit, good reproducibility, stability, and anti-interference ability, which provided a promising platform for sensing of this anti-TB drug. This study has also demonstrated the practical analytical efficiency of the prepared modified electrode for the determination of RIF in human urine samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Protein Technology Research Center (PTRC) of Shahid Beheshti University of Medical Science for financial support of this project.References
- C. Roger, Perspect. in Public Health, 2010, 130(5), 240 CrossRef.
- D. G. Storla, S. Yimer and G. A. Bjune, BMC Public Health, 2008, 8, 1 CrossRef PubMed.
- A. L. Allanson, M. M. Cotton, J. N. A. Tettey and A. C. Boyter, J. Pharm. Biomed. Anal., 2007, 44, 963 CrossRef CAS PubMed.
- M. Tatarczak, J. Flieger and H. Szumiło, J. Planar Chromatogr.--Mod. TLC, 2005, 18, 207 CrossRef CAS.
- D. Bao, T. T. Truong, P. J. Renick, M. E. Pulse and W. J. Weiss, J. Pharm. Biomed. Anal., 2008, 46, 723 CrossRef CAS PubMed.
- C. Zent and P. Smith, Tuber. Lung Dis., 1995, 76, 109 CrossRef CAS PubMed.
- S. Oswald, J. Peters, M. Venner and W. Siegmund, J. Pharm. Biomed. Anal., 2011, 55, 194 CrossRef CAS PubMed.
- I. Calleja, M. J. Blanco-Príeto, N. Ruz, M. J. Renedo and M. C. Dios-Viéitez, J. Chromatogr. A, 2004, 1031, 289 CrossRef CAS PubMed.
- P. F. Fang, H. L. Cai, R. H. Zhu, Q. Y. Tan, W. Gao, P. Xu, Y. P. Liu, W. Y. Zhang, Y. C. Chen and F. Zhang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2286 CrossRef CAS PubMed.
- R. Fernandez-Torres, M. A. Bello-López, M. Callejón-Mochón and J. C. Jiménez-Sánchez, Anal. Chim. Acta, 2008, 608, 204 CrossRef CAS PubMed.
- A. P. Argekar, S. S. Kunjir and K. S. Purandare, J. Pharm. Biomed. Anal., 1996, 14, 1645 CrossRef CAS PubMed.
- S. Rastgar and S. Shahrokhian, Talanta, 2014, 119, 156 CrossRef CAS PubMed.
- S. Gutiérrez-Fernández, M. C. Blanco-López, M. J. Lobo-Castañón, A. J. Miranda-Ordieres and P. Tuñón-Blanco, J. Electroanal., 2004, 16, 1660 CrossRef.
- M. A. A. Lomillo, O. D. Renedo and M. J. A. Martínez, Electrochim. Acta, 1807, 2005, 50 Search PubMed.
- K. Tyszczuk and M. Korolczuk, J. Electroanal., 2009, 21, 101 CrossRef CAS.
- P. Daneshgar, P. Norouzi, F. Dousty, M. R. Ganjali and A. A. Moosavi-Movahedi, Curr. Pharm. Anal., 2009, 5, 246 CrossRef CAS.
- K. Asadpour-Zeynali and F. Mollarasouli, Biosens. Bioelectron., 2017, 92, 509 CrossRef CAS PubMed.
- Y. L. Su and S. H. Cheng, Anal. Chim. Acta, 2015, 901, 41 CrossRef CAS PubMed.
- S. Cotchim, P. Thavarungkul, P. Kanatharana and W. Limbut, Electrochim. Acta, 2015, 184, 102 CrossRef CAS.
- F. Tadayon, S. Vahed and H. Bagheri, Mater. Sci. Eng., C, 2016, 68, 805 CrossRef CAS PubMed.
- P. Rameshkumar, R. Praveen and R. Ramaraj, J. Electroanal. Chem., 2015, 754, 118 CrossRef CAS.
- L. Li, E. Liu, X. Wang, J. Chen and X. Zhang, Mater. Sci. Eng., C, 2015, 53, 36 CrossRef CAS PubMed.
- L. Zhu, Y. Liu, P. Yang and B. Liu, Electrochim. Acta, 2015, 172, 88 CrossRef CAS.
- A. Benvidi, A. Dehghani-Firouzabadi, M. Mazloum-Ardakani, B. B. F. Mirjalili and R. Zare, J. Electroanal. Chem., 2015, 736, 22 CrossRef CAS.
- S. Koçak and B. Aslışen, Sens. Actuators, B, 2014, 196, 610 CrossRef.
- G. G. Wildgoose, C. E. Banks and R. G. Compton, Small, 2006, 2, 182 CrossRef CAS PubMed.
- H. Bagheri, R. P. Talemi and A. Afkhami, RSC Adv., 2015, 5, 58491 RSC.
- J. Li and X. Lin, Sens. Actuators, B, 2007, 126, 527 CrossRef CAS.
- T. Li, J. Xu, L. Zhao, S. Shen, M. Yuan, W. Liu, Q. Tu, R. Yu and J. Wang, Talanta, 2016, 159, 356 CrossRef CAS PubMed.
- S. Baskar, C. W. Liao, J. L. Chang and J. M. Zen, Electrochim. Acta, 2013, 88, 1 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, New York, 2001 Search PubMed.
- L. Fotouhi, M. Nemati and M. M. Heravi, J. Appl. Electrochem., 2011, 41, 137 CrossRef CAS.
- M. Baccarin, B. C. Janegitz, R. Berté, F. C. Vicentini, C. E. Banks, O. Fatibello-Filho and V. Zucolotto, Mater. Sci. Eng., C, 2016, 58, 97 CrossRef CAS PubMed.
- H. Bagheri, A. Afkhami, H. Khoshsafar, A. Hajian and A. Shahriyari, Biosens. Bioelectron., 2017, 89, 829 CrossRef CAS PubMed.
- H. Bagheri, E. Ranjbari, M. Amiri-Aref, A. Hajian, Y. Hosseinzadeh Ardakani and S. Amidi, Biosens. Bioelectron., 2016, 85, 814 CrossRef CAS PubMed.
- P. Hashemi, A. Afkhami, H. Bagheri, S. Amidi and T. Madrakian, Anal. Chim. Acta, 2017 DOI:10.1016/j.aca.2017.06.038.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04865c |
| This journal is © The Royal Society of Chemistry 2017 |