
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel protostane-type triterpenoids with inhibitory human carboxylesterase 2 activities†
Zhi-Jie Zhang‡
a,
Xiao-Kui Huo‡b,
Xiang-Ge Tian‡c,
Lei Fengb,
Jing Ningb,
Xin-Yu Zhaob,
Cheng-Peng Sun *b,
Chao Wangb,
Sa Dengb,
Bao-Jing Zhangb,
Hou-Li Zhangb and
Yong Liu*a
*b,
Chao Wangb,
Sa Dengb,
Bao-Jing Zhangb,
Hou-Li Zhangb and
Yong Liu*a
aClincial Laboratory, Shengjing Hospital of China Medical University, Sanhao Street, Shenyang 110004, China. E-mail: liuy@sj-hospital.org
bCollege of Pharmacy, Dalian Medical University, South Road of Lvshun, Dalian 116044, China. E-mail: suncp146@163.com
cBasic Medical College, Dalian Medical University, South Road of Lvshun, Dalian 116044, China
First published on 31st May 2017
Abstract
The rhizomes of Alisma orientalis have been used for centuries in China and other Asian countries as an effective herbal remedy. The phytochemical investigation of A. orientalis and biotransformation of two major triterpenoids alisols A (11) and B 23-acetate (13) by Cunninghamella elagans AS 3.2028 and Penicillium janthinellum AS 3.510 have led to the isolation of ten new protostane-type triterpenoids (1–5 and 18–22), including one novel 26-nor-protostane (1) and one unusual 17-nor-protostane (2), together with twelve known analogues. Their structures were determined by 1D and 2D NMR, and HRESIMS spectroscopic analyses. All the isolated compounds were assayed for their inhibitory activities against human carboxylesterase 2 (HCE-2). Compounds 1, 3–9, 12, 14–16, 19, and 20 showed significant inhibitory activities on HCE-2 with IC50 values from 0.51 ± 0.09 μM to 9.45 ± 0.73 μM. The inhibition kinetics of compound 5 toward HCE-2 were established, and its Ki value was determined as 0.57 μM. The interaction of compound 5 with HCE-2 was investigated using molecular docking.
Introduction
The genus Alisma (Alismataceae) are mainly distributed in the subtropical and temperate regions all over the world, including 11 species.1 The rhizome of Alisma orientalis (Sam.) Juzep. (Chinese name “Zexie”) is a traditional Chinese medicine, and divided into Jian Zexie (Fujian Province, China) and Chuan Zexie (Sichuan Province, China) based on their original places. This has been widely used as a diuretic, for detumescence and to treat obesity, diabetes and hyperlipidemia in China,2 and is also utilized to cure analogous illnesses in Japan and Korea.3 Phytochemical investigations of the genus Alisma have indicated that protostane-type triterpenoids are the major constituents and display remarkable pharmacological effects,3–5 including hepatoprotective,6 anti-tumor,7,8 anti-inflammatory,9 immuno-enhancing,10,11 and hypolipidemic activities,12,13 as well as inhibition of human carboxylesterase 2.3,14Human carboxylesterases (HCE-1 and HCE-2) are the important enzymes to hydrolyze chemicals with such functional groups as a carboxylic acid ester and amide, and they are known to play vital roles in drug metabolism and insecticide detoxication.15 HCE-1 is abundantly expressed in the liver, whereas HCE-2 is predominately expressed in the gastrointestinal tract. HCE-2, as a major mediator, could reduce drug toxicity and enhance drug bioavailability in drug metabolism, thus, it has been paid more attention.3,14,16,17
As part of our ongoing research on the genus Alisma to discovery a series of potential inhibitors of HCE-2,3,14,18 the 80% EtOH extracts of rhizomes of A. orientalis were investigated to afford five new protostane-type triterpenoids (1–5), including two novel 26-nor and 17-nor protostanes (1 and 2) together with twelve known ones (6–17, Fig. 1). In order to enrich the chemical structures of protostane-type triterpenoids, two major constituents alisols A (11) and B 23-acetate (13) isolated from A. orientalis were biotransformed by Cunninghamella elagans AS 3.2028 and Penicillium janthinellum AS 3.510, respectively, to provide five new metabolites (18–22, Fig. 1). Their structures were determined by 1D and 2D NMR, and HRESIMS analyses. All these isolated compounds were evaluated for their inhibitory effects against HCE-2.
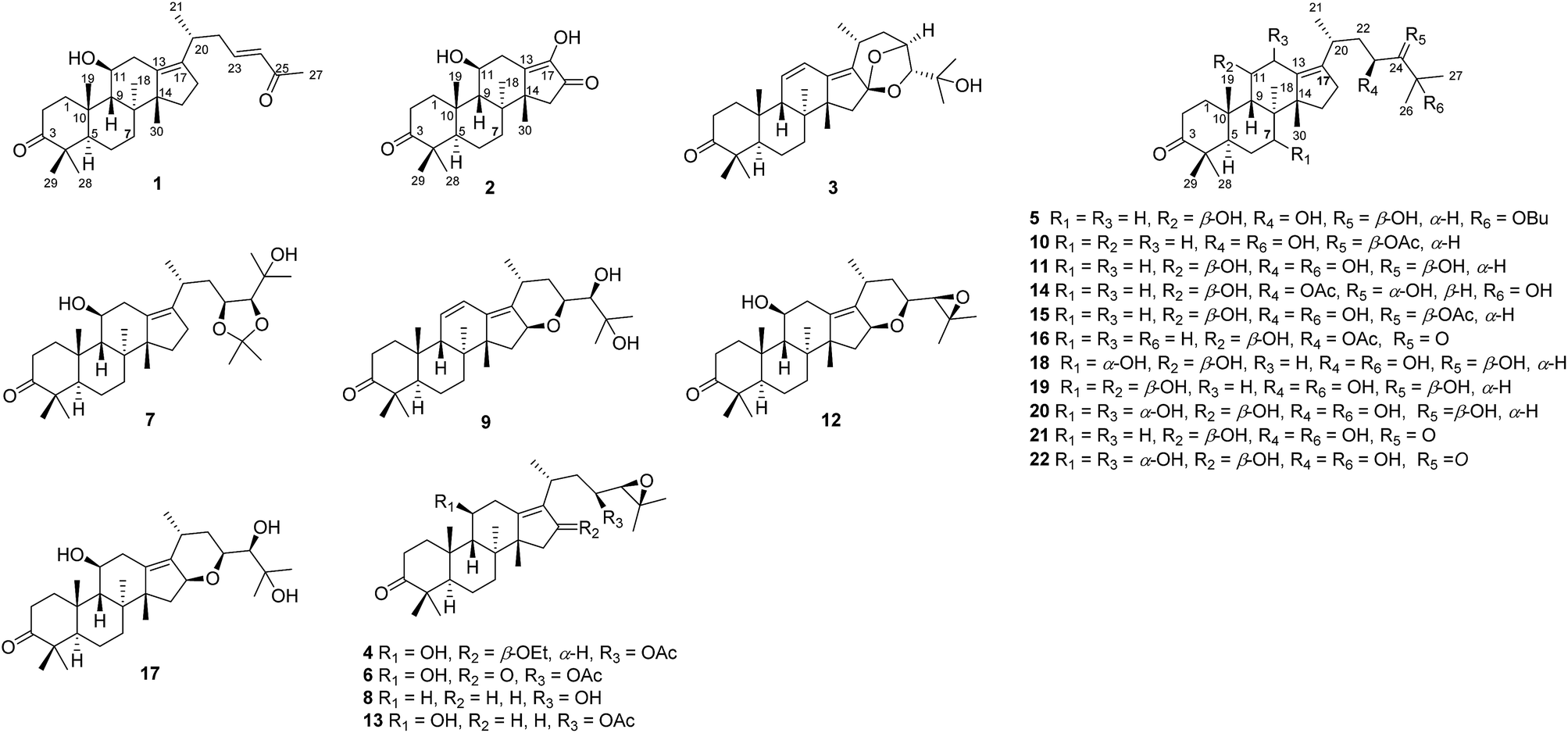 | ||
| Fig. 1 Chemical constituents (1–17) of A. orientalis and biotransformed products (18–22) of alisols A (11) and B 23-acetate (13). | ||
Results and discussion
Compound 1 has the molecular formula C29H44O3 by HRESIMS (m/z 458.3631 [M + NH4]+, calcd for C29H48NO3, 458.3634) and 13C NMR data, indicating 8 degrees of unsaturation. The 1H NMR spectrum of 1 showed signals of two olefinic protons at δH 6.77 (1H, dt, J = 15.8, 7.2 Hz, H-23) and 6.00 (1H, d, J = 15.8 Hz, H-24) in combination with characteristic carbons at δC 201.4 (C-25), 150.2 (C-23), and 132.8 (C-24), indicating the presence of an α,β-unsaturated carbonyl group. In the high field of 1H NMR spectrum, the signals of seven methyl were observed at δH 2.21 (3H, s, Me-27), 1.13 (3H, s, Me-30), 1.07 (3H, s, Me-28), 1.06 (3H, d, J = 6.7 Hz, Me-21), 1.04 (3H, s, Me-19), 1.04 (3H, s, Me-29), and 1.04 (3H, s, Me-18). The 13C NMR spectrum of 1 displayed 29 carbon resonances, including one carbonyl carbon (δC 223.7), two olefinic carbons (δC 139.2 and 136.1), and an oxygenated carbon (δC 70.5). All the above-mentioned data suggest that 1 was a nor-protostane type triterpenoid.3,14,18 A comparison of 1H and 13C NMR data of 1 and alisol A (11)19 suggested that their difference was the side chain of C-17. The signals of three oxygenated carbons [δC 77.6 (C-24), 74.1 (C-25), and 69.5 (C-23)] and one methyl carbon [δC 27.6 (C-26)] in 11 were absent, and signals of one carbonyl carbon [δC 201.4 (C-25)] and two olefinic carbons [δC 150.2 (C-23) and 132.8 (C-24)] in 1 were present, which indicated the presence of a Δ23,24 double bond and a carbonyl group in the C-17 side chain. The deduction was supported by an HMBC experiment in which showed correlations of Me-2 with C-17/C-20, H-23 with C-20/C-24/C-25, and Me-27 with C-24/C-25 (Fig. 2). The relative configuration of 1 was determined by NOESY correlations between H-11 and Me-18, requiring a β-orientation of OH-11 (Fig. 3). The Δ23,24 double bond was deduced to be transformed on a basis of large coupling constants [δH 6.77 (1H, dt, J = 15.8, 7.2 Hz, H-23) and 6.00 (1H, d, J = 15.8 Hz, H-24)]. Thus, 1 was elucidated as alismanol H.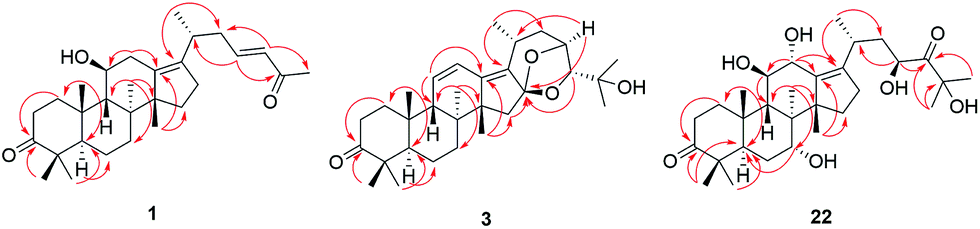 | ||
| Fig. 2 Selected HMBC correlations of compounds 1, 3, and 22. | ||
 | ||
| Fig. 3 Selected NOESY correlations of compounds 1, 3, and 22. | ||
Compound 2 was obtained as a white powder, and its molecular formula was defined as C22H32O4 based on HRESIMS (m/z 383.2188 [M + Na]+, calcd for C22H32O4Na, 383.2198) and 13C NMR data. The 1H NMR spectrum of 2 exhibited the presence of an oxygenated methine [δH 3.98 (1H, m, H-11)] and five methyls [δH 1.33 (3H, s, Me-30), 1.08 (3H, s, Me-19), 1.08 (3H, s, Me-28), 1.05 (3H, s, Me-29), and 0.95 (3H, s, Me-30)]. The 13C NMR spectrum of 2 revealed 22 carbons, including two carbonyl carbons (δC 223.3 and 205.2), two olefinic carbons (δC 152.4 and 148.3), and an oxygenated carbon (δC 69.8). Compared with 20-hydroxy alisol C isolated from A. orientalis,3 signals of the C-17 side chain in 20-hydroxyanisole C were absent, which suggested that 2 was a 17-nor protostane-type triterpenoid. In the HMBC spectrum of 2, the correlations of H-12a with C-13, H-13b with C-13/C-14, and H-15a with C-13/C-14/C-16 confirmed the conclusion. The β-orientation of OH-11 was determined by an NOESY correlation from H-11 to Me-18. Thus, 2, as a first 17-nor protostane, was defined as alismanol I.
Compound 3, as a white powder, possessed a molecular formula of C30H44O4 established by HRESIMS (m/z 469.3319 [M + H]+, calcd for C30H45O4, 469.3318). The 1H and 13C NMR data of 3 were similar to those of 24-deacetyl alisol O (9) isolated from A. orientalis,20 except for chemical shifts of C-16 and C-24 deshielded from δC 80.5 and 76.3 in 9 to δC 119.1 and 86.2 in 3 and the absence of an oxygenated methine [δH 4.57 (1H, ddd, J = 7.9 Hz, H-16)] in 9. The HMBC experiment of 3 displayed correlations of H-15a/H-15b with C-16, H-24 with C-16, and H-23 with C-16 (Fig. 2), requiring the presence of a C(16)–O–C(23) unit and a C(16)-O-C(24) moiety. The relative configuration of 3 was established by NOESY correlations from H-20 to H-24 and H-23 to Me-26 (Fig. 3), which indicated an α-orientation of H-23 and a β-orientation of H-24. Accordingly, 3 was established as alismanol J.
Compound 4 was assigned the molecular formula C34H56O6 by HRESIMS (m/z 581.3813 [M + Na]+, calcd for C34H54O6Na, 581.3818) and 13C NMR data. The 1H and 13C NMR data of 4 closely resembled those of alisol B 23-acetate (13),21 except for the presence of an ethoxy group [δH 3.52 (1H, m, H-1′a), 3.35 (1H, m, H-1′b), and 1.16 (3H, t, J = 7.0 Hz, Me-2′); δC 65.6 (C-1′) and 16.5 (C-2′)] and an oxygenated methine [δH 4.43 (1H, dd, J = 8.0, 4.7 Hz, H-16)] in 4, and the downfield shifts of C-16 from δC 29.1 in 13 to δC 86.5 in 4, indicating the presence of an ethoxy moiety at C-16. The deduction was confirmed by HMBC correlations from H-1′a/H-1′b to C-16/C-2′. In the NOESY spectrum of 4, cross-peaks of Me-18 with H-11/H-16 suggested the β-configurations of OH-11 and OEt-16. In addition, the NOESY cross-peak from H-24 to Me-27 and the characteristic NMR data [δH 4.74 (1H, ddd, J = 10.6, 8.5, 2.0 Hz, H-23) and 2.73 (1H, d, J = 8.5 Hz, H-24); δC 73.7 (C-23), 67.1 (C-24), and 60.5 (C-25)] suggested that 4 possessed the same side chain at C-17 as alisol B series.3,18 Therefore, 4 was assigned as 16β-ethoxy alisol B 23-acetate.
The molecular formula of 5 was established as C34H58O5 according to HRESIMS (m/z 569.4181 [M + Na]+, calcd for C34H56O5Na, 569.4182) spectrum. Comparison of the 1H and 13C NMR data of 7 and alisol A (11)19 indicated the presence of a butyl group [δH 3.41 (2H, m, H2-1′), 1.49 (2H, m, H2-2′), 1.37 (2H, m, H2-3′), and 0.93 (3H, t, J = 7.4 Hz, Me-4′); δC 62.3 (C-1′), 33.9 (C-2′), 20.7 (C-3′), and 14.5 (C-4′)] in 5. The HMBC correlation from H-1′ to C-25 and COSY cross-peaks of H-1′ with H-2′, H-2′ with H-3′, and H-3′ with Me-4′ suggested that the butyl moiety was attached to OH-25. Thus, 5 was identified as 25-O-butyl alisol A.
HRESIMS spectrum of 18 showed a molecular ion peak at m/z 529.3507 [M + Na]+ (calcd for C30H50O6Na, 529.3505), which was the molecular formula C30H50O6. Compared with alisol A (11),19 the signal of an oxygenated methine [δH 4.17 (1H, dd, J = 6.4, 1.1 Hz, H-7)] was present, and the chemical shift value of C-7 was deshielded from δC 34.6 in 11 to δC 67.9 in 18. The above data required the location of a hydroxy group at C-7, which was further confirmed by HMBC correlations of H-7 with C-5/C-8/C-14 and COSY correlations from H-5 to H-6a/H-6b and H-6a/H-6b with H-7. The NOESY spectrum of 18 exhibited cross-peaks between H-7 and H-9/Me-30, indicating that OH-7 was α-oriented. Thus, 18 was established as 7α-hydroxy alisol A.
HRESIMS data of 19 suggested that 19 possessed the same molecular formula as 18. The 1H and 13C NMR spectroscopic data of 19 were similar to those of 18, with the exception that the signal of H-7 was shielded from δH 4.17 (1H, dd, J = 6.4, 1.1 Hz) in 18 to δH 3.75 (1H, dd, J = 13.7, 6.4 Hz) in 19, and the chemical shift of C-7 was deshielded from δC 69.7 in 18 to δC 81.8 in 19, which suggested that their difference was the configuration of C-7. The deduction was confirmed by an NOESY correlation of H-7 with Me-18, indicating a β-orientation of OH-7. Accordingly, 19 was defined as 7β-hydroxy alisol A.
HRESIMS spectrum of 20 observed a molecular ion peak at m/z 527.3343 [M + Na]+ (calcd for C30H48O6Na, 527.3349), which suggested that its molecular formula was C30H50O7. A comparison of NMR data of 20 and 18 revealed that the chemical shift value of C-12 was deshielded from δC 35.1 in 18 to δC 67.6 in 20, requiring the location of a hydroxy group at C-12. In the HMBC spectrum of 20, the long-range correlations from H-9/H-11 to C-12 and H-12 to C-9/C-11/C-13/C-17 confirmed the deduction. The NOESY spectrum of 20 observed the cross-peaks of H-12 with H-9/Me-19, indicating an α-orientation of OH-12. Therefore, 20 was defined as 7α,12α-dihydroxy alisol A.
Compound 21 was obtained as a white powder. Its molecular formula was assigned as C30H50O7 established by HRESIMS (m/z 545.3457 [M + Na]+, calcd for C30H50O7Na, 545.3454) data. The 1D NMR data indicated that 21 was a protostane-type triterpenoid with a C-3 carbonyl group,3,14,18 a C-13/C-17 cyclic double bond, and oxygenated carbons (C-7, C-11, C-23, C-25), which suggested that the structure of 21 was similar to that of 19, except for C-24. The C-24 carbonyl moiety was established through an HMBC experiment in which showed long-range correlations of H-23 with C-24/C-25 and Me-26/Me-27 with C-24/C-25. The α-orientation of OH-7 was established by NOESY cross-peak of H-7 with Me-30. Therefore, 21 was defined as 7α-hydroxy-24-oxo alisol A.
Analysis of HRESIMS and 1D NMR data of 22 and 21 revealed that their differences were in ring C since signals of an oxygenated methine were observed at δH 4.55 (1H, d, J = 5.0 Hz, H-12) and δC 67.7 (C-12). This conclusion was supported by HMBC correlations from H-9 to C-12 and H-12 to C-9/C-11/C-13/C-17 (Fig. 2). The configurations of OH-7 and OH-12 were assigned as an α-orientation based on the correlations of H-7 with Me-30 and H-12 with H-9/Me-30 in the NOESY experiment (Fig. 3). Accordingly, 22 was established as 7α,12α-dihydroxy-24-oxo alisol A.
In addition, eleven known compounds were isolated from the dried rhizomes of A. orientalis, and their structures were elucidated to be alisol C 23-acetate (6),19 alisol A 23,24-acetonide (7),5 11-deoxy alisol B (8),19 24-deacetyl alisol O (9),20 11-deoxy alisol A (10),19 alisol A (11),19 16,23-oxido alisol B (12),19 alisol B 23-acetate (13),21 alisol E 23-acetate (14),22 alisol A 24-acetate (15),19 alisol B monoacetate (16),23 and alisol F (17).22
Previous investigation of the biotransformation of protostane-type triterpenoid alisol G by P. janthinellum AS 3.510 led to four new metabolites, which demonstrated that microbial transformation is an important approach to rich structural diversity of protostane-type triterpenoids.14 In this work, the biotransformation of alisols A (11) and B 23-acetate (13) by C. elagans AS 3.2028 and P. janthinellum AS 3.510, respectively, yielded five new metabolites (18–22). According to the biotransformed results, C. elagans AS 3.2028 and P. janthinellum AS 3.510 possessed the hydroxylated and oxidized capabilities, while hydroxylation is still its main reaction, and their biotransformation sites could be at C-7 and C-12.
In drug metabolism, HCE-2 plays an important role in reducing drug toxicity and enhancing drug bioavailability.3,14,16,17 Previous studies have indicated that protostane-type triterpenoids possess inhibitory activities on HCE-2.3,14 Therefore, all the isolated compounds were assayed for inhibitory HCE-2 effects. As shown in Table 4, compound 5 exerted the most significant inhibitory activities with IC50 of 0.51 ± 0.09 μM, and others 1, 3, 4, 6–9, 12, 14–16, 19, and 20 displayed significant inhibitory activities against HCE-2 with IC50 values from 2.58 ± 0.51 μM to 9.45 ± 0.73 μM. A comparison of the inhibitory efficiency of 5, 11, and 15 indicated that acetyl and butyl groups linked at OH-24 and OH-25 were benefit for inhibitory activities. The biotransformed products (18–22) displayed more significant inhibitory effects than the substrates (11 and 13). Compound 18 showed moderate inhibitory effect with the IC50 value of 18.05 ± 1.53 μM, whereas IC50 values of compounds 19 and 20 were 7.39 ± 1.21 and 3.73 ± 0.76 μM, respectively, which suggested that the β-configuration of OH-7 and the oxidization of OH-24 were in favour of inhibition on HCE-2. The results indicated that biotransformation was an effective method to rich the structure of protostane-type triterpenoids and improve their bioactivities.
The inhibition kinetics and Ki value were further investigated (Fig. 4) in order to indicate the potential inhibition of HCE-2 by compound 5. As shown in Fig. 4, compound 5 displayed concentration-dependent inhibition of HCE-2, and the Ki value was calculated as 0.57 μM for the activity of HCE-2.
 | ||
| Fig. 4 Evaluation of the inhibition of recombinant HCE-2 by compound 5. (A) Compound 5 exhibited concentration-dependent inhibition of HCE-2. (B) The inhibition kinetics of compound 5. | ||
The interaction mechanism between compound 5 and HCE-2 was also investigated. As shown in Fig. 5, compound 5 could be well docked into the catalytic site of HCE-2, while the carbonyl group of compound 5 was near to the catalytic site of HCE-2 (Ser-221, Glu-220, and His-140). As a result, the docking in the best binding mode gave rise to the lowest binding free energy (−5.97 kcal mol−1), indicating that compound 5 had high affinity for HCE-2, which was in agreement with our results.
 | ||
| Fig. 5 Possible binding modes of the amino acid residues Ser-221, Glu-220, and His-140 in an active site triad of HCE-2 with compound 5. White, nonpolar amino acid; green, polar amino acid; red, acidic amino acid. | ||
Conclusions
Investigation on the rhizomes of A. orientalis led to the isolation and structure elucidation of five new triterpenoids including two novel nor-protostanes (1 and 2) as well as thirteen analogues. It was first reported about the presence of 17-nor protostane-type triterpenoid (2) in nature. In addition, two major constituents of A. orientalis, alisols A (11) and B 23-acetate (13) were transformed by C. elagans AS 3.2028 and P. janthinellum AS 3.510, respectively, yielding five new metabolites (18–22). The results indicated that C. elagans AS 3.2028 and P. janthinellum AS 3.510 mainly possessed the hydroxylated and oxidized activities in these biotransformation processes, and their hydroxylated sites could be at C-7 and C-12. Compounds 1, 3–9, 12, 14–16, 19, and 20 showed significant inhibitory activities on HCE-2 with IC50 values from 0.51 ± 0.09 μM to 9.45 ± 0.73 μM. The inhibition kinetics of compound 5 toward HCE-2 were established, and the Ki value was determined as 0.57 μM. The interaction of compound 5 with HCE-2 was validated using the molecular docking.Experimental section
General methods and materials
Optical rotations were measured on a Perkin-Elmer 241 polarimeter. UV spectra were recorded on a JASCO V-650 spectrophotometer. The NMR spectra were recorded on a Bruker-600 spectrometer, using tetramethylsilane (TMS) as internal standard. Chemical shifts are in δ (ppm), and coupling constants (J) in Hz. HRESIMS spectra were measured on an Agilent 1100 series LC/MSD ion trap mass spectrometer. High-performance liquid chromatography (HPLC) analyses were performed on a UItimate 3000 HPLC system equipped with a photodiode array detector and a quaternary pump system and a column compartment. Preparative HPLC was performed on an Elite P2300 instrument with an Elite UV2300 detector and a Thermo C18 column (250 mm × 10 mm, 5 μm). All solvents were obtained from Tianjin Kemiou Chemical Reagent Company (Tianjing, China), CH3CN and MeOH for HPLC analysis were chromatographic grade (Merck, Darmstadt, Germany). Silica gel (200–300 mesh) for column chromatography (CC) were purchased from Qingdao Marine Chemical Factory (Qingdao, People's Republic of China).Plant material
Dried rhizomes of A. orientalis were purchased in January 2013 from Beijing Tongrentang Co., Ltd., China, and identified by Prof. Jing-Ming Jia, Shenyang Pharmaceutical University. A voucher specimen (301114120P) has been deposited in the herbarium of the Department of Medicinal Chemistry, Dalian Medical University.Extraction and isolation
The dried rhizomes of A. orientalis (4.0 kg) were extracted with 80% EtOH (3 × 2 h × 10 L) to afford a residue after solvent removal in vacuo. The residue was suspended in H2O (5 L), and extracted with petroleum ether (3 × 5 L), CHCl3 (3 × 5 L), EtOAc (3 × 5 L), and n-BuOH (3 × 5 L), successively.The CHCl3 extract (240 g) was separated by a silica gel column, eluted with CHCl3–MeOH (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–4:1), to afford fractions 1–24. Fr. 6 (7.8 g) was purified by an ODS column with CH3CN–H2O (20–60%) and preparative HPLC (CH3CN–H2O, 25%) to obtain compounds 7 (9 mg) and 16 (3 mg).
:1–4:1), to afford fractions 1–24. Fr. 6 (7.8 g) was purified by an ODS column with CH3CN–H2O (20–60%) and preparative HPLC (CH3CN–H2O, 25%) to obtain compounds 7 (9 mg) and 16 (3 mg).
The EtOAc extracts (43 g) were subjected to silica gel CC eluted with CH2Cl2–MeOH (100:1–1:1) to afford fractions 1–27. Fr. 7 (2.5 g) was separated by an ODS column eluted with MeOH–H2O (50–80%), and purified by preparative HPLC (CH3CN–H2O, 40–60%) to afford compounds 1 (2 mg), 4 (22 mg), 6 (2 mg), 11 (510 mg), 13 (450 mg), and 17 (9 mg). Fr. 9 (1.6 g) was separated through ODS CC eluted with MeOH–H2O (60–80%), and purified by preparative HPLC (CH3CN–H2O, 40–55%) to afford compounds 10 (2 mg), 14 (15 mg), and 15 (9 mg).
The n-BuOH extracts (86 g) were subjected to D101 resin CC eluted with EtOH–H2O (0–95%) to afford four fractions 1–4. Fr. 4 (3.0 g) was separated by a silica gel column eluted with petroleum ether–acetone (10:1–1:1), yielding four subfractions Fr. 41–Fr. 44. The purification of Fr. 44 (2.0 g) by HPLC eluted with MeOH–H2O (75–85%) has led to the isolation of compounds 2 (2 mg), 3 (41 mg), 5 (1.9 mg), 8 (4 mg), 9 (2 mg), and 12 (3 mg).
ε) 222 (4.0) nm; 1H (600 MHz, MeOH-d4) and 13C NMR (150 MHz, MeOH-d4) data, see Tables 1 and 3; HRESIMS m/z 458.3631 [M + NH4]+ (calcd for C29H48NO3, 458.3634).
| No. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 2.30 m | 2.27 m | 2.13 m | 2.36 m | 2.31 m |
| 2.09 m | 2.10 m | 1.67 m | 2.06 m | 2.10 m | |
| 2 | 2.81 m | 2.82 m | 2.84 m | 2.79 m | 2.82 m |
| 2.23 m | 2.24 m | 2.20 m | 2.22 m | 2.25 m | |
| 5 | 2.17 m | 2.19 br d (11.7) | 2.42 d (12.2) | 2.18 br d (11.2) | 2.21 m |
| 6 | 1.48 m | 1.54 m | 1.53 m | 1.49 m | 1.37 m |
| 1.30 m | 1.39 m | 1.40 m | 1.31 m | 1.05 m | |
| 7 | 2.06 m | 2.31 m | 1.90 m | 2.06 m | 2.07 m |
| 1.27 m | 1.31 m | 1.32 m | 1.27 m | 1.29 m | |
| 9 | 1.78 d (10.9) | 1.89 d (10.6) | 2.27 br s | 1.77 d (10.9) | 1.79 d (10.7) |
| 11 | 3.80 m | 3.98 m | 5.77 br d (10.3) | 3.69 m | 3.81 m |
| 12 | 2.69 m | 3.14 m | 6.53 d (10.3) | 2.49 m | 2.79 m |
| 1.98 m | 2.26 m | 1.99 m | 2.05 m | ||
| 15 | 1.93 m | 2.43 d (19.3) | 2.19 d (14.5) | 2.31 dd (14.0, 8.0) | 1.95 m |
| 1.34 m | 1.87 d (19.3) | 1.66 d (14.5) | 1.20 dd (14.0, 4.7) | 1.34 m | |
| 16 | 2.26 m | 4.43 dd (8.0, 4.7) | 2.24 m | ||
| 2.19 m | 2.19 m | ||||
| 18 | 1.04 s | 0.95 s | 1.02 s | 0.91 s | 1.06 s |
| 19 | 1.04 s | 1.08 s | 0.90 s | 1.03 s | 1.05 s |
| 20 | 2.78 m | 2.81 m | 2.72 m | 2.84 m | |
| 21 | 1.06 d (6.7) | 1.28 d (6.7) | 1.06 d (6.9) | 1.02 d (7.0) | |
| 22 | 2.29 m | 1.68 m | 2.07 m | 1.59 m | |
| 2.24 m | 1.54 m | 1.68 m | 1.47 m | ||
| 23 | 6.77 dt (15.8, 7.2) | 4.40 br s | 4.74 ddd (10.6, 8.5, 2.0) | 3.75 dd (10.2, 2.0) | |
| 24 | 6.00 d (15.8) | 3.72 br s | 2.73 d (8.5) | 3.06 br s | |
| 26 | 1.18 s | 1.26 s | 1.20 s | ||
| 27 | 2.21 s | 1.10 s | 1.35 s | 1.20 s | |
| 28 | 1.07 s | 1.08 s | 1.09 s | 1.07 s | 1.08 s |
| 29 | 1.04 s | 1.05 s | 1.04 s | 1.03 s | 1.05 s |
| 30 | 1.13 s | 1.33 s | 1.06 s | 1.24 s | 1.18 s |
| 16-OEt | 3.35 m | ||||
| 1.16 t (7.0) | |||||
| 23-OAc | 2.08 s | ||||
| 3.52 m | |||||
| 25-OBu | 3.41 m | ||||
| 1.49 m | |||||
| 1.37 m | |||||
| 0.93 t (7.4) |
ε) 267 (3.9) nm; 1H (600 MHz, MeOH-d4) and 13C NMR (150 Mz, MeOH-d4) data, see Tables 1 and 3; HRESIMS m/z 383.2188 [M + Na]+ (calcd for C22H32O4Na, 383.2198).ε) 250 (3.9) nm; 1H (600 MHz, MeOH-d4) and 13C NMR (150 MHz, MeOH-d4) data, see Tables 1 and 3; HRESIMS m/z 469.3319 [M + H]+ (calcd for C30H45O4, 469.3318).ε) 203 (4.0) nm; 1H (600 MHz, MeOH-d4) and 13C NMR (150 MHz, MeOH-d4) data, see Tables 1 and 3; HRESIMS m/z 581.3813 [M + Na]+ (calcd for C34H54O6Na, 581.3818).ε) 202 (3.9) nm; 1H (600 MHz, MeOH-d4) and 13C NMR (150 MHz, MeOH-d4) data, see Tables 1 and 3; HRESIMS m/z 569.4181 [M + Na]+ (calcd for C34H56O5Na, 569.4182).Biotransformation of alisols A (11) and B 23-acetate (13)
Aspergillus niger AS 3.739, A. niger AS 3.4627, A. niger AS 3.795, A. niger AS 3.1858, Chaetomium blakesleana AS 3.910, C. blakesleana AS 3.970, Cunninghamella elegans AS 3.1207, C. echinulata AS 3.2004, C. elegans AS 3.2028, C. echinulata AS 3.3400, Syncephalastrum racemosum AS 3.264, Fusarium avenaceum AS 3.4594, Penicillium melinii AS 3.4474, P. janthinellum AS 3.510, and Mucor rouxianus AS 3.3447 were purchased from Chinese General Microbiological Culture Collection Center in Beijing, China. All the culture and biotransformation experiments were performed as the previous method.24 Preliminary screenings were performed in 250 mL Erlenmeyer flasks containing potato medium (100 mL). After incubation for 24 h, 0.2 mL of alisols A (11) or B 23-acetate (13) (10 mg mL−1, in MeOH) was added to each flask for preliminary screening and incubated for 3 days. Both substrate and organism controls were incubated under the same conditions to prove the stability of the substrate in the control (blank) culture. Similarly, preparative experiments were carried out in 1000 mL Erlenmeyer flasks with 400 mL of potato dextrose medium, and the microorganism was pre-cultured under the above culture conditions for 24 h. Then, 1.0 mL of 11 or 13 (10 mg mL−1, in MeOH) was added to each flask. 11 (200 mg) and 13 (200 mg) were transformed by C. elegans AS 3.2028 and P. janthinellum AS 3.510, respectively.The cultures of 11 were pooled and filtered, and the filtrates were extracted with EtOAc (3 × 8 L) to afford the residue. The residue (6 g) was subjected to silica gel CC eluted with CHCl3–MeOH (from 40:1 to 10:1) to afford five fraction 1–5. Separation of Fr. 5 (101 mg) by preparative HPLC (MeOH–H2O, 55%) led to the isolation of compounds 18 (24 mg), 19 (16 mg), and 20 (6 mg).
The cultures of 13 were conducted as the same method as 11. The residue (1.6 g) was subjected to silica gel CC eluted with CHCl3–MeOH (from 50:1 to 10:1) to afford fraction 1–5. Fr. 4 (121 mg) was purified by preparative HPLC (MeOH–H2O, 50%), yielding compound 21 (30 mg). Fr. 5 (62 mg) was separated by preparative HPLC (MeOH–H2O, 52%) to obtain compound 22 (12 mg).
ε) 202 (4.2) nm; 1H (600 MHz, MeOH-d4) and 13C NMR (150 MHz, MeOH-d4) data, see Tables 2 and 3; HRESIMS m/z 529.3507 [M + Na]+ (calcd for C30H50O6Na, 529.3505).
| No. | 18 | 19 | 20 | 21 | 22 |
|---|---|---|---|---|---|
| 1 | 2.26 m | 2.28 m | 2.30 m, | 2.28 m | 2.30 m |
| 2.11 m | 2.19 m | 2.20 m | 2.19 m | 2.20 m | |
| 2 | 2.81 m | 2.82 m | 2.82 m | 2.82 m | 2.82 m |
| 2.21 m | 2.22 m | 2.23 m | 2.22 m | 2.23 m | |
| 5 | 2.45 dd (13.4, 1.5) | 2.45 m | 2.42 dd (13.0, 1.5) | 2.45 m | 2.43 dd (13.5, 2.0) |
| 6 | 1.71 m | 1.71 m | 1.72 m | 1.72 m | 1.76 m |
| 1.38 m | 1.47 m | 1.39 m | 1.39 m | 1.40 m | |
| 7 | 4.17 dd (6.4, 1.1) | 3.75 dd (13.7, 6.4) | 4.23 br d (6.0) | 4.18 d (5.0) | 4.25 br d (5.5, 1.5) |
| 9 | 1.61 d (10.2) | 2.08 d (10.4) | 1.85 d (11.5) | 1.61 d (10.7) | 1.86 d (11.0) |
| 11 | 3.86 m | 3.84 m | 3.91 dd (11.0, 4.5) | 3.87 m | 3.91 dd (11.0, 5.0) |
| 12 | 2.79 m | 2.79 m | 4.53 d (4.5) | 2.79 m | 4.55 d (5.0) |
| 2.02 m | 2.02 m | 2.03 m | |||
| 15 | 2.26 m | 2.22 m | 2.26 m | 2.27 m | 2.51 m |
| 1.40 m | 1.68 m | 2.21 m | 1.41 m | 2.22 m | |
| 16 | 2.21 m | 2.27 m | 2.30 m | 2.45 m | 2.26 m |
| 2.19 m | 2.13 m | 2.16 m | 2.20 m | ||
| 18 | 1.00 s | 1.02 s | 1.00 s | 1.02 s | 1.00 s |
| 19 | 1.03 s | 1.10 s | 1.04 s | 1.04 s | 1.04 s |
| 20 | 2.82 m | 2.82 m | 2.99 m | 2.97 m | 3.10 m |
| 21 | 1.03 d (6.8) | 1.03 d (6.8) | 1.04 d (7.0) | 1.04 d (6.8) | 1.05 d (7.0) |
| 22 | 1.61 m | 1.61 m | 1.83 m | 1.97 m | 2.04 m |
| 1.50 m | 1.50 m | 1.37 m | 1.27 m | 1.36 m | |
| 23 | 3.73 dd (10.2, 2.0) | 3.73 dd (10.7, 1.2) | 3.78 dt (10.5, 1.5) | 4.46 dd (10.7, 2.0) | 4.61 dd (11.0, 2.0) |
| 24 | 3.02 d (1.2) | 3.03 d (1.2) | 3.07 d (1.0) | ||
| 26 | 1.20 s | 1.20 s | 1.19 s | 1.28 s | 1.29 s |
| 27 | 1.21 s | 1.21 s | 1.22 s | 1.32 s | 1.33 s |
| 28 | 1.08 s | 1.09 s | 1.08 s | 1.08 s | 1.09 s |
| 29 | 1.03 s | 1.08 s | 1.04 s | 1.03 s | 1.04 s |
| 30 | 1.18 s | 1.02 s | 1.30 s | 1.23 s | 1.34 s |
| No. | 1 | 2 | 3 | 4 | 5 | 18 | 19 | 20 | 21 | 22 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 32.1 | 32.0 | 33.5 | 31.9 | 32.1 | 32.2 | 32.5 | 33.0 | 32.3 | 33.0 |
| 2 | 34.8 | 34.7 | 34.6 | 34.7 | 34.8 | 30.8 | 29.9 | 35.0 | 35.2 | 35.0 |
| 3 | 223.7 | 223.3 | 222.8 | 223.5 | 223.8 | 223.6 | 223.2 | 223.5 | 223.7 | 223.7 |
| 4 | 48.3 | 48.3 | 48.3 | 48.2 | 48.3 | 47.9 | 48.1 | 48.0 | 47.9 | 48.0 |
| 5 | 49.8 | 49.8 | 47.5 | 49.6 | 49.7 | 43.9 | 47.9 | 44.2 | 43.9 | 44.2 |
| 6 | 21.2 | 21.1 | 20.5 | 21.2 | 20.7 | 32.7 | 32.2 | 32.6 | 32.7 | 32.6 |
| 7 | 35.5 | 36.1 | 32.4 | 35.5 | 35.5 | 69.7 | 81.8 | 69.0 | 69.6 | 69.0 |
| 8 | 41.8 | 41.8 | 39.2 | 41.8 | 42.0 | 46.9 | 48.8 | 45.9 | 46.9 | 46.5 |
| 9 | 50.7 | 49.9 | 48.4 | 50.3 | 50.7 | 50.8 | 51.1 | 46.4 | 50.8 | 46.0 |
| 10 | 38.3 | 38.4 | 37.0 | 38.3 | 38.3 | 38.5 | 38.7 | 38.1 | 38.5 | 38.3 |
| 11 | 70.5 | 69.8 | 132 | 70.5 | 70.7 | 70.7 | 70.5 | 71.8 | 70.7 | 72.0 |
| 12 | 35.1 | 34.0 | 124.1 | 35.5 | 35.2 | 35.1 | 35.0 | 67.6 | 35.2 | 67.7 |
| 13 | 139.2 | 152.4 | 139.6 | 144.6 | 139.2 | 139.2 | 140.9 | 141.5 | 139.9 | 142.2 |
| 14 | 58.4 | 47.2 | 52.5 | 56.4 | 58.4 | 58.1 | 58.0 | 57.2 | 58.2 | 57.6 |
| 15 | 31.7 | 44.0 | 39.8 | 41.2 | 31.9 | 32.3 | 34.5 | 34.5 | 32.3 | 29.5 |
| 16 | 30.4 | 205.2 | 119.1 | 86.5 | 30.4 | 30.4 | 30.8 | 32.7 | 30.6 | 29.4 |
| 17 | 136.1 | 148.3 | 136 | 135.5 | 137.1 | 137.3 | 136.1 | 144.5 | 136.5 | 143.7 |
| 18 | 24.6 | 23.5 | 23.1 | 24.3 | 24.6 | 14.9 | 24.6 | 15.2 | 14.9 | 15.1 |
| 19 | 26.2 | 26.1 | 25.2 | 26.1 | 26.2 | 26.0 | 26.1 | 26.1 | 26.0 | 25.9 |
| 20 | 33.2 | 29.0 | 29.2 | 29.8 | 30.0 | 29.9 | 29.7 | 29.9 | 29.7 | |
| 21 | 20.2 | 20.8 | 19.9 | 21.0 | 20.9 | 20.9 | 20.5 | 20.8 | 20.3 | |
| 22 | 39.6 | 40.2 | 39.1 | 42.1 | 42.0 | 42.0 | 42.1 | 40.5 | 40.5 | |
| 23 | 150.2 | 75.5 | 73.7 | 69.8 | 70.5 | 70.5 | 69.2 | 73.1 | 72.5 | |
| 24 | 132.8 | 86.2 | 67.1 | 79.8 | 79.6 | 79.7 | 79.7 | 219.2 | 218.4 | |
| 25 | 201.4 | 72.3 | 60.3 | 79.7 | 74.8 | 74.8 | 74.6 | 77.8 | 77.8 | |
| 26 | 26.9 | 25.1 | 23.5 | 27.3 | 27.3 | 27.1 | 28.6 | 28.1 | ||
| 27 | 26.7 | 24.2 | 20.3 | 21.7 | 26.7 | 26.8 | 26.2 | 27.8 | 27.7 | |
| 28 | 30.0 | 30.0 | 29.7 | 30.0 | 30.0 | 29.6 | 29.9 | 29.1 | 29.6 | 29.5 |
| 29 | 20.6 | 20.6 | 19.9 | 20.7 | 20.6 | 20.8 | 20.8 | 20.8 | 20.7 | 20.8 |
| 30 | 23.6 | 23.3 | 23.8 | 25.5 | 23.9 | 24.4 | 26.5 | 25.6 | 24.4 | 26.1 |
| 16-OEt | 65.6 | |||||||||
| 16.5 | ||||||||||
| 23-OAc | 172.4 | |||||||||
| 21.6 | ||||||||||
| 25-OBu | 62.3 | |||||||||
| 33.9 | ||||||||||
| 20.7 | ||||||||||
| 14.5 |
| Compound | IC50 (μM) | Compound | IC50 (μM) |
|---|---|---|---|
| a No determined.b Control drug. | |||
| 1 | 2.67 ± 0.04 | 13 | 35.61 ± 1.21 |
| 2 | 75.64 ± 3.86 | 14 | 3.78 ± 0.21 |
| 3 | 7.44 ± 0.41 | 15 | 6.11 ± 0.46 |
| 4 | 7.38 ± 0.18 | 16 | 4.18 ± 0.48 |
| 5 | 0.51 ± 0.09 | 17 | 49.43 ± 1.79 |
| 6 | 9.45 ± 0.73 | 18 | 18.05 ± 1.53 |
| 7 | 2.99 ± 0.60 | 19 | 7.39 ± 1.21 |
| 8 | 3.39 ± 0.81 | 20 | 3.73 ± 0.76 |
| 9 | 2.58 ± 0.51 | 21 | 21.89 ± 1.34 |
| 10 | —a | 22 | 13.74 ± 0.09 |
| 11 | 99.65 ± 2.81 | Loperamideb | 1.26 ± 0.03 |
| 12 | 2.66 ± 0.59 | ||
ε) 201 (4.1) nm; 1H (600 MHz, MeOH-d4) and 13C NMR (150 MHz, MeOH-d4) data, see Tables 2 and 3; HRESIMS m/z 529.3517 [M + Na]+ (calcd for C30H50O6Na, 529.3505).ε) 201 (3.9) nm; 1H (600 MHz, MeOH-d4) and 13C NMR (150 MHz, MeOH-d4) data, see Tables 2 and 3; HRESIMS m/z 527.3343 [M + Na]+ (calcd for C30H48O6Na, 527.3349).ε) 200 (3.9) nm; 1H (600 MHz, MeOH-d4) and 13C NMR (150 MHz, MeOH-d4) data, see Tables 2 and 3; HRESIMS m/z 545.3457 [M + Na]+ (calcd for C30H50O7Na, 545.3454).ε) 201 (3.8) nm; 1H (600 MHz, MeOH-d4) and 13C NMR (150 MHz, MeOH-d4) data, see Tables 2 and 3; HRESIMS m/z 543.3292 [M + Na]+ (calcd for C30H48O7Na, 543.3298).Inhibitory HCE-2 bioassay
All the isolated and biotransformed compounds were assayed for inhibitory HCE-2 effects as our pervious method.3,14 Compounds 1–22 were dissolved in DMSO and diluted to final concentrations of 0.1, 1.0, 5.0, 10.0, 20.0, 50.0, 100.0, and 200.0 μM. All compounds were hydrolyzed by HCE-2 at 37 °C with the probe substrate 4-benzoyl-N-butyl-1,8-naphthalimide (MPN) in a 96-well plate, then the fluorescence signal was detected at 564 nm. The probe substrate groups (without evaluated compounds) were used as control. Loperamide was used as the control drug.Molecular modeling
The initial structure of HCE-2 was modeled in terms of the protein structure of human liver carboxylesterase 1 (HCE-1, PDB ID: 1YA4) using the modeller (version 9) program.25 The protein structure of HCE-2 was further refined by performing molecular dynamics (MD) simulations using NAMD 2.9 software.26 The long-range electrostatic interactions were treated using the PME method. The integration time step is 2 fs. The system was first energy minimized for 10000 steps with the protein structure fixed. Subsequently, the system was heated from 0 to 310 K in 100 ps, followed by an equilibration at 310 K for 100 ps. Langevin dynamics and Langevin piston methods were used to maintain the temperature at 310 K and pressure at 1 bar. The production MD simulations were run for 10 ns. The final 10 ns protein structure was used for docking with AutoDock software.27 The ligand parameters were obtained using the PRODRG Server (http://davapc1.bioch.dundee.ac.uk/cgibin/prodrg). The Gasteiger charge was calculated for the HCE-2 protein structure. The grid box was sufficiently large to cover the whole protein, and compound 5 was blindly docked into the protein using the Lamarckian generic algorithm. The other default parameters in AutoDock were applied for docking.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 81503201), Dalian Outstanding Youth Science and Technology Talent (2015J12JH201), Liaoning BaiQianWan Talents Program, and Innovation Team of Dalian Medical University.Notes and references
- Flora of China Committee, Flora of China, Science Press, Beijing, 1992, pp. 140–142 Search PubMed.
- Pharmacopoeia Commission of People's Republic of China, Pharmacopoeia of the People's Republic of China (Part 1), Chinese Medical Science and Technology Press, Beijing, 2010, p. 212 Search PubMed.
- Z. P. Mai, K. Zhou, G. B. Ge, C. Wang, X. K. Huo, P. P. Dong, S. Deng, B. J. Zhang, H. L. Zhang, S. S. Huang and X. C. Ma, J. Nat. Prod., 2015, 78, 2372–2380 CrossRef CAS PubMed.
- T. Murata, Y. Imai, T. Hirata and M. Miyamoto, Chem. Pharm. Bull., 1970, 18, 1347–1353 CrossRef CAS PubMed.
- T. Murata and M. Miyamoto, Chem. Pharm. Bull., 1970, 18, 1354–1361 CrossRef CAS.
- X. Z. Hong, H. Q. Tang, L. M. Wu and L. D. Li, J. Pharm. Pharmacol., 2006, 58, 1391–1398 CrossRef CAS PubMed.
- W. F. Fong, C. Wang, G. Y. Zhu, C. H. Leung, M. S. Yang and H. Y. Cheung, Phytomedicine, 2007, 14, 160–165 CrossRef CAS PubMed.
- B. Y. Law, M. Wang, D. L. Ma, F. Al-Mousa, F. Michelangeli, S. H. Cheng, M. H. Ng, K. F. Mok, A. Y. Ko, R. Y. Lam, S. K. Chen, F. Che, C. M. Chiu and B. C. P. Ko, Mol. Cancer Ther., 2010, 9, 718–730 CrossRef CAS PubMed.
- H. Matsuda, T. Kageura, I. Toguchida, T. Murakami, A. Kishi and M. Yoshikawa, Bioorg. Med. Chem. Lett., 1999, 9, 3081–3086 CrossRef CAS PubMed.
- J. H. Lee, O. S. Kwon, H. G. Jin, E. R. Woo, Y. S. Kim and H. P. Kim, Biol. Pharm. Bull., 2012, 35, 1581–1587 CAS.
- M. Kubo, H. Matsuda, N. Tomohiro and M. Yoshikawa, Biol. Pharm. Bull., 1997, 20, 511–516 CAS.
- H. Dan, J. Wu, M. Peng, X. F. Hu, C. W. Song, Z. W. Zhou, S. G. Yu and N. B. Fang, Saudi Med. J., 2011, 32, 701–707 Search PubMed.
- S. S. Wu, G. G. Guo, H. Shi, H. Wang and L. Davia, China J. Tradit. Chin. Med. Pharm., 2007, 22, 475–477 Search PubMed.
- Z. P. Mai, X. L. Xin, N. Zhang, S. S. Huang, C. Wang, L. Chen, Y. Li, X. K. Huo and G. J. Fan, Phytochem. Lett., 2015, 13, 228–233 CrossRef CAS.
- D. F. Yang, R. E. Pearce, X. L. Wang, R. Gaedigk, Y. J. Yvonne Wan and B. F. Yan, Biochem. Pharmacol., 2009, 77, 238–247 CrossRef CAS PubMed.
- K. J. P. Yoon, J. L. Hyatt, C. L. Morton, R. E. Lee, P. M. Potter and M. K. Danks, Mol. Cancer Ther., 2004, 3, 903–909 CAS.
- M. J. Hatfield and P. M. Potter, Expert Opin. Ther. Pat., 2011, 21, 1159–1171 CrossRef CAS PubMed.
- J. Cang, C. Wang, X. K. Huo, X. G. Tian, C. P. Sun, S. Deng, B. J. Zhang, H. L. Zhang, K. X. Liu and X. C. Ma, Phytochem. Lett., 2017, 19, 83–88 CrossRef CAS.
- Y. Nakajima, Y. Satoh, M. Katsumata, K. Tsujiyama, Y. Ida and J. Shoji, Phytochemistry, 1994, 36, 119–127 CrossRef CAS.
- A. C. Zhou, C. F. Zhang and M. Zhang, Chin. J. Nat. Med., 2008, 6, 109–111 CrossRef CAS.
- G. P. Peng and F. C. Lou, Nat. Prod. Res. Dev., 2011, 13, 1–4 Search PubMed.
- M. Yoshikawa, S. Hatakeyama, N. Tanaka, Y. Fukuda, J. Yamahara and N. Murakami, Chem. Pharm. Bull., 1993, 41, 1948–1954 CrossRef CAS.
- T. Murata, M. Shinohara, T. Hirata, K. Kamiya, M. Nishikawa and M. Miyamoto, Tetrahedron Lett., 1968, 1, 103–108 CrossRef.
- X. L. Xin, Y. Wang, G. J. Fan, L. Chen and C. P. Sun, Phytochem. Lett., 2017, 19, 210–214 CrossRef CAS.
- A. Šali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: copies of HRESIMS and NMR spectra for all new compounds. See DOI: 10.1039/c7ra04841f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |