
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicellization and gelatinization in aqueous media of pH- and thermo-responsive amphiphilic ABC (PMMA82-b-PDMAEMA150-b-PNIPAM65) triblock copolymer synthesized by consecutive RAFT polymerization†
Ye Huanga,
Ping Yonga,
Yan Chena,
Yuting Gaoa,
Weixiong Xua,
Yongkang Lva,
Liming Yanga,
Rui L. Reis b,
Rogério P. Pirracob and
Jie Chen*a
b,
Rogério P. Pirracob and
Jie Chen*a
aDepartment of Chemical Engineering, School of Environmental and Chemical Engineering, Shanghai University, Shangda Road 99, Shanghai 200444, P. R. China. E-mail: jchen@shu.edu.cn; Tel: +86 21 66137482
b3B's Research Group – Biomaterials, Biodegradables and Biomimetics, University of Minho, Portugal
First published on 30th May 2017
Abstract
The multi-stimuli-responsive amphiphilic ABC triblock copolymer of poly(methyl methacrylate)-block-poly[N,N-(dimethylamino) ethyl methacrylate]-block-poly(N-isopropylacrylamide) (PMMA-b-PDMAEMA-b-PNIPAM) was synthesized by sequential reversible addition–fragmentation chain transfer (RAFT) polymerizations. Due to the pH- and thermo-responsive blocks of PDMAEMA and thermo-responsive blocks of PNIPAM, the copolymer solution properties can be manipulated by changing the parameters such as temperature and pH, and it formed diverse nanostructures and had gel behavior in different conditions. In detail, when the pH was below 7.3, the pKa of DMAEMA, tertiary amino groups were protonated, the polymer micellar solution like weak gel changed into free-standing gel at around 40 °C even at a low concentration of 2 wt%. Further, the gel behavior and morphology of the system were studied by rheology, turbidimetry measurements, transmission electron microscopy (TEM) and scanning electron microscopy (SEM), respectively. When the pH was above the pKa, the triblock copolymer self-assembled into diverse micellar structures including shell–core, corona–shell–core and shell–shell–core nanoparticles as the temperature increased, but no free-standing gelation. The two-step thermo-responsive behavior, corresponding to the different LCSTs of PNIPAM block and DMAEMA block, was evidenced by turbidity analysis. The assembled structures rapidly collapsed due to the enhanced hydrophobic interaction when the temperature further increased. Dynamic light scattering (DLS) was used to confirm the transitions.
Introduction
Hydrogels are water-swollen polymeric networks.1 They are widely used in coatings,2 cosmetics,3 drug delivery,4,5 tissue engineering,6 biosensors and separation processes,7 and other biomedical applications.8,9 During the past decade, hydrogels which can switch between free-flowing liquid and free-standing gel states have attracted much interest.10 In particular, great attention has been focused on the intelligent ones, which have broadly tunable characteristics, as they can exhibit a sol–gel transition in response to various external stimuli, such as pH,11 temperature,12 and ionic strength,3 magnetic fields, etc.13–15 Compared to chemical hydrogels, physical hydrogels formed by physical mixture, hydrophobic interaction, hydrogen bonding or ionic interactions, etc. are particularly more appealing.16,17Environment-sensitive polymers play an important role in the development of smart hydrogels.11,18,19 Thermo-responsive polymers can become hydrophobic from hydrophilic when the temperature is above a critical point, where a soluble-to-insoluble phase transition occurs, known as lower critical solution temperature (LCST).20 Poly(N-isopropylacrylamide) (PNIPAM) is a water soluble polymer that exhibits phase separation at the LCST of around 32 °C, depending upon the detailed microstructure of the macromolecule.21 Namely, below 32 °C, PNIPAM dissolves well in water, in contrast, changes into hydrophobic aggregation and may deposit from the aqueous because of the breakdown of the hydrogen bonds presented at a lower temperature.4 Because the LCST is between room temperature(25 °C) and physiological temperature (37 °C), it has been widely employed in many biomedical fields especially in on/off drug release and the reversible attachment/detachment of cultured cells.22
Besides the temperature sensitive hydrogels based on PNIPAM, another type of typical stimuli sensitive hydrogels is the pH sensitive hydrogels, which may change their shapes and volumes with the variation of external pH.23 Poly(dimethylaminoethyl methacrylate) (PDMAEMA) is a pH-sensitive polymer with a pKa of 7.3 in water. Moreover, PDMAEMA is thermos-sensitive, and the LCST is hinged upon its molecular weight and pH. In detail, the LCST strongly decreased with increasing pH of the solution.24 It has been widely used for the producing of multi-sensitive materials.11,25
The copolymerization of DMAEMA and PNIPAM is supposed to have some interesting properties since both monomers are thermos-sensitive and only DMAEMA is pH-sensitive. Wang et al.26 prepared hydrogels based on NIPAM and DMAEMA via free radical polymerization, the resulted P(NIPAM-co-DMAEMA) hydrogels have the strong desire to respond to external temperature and pH stimuli. The synthesis of terpyridine end-capped PDMAEMA-block-PNIPAM diblock copolymers via controlled radical copolymerization has been reported by Jeremy Brassinne and his co-workers.27
Over the last few years, hydrogel formation by ABA triblock copolymers containing hydrophilic mid-blocks and hydrophobic end blocks has been extensively investigated.28–31 A triblock polymer (PPO43–PMPC160–PNIPAM81) was synthesized by ATRP and its gelation efficiency in water was evaluated by Li and his co-workers.32 However, almost 20 wt% polymer was required to be gel, the gelation efficiency was sufficiently lower than that observed in the PNIPAM–PMPC–PNIPAM triblock copolymers. Besides high critical gelation concentration, these hydrogels often exhibit broad or slow sol–gel transitions.32,33 Comparing to ABA model, ABC triblock copolymers, which the C block is hydrophobic and the B and A blocks are water-soluble below the sol–gel transition temperature, are more efficient to realize the sol–gel transition.18,19 Because the three blocks are mutually immiscible, they can form diverse nanostructures in aqueous solution in response to the change of the environment.1,32
Reversible addition–fragmentation chain transfer (RAFT) polymerization which is an attractive controlled/living radical polymerization method for the synthesis of block copolymers has arouse great interest in the past decade.34,35 Since it is a versatile and facile technique, a variety of chain transfer agents are used without transition metal catalysts.36,37 This process leads to the production of materials with controlled molecular weights and low polydispersity.38
When thermo-responsive segments are contained in a single polymer chain, the copolymers may show multiple LCSTs.39 That the combination of different functions within a single polymer can contribute to improvement of carrier efficiency is reported in many researches.40 Block copolymers containing PDMAEMA block and PNIPAM segment provide a flexible control to the polymer phase behavior, resulting in multifunctional smart materials. The lack of data of ABC triblock copolymers relating to the RAFT copolymerization of DMAEMA and PNIPAM motivated us to investigate this system.
In this study, we synthesized a novel pH- and dual-thermo-sensitive amphiphilic ABC triblock copolymer composed of PMMA, PDMAEMA and PNIPAM by RAFT polymerization. The hydrophilic triblock copolymer molecularly could be dissolved in aqueous solution and was a viscous liquid like weak gel with shell–core micelle at room temperature. In acidic aqueous solution, that the pH was below the pKa of the PDMAEMA block, the free-flowing liquid changed into free-standing gel with the elevated temperatures. While in alkaline solutions, the sturdy gelation structure would be destroyed with doubly thermo-responsive steps, due to the wide difference of the LCSTs of PNIPAM and PDMAEMA chains. Indeed, it formed diverse nanoparticles such as corona–shell–core and shell–shell–core, finally precipitated from aqueous solution as the temperature increased (Fig. 1). The thermo- and pH-responsive copolymer may boast a great potential and value in biomedical areas, such as tissue engineering and controlled drug delivery and release.
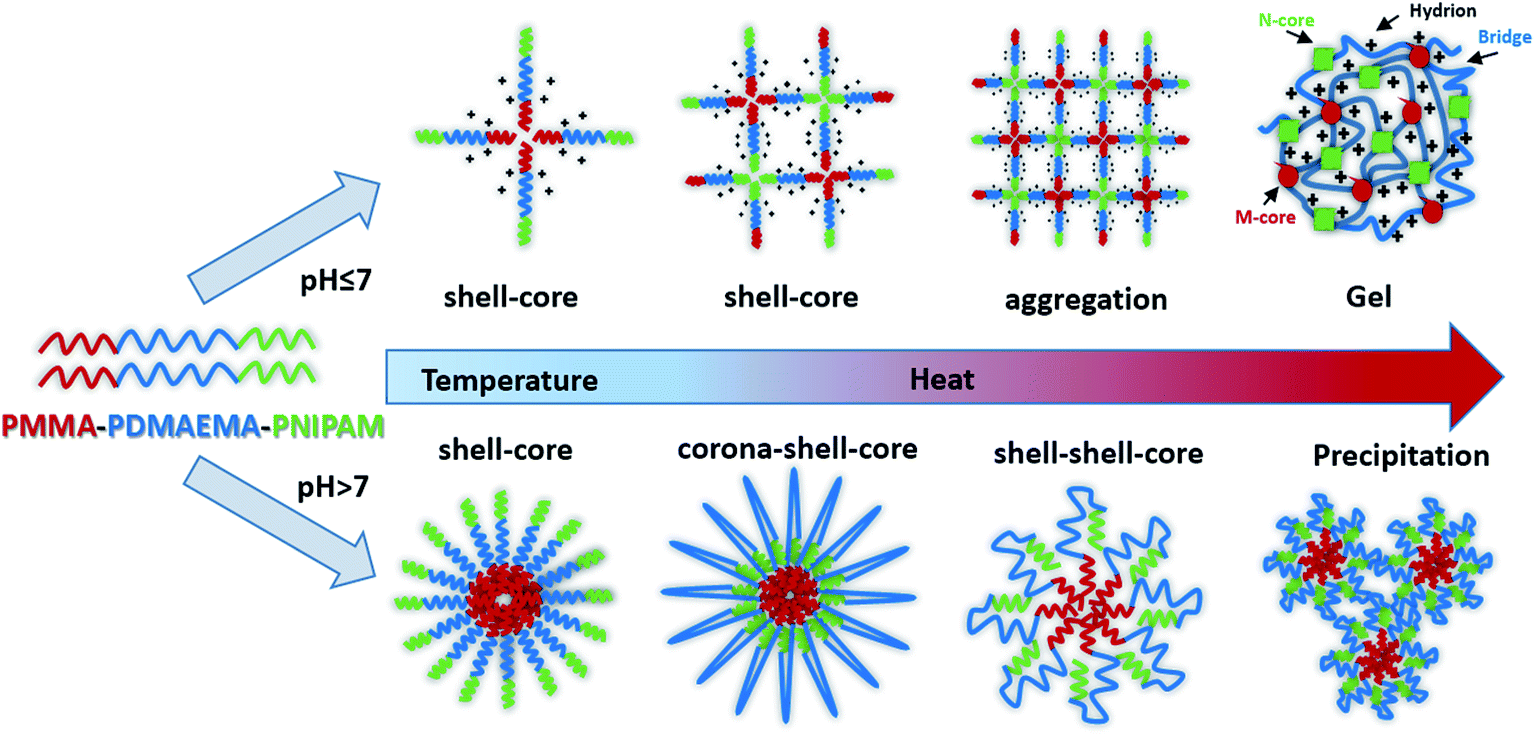 | ||
| Fig. 1 Schematic illustration of the PMMA-b-PDMAEMA-b-PNIPAM nanoparticles dispersed in water with temperature increased in different pH. The PMMA cores, ionized PDMAEMA chains and PNIPAM cores are marked M-core, bridge and N-cores respectively. | ||
Experimental
Materials
All reagents were purchased from Aladdin and used as received unless otherwise stated. Methyl methacrylate (MMA, 99%) were filtered before utilization through a basic alumina column to remove the radical inhibitor. N,N-(Dimethylamino) ethyl methacrylate (DMAEMA, 99%) was dried with CaH2 and vacuum distilled before use. N-Isopropylacrylamide (NIPAM, 97%) was recrystallized from mixtures of toluene and hexane. 2,2′-Azobis (isobutyronitrile) (AIBN) was recrystallized from methanol. The chain transfer agents (CTA) 4-cyanopentanoic acid dithiobenzoate (CPADB) was synthesized according to the reported procedures.41 Other chemical reagents, such as toluene, petroleum ether, 1,4-dioxane and CH2Cl2 were purified by standard methods before use. Deionized water was used in current experiments.Synthesis of PMMA-b-PDMAEMA-b-PNIPAM by sequential RAFT polymerization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 831 g mol−1, DpDMAEMA = 150.186 g mol−1, DpNIPAM = 65. The overall experiment procedure is illustrated in Scheme 1.
831 g mol−1, DpDMAEMA = 150.186 g mol−1, DpNIPAM = 65. The overall experiment procedure is illustrated in Scheme 1.
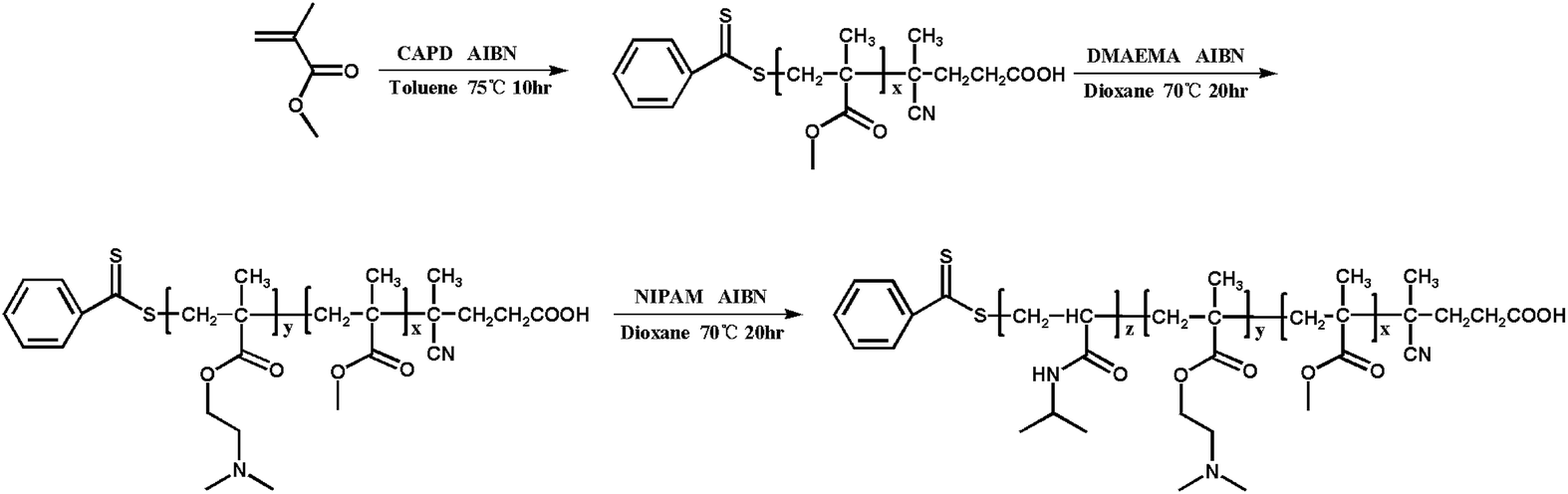 | ||
| Scheme 1 Schematic illustration of the synthesis of PMMA-b-PDMAEMA-b-PNIPAM by sequential RAFT polymerization. | ||
Characterizations
The Fourier transform infrared (FT-IR) spectra were obtained on an AVATAR 370 FT-IR spectrometer (Nicolet, USA). Nuclear magnetic resonance (NMR) spectra were collected on a Bruker AV 500 MHz spectrometer using CDCl3 as solvent. Gel permeation chromatography (GPC) was performed on a Waters Alliance e2695 GPC system. The turbidity behavior was conducted for 0.2 wt% polymer solutions on UV-visible spectrophotometer (UV-vis, Agilent, USA) at a wavelength of 500 nm, the temperature ramping rate was 2 °C min−1, and the solution was held for 2 min before measurement was taken under various temperature conditions. Cloud point (CP) is usually approximated as the LCST of the solution, and is determined as the temperature at which the transmittance started to drop dramatically resulting from phase separation of the polymer.22 Dynamic light scattering (DLS) measurements were performed on Zetasizer Nano ZS90 (Malvern Instruments Ltd., Malvern, U.K.) equipped with a He–Ne laser (633 nm, 4 mW), at a detection angle of 90°, in which size distribution was determined by intensity following the CONTIN method. Z-Average diameter was analyzed using the Malvern software. Variable-temperature DLS was conducted at an interval of 5 °C using the built-in Peltier thermal control. At each temperature the sample was thermally equilibrated for 2 min. The viscosity measurements were carried out using a LVDV-11 DV-II+Pro viscometer (Brookfield, USA) with 0.5 RPM and 64# spindle. The rheological measurements were performed on a Malvern Kinexus Pro rheometer in the temperature range of 20–80 °C. Temperature dependent dynamic shear moduli (storage moduli G′ and loss moduli G′′) of the viscous aqueous solution were recorded with a temperature ramping rate of 1 °C min−1 at a frequency of 0.16 Hz. The transmission electron microscopy (TEM) samples of 0.2 mg mL−1 (0.02 wt%) were prepared by dipping a carbon-coated copper grid (400 mesh) into the copolymer solutions preheated at different temperatures. Also, during sample preparation, all used tools must be preheated to the corresponding temperature. The TEM analysis was then carried out on a JEOL JEM-2100 (Japan) instrument at an acceleration voltage of 200 kV to examine the particle characteristics. The solution and gelation morphology were investigated using a LEO 1530 VP Scanning Electron Microscope (SEM). The samples were prepared by maintaining the polymer solution of 30 mg mL−1 (3 wt%) at 25 °C, 50 °C and 75 °C for 30 min, respectively, then the samples were frozen in liquid nitrogen and freeze-dried immediately. Finally, the dried samples were sprayed with gold to their surface and subsequently subjected to SEM.42 The solutions of multi-responsive triblock copolymer, including dilute and concentrated solutions used to morphology and rheology and turbidity study were prepared by ultrasonic-standing method in acid or alkaline aqueous media.Results and discussion
Synthesis of PMMA-b-PDMAEMA-b-PNIPAM terpolymer polymers
Many papers have reported the synthesis of different block copolymers using different chain transfer agents (CTA) by consecutive RAFT polymerization.38,43 In this study, 4-cyanopentanoic acid dithiobenzoate (CPADB) was used as the CTA. The synthetic procedure has been described in the Experimental section. According to the order: PMMA, PMMA-b-PDMAEMA, PMMA-b-PDMAEMA-b-PNIPAM, each purified product was characterized by FT-IR, 1H NMR and GPC.FT-IR spectra of the polymers are shown in Fig. 2. There are some strong absorption peaks around 2900 cm−1, which could be attributed to the stretching vibrations of –CH3 and –CH2– groups.42 All spectra exhibit an absorption band at 1730 cm−1 from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, which is assigned to the carbonyl stretching vibration of the ester group. Furthermore, they show a peak at 1147 cm−1 corresponding to the C–O stretch. The absorption peak at 1063 cm−1 is the typical stretching vibrations of CS groups in CTA.44 The FT-IR spectrum (B) of the PMMA-b-PDMAEMA contain the characteristic band for C–N stretching of DMAEMA at 1020 cm−1.44 For PMMA-b-PDMAEMA-b-PNIPAM (spectra C), the peak at 1650 cm−1 and 1544 cm−1 are attributed to CO and N–H vibrational bands from –CONH of NIPAM, respectively.45 Fig. 3 displays the 1H NMR spectrum of PMMA-b-PDMAEMA-b-PNIPAM in CDCl3. The structure signals have already been mentioned in Experimental part.46,47 The 1H NMR spectra of PMMA and PMMA-b-PDMAEMA are also detected (Fig. S1 and S2†).45,48,49 From structure signals, we calculated the ratio of the integral peak area between the methylic protons of PMMA (marked c) and the para-C6H5 protons of CPADB (marked 1), the Dp of PMMA was estimated to be about 82. Similarly, the Dp of other polymers could be also roughly calculated.42 GPC traces of the block copolymers are shown in Fig. 4. In particular, with the addition of the monomers, the clear shift to short elution time of the GPC peak without any other peaks indicates that the chain extension has occurred. GPC was also used to characterize the molecular weight of the polymers, and the results are summarized in Table 1. The molecular weight of polymers synthesized with processing in every step increased significantly. The molecular weight calculated by 1H NMR is approximate with the average molecular mass of polymers characterized by GPC, and the PDI is broadening. A possible explanation is as follows: the overall RAFT process provides insertion of monomer units into the C–S bond of the RAFT agent structure, polymer chains lengthened constantly with the polymerization processing. In order to ensure higher monomer conversion rate, we extended for several hours. While long polymerization time may cause the reactive end-groups of RAFT agents fall off under the synthetic circumstances, leading to chain termination because of the potential lability of the C–S bond in the RAFT end-group. Therefore, the polymer was contaminated, displayed as broadening PDI detected by GPC. In short, all of the results above indicate that the target triblock copolymer has been successfully synthesized.
O stretching vibration, which is assigned to the carbonyl stretching vibration of the ester group. Furthermore, they show a peak at 1147 cm−1 corresponding to the C–O stretch. The absorption peak at 1063 cm−1 is the typical stretching vibrations of CS groups in CTA.44 The FT-IR spectrum (B) of the PMMA-b-PDMAEMA contain the characteristic band for C–N stretching of DMAEMA at 1020 cm−1.44 For PMMA-b-PDMAEMA-b-PNIPAM (spectra C), the peak at 1650 cm−1 and 1544 cm−1 are attributed to CO and N–H vibrational bands from –CONH of NIPAM, respectively.45 Fig. 3 displays the 1H NMR spectrum of PMMA-b-PDMAEMA-b-PNIPAM in CDCl3. The structure signals have already been mentioned in Experimental part.46,47 The 1H NMR spectra of PMMA and PMMA-b-PDMAEMA are also detected (Fig. S1 and S2†).45,48,49 From structure signals, we calculated the ratio of the integral peak area between the methylic protons of PMMA (marked c) and the para-C6H5 protons of CPADB (marked 1), the Dp of PMMA was estimated to be about 82. Similarly, the Dp of other polymers could be also roughly calculated.42 GPC traces of the block copolymers are shown in Fig. 4. In particular, with the addition of the monomers, the clear shift to short elution time of the GPC peak without any other peaks indicates that the chain extension has occurred. GPC was also used to characterize the molecular weight of the polymers, and the results are summarized in Table 1. The molecular weight of polymers synthesized with processing in every step increased significantly. The molecular weight calculated by 1H NMR is approximate with the average molecular mass of polymers characterized by GPC, and the PDI is broadening. A possible explanation is as follows: the overall RAFT process provides insertion of monomer units into the C–S bond of the RAFT agent structure, polymer chains lengthened constantly with the polymerization processing. In order to ensure higher monomer conversion rate, we extended for several hours. While long polymerization time may cause the reactive end-groups of RAFT agents fall off under the synthetic circumstances, leading to chain termination because of the potential lability of the C–S bond in the RAFT end-group. Therefore, the polymer was contaminated, displayed as broadening PDI detected by GPC. In short, all of the results above indicate that the target triblock copolymer has been successfully synthesized.
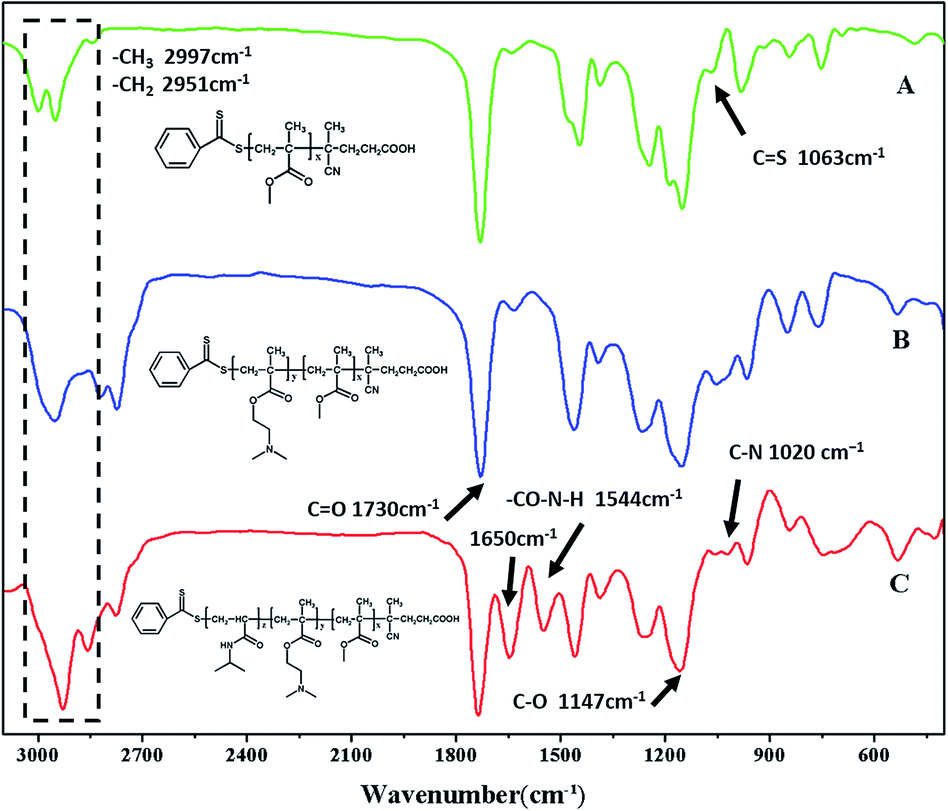 | ||
| Fig. 2 FT-IR spectra of polymers: (A) PMMA (B) PMMA-b-PDMAEMA (C) PMMA-b-PDMAEMA-b-PNIPAM. | ||
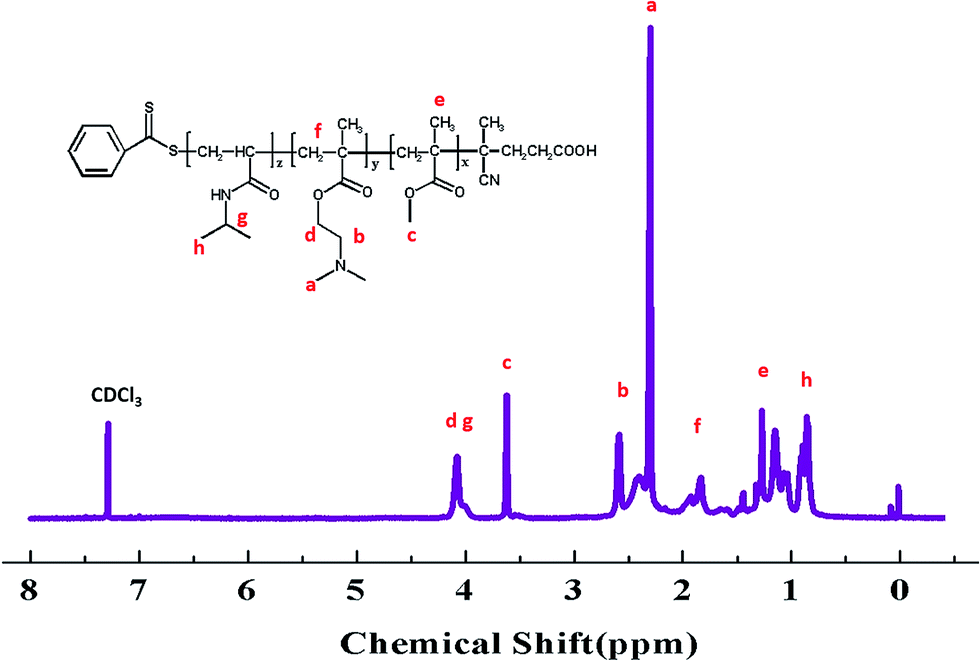 | ||
| Fig. 3 1H NMR spectrum of PMMA-b-PDMAEMA-b-PNIPAM in CDCl3. | ||
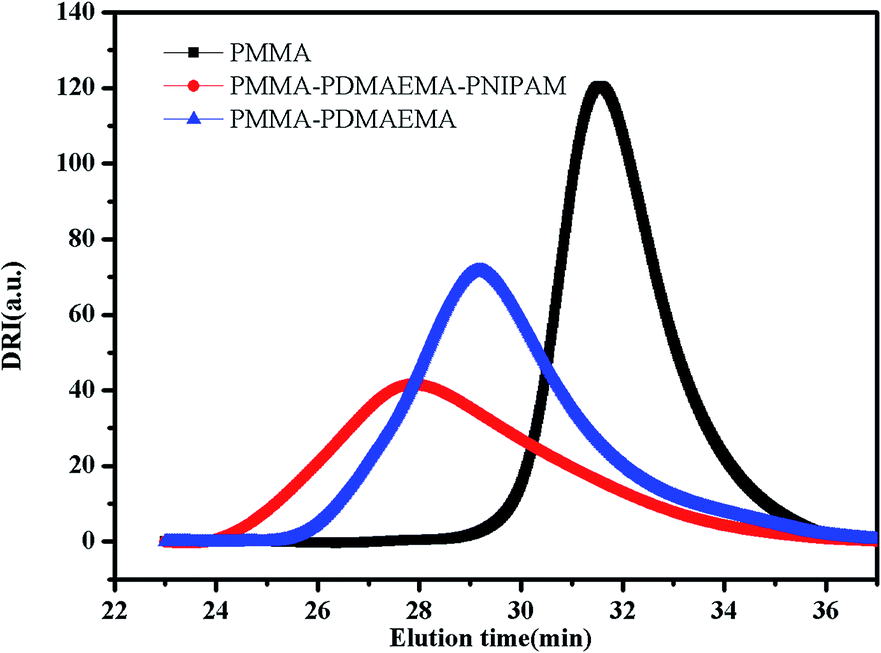 | ||
| Fig. 4 The GPC traces of the PMMA, PMMA-b-PDMAEMA and PMMA-b-PDMAEMA-b-PNIPAM copolymers. | ||
| Mn, NMRa (g mol−1) | Mn, GPCb (g mol−1) | PDIc (Mw/Mn) | |
|---|---|---|---|
| a The molecular weight by 1H NMR analysis.b The number-average molecular weight by GPC analysis.c The PDI or the Mw/Mn values determined by GPC analysis. | |||
| PMMA macro-CTA | 8217 | 8386 | 1.25 |
| PMMA-b-PDMAEMA macro-CTA | 31831 |
32802 |
1.43 |
| PMMA-b-PDMAEMA-b-PNIPAM | 39186 |
41152 |
1.67 |
The gelatinization of the PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer in acidic solutions
In PMMA-b-PDMAEMA-b-PNIPAM segment, PMMA is hydrophobic, PNIPAM is a water soluble polymer below the LCST around 32 °C, PDMAEMA, a classical pH- and thermo-sensitive weak polybase polymer, generally has excellent solubility in neutral and acidic media because of the protonated tertiary amino groups. As a consequence, the terpolymer PMMA–PDMAEMA–PNIPAM firstly conformed a shell–core (PDMAEMA–PNIPAM)–(PMMA) structure below LCST. With the increasing of temperature, the sticky polymer micellar solution changed into free-standing gel, and the macroscopic gelation could be seen clearly in acidic conditions. When the temperature decreased to the room temperature, the solution was a partly transparent liquid that could be free-flowing when tilted.Typical viscosity vs. temperature plots obtained from pH = 1 to pH = 6 aqueous solutions of the PMMA-b-PDMAEMA-b-PNIPAM copolymer are shown in Fig. 5(A). The viscosity of PMMA-b-PDMAEMA-b-PNIPAM copolymer first increased slowly (20–30 °C), then rose sharply (30–50 °C), and decreased gradually (50–75 °C), finally was steady. The maximum rate of change was about 40 °C, it is higher than the LCST of NIPAM (32 °C), because the ionization degree of the PDMAEMA block can influence the phase transition temperature of not only PDMAEMA but also the neighbour PNIPAM block due to the weakened hydrophobic aggregation force.27,50 The impact on the thermos-sensitivity of polymerized hydrogel with the incorporation of pH sensitivity has been widely reported.26 At room temperature, the viscosity of the micellar solution exceeded 100 Pa s, it was a viscoelastic liquid like weak gel, which was attributed to the effect of hydrogen bonds between NIPAM and DMAEMA. When the temperature was increased to 50 °C, PNIPAM turns to hydrophobic, the terpolymer bears at both ends hydrophobic blocks that should associate forming a stronger network because of the increasing bridging, furthermore, the free amino groups in the PMMA-b-PDMAEMA-b-PNIPAM are ionized in acidic solutions, which might generate the electrostatic repulsion among polymer chains. Therefore, the free-standing gel formed.51 The phase transition corresponding to gelation with high concentration mainly attributed to the hydrophobic PMMA and thermo-sensitive PNIPAM moiety.52 Above 50 °C, the viscosity decreased gradually strangely. A tentative explanation for this unexpected effect is as follows, it might be the relaxation time which increases upon heating in this regime.53 Nevertheless, the gel was still free-standing. The same phenomenon was also detected in the rheological experiments (Fig. S3†). The gel was strengthening with the increasing temperature, and exhibiting maximum at around 55 °C, though there is a deviation of 5 °C comparing to the viscosity experiment, the trend of the modulus were in strong agreement with the viscosity in Fig. 5(A). Interestingly, the viscosity of the free-standing gel was different at the same temperature in various pH, which was higher at pH = 4, probably due to the degree of the protonation of the amine functions of the PDMAEMA block, at pH 4, the degree of ionization of the central PDMAEMA chains is about 90%, this is the reason of the viscosity maximum since it favors a 3D network formation.24,54 In detail, when pH > 4, a gradual deprotonation of the monomeric units will decrease the degree of ionization and increase chain flexibility, making bridge to loop transitions and hence viscosity decrease when pH increased. While pH < 4, lowering pH will make the ionic strength of the solution increase, and lead to electrostatic screening effects that result in viscosity decrease. The ABA triblock copolymers (PMMA-b-PDMAEMA-b-PMMA) synthesized by Frederic Bossard and his coworkers have the similar properties.53
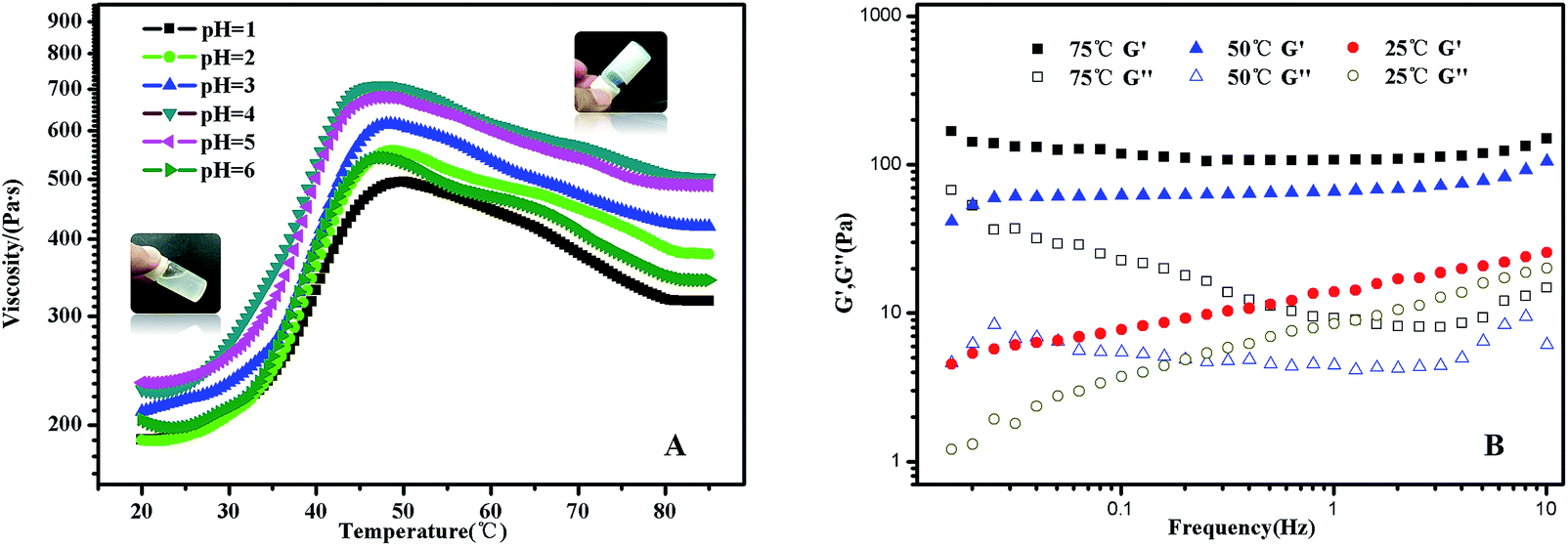 | ||
| Fig. 5 (A) Viscosity curves of PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer solution at different temperatures in acidic conditions. The viscosity measurements were carried out using 64# spindle with a constant rotation speed of 0.5 RPM; (B) frequency dependences of the storage modulus (G′) and loss modulus (G′′) at a constant strain of 0.5% for PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer at 25 °C, 50 °C and 75 °C. Both measurements were carried at the concentration of 30 mg mL−1 (3 wt%). | ||
The transition from a viscoelastic liquid to a 3-dimensional network gel was also evidenced by the data collected from frequency sweep experiments (Fig. 5(B)). The frequency dependences of G′ and G′′ of the polymer solution at selected temperatures of 25 °C, 50 °C and 75 °C were obtained by frequency sweep tests from 0.016 Hz to 10 Hz. At 25 °C, the solution was a transparent liquid that can flow when tilted. G′ and G′′ were of similar magnitudes in the frequency range of 1–10 Hz, this is the signature of the transition between liquid-like and solid-like behavior.28 Above 50 °C, the sample was a free-standing gel. G′ is significantly greater than G′′ and is nearly independent of frequency in the selected frequency range, which was a characteristic of solid-like behaviour.55 Further, we investigated the mechanical properties of the gel states to explore the gelation at 50 °C.
The concentration of polymer has a significant impact on gelation property. Therefore, we studied the gelation behaviour in different concentrations at the selected of pH = 4, which afforded total protonation of the amine functions (Fig. 6). The viscosity of triblock copolymer system increased gradually with the increasing of the concentration of polymer, indicating stronger interaction among polymer chains at higher concentrations. The sticky triblock copolymer solution could form free-standing gel at around 40 °C even at a quite low concentration (2 wt% in water). Generally, the gelation of such systems is efficient in the sense of the minimum gelation concentrations.1 The tube was inverted to determine if a physical gel was formed, three times repeated heating and cooling experiments indicated that the transition is fully reversible (Fig. S4†), there was a deviation about 5 °C in each cycle, which may be caused by a lower rate of heat transfer of gel than sol. Fig. 7 shows the strain dependence of G′ and G′′. The polymer solution exhibits a linear viscoelastic response below a critical shear strain 1.2%, where G′ > G′′. Above this linear viscoelastic regime, G′ decreases as shear strain increases whereas G′′ increases, then they reach a equivalence at 18%, that means the system had lower mechanical strength when the strains increased and began flow at strains of 18%.
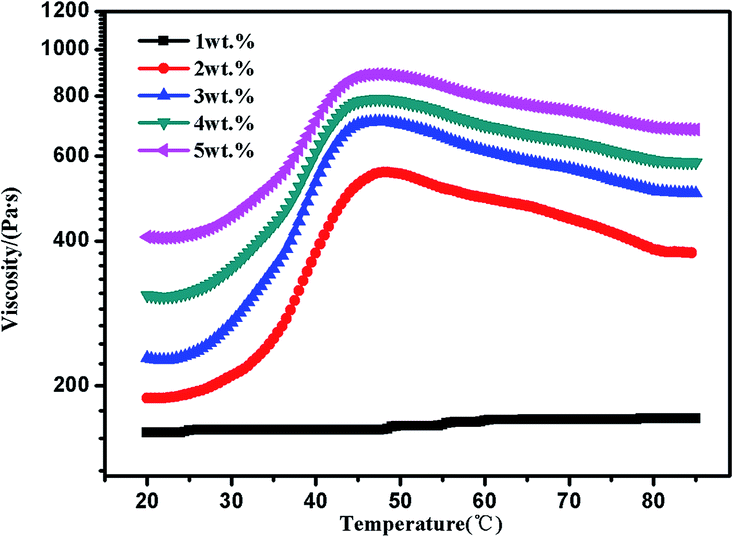 | ||
| Fig. 6 Viscosity curves of PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer solution at different concentrations at pH = 4. | ||
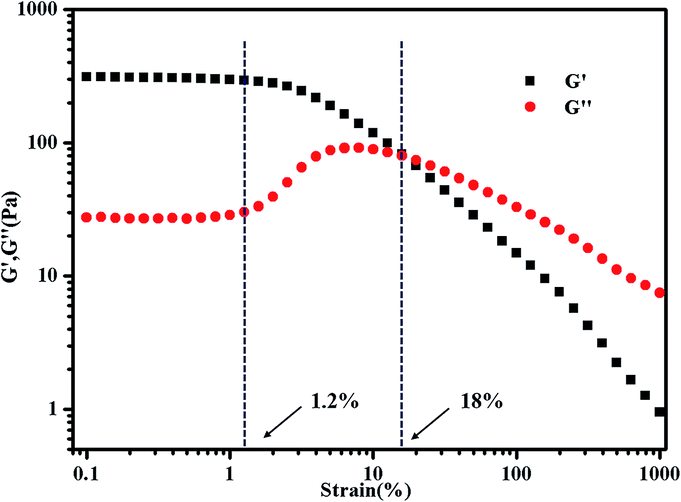 | ||
| Fig. 7 Strain dependence of G′ and G′′ for 3 wt% PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer at 50 °C. The frequency was held constant at 1 Hz. | ||
Moreover, the 3-dimensional network gels were evidenced by SEM images. The formation of 3-dimensional network structure of the gel is shown in Fig. 8 and S5.† With the increasing temperature, PNIPAM becomes hydrophobic, the net points which contained PMMA and PNIPAM cores were bridged by ionized PDMAEMA chains (Fig. 1), and more triblock polymer chains associated forming a stronger network structure. In addition, with the help of electrostatic interaction in acid media, the 3-dimensional network became regular and dense.
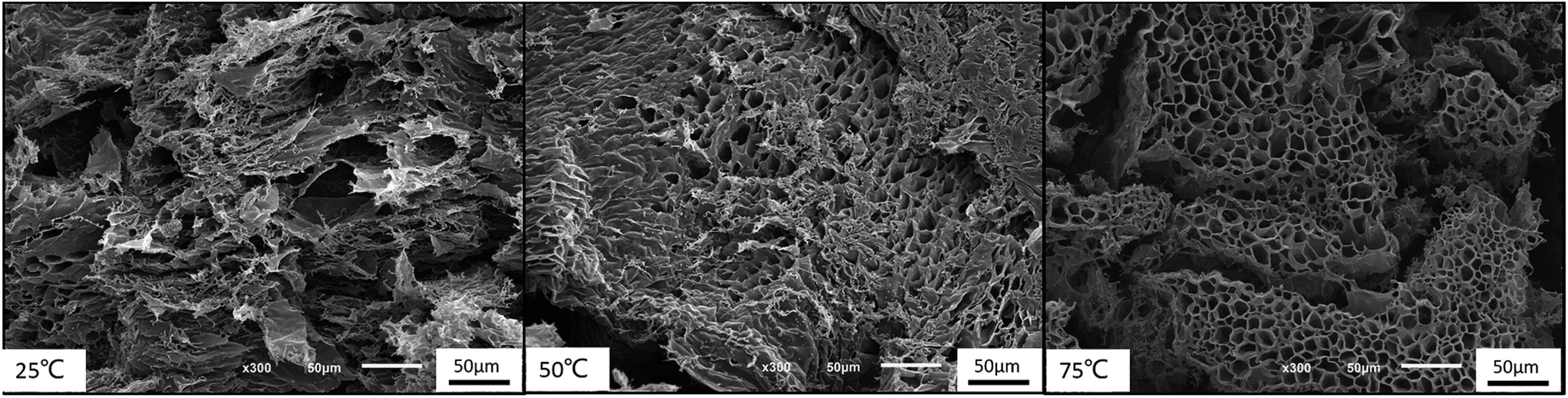 | ||
| Fig. 8 SEM images of gelation behavior at different temperatures at pH = 4. | ||
The micellization of the PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer in acidic solutions
The copolymer dissolve in acidic aqueous solution at room temperature, and form micellar solution. The nanoparticle has a shell–core structure with the hydrophobic PMMA block as the core, hydrophilic PNIPAM block and protonated PDMAEMA as the shell. When the temperature is above the LCST of NIPAM 32 °C, the shell–core structure nanoparticles have two kinds of core, one is PMMA block core, and the other is PNIPAM core. Upon continuous heating, there will be many more hydrophobic cores forming and aggregating. To further study the micro-properties, the PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer were investigated by UV-vis absorbance spectroscopy and dynamic light scattering (DLS). Fig. 9 shows the temperature dependent transmittance at a wavelength of 500 nm obtained for aqueous solutions of PMMA-b-PDMAEMA-b-PNIPAM micelles at different pH values. The transmittance started to decrease at around 35 °C. The LCST of the copolymer solutions ranged from 40 °C to 45 °C, with a middle point at around 42 °C, corresponding to the result of viscosity and rheological measurements. It was found that the micelles were very stable and could keep suspending in water at much higher temperatures than the LCST of polymer, which was ascribed to the hydrophobic NIPAM and hydrophilic PDMAEMA block.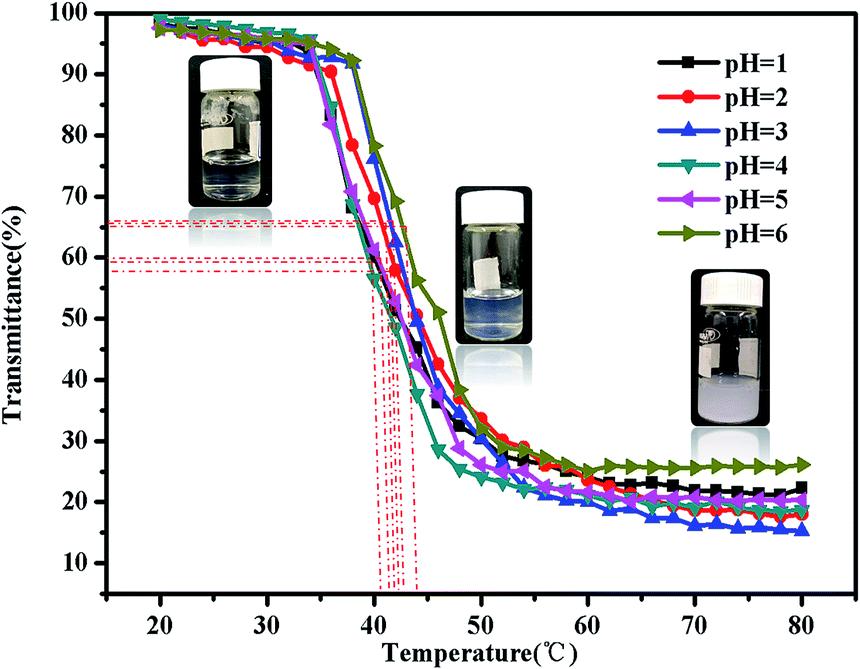 | ||
| Fig. 9 Turbidity curves of the PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer solution (2 mg mL−1) at various temperatures in acidic condition. | ||
In addition, DLS is a powerful tool to get detailed information about the size distribution of the aggregates. Fig. 10 shows the hydrodynamic diameter of PMMA-b-PDMAEMA-b-PNIPAM in 0.15 mg mL−1 aqueous solutions. The hydrodynamic diameter distribution of the triblock copolymer nanoparticles is narrowly dispersed. It can be clearly monitored that the peak of intensity spectra increases and shifts to higher aggregate size, for 79 nm at 25 °C with the polydispersity index (PDI) of 0.328, 122 nm at 50 °C with the PDI of 0.217 and 142 nm at 75 °C with the PDI of 0.174. That means the sizes of micelles become much bigger when temperature increased, especially from 25 °C to 50 °C. It is because of the progressive hydrophobic transformation of PNIPAM blocks in the micellar corona, and some NIPAM blocks may also shrink to form new cores when the temperature increased above the LCST, many micelles aggregated, leading to clusters of micelles at low concentrations and strengthening the gel at high concentrations.
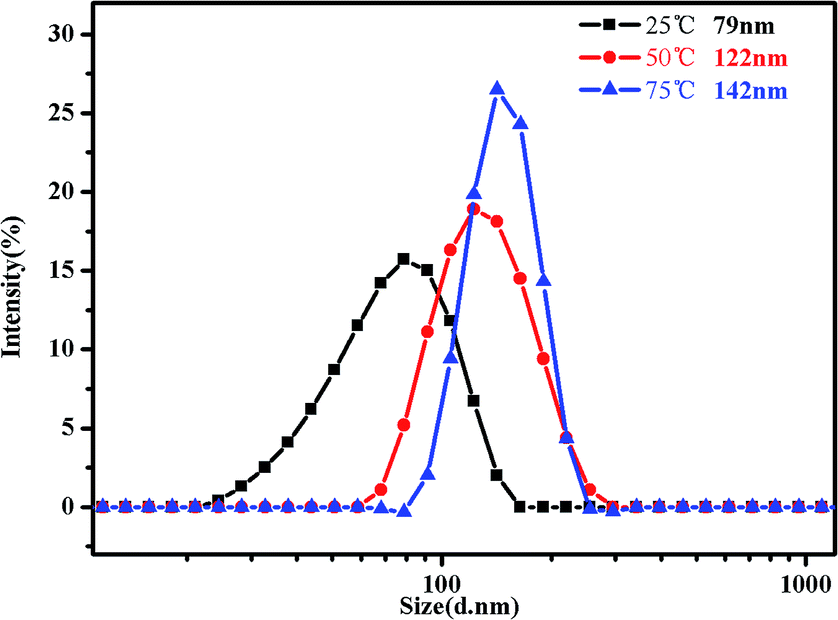 | ||
| Fig. 10 Intensity distribution of hydrodynamic diameter of the PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer solution (0.15 mg mL−1) at pH = 4 at 25 °C, 50 °C and 75 °C, respectively. | ||
To study the trend of Z-average diameter as the temperature increased, 0.5 mg mL−1 of PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer solution at pH = 4 was used for the demonstration (Fig. 11). It was observed that Z-average diameter started to increase at around 30 °C and stabilized at around 50 °C. Moreover, the derived count rate gradually increased. It is a measurement of light scattering intensity, which varied directly with the production of particle size and concentration.56 The increasing light scattering intensity until 50 °C suggested that the aggregation of shell–core nanoparticles and tight structure. The Z-average diameter of other research groups were also measured (Fig. S6†). Interestingly, we found that at the pH of 3–5, the Z-average diameters were bigger than that of other conditions at one temperature, the same tendency was observed in the viscosity test and the specific reason has already been elucidated before.
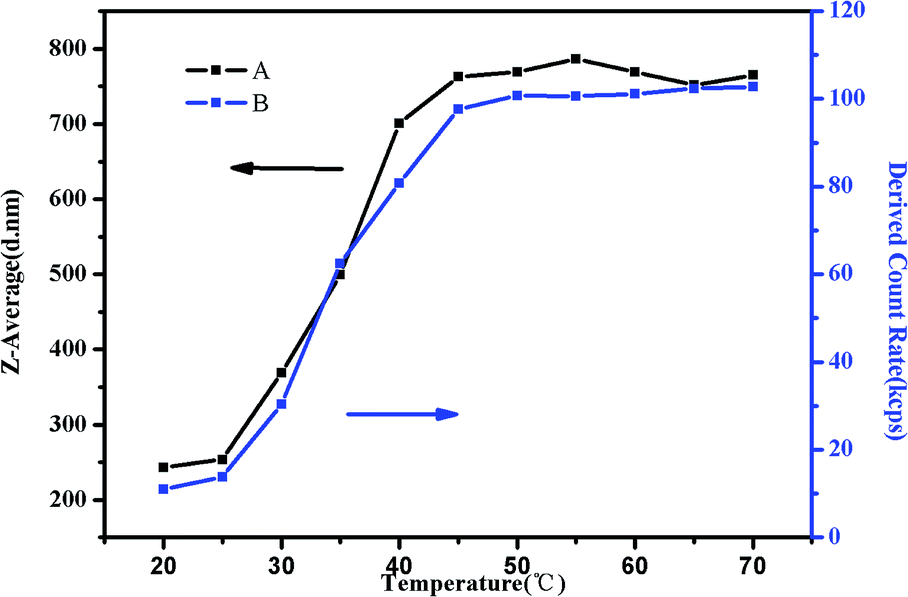 | ||
| Fig. 11 (A) The Z-average diameter and (B) light scattering intensity as a function of temperature in a DLS study of 0.5 mg mL−1 PMMA–PDMAEMA–PNIPAM solution at pH 4. | ||
The morphology of the core–shell micelles is further confirmed by TEM observation. TEM images (Fig. 12) show the micelles were well dispersed in the solution at room temperature, the structure has a change from loose spherical micelle to tight aggregation upon increasing temperature, and the trends of size and distribution are similar with DLS. On the TEM images, we found that the values of the hydrodynamic diameter of the micelles observed by TEM are smaller than those measured by DLS. Firstly, this is because TEM observation shows the diameter of the dried aggregates, whereas the DLS analysis detects the solvated nanoparticles. Moreover, the electrostatic repulsion formed by ionized PDMAEMA in dried micelles will disappear.12,42 Secondly, the intensity-weighted diameter analyzed by DLS is always oversized to the number-averaged diameter observed by TEM.46
 | ||
| Fig. 12 TEM images of triblock copolymer nanoparticles of PMMA-b-PDMAEMA-b-PNIPAM (0.2 mg mL−1) at different temperatures at pH = 4. | ||
The gelatinization of the PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer in neutral and alkaline solutions
PDMAEMA has a pKa about 7.3 in aqueous. At the pH of 7, the electrostatic repulsive forces among protonated PDMAEMA segments weakened, the weak electrostatic repulsion among polymer chains is not strong enough to form free-standing gel. As the pH increased, the tertiary amino groups of PDMAEMA are almost completely deprotonated, resulting in a lower LCST, and above the LCST, the PDMAEMA block becomes particularly hydrophobic.25 The LCST of PDMAEMA ranges from 30 °C to more than 60 °C in aqueous solution, depending on molecular weight, pH, and concentration.25 In alkaline conditions, we found that the viscosity tested by viscometer decreased with the increasing temperature, besides the LCST of PNIPAM, there were two stages which could be discerned roughly (see Fig. 13(A)). At room temperature, the solution was a viscous liquid like soft gel, then became weak and could flow freely when temperature increased. Firstly, it is probably attributed to the deprotonation of amino groups of PDMAEMA. Since a lot of complex hydrogen bonds would exist among the free amino groups, which would restrict the movement or relaxation of network chains. A lower swelling ratio results in the lower viscosity.26 Secondly, that possibly caused by hydrophobic effect.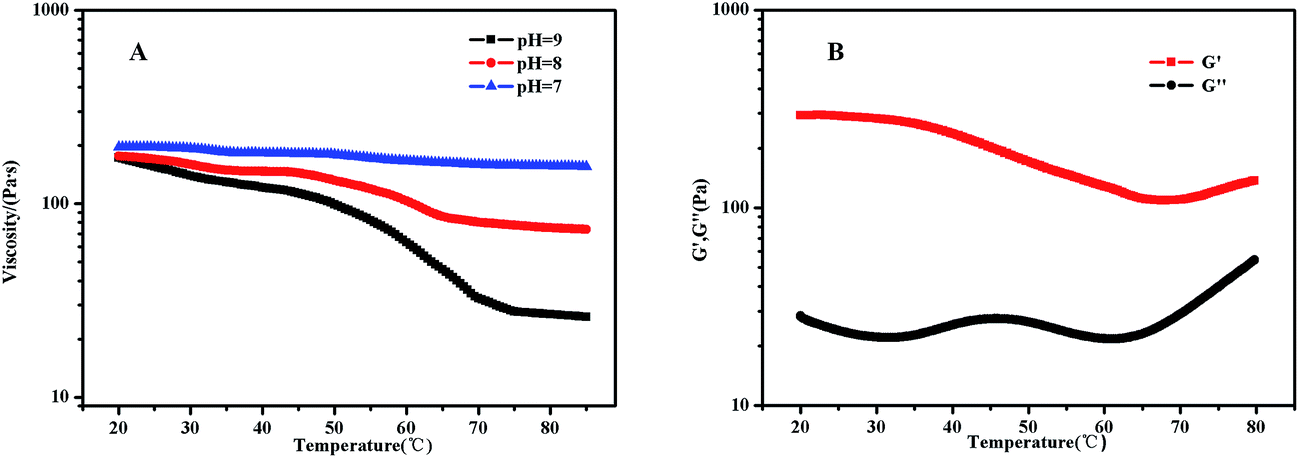 | ||
| Fig. 13 (A) Viscosity curves of 3 wt% PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer solution at different temperatures; (B) temperature dependence of the storage modulus (G′) and loss modulus (G′′) at a constant strain of 0.5% for 3 wt% PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer at the frequency of 0.16 Hz at pH = 9. | ||
Fig. 13(B) shows that the storage modulus (G′) is greater than storage modulus (G′′), since the micellar solution was a viscous liquid like soft gel at room temperature. It was noteworthy that the gap between G′ and G′′ became smaller with the increasing of temperature, indicating that the viscosity decreased and became weak gel with increasing temperature. The loss modulus has a slight fluctuation with the increasing temperature, probably caused by the hydrophobic effect of NIPAM and DMAEMA when the temperature is above their LCSTs, respectively. Physical network was formed through an association mechanism between hydrophobic end groups, the sticky property results in the little effect on loss modulus, while the hydrophobic interaction among the three blocks upon increasing temperature leads to the decreasing elastic modulus. The modulus increased when the temperature was above 70 °C, which means that both PNIPAM and PDMAEMA chains could completely reswollen.57 SEM images (Fig. 14 and S7†) were measured at 30 mg mL−1. The loose 3D net structure was found at 25 °C since the polymer solution was a viscous liquid, and the structure was collapsed gradually with the increasing temperature. These observations are highly consistent with the conclusion obtained from rheology measurement previously.
 | ||
| Fig. 14 SEM images of gelation behavior at different temperatures at pH = 9. | ||
The micellization of the PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer in neutral and alkaline solutions
There are some interesting aspects in current study. Firstly, in alkaline conditions, the thermo-induced triblock copolymer assembled into micellar nanostructures at room temperature, with PMMA block as the core, and PDMAEMA and PNIPAM blocks as the shell. When the temperature increased above the first LCST of the triblock copolymer, PNIPAM block becomes hydrophobic, some of the hydrophobic PNIPAM blocks form new cores, while the rest of them would be deposited onto the PMMA cores to form corona–shell–core nanoparticles, in which the hydrophobic PMMA block forms the core, the dehydrated PNIPAM forms the shell and the looped and hydrophilic PDMAEMA forms the corona. Upon continuous heating through the second LCST, the PDMAEMA block becomes hydrophobic, some of them form new cores, and others dehydrate to form a shell–shell–core structure. When the temperature is further increased, the triblock copolymer nanoparticles aggregate, then precipitation occurs because of the three hydrophobic blocks (see Fig. 1).46Furthermore, the thermo-response of the PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer nanoparticles dispersed in water was checked by UV-vis absorbance spectroscopy and dynamic light scattering (DLS). As shown in Fig. 15, the triblock copolymer exhibits two-stage thermally induced collapse, corresponding to the LCST of PNIPAM and PDMAEMA blocks, respectively. The first-stage collapse, which can be ascribed to the shrinking of the PNIPAM chains, the second-stage collapse, is attributed to the shrinking of PDMAEMA chains. At pH = 8, the first-stage collapse ranges from 29 °C to 39 °C, with a middle point at 34 °C, and the second-stage collapse ranges from 52 °C to 60 °C, with a middle point at 56 °C. While at pH = 9, the first-stage collapse ranges from 34 °C to 40 °C, with a middle point at 37 °C, and the second-stage collapse ranges from 44 °C to 60 °C, with a middle point at 52 °C. At pH = 7, there is a broadening transition ranging from 34 °C to 62 °C, with a middle point at 48 °C. This phenomenon is probably caused by the following two factors: firstly, DMAEMA is a cationic hydrophilic polymer as its amine groups are protonated below the pKa value of 7.3, which is the reason why the turbidity curve is similar to that in acidic conditions.54 Moreover, PDMEMA is water-soluble over the whole pH range below the LCST, but the LCST of PDMAEMA decreases with the pH increasing. Secondly, the strong inter-chain interactions are presented among the polymer chains.58,59 This predication have already been confirmed by some experimental results.60 Nevertheless, the results were lower than the triblock copolymers synthesized by Li probably because of the different molecular weight.46
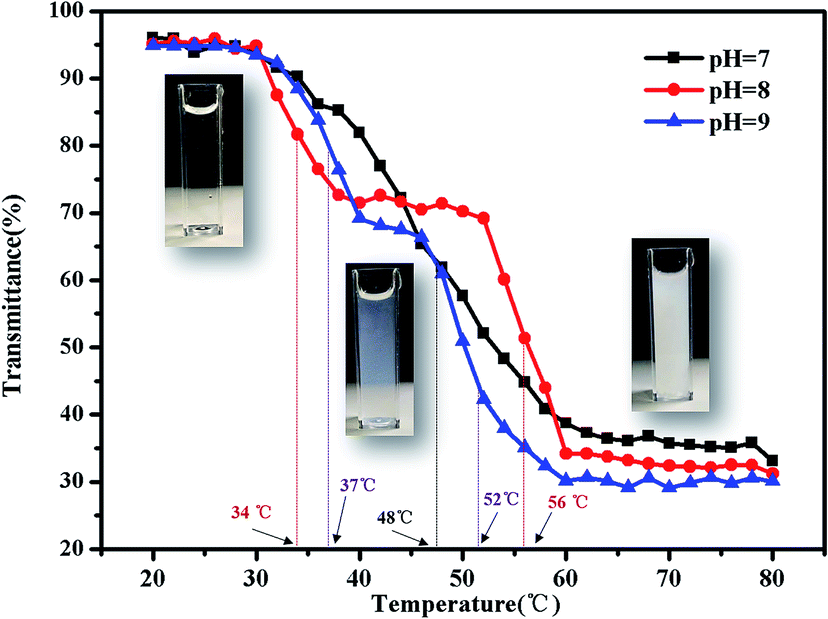 | ||
| Fig. 15 Turbidity curves of the PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer solution (2 mg mL−1) at various temperatures. | ||
Fig. 16 shows the evolution of the size distribution of the PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer solution as the temperature changed. When the temperature is increased from 20 °C to 50 °C, the evolution of the size distribution is similar to that in acidic conditions. However, the difference occurs when the temperature increases from 50 °C to 75 °C, which means, above the second LCST of the triblock copolymer, the peak of the intensity decreased as the temperature increased.
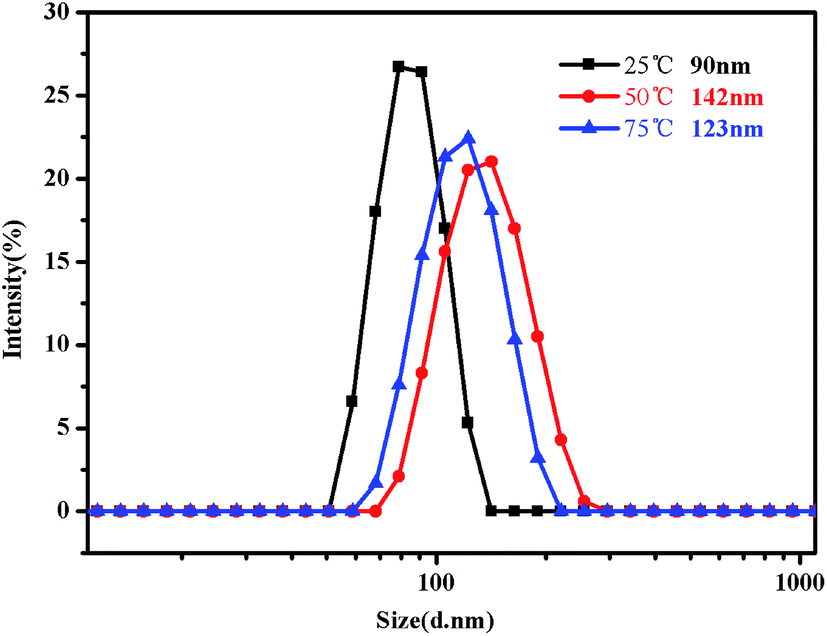 | ||
| Fig. 16 Intensity distribution of hydrodynamic diameter of the PMMA-b-PDMAEMA-b-PNIPAM triblock copolymer solution (0.15 mg mL−1) at pH = 9 at 25 °C, 50 °C and 75 °C, respectively. | ||
As shown in Fig. 17(A), there is a significant rising of the Z-average diameter when the temperature is increased from 30 °C to 50 °C. Above 32 °C, some of NIPAM blocks may collapse on the PMMA core and shrink to form corona–shell–core structures, the hydrodynamic diameter of the micelles would decrease. However, many more NIPAM blocks may form new cores to be shell–core nanoparticles. Besides shell–core nanoparticles with PMMA as cores, aggregation occurs due to the hydrophobic interactions among hydrophobic groups, which could enlarge the diameter.57,61 When the temperature is above 50 °C, the Z-average diameter starts to decrease. This is because the polymer precipitates from the solution when the temperature is raised sequentially which could be further attributed to the hydrophobic interaction among the three blocks above the second LCST.46 As we can see in Fig. 17(B), the intensity of scattered light, which depends on the concentration of light scatters, starts to decrease gradually at around 35 °C due to the hydrophobic block of NIPAM. Although aggregation occurred as a result of hydrophobic interaction, the high temperature would make the structure loose and viscosity low till precipitation.
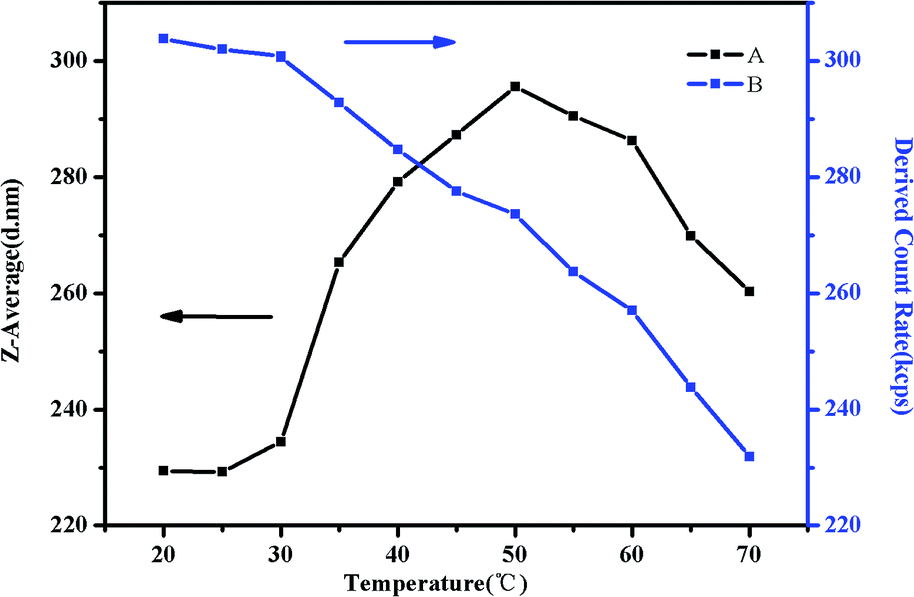 | ||
| Fig. 17 The Z-average diameter (A) and light scattering intensity (B) as a function of temperature in a DLS study of 0.5 mg mL−1 PMMA-b-PDMAEMA-b-PNIPAM solution at pH 9. | ||
The TEM images (Fig. 18) confirm the formation of micelles and the temperature-induced collapse of the triblock copolymer system, which were formed by three hydrophobic blocks. It is shown in TEM images that micelles formed were well dispersed as nanoscale micelles at room temperature, and the transition of the aggregating micellar nanoparticles with the increasing of the temperature can be found, and finally precipitation occurred at 75 °C. However, it is very difficult to identify the core layer, shell layer and corona layer of core–shell–corona micelles, the nanoscale micelles is usually observed to be a sphere by TEM, which is just like a core–shell one.38
 | ||
| Fig. 18 TEM images of triblock copolymer nanoparticles of PMMA-b-PDMAEMA-b-PNIPAM (0.2 mg mL−1) at different temperatures at pH = 9. | ||
Conclusions
In summary, a novel pH- and dual-thermo-sensitive amphiphilic ABC triblock copolymer composed of PMMA, PDMAEMA and PNIPAM was successfully synthesized by sequential RAFT polymerization. The resultant PMMA-b-PDMAEMA-b-PNIPAM was characterized by GPC, 1H NMR and FT-IR spectroscopy. The micellar solution was a viscous liquid in 0.3 mg mL−1 dilute solution at room temperature, the self-assembly behaviour in aqueous solution was explored by TEM and DLS. Rheology measurements, SEM and turbidity analysis revealed that the triblock polymers exhibit a transition and can form free-standing gel in response to the temperature in acidic conditions. The transition temperature point was about 40 °C and the critical gelation concentration was 2 wt% in water. At pH = 7, the solution was soft gel. While in alkaline conditions, the solution was weak gel at 25 °C and no free-standing gel formed with the increasing temperature. The double temperature responsive behavior was successfully proven by cloud point measurements and the formation of micelles was demonstrated by TEM. At pH = 8, the LCSTs are 34 °C and 56 °C, at pH = 9, the LCSTs are 37 °C and 52 °C. Upon the temperature increasing above the first LCST, the shell–core nanoparticles convert into corona–shell–core ones. When above the second LCST, the nanoparticles changed into shell–shell–core, and then precipitated completely from aqueous solution due to the hydrophobic interaction among the three blocks. Based on aforementioned studies, this ABC amphiphilic triblock copolymer may be anticipated to possess large potential value in biomedical applications. We hope all works may provide some theoretical references in fabricating multi-responsive ABC copolymers.Acknowledgements
The authors are grateful for assistance of Instrumental Analysis and Research Center of Shanghai University, and University of Minho, Portugal for the invaluable assistance obtaining the SEM images used in this work.Notes and references
- C. Zhou, M. A. Hillmyer and T. P. Lodge, J. Am. Chem. Soc., 2012, 134, 10365–10368 CrossRef CAS PubMed.
- Y. Dahman, J. E. Puskas, A. Margaritis, Z. Merali and M. Cunningham, Macromolecules, 2003, 36, 2198–2205 CrossRef CAS.
- M. A. Stuart, W. T. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- H. Wei, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, Prog. Polym. Sci., 2009, 34, 893–910 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- S. Reinicke, J. Schmelz, A. Lapp, M. Karg, T. Hellweg and H. Schmalz, Soft Matter, 2009, 5, 2648–2657 CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- T. Diaz, A. Fischer, A. Jonquieres, A. Brembilla and P. Lochon, Macromolecules, 2003, 36, 2235–2241 CrossRef CAS.
- K. Matyjaszewski, Prog. Polym. Sci., 2005, 30, 858–875 CrossRef CAS.
- X. Han, X. Zhang, H. Zhu, Q. Yin, H. Liu and Y. Hu, Langmuir, 2013, 29, 1024–1034 CrossRef CAS PubMed.
- J. Chen, M. Liu, H. Gong, Y. Huang and C. Chen, J. Phys. Chem. B, 2011, 115, 14947–14955 CrossRef CAS PubMed.
- C. Tsitsilianis, Soft Matter, 2010, 6, 2372–2388 RSC.
- K. Zhang and X. Y. Wu, Biomaterials, 2004, 25, 5281–5291 CrossRef CAS PubMed.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- C. He, S. W. Kim and D. S. Lee, J. Controlled Release, 2008, 127, 189–207 CrossRef CAS PubMed.
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2012, 64, 154–162 CrossRef.
- S. Sugihara, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2601–2611 CrossRef CAS.
- M. A. Ward and T. K. Georgiou, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2850–2859 CrossRef CAS.
- M. E. Alf, T. A. Hatton and K. K. Gleason, Polymer, 2011, 52, 4429–4434 CrossRef CAS.
- J. Xu, J. Ye and S. Liu, Macromolecules, 2007, 40, 9103–9110 CrossRef CAS.
- T. Qu, A. Wang, J. Yuan, J. Shi and Q. Gao, Colloids Surf., B, 2009, 72, 94–100 CrossRef CAS PubMed.
- T. Qu, A. Wang, J. Yuan and Q. Gao, J. Colloid Interface Sci., 2009, 336, 865–871 CrossRef CAS PubMed.
- F. A. Plamper, M. Ruppel, A. Schmalz, O. Borisov, M. Ballauff and A. H. E. Müller, Macromolecules, 2007, 40, 8361–8366 CrossRef CAS.
- A. Emileh, E. Vasheghani-Farahani and M. Imani, Eur. Polym. J., 2007, 43, 1986–1995 CrossRef CAS.
- B. Wang, X. D. Xu, Z. C. Wang, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Colloids Surf., B, 2008, 64, 34–41 CrossRef CAS PubMed.
- J. Brassinne, E. Poggi, C.-A. Fustin and J.-F. Gohy, Macromol. Rapid Commun., 2015, 36, 610–615 CrossRef CAS PubMed.
- T. G. O'Lenick, X. Jiang and B. Zhao, Langmuir, 2010, 26, 8787–8796 CrossRef PubMed.
- Z. Lin, S. Cao, X. Chen, W. Wu and J. Li, Biomacromolecules, 2013, 14, 2206–2214 CrossRef CAS PubMed.
- Y. He and T. P. Lodge, Macromolecules, 2008, 41, 167–174 CrossRef CAS.
- Y.-L. Luo, W. Yu, F. Xu and L.-L. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2053–2067 CrossRef CAS.
- C. Li, N. J. Buurma, I. Haq, C. Turner, S. P. Armes, V. Castelletto, I. W. Hamley and A. L. Lewis, Langmuir, 2005, 21, 11026–11033 CrossRef CAS PubMed.
- S. Xuan, C. U. Lee, C. Chen, A. B. Doyle, Y. Zhang, L. Guo, V. T. John, D. Hayes and D. Zhang, Chem. Mater., 2016, 28, 727–737 CrossRef CAS PubMed.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2013, 4, 873–881 RSC.
- C. Gao, Q. Li, Y. Cui, F. Huo, S. Li, Y. Su and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2155–2165 CrossRef CAS.
- N. Zammarelli, M. Luksin, H. Raschke, R. Hergenroder and R. Weberskirch, Langmuir, 2013, 29, 12834–12843 CrossRef CAS PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- J. Chen, M. Liu, C. Gao, S. Lü, X. Zhang and Z. Liu, RSC Adv., 2013, 3, 15085–15093 RSC.
- Q. Li, F. Huo, Y. Cui, C. Gao, S. Li and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2266–2278 CrossRef CAS.
- R. I. Moustafine, A. R. Salachova, E. S. Frolova, V. A. Kemenova and G. Van den Mooter, Drug Dev. Ind. Pharm., 2009, 35, 1439–1451 CrossRef CAS PubMed.
- C. Boyer, J. Teo, P. Phillips, R. B. Erlich, S. Sagnella, G. Sharbeen, T. Dwarte, H. T. T. Duong, D. Goldstein, T. P. Davis, M. Kavallaris and J. McCarroll, Mol. Pharmaceutics, 2013, 10, 2435–2444 CrossRef CAS PubMed.
- P. Yong, Y. Yang, Z. Wang, L. Yang and J. Chen, RSC Adv., 2016, 6, 88306–88314 RSC.
- J. Chen, M. Liu, H. Gong, G. Cui, S. Lü, C. Gao, F. Huang, T. Chen, X. Zhang and Z. Liu, Polym. Chem., 2013, 4, 1815–1825 RSC.
- J. N. Shen, Y. F. Ye, G. N. Zeng and J. H. Qiu, Adv. Mater. Res., 2011, 284–286, 1717–1723 CrossRef CAS.
- A. E. Smith, X. Xu, S. E. Kirkland-York, D. A. Savin and C. L. McCormick, Macromolecules, 2010, 43, 1210–1217 CrossRef CAS.
- Q. Li, C. Gao, S. Li, F. Huo and W. Zhang, Polym. Chem., 2014, 5, 2961–2972 RSC.
- A. I. Triftaridou, M. Vamvakaki and C. S. Patrickios, Polymer, 2002, 43, 2921–2926 CrossRef CAS.
- J. C. P. de Souza, A. F. Naves and F. H. Florenzano, Colloid Polym. Sci., 2012, 290, 1285–1291 Search PubMed.
- C. N. Toledo, F. H. Florenzano and J. M. Schneedorf, Int. J. Electrochem., 2014, 2014, 1–7 CrossRef.
- A. E. Smith, X. Xu, T. U. Abell, S. E. Kirkland, R. M. Hensarling and C. L. McCormick, Macromolecules, 2009, 42, 2958–2964 CrossRef CAS.
- L. Hou, Q. Chen, Z. An and P. Wu, Soft Matter, 2016, 12, 2473–2480 RSC.
- H. Yu and D. W. Grainger, J. Appl. Polym. Sci., 1993, 49, 1553–1563 CrossRef CAS.
- F. d. r. Bossard, T. Aubry, G. Gotzamanis and C. Tsitsilianis, Soft Matter, 2006, 2, 510 RSC.
- G. T. Gotzamanis, C. Tsitsilianis, S. C. Hadjiyannakou, C. S. Patrickios, R. Lupitskyy and S. Minko, Macromolecules, 2006, 39, 678–683 CrossRef CAS.
- K. Nishinari, Colloid Polym. Sci., 1997, 275, 1093–1107 CAS.
- Q. Jin, L.-P. Lv, G.-Y. Liu, J.-P. Xu and J. Ji, Polymer, 2010, 51, 3068–3074 CrossRef CAS.
- J. Li, W.-D. He, S.-c. Han, X.-l. Sun, L.-y. Li and B.-y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 786–796 CrossRef CAS.
- B. Zhao and W. J. Brittain, Prog. Polym. Sci., 2000, 25, 677–710 CrossRef CAS.
- A. Halperin, M. Tirrell and T. P. Lodge, Adv. Polym. Sci., 1991, 100, 31–71 Search PubMed.
- P. W. Zhu and D. H. Napper, J. Colloid Interface Sci., 1994, 164, 489–494 CrossRef CAS.
- M. Taha, B. S. Gupta, I. Khoiroh and M.-J. Lee, Macromolecules, 2011, 44, 8575–8589 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–S7: 1H NMR spectra, rheology, SEM image of triblock copolymer. See DOI: 10.1039/c7ra04351a |
| This journal is © The Royal Society of Chemistry 2017 |