
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA Ni-containing decaniobate incorporating organic ligands: synthesis, structure, and catalysis for allylic alcohol epoxidation†
Li Li,
Yanjun Niu,
Kaili Dong,
Pengtao Ma,
Chao Zhang,
Jingyang Niu * and
Jingping Wang*
* and
Jingping Wang*
Henan Key Laboratory of Polyoxometalate Chemistry, Institute of Molecular and Crystal Engineering, College of Chemistry and Chemical Engineering, Henan University, Kaifeng 475004, Henan, China. E-mail: jyniu@henu.edu.cn; jpwang@henu.edu.cn; Fax: +86-371-23886876
First published on 31st May 2017
Abstract
An organic–inorganic hybrid polyoxoniobate, Na8{Ni[Ni(en)]2Nb10O32}·28H2O (1) (en = ethanediamine), has been synthesized and characterized. It represents the first example of a trinuclear nickel-containing polyoxoniobate. The catalysis of 1 for allylic alcohol epoxidation was investigated at room temperature in aqueous solution, and was found to catalyze the epoxidation of 3-methyl-2-buten-1-ol with high conversion (98%) and selectivity (94%). Furthermore, magnetic measurements showed that the compound exhibits ferromagnetic interactions.
Introduction
Polyoxoniobates (PONbs), an important subclass of polyoxometalates (POMs), have been investigated since the last century due to their unique characteristics of high basicity and high surface charge endowing them with spectroscopic, magnetic, and catalytic properties.1,2 Nevertheless, in comparison with other POMs, the development of PONbs is still in its infancy owing to difficulties in controlling their synthesis, especially the narrow working pH range of PONbs.3 Generally, most of the reported PONbs focus on heteropolyoxoniobates with the typical examples [XNb12O40]16− (X = Si, Ge),4a [Ti2O2][XNb12O40]12− (X = Si, Ge),4b,c [Nb2O2][SiNb12O40]10−,4d and [SiNb18O54]14−.4e Up to now, isopolyoxoniobates including [Nb10O28]6−,5a [Nb20O54]8−,5b [Nb24O72H9]15−,5c [HNb27O76]16−, [H10Nb31O93(CO3)]23−,5d and [Nb32O96H28]4− (ref. 2b) have been isolated. In addition, a number of transition-metal (TM)-containing PONbs have also been obtained. The few examples of the PONbs are {Ti2Nb8},6a {TiNb9},6b {Ti12Nb6},6c {V3Nb12},7a {V4Nb6},7b {V4Nb10},7c {PV2Nb12},7d {XV8Nb8} (X = P, V),7e {PV6Nb12},7f {VNb14},7g {V8Nb48},7h {Cu24Nb56}, {Cu25.5Nb56},8a {CuNb11},8b {Co14Nb36},9a {Co8Nb24},9b {NiNb12},10a {NiNb9},10b {Ni10Nb32}.10c Introducing TMs with d electrons into the reaction can enrich the diversity of structures and properties in polyoxoniobates.On the other hand, POMs have been widely used as catalysts for epoxidation reactions with hydrogen peroxide in the past few years.11a,b Epoxide alcohols, as an important intermediate and final product of the organic chemical industry, have already been applied in the field of fine chemicals, such as components of cosmetic formulations for moister and cleaning of skin. Traditionally, one of the useful approaches for the synthesis of required epoxide alcohols is the epoxidation reactions of allylic alcohols in the presence of H2O2 as the oxidation agent.11c–e Therefore, we focus on the synthesis of TM-containing PONbs and expect to discover good efficiency in the catalytic epoxidation of allylic alcohols.
Herein, the sodium salt of nickel-decaniobate derivative Na8{Ni[Ni(en)]2Nb10O32}·28H2O (1) was obtained by the hydrothermal and diffusional method, which was composed of a {Ni[Ni(en)]2} cluster coordinated by two monolacunary [Nb5O18]11− units. Furthermore, the Ni2+ ions at both ends of the {Ni[Ni(en)]2} cluster are functionalized by one en molecule respectively, resulting in a pseudosandwich-type PONb-based organic–inorganic hybrid compound. Most notably, it represents the first example of trinuclear Ni-containing PONb and extends into a one-dimensional (1D) chain by high-nuclear sodium-clusters. Furthermore, the catalytic epoxidation of allylic alcohols has also been preliminary studied. To the best of our knowledge, this is the first example of the epoxidation of allylic alcohols using PONb as an efficient catalyst at room temperature in aqueous solution.
Results and discussion
Synthesis
An interesting TM-substituted PONb Na8{Ni[Ni(en)]2Nb10O32}·28H2O (1) was synthesized using the hydrothermal synthesis method by reaction of the SeO2, Ni(NO3)2·6H2O, and K7HNb6O19·13H2O with the presence of en at the pH value of 12.3–12.8 (Scheme 1). Parallel experiments show that the final product depend on three key factors: (a) the introduction of en ligand. Its presence during the course of the reaction is crucial because the polyoxoanion could not be obtained if 1,2-diaminopropane or 1,3-diaminopropane was used to replace it or in the absence of it; (b) using NaOH solution to adjust pH value. Its presence is necessary to obtain pure bulk samples and the crystals could not be gained when NaOH solution was replaced by LiOH, KOH, or CsOH solution. The results confirm that counter cations can have significant effects on the synthesis; (c) the presence of SeO2. Notably, although SeO2 was used as a staring material in the system, no selenium atom was observed in the compound. When SeO2 was removed from the reaction, no crystals were afforded. The specific role of SeO2 was not well understood, and SeO2 may play a synergistic action with other materials in the reaction and the similar phenomena have been previously reported.12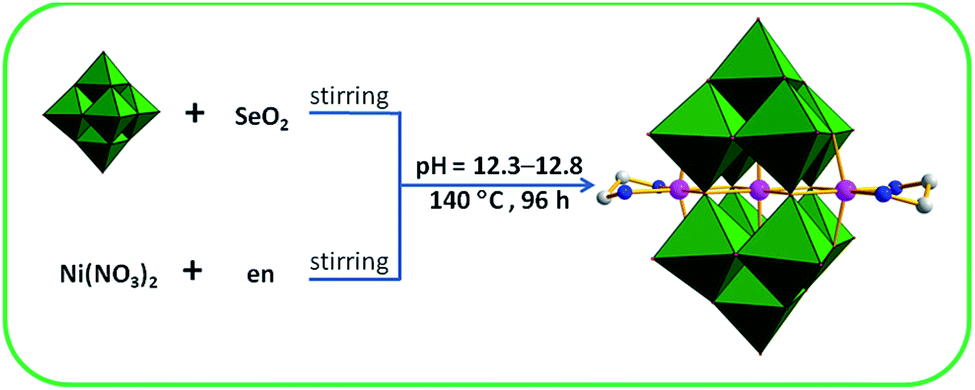 | ||
| Scheme 1 The preparation process of 1. | ||
Crystal structure
Single-crystal X-ray diffraction analysis indicates that the structure of compound 1 possesses a cluster of {Ni[Ni(en)]2Nb10O32} along with eight Na+ cations and twenty-eight water molecules. Compound 1 crystallizes in the triclinic molecular symmetry, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The {Ni[Ni(en)]2Nb10O32} cluster is constructed by a {Ni[Ni(en)]2} cluster sandwiched into the novel {Nb10O32} fragment (Fig. 1a). And the {Nb10O32} unit can be described as {W10O32} reported by Fuchs et al.13 It consists of two equivalent lacunary [Nb5O18]11− subunits fused together by four bridging oxygen atoms and the [Nb5O18]11− unit is obtained by removal of one {NbO6} octahedron from well-known Lindqvist anion [Nb6O19]18− (Fig. S1†). There are four types of oxygen atoms in the {Ni[Ni(en)]2Nb10O32} unit: ten terminal oxygen atoms (Ot), twelve bridging μ2-oxygen atoms, four bridging μ3-oxygen atoms, four bridging μ4-oxygen atoms, and two central μ6-oxygen atom. The bond lengths of Nb–Ot, Nb–μ2-O, Nb–μ3-O, Nb–μ4-O and Nb–Oc in {Ni[Ni(en)]2Nb10O32} are in the range of 1.766(5)–1.773(5) Å, 2.029(4)–2.032(4) Å, 2.052(4)–2.098(4) Å, 1.929(4)–2.012(4) Å and 2.350(4)–2.410(4) Å. In the {Ni[Ni(en)]2} cluster, three nickel ions constitute a linear {Ni3} cluster via four bridging oxygen atoms (Fig. 1b). The Ni ions in {Ni[Ni(en)]2} cluster are all in the octahedral coordination environment, Ni1 ion is coordinated by four μ2-O and two central oxygen atoms from two [Nb5O18]11− units (Fig. 1c) and Ni2 ions are completed by two N atoms from an en ligand and four O oxygen atoms from two [Nb5O18]11− skeletons (Fig. 1d).
space group. The {Ni[Ni(en)]2Nb10O32} cluster is constructed by a {Ni[Ni(en)]2} cluster sandwiched into the novel {Nb10O32} fragment (Fig. 1a). And the {Nb10O32} unit can be described as {W10O32} reported by Fuchs et al.13 It consists of two equivalent lacunary [Nb5O18]11− subunits fused together by four bridging oxygen atoms and the [Nb5O18]11− unit is obtained by removal of one {NbO6} octahedron from well-known Lindqvist anion [Nb6O19]18− (Fig. S1†). There are four types of oxygen atoms in the {Ni[Ni(en)]2Nb10O32} unit: ten terminal oxygen atoms (Ot), twelve bridging μ2-oxygen atoms, four bridging μ3-oxygen atoms, four bridging μ4-oxygen atoms, and two central μ6-oxygen atom. The bond lengths of Nb–Ot, Nb–μ2-O, Nb–μ3-O, Nb–μ4-O and Nb–Oc in {Ni[Ni(en)]2Nb10O32} are in the range of 1.766(5)–1.773(5) Å, 2.029(4)–2.032(4) Å, 2.052(4)–2.098(4) Å, 1.929(4)–2.012(4) Å and 2.350(4)–2.410(4) Å. In the {Ni[Ni(en)]2} cluster, three nickel ions constitute a linear {Ni3} cluster via four bridging oxygen atoms (Fig. 1b). The Ni ions in {Ni[Ni(en)]2} cluster are all in the octahedral coordination environment, Ni1 ion is coordinated by four μ2-O and two central oxygen atoms from two [Nb5O18]11− units (Fig. 1c) and Ni2 ions are completed by two N atoms from an en ligand and four O oxygen atoms from two [Nb5O18]11− skeletons (Fig. 1d).
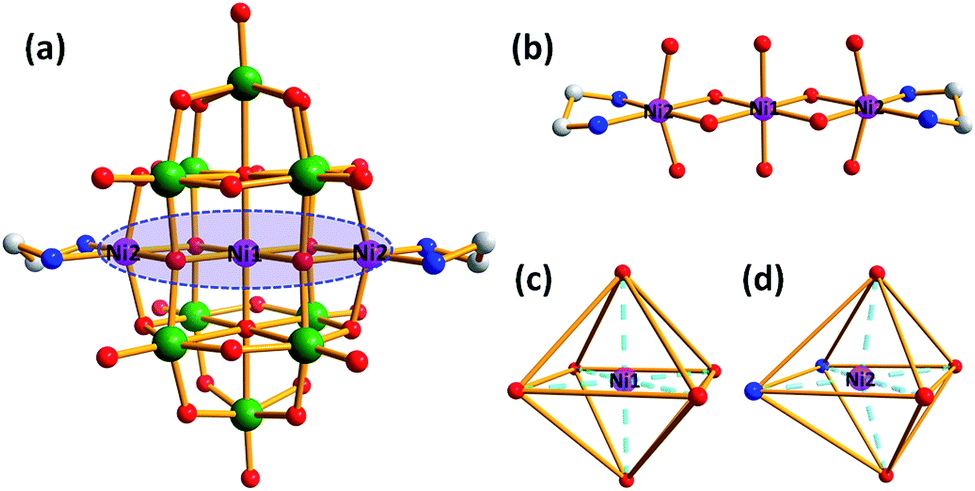 | ||
| Fig. 1 (a) Ball-and-stick representation of the {Ni[Ni(en)]2Nb10O32} unit; (b) view of the {Ni[Ni(en)]2} cluster in 1; (c) the coordinated environment of Ni1 in 1; (d) the coordinated environment of Ni2 in 1. All hydrogens, sodium ions, and lattice water molecules are omitted for clarity. Colour code: Nb (green), Ni (pink), N (blue), C (gray) and O (red). | ||
As shown in Fig. 2, the adjacent cluster anions in 1 are linked alternately via {Na8} clusters into one-dimensional (1D) chain. It represents the first example of a high-nuclear sodium-cluster-containing in PONb. The Na+ ions in the {Na8} cluster could be divided into four groups according to the difference between their coordinated environments. The Na1, Na2 and Na3 ions are all embedded in octahedral geometries with different coordination environments whereas Na4 ions exhibit a square pyramid geometry. On the one hand, Na1 and Na2 ions links each other by sharing two μ2-O atoms forming a type of [Na2]2+ dimeric clusters in the edge-sharing mode, meanwhile the adjacent [Na2]2+ clusters are connected by two μ2-O atoms generating a [Na4]4+ tetrameric cluster. On the other hand, Na3 and Na4 ions also join together by sharing two μ2-O atoms forming another type of [Na2]2+ dimeric clusters. Moreover, one [Na4]4+ and two [Na2]2+ dimeric clusters are connected by sharing four μ2-O atoms forming the {Na8} cluster and displaying a unique cruciform structure.
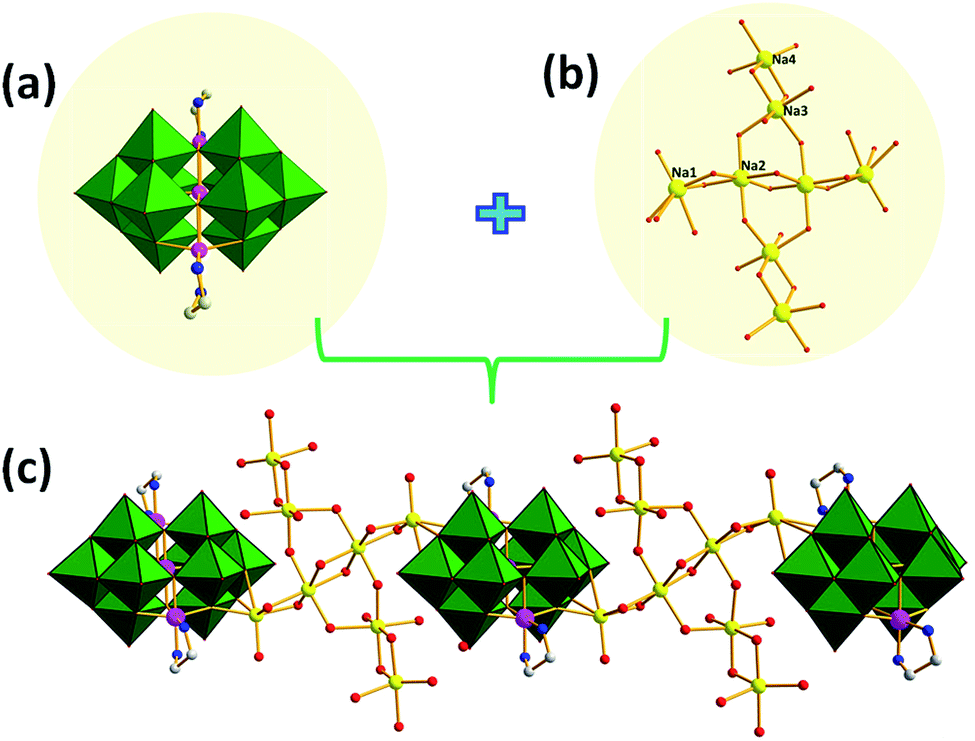 | ||
| Fig. 2 (a) Combined polyhedral/ball-and-stick representation of the {Ni[Ni(en)]2Nb10O32} unit; (b) ball-and-stick representation of the {Na8} cluster; (c) combined polyhedral/ball-and-stick representation of one-dimensional (1D) of 1. All hydrogens, and lattice water molecules are omitted for clarity. Colour code: Nb (green), Ni (pink), N (blue), C (gray), Na (yellow) and O (red) respectively. | ||
The phase purity of 1 was characterized by the powder X-ray diffraction (PXRD) pattern of the bulk products (Fig. S3†). The PXRD pattern is in good agreement with the simulated XRPD patterns resulted from single-crystal X-ray diffraction, which confirms that the phase is pure. And the difference in intensity may be due to the preferred orientation of the powder samples. The band valence sum (BVS) calculations of compound 1 confirm that the oxidation states of the Ni and Nb atoms are +2, +5, respectively.14 The BVS values for all oxygen atoms and nickel atoms in 1 are also listed in Tables S1 and S2.†
IR and UV spectra
The infrared spectrum of compound 1 was recorded between 4000 and 400 cm−1 with KBr pellet. The IR spectra shows seven characteristic vibration bands in the low-wavenumber region (v < 1000 cm−1) belonging to the Lindqvist hexaniobate structure, the peak at 867 cm−1 can be assigned to terminal Nb–Ot characteristic vibration, as well as peaks at 667–419 cm−1 can be attributed to bridging Nb–Ob vibration. In comparison with the precursor K7HNb6O19·13H2O, the bridging Nb–Ob vibration band splits into more components and shows slightly red shifts, which may originate from the coordination of Ni2+ with the bridging oxygen atoms of the PONb anion.15 Moreover, the peaks at 1025 cm−1 can be assigned to C–N bond characteristic vibration of organic-ligand en (Fig. S4†).UV-vis absorption spectrum of compound 1 (3 × 10−6 mol L−1) in the aqueous solution displays one strong absorption band at 192 nm and one wide absorption band at 225 nm (Fig. S5 and S6†), which can be tentatively assigned to the pπ–dπ charge-transfer transitions of oxygen-to-niobium bonds.12b In comparison with UV spectra of the precursor K7HNb6O19·13H2O, the absorption band at 225 nm shows slightly blue shifts, which results from the coordination of Ni2+ with the oxygen atoms of the PONb anion.15
ESI-MS studies
During the course of this study, negative electrospray ionization mass spectrometry (ESI-MS) allowed us to identify intact polyanions {Ni[Ni(en)]2Nb10O32}.16 The compound 1 was dissolved in a minimum amount of water, and a few drops of this aqueous solution were mixed with acetonitrile (V(H2O)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V(CH3CN) = 1:1). ESI-MS data shows two dominant series of multiply charged peaks which can be attributed to series of peaks for −3 and −2 charged ions (Fig. 3). The series of −3 and −2 ion peaks can be attributed to {NaxHyNi[Ni(en)]2Nb10O32}3− (x + y + 3 = 8), {NaxHyNi[Ni(en)]2Nb10O32}2− (x + y + 2 = 8) polyanions on the basis of their m/z values, showing the presence of intact cluster in the mixture solution. Primary peaks centered at m/z 587.95 and 904.45 are clearly observed in the m/z 0–1500 range (Fig. 3, S7 and S8†). The extended peaks at around m/z 587.95 and 904.45 are provided in the ESI (Tables S3 and S4†).
:V(CH3CN) = 1:1). ESI-MS data shows two dominant series of multiply charged peaks which can be attributed to series of peaks for −3 and −2 charged ions (Fig. 3). The series of −3 and −2 ion peaks can be attributed to {NaxHyNi[Ni(en)]2Nb10O32}3− (x + y + 3 = 8), {NaxHyNi[Ni(en)]2Nb10O32}2− (x + y + 2 = 8) polyanions on the basis of their m/z values, showing the presence of intact cluster in the mixture solution. Primary peaks centered at m/z 587.95 and 904.45 are clearly observed in the m/z 0–1500 range (Fig. 3, S7 and S8†). The extended peaks at around m/z 587.95 and 904.45 are provided in the ESI (Tables S3 and S4†).
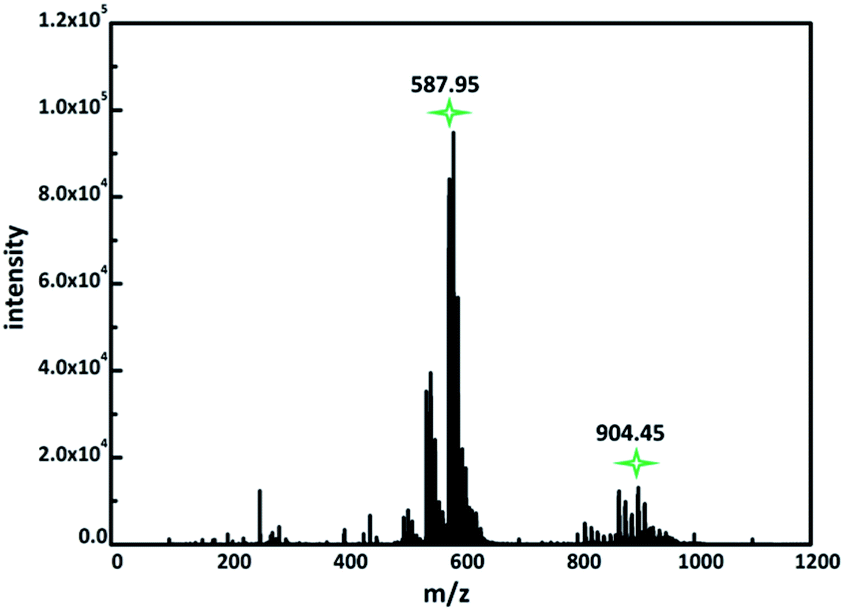 | ||
| Fig. 3 The ESI mass spectrum of {Ni(Nien)2Nb10O32} in the range of m/z = 0−1500. | ||
Magnetic properties
The magnetic susceptibility for 1 was measured at 2–300 K under a 1000 Oe magnetic field. Their plots of χMT and χM−1 versus T are illustrated in Fig. 4. Along with the temperature decreased, the χMT product of 1 increased from 5.355 emu K mol−1 at 300 K to a maximum of 8.622 emu K mol−1 at 5 K. This characteristic thermal behavior is indicative of an overall ferromagnetic coupling between nickel ions.17 Upon the temperature lower than 5 K, the rapid drop of the χMT products may also be assigned to zero-field splitting. The experimental data have been well fitted by the Curie Weiss law (χM = C/(T − θ)) above 1.8 K with the following Curie and Weiss constants: 5.234 emu mol−1 K and 5.317 K, respectively (Fig. 4 and S9†). The positive Weiss constant confirms the presence of the ferromagnetic couplings between Ni2+ centers.18 When the experimental χMT value of 5.355 emu K mol−1 at room temperature (300 K) is compared with that of the spin-only value of 3 emu K mol−1 for 3 non-interacting Ni2+ ions (S = 1) with g = 2.0, we can see that the experimental χMT value is higher than the calculate χMT value. So there exist spin–orbit coupling and ferromagnetic couplings among Ni2+ ions.9b,10c | ||
| Fig. 4 The plots of χMT and χM−1 versus T for 1 between 2 and 300 K. (outside: χMT; inside: 1/χM−1). | ||
Catalytic properties
Compound 1 was investigated as a heterogeneous catalyst in allylic alcohols epoxidation with H2O2 and allylic alcohols can be transform to epoxy alcohols as major products and the corresponding aldehydes and polyols were formed together as by-products (Table S5†). Initial experiments were carried out by using 3-methyl-2-buten-1-ol as a model substrate with the assistance of H2O2. Furthermore, the results of a series of parallel experiments showed that the conversion increased as the catalyst or H2O2 dosage increased (Table 1, entries 1–3 and 6), and didn't change obviously with the increase of reaction temperature and reaction time. As shown in Table 1, 3-methyl-2-buten-1-ol was oxidized to epoxy alcohol with 98% conversion and 94% selectivity at a temperature of 25 or 35 °C for 5 min (entries 3 and 7). This fact indicated that the conversion and selectivity of transformation to epoxide cannot achieve an obviously higher level as expected when the temperature is significantly higher than 25 °C. Moreover, the performance of epoxidation under low temperature ensures better security and allows the reduction of operating costs, so the temperature of 25 °C is the optimal reaction temperature. On the basis of these results, the catalyst and H2O2 dosage were the crucial factors for the conversion of epoxy alcohols. In contrast to catalyst 1, a blank experiment without 1 (Table 1, entry 5) indicated that the epoxidation scarcely happened and the epoxidation reaction with the precursor K7HNb6O19·13H2O indicated that the conversion was 95% (Table S6,† entry 3). As a result, the conversion of allylic alcohols epoxidation reactions of 1 was better than that of K7HNb6O19·13H2O, which suggests that Nb atoms play the main role in the process of the epoxidation reactions. Under optimal conditions (12 μmol catalyst, 5 mmol substrate and 8 mmol H2O2), 3-methyl-2-buten-1-ol was oxidized to epoxide with 98% conversion and 94% selectivity at 25 °C for 5 min (Table 1, entry 3). After the epoxidation reaction, the product was separated from the aqueous phase by extraction with ethyl acetate. And the catalyst still existed in the aqueous solution after extraction, so the aqueous phase could be used for the next run.19 The catalytic activity of compound 1 was not obviously changed upon completion of the sixth run (Fig. S10†), but the catalytic activity of K7HNb6O19·13H2O was decreased upon the process of duplicate operation (Fig. S11†). After epoxidation was completed, the catalyst was retrieved from the solvent/oxidant/substrate system by centrifugation. According to the comparisons of IR spectra of the fresh catalyst and the used catalyst after the sixth-run reaction (Fig. S12†), the noticeable differences of the IR spectrum are at the peaks of 1388 and 668 cm−1. The results suggested that the framework structure of the catalyst was partially decomposed. The reason for the differences in IR spectra may be that the recovered catalyst has been partly peroxidated by H2O2.20
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Product | 30%H2O2 (mmol) | Time (min) | Con.a (%) | Sel. (%) |
| a Conversion determined by GC with dodecane as an internal standard. The products were identified by GC-MS.b Reaction conditions for the entries 1 to 5: catalyst (12 μmol), substrate (5 mmol) and H2O (3 mL) at 25 °C.c Blank experiment, the reaction was carried out without catalyst.d Reaction conditions: catalyst (6 μmol), substrate (5 mmol) and H2O (3 mL) at 25 °C.e Reaction conditions for the entries 7 to 10: catalyst (12 μmol), substrate (5 mmol) and H2O (3 mL) at 35 °C.f cis- and trans-mixture. | ||||||
| 1b | 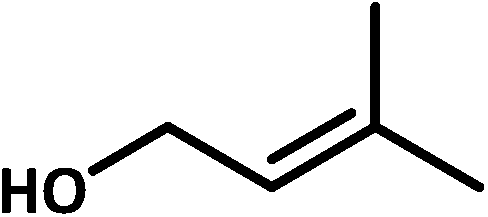 |
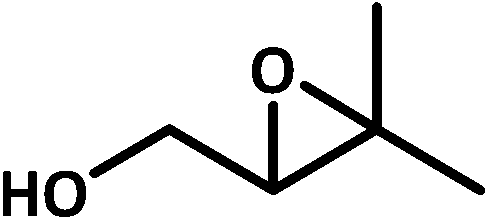 |
3 | 5 | 36 | 94 |
| 2 | 5 | 5 | 80 | 94 | ||
| 3 | 8 | 5 | 98 | 94 | ||
| 4 | 8 | 15 | 97 | 94 | ||
| 5c | 8 | 5 | 5 | 60 | ||
| 6d | 8 | 5 | 84 | 93 | ||
| 7e | 8 | 5 | 98 | 94 | ||
| 8 | 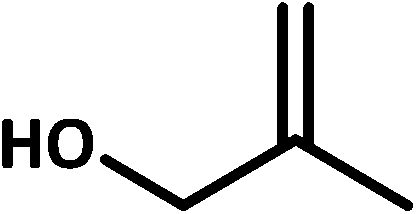 |
 |
8 | 60 | 36 | 94 |
| 9 |  |
 |
8 | 60 | 41 | 93 |
| 10f |  |
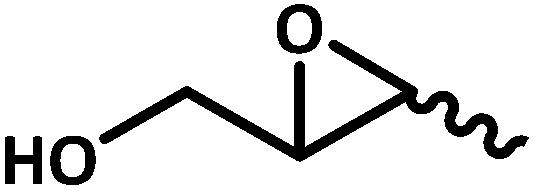 |
8 | 60 | 71 | 81 |

The catalytic activities of other allylic alcohols like 2-methyl-2-propen-1-ol, trans-2-hexen-1-ol and crotyl alcohol were also tested under the same reaction conditions (Tables 1 and S8†). The results showed that 1 was active for these allylic alcohols as well (Table 1, entries 8–10). 3-Methyl-2-buten-1-ol affords the highest epoxide yields and selectivity, while 2-methyl-2-propen-1-ol and trans-2-hexen-1-ol are less reactive corresponding to 36% conversion and 94% selectivity (Table 1, entry 8) and 41% conversion and 93% selectivity (Table 1, entry 9), respectively. In addition, crotyl alcohol gave 71% conversion and 81% selectivity (Table 1, entry 10). This result suggests that 1 can efficiently catalyze allylic alcohols epoxidation. Comparing with the reactivity for other allylic alcohols of catalyst 1 and K7HNb6O19·13H2O, the reactivity of K7HNb6O19·13H2O is slightly higher than that of catalyst 1 (Table S8†). After epoxidation was completed, hydrogen peroxide consumption was determined iodometrically,21 and the results indicated that the conversion of transformation to epoxy alcohols is related to the consumption of hydrogen peroxide (Table S9†).
Experimental section
Materials and methods
The raw material K7HNb6O19·13H2O were prepared as described in the literature and characterized by IR spectrum.22 All chemicals were purchased from commercial sources and used as received. Elemental analyses (C, H, N) were performed on an Elementar Vario EL cube CHNS analyzer. The IR spectrum was recorded on a Bruker-Vertex 70 FT-IR spectrometer using KBr pellets in a range of 4000–400 cm−1. The UV spectra were recorded with a U1900 spectrometer (distilled water as solvent) in the range of 800–190 nm. Powder X-ray diffraction (PXRD) data were recorded on a Bruker AXS D8 Advance instrument with Cu Kα radiation (λ = 1.5418 Å) in the angular range 2θ = 5–50° at 293 K. Variable-temperature magnetic susceptibility data were obtained on Quantum Design MPMS3 magnetometer in the temperature range of 1.8–300 K with an applied field of 1000 Oe. The mass spectrometer used for the measurements was AB SCIEX Triple TOF 4600. The epoxidation products were identified by Agilent Technologies 7890B/5977B GC-MS instrument, and the conversion determined by BRUKER 450-GC instrument.:3) was carefully layered onto the resulting amaranthine solution, and then the pure alcohol was layered onto the ethanol/water phase. After about one month the light-blue crystals are found in the test tube. Yield: 54% (based on K7HNb6O19·13H2O). Elemental analysis (%) calculated for compound 1: C 1.98, H 2.99, N 2.31; found for C 1.83, H 2.94, N 2.28. Selected IR (KBr, cm−1): 3248 (vs.), 2174 (m), 1663 (s), 1585 (w), 1458 (w), 1023 (s), 867 (s), 775 (w), 667 (s), 621 (w), 546 (m), 483 (m), 419 (m).X-ray crystallography
A suitable single crystal was selected from its mother liquor and sealed in a thin glass tube since the crystal is easily efflorescent. X-ray diffraction intensity data collection were mounted on a Bruker Apex-II CCD diffractometer using graphite-monochromatized Mo Kα radiation (λ = 0.71073 Å) at 293(2) K (Table 2). Routine Lorentz and polarization corrections were applied, and a multi-scan absorption correction was performed using the SADABS program. The structure was solved by direct methods and refined by the full-matrix least-squares method on F2 using the SHELXS-97 and SHELXL-2014 program suite.23 In the final refinement, the Nb and Na atoms were refined anisotropically; the O, C and N atoms were refined isotropically. The hydrogen atoms of the organic groups were placed in calculated positions and then refined using a riding model with a uniform value of Uiso = 1.2 or 1.5Ueq. All H atoms on water molecules were directly included in the molecular formula.| Formula | C4N4Nb10Ni3O60H72Na8 |
| Mr (g mol−1) | 2425.80 |
| T (K) | 293(2) |
| Space group | P |
| Crystal system | Triclinic |
| a (Å) | 12.5635(8) |
| b (Å) | 12.9279(9) |
| c (Å) | 13.1370(9) |
| α (deg) | 61.8590(10) |
| β (deg) | 63.6420(10) |
| γ (deg) | 61.2230(10) |
| V (Å3) | 1582.83(19) |
| Z | 1 |
| Dc (g cm−3) | 2.545 |
| μ (mm−1) | 2.783 |
| F (000) | 1186.0 |
| 2θ range (deg) | 3.664 to 50.196 |
| Data/restraints/parameters | 5578/0/238 |
| Reflections collected | 8189 |
| Index ranges | −14 ≤ h ≤ 14, −15 ≤ k ≤ 15, −13 ≤ l ≤ 15 |
| GOF on F2 | 1.021 |
| R1, wR2 [I > 2σ(I)] | R1 = 0.0411, wR2 = 0.0992 |
| R1, wR2 [all data] | R1 = 0.0545, wR2 = 0.1077 |
Conclusions
In summary, we have synthesized and characterized a novel organic–inorganic hybrid PONb which contains trinuclear nickel cluster. Furthermore, compound 1 shows catalytic activity for the epoxidation of various allylic alcohols with hydrogen peroxide. The successful synthesis of compound 1 enriches the structural diversity of PONbs and further proves that the use of metal–ligand cations could provide new way to synthesize diverse PONb anion architecture. In the following work, we will concentrate on improving the activity and selectivity for allylic alcohols epoxidation and the synthesis of novel PONb derivatives.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21371048), Key Lab of Polyoxometalate Science of Ministry of Education, the Natural Science Foundation of Henan Province for financial support.Notes and references
- (a) C. A. Ohlin, E. M. Villa, J. C. Fettinger and W. H. Casey, Angew. Chem., Int. Ed., 2008, 47, 8251 CrossRef CAS PubMed; (b) A. Flemming and M. Köckerling, Angew. Chem., Int. Ed., 2009, 48, 2605 CrossRef CAS PubMed; (c) D.-Y. Du, J.-S. Qin, S.-L. Li, Z.-M. Su and Y.-Q. Lan, Chem. Soc. Rev., 2014, 43, 4615 RSC.
- (a) L. Jin, X.-X. Li, Y.-J. Qi, P.-P. Niu and S.-T. Zheng, Angew. Chem., Int. Ed., 2016, 55, 13793 CrossRef CAS PubMed; (b) P. Huang, C. Qin, Z.-M. Su, Y. Xing, X.-L. Wang, K.-Z. Shao, Y.-Q. Lan and E.-B. Wang, J. Am. Chem. Soc., 2012, 134, 14004 CrossRef CAS PubMed; (c) J.-Q. Shen, Q. Wu, Y. Zhang, Z.-M. Zhang, Y.-G. Li, Y. Lu and E.-B. Wang, Chem.–Eur. J., 2014, 20, 2840 CrossRef CAS PubMed; (d) D. Zhang, F. Cao, P. Ma, C. Zhang, Y. Song, Z. Liang, X. Hu, J. Wang and J. Niu, Chem.–Eur. J., 2015, 21, 17683 CrossRef CAS PubMed.
- M. Nyman, Dalton Trans., 2011, 40, 8049 RSC.
- (a) M. Nyman, F. Bonhomme, T. M. Alam, J. B. Parise and G. M. B. Vaughan, Angew. Chem., Int. Ed., 2004, 43, 2787 CrossRef CAS PubMed; (b) M. Nyman, F. Bonhomme, T. M. Alam, M. A. Rodriguez, B. R. Cherry, J. L. Krumhansl, T. M. Nenoff and A. M. Sattler, Science, 2002, 297, 996 CrossRef CAS PubMed; (c) W. Guo, H. Lv, K. P. Sullivan, W. O. Gordon, A. Balboa, G. W. Wagner, D. G. Musaev, J. Bacsa and C. L. Hill, Angew. Chem., Int. Ed., 2016, 55, 7403 CrossRef CAS PubMed; (d) Z. Zhang, Q. Lin, D. Kurunthu, T. Wu, F. Zuo, S.-T. Zheng, C. J. Bardeen, X. Bu and P. Feng, J. Am. Chem. Soc., 2011, 133, 6934 CrossRef CAS PubMed; (e) Y. Hou, M. Nyman and M. A. Rodriguez, Angew. Chem., Int. Ed., 2011, 50, 12514 CrossRef CAS PubMed.
- (a) E. J. Graeber and B. Morosin, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 2137 Search PubMed; (b) M. Maekawa, Y. Ozawa and A. Yagasaki, Inorg. Chem., 2006, 45, 9608 CrossRef CAS PubMed; (c) R. P. Bontchev and M. Nyman, Angew. Chem., Int. Ed., 2006, 45, 6670 CrossRef CAS PubMed; (d) R. Tsunashima, D.-L. Long, H. N. Miras, D. Gabb, C. P. Pradeep and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 113 CrossRef CAS PubMed.
- (a) M. Nyman, L. J. Criscenti, F. Bonhomme, M. A. Rodriguez and R. T. Cygana, J. Solid State Chem., 2003, 176, 111 CrossRef CAS; (b) C. A. Ohlin, E. M. Villa and J. C. Fettinger, Dalton Trans., 2009, 15, 2677 RSC; (c) C. A. Ohlin, E. M. Villa, J. C. Fettinger and W. H. Casey, Angew. Chem., Int. Ed., 2008, 47, 5634 CrossRef CAS PubMed.
- (a) G. Guo, Y. Xu, J. Cao and C. Hu, Chem. Commun., 2011, 47, 9411 RSC; (b) G. Guo, Y. Xu, J. Cao and C. Hu, Chem.–Eur. J., 2012, 18, 3493 CrossRef CAS PubMed; (c) P. Huang, C. Qin, X.-L. Wang, C.-Y. Sun, G.-S. Yang, K.-Z. Shao, Y.-Q. Jiao, K. Zhou and Z.-M. Su, Chem. Commun., 2012, 48, 103 RSC; (d) J.-H. Son, C. A. Ohlin, R. L. Johnson, P. Yu and W. H. Casey, Chem.–Eur. J., 2013, 19, 5191 CrossRef CAS PubMed; (e) J.-Q. Shen, Q. Wu, Y. Zhang, Z.-M. Zhang, Y.-G. Li, Y. Lu and E.-B. Wang, Chem.–Eur. J., 2014, 20, 2840 CrossRef CAS PubMed; (f) J.-Q. Shen, Y. Zhang, Z.-M. Zhang, Y.-G. Li, Y.-Q. Gao and E.-B. Wang, Chem. Commun., 2014, 50, 6017 RSC; (g) P. Huang, E.-L. Zhou, X.-L. Wang, C.-Y. Sun, H.-N. Wang, Y. Xing, K.-Z. Shao and Z.-M. Su, CrystEngComm, 2014, 16, 9582 RSC; (h) Y.-T. Zhang, C. Qin, X.-L. Wang, P. Huang, B.-Q. Song, K.-Z. Shao and Z.-M. Su, Inorg. Chem., 2015, 54, 11083 CrossRef CAS PubMed.
- (a) J. Niu, P. Ma, H. Niu, J. Li, J. Zhao, Y. Song and J. Wang, Chem.–Eur. J., 2007, 13, 8739 CrossRef CAS PubMed; (b) J.-Y. Niu, G. Chen, J.-W. Zhao, P.-T. Ma, S.-Z. Li, J.-P. Wang, M.-X. Li, Y. Bai and B.-S. Ji, Chem.–Eur. J., 2010, 16, 7082 CrossRef CAS PubMed.
- (a) J. Niu, F. Li, J. Zhao, P. Ma, D. Zhang, B. Bassil, U. Kortz and J. Wang, Chem.–Eur. J., 2014, 20, 9852 CrossRef CAS PubMed; (b) Z. Liang, D. Zhang, P. Ma, J. Niu and J. Wang, Chem.–Eur. J., 2015, 21, 8380 CrossRef CAS PubMed.
- (a) C. M. Flynn Jr and G. D. Stucky, Inorg. Chem., 1969, 8, 332 CrossRef; (b) J.-H. Son, C. A. Ohlin and W. H. Casey, Dalton Trans., 2013, 42, 7529 RSC; (c) Z. Liang, D. Zhang, H. Wang, P. Ma, Z. Yang, J. Niu and J. Wang, Dalton Trans., 2016, 45, 16173 RSC.
- (a) P. Liu, H. Wang, Z. Feng, P. Ying and C. Li, J. Catal., 2008, 256, 345 CrossRef CAS; (b) W. Qi, Y. Wang, W. Li and L. Wu, Chem.–Eur. J., 2010, 16, 1068 CrossRef CAS PubMed; (c) A. Wróblewska, E. Ławro and E. Milchert, Ind. Eng. Chem. Res., 2006, 45, 7365 CrossRef; (d) A. Wróblewska, Appl. Catal., A, 2006, 309, 192 CrossRef; (e) A. Wróblewska and E. Makuch, React. Kinet. Catal. Lett., 2012, 105, 451 CrossRef; (f) K. Kamata, T. Hirano, S. Kuzuya and N. Mizuno, J. Am. Chem. Soc., 2009, 131, 6997 CrossRef CAS PubMed.
- (a) Z.-M. Zhang, Y.-G. Li, S. Yao, E.-B. Wang, Y.-H. Wang and R. Clérac, Angew. Chem., Int. Ed., 2009, 48, 1581 CrossRef CAS PubMed; (b) J. Niu, G. Wang, J. Zhao, Y. Sui, P. Ma and J. Wang, Cryst. Growth Des., 2011, 11, 1253 CrossRef CAS.
- J. Fuchs, H. Hartl, W. Schiller and U. Gerlach, Acta Crystallogr., Sect. B, 1976, B32, 740 CrossRef CAS.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- P. Ma, G. Chen, G. Wang and J. Wang, Russ. J. Coord. Chem., 2011, 37, 772 CrossRef CAS.
- (a) M. N. Sokolov, S. A. Adonin, D. A. Mainichev, C. Vicent, N. F. Zakharchuk, A. M. Danilenko and V. P. Fedin, Chem. Commun., 2011, 47, 7833 RSC; (b) M. N. Corella-Ochoa, H. N. Miras, A. Kidd, D.-L. Long and L. Cronin, Chem. Commun., 2011, 47, 8799 RSC.
- (a) L. Yang, X. Ma, P. Ma, J. Hua and J. Niu, Cryst. Growth Des., 2013, 13, 2982 CrossRef CAS; (b) W. Huang, D. Wu, P. Zhou, W. Yan, D. Guo, C. Duan and Q. Meng, Cryst. Growth Des., 2009, 9, 1361 CrossRef CAS.
- H. Hou, G. Li, L. Li, Y. Zhu, X. Meng and Y. Fan, Inorg. Chem., 2003, 42, 428 CrossRef CAS PubMed.
- W. Zhao, C. Yang, Z. Cheng and Z. Zhang, Green Chem., 2016, 18, 995 RSC.
- (a) L. Huang, S.-S. Wang, J.-W. Zhao, L. Cheng and G.-Y. Yang, J. Am. Chem. Soc., 2014, 136, 7637 CrossRef CAS PubMed; (b) Y. Huo, Z. Huo, P. Ma, J. Wang and J. Niu, Inorg. Chem., 2015, 54, 406 CrossRef CAS PubMed.
- W. F. Brill, J. Am. Chem. Soc., 1963, 85, 141 CrossRef CAS.
- M. Filowitz, R. K. C. Ho, W. G. Klemperer and W. Shum, Inorg. Chem., 1979, 18, 93 CrossRef CAS.
- (a) G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Bond valence sum calculations of O and Ni (Tables S1 and S2), additional structural figures (Fig. S1 and S2), additional measurements (Fig. S3–S8, Tables S3 and S4), catalytic properties (Fig. S9, S10, Tables S5 and S6). CCDC 1526285 for 1. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra03254d |
| This journal is © The Royal Society of Chemistry 2017 |