
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMono- and double carbonylation of aryl iodides with amine nucleophiles in the presence of recyclable palladium catalysts immobilised on a supported dicationic ionic liquid phase†
M. Pappa,
P. Szabó‡
b,
D. Srankóc,
G. Sáfránd,
L. Kolláre and
R. Skoda-Földes *a
*a
aUniversity of Pannonia, Institute of Chemistry, Department of Organic Chemistry, P. O. Box 158, H-8201 Veszprém, Hungary. E-mail: skodane@almos.uni-pannon.hu; Fax: +36-88-624469; Tel: +36-88-624719
bUniversity of Pannonia, Department of Analytical Chemistry, Hungary
cHungarian Academy of Sciences, Centre for Energy Research, Department of Surface Chemistry and Catalysis, P. O. Box 49, H-1525 Budapest 114, Hungary
dHungarian Academy of Sciences, Research Institute for Technical Physics and Materials Science, P. O. Box 49, H-1525 Budapest, Hungary
eUniversity of Pécs, Department of Inorganic Chemistry, MTA-PTE Research Group for Selective Chemical Syntheses, Ifjúság u. 6. (P. O. Box 266), H-7624 Pécs, Hungary
First published on 15th September 2017
Abstract
Silica modified with organic dicationic moieties proved to be an excellent support for palladium catalysts used in the aminocarbonylation of aryl iodides. By an appropriate choice of the reaction conditions, the same catalyst could be used for selective mono- or double carbonylations leading to amide and α-ketoamide products, respectively. The best catalyst could be recycled for at least 10 consecutive runs with a loss of palladium below the detection limit. By the application of the new support, efficient catalyst recycling could be achieved under mild reaction conditions (under low pressure and in a short reaction time). Palladium-leaching data support a mechanism with dissolution—re-precipitation of the active palladium species.
Introduction
Palladium-catalysed aminocarbonylation involves the reaction of aryl/alkenyl halides (or halide equivalents) with amines using carbon monoxide as carbonyl source.1–5 The process leads to amides and α-ketoamides via mono- and double carbonylation, respectively. Both the amide and α-ketoamide functionalities are important motifs in a great variety of natural and non-natural biologically active compounds.6,7 Also, α-ketoamides can be used as starting material in Mannich-type reactions and in the synthesis of heterocycles.7In the past few years, there is an increasing interest in the development of new, heterogeneous catalysts in order to reduce the metal content of the products and to make catalyst recycling possible. There is an intense ongoing research aiming at the synthesis of carboxamides via monocarbonylation reactions. Some recent examples include the use of palladium nanoparticles dispersed on metal–organic8 or zeolitic imidazole frameworks,9,10 carbon nanotubes11 or on a siliceous mesocellular foam.12 Several carboxamides were synthesised using palladium complexes, such as palladium–phosphine,13 palladium–Schiff base14 or palladium–bisoxazoline15 derivatives anchored on polymers14,15 or silica,13 as well as in the presence of a palladium-1,10-phenanthroline complex encaged in Y zeolite.16 Palladium immobilised on a silica support with grafted imidazolium ions was also shown to be suitable catalyst by ourselves17 and others.18 Although some catalysts showed good activity even under atmospheric conditions,8,12,17 effective recycling could usually be achieved using 2–20 bar CO pressure.
Much fewer examples have been reported for heterogeneous double carbonylation reactions. A silica-supported polytitazane–palladium (Ti–N–Pd) compound,19 a Pd/C + PPh3 catalyst system20 and palladium–phosphine complexes grafted onto mesoporous silica (SBA-15)21 were proved to be selective catalysts. In recent studies, heterogeneous double carbonylation was carried out in the presence of palladium nanoparticles that were supported on a cross-linked functional polymer22 or on a triazine framework.23 With the exception of the last example,23 when atmospheric conditions were used, selective double carbonylation was achieved at high pressure (30–40 bar).19–22
Beside CO pressure, the selectivity of the aminocarbonylation is affected by a number of other parameters, such as temperature, solvent and added base, as well as by the structure of the reaction partners. Under homogeneous conditions, it is also possible to use the same catalyst either for mono- or double carbonylation by a simple change in the temperature of the reaction.24
In case of supported catalysts, there are only two reports demonstrating the applicability of the same catalysts in the selective production of either amides or ketoamides: with palladium immobilised on a mesoporous poly-melamine-formaldehyde material25 or on silica functionalised with imidazolium ions.26 Although both catalysts showed good recyclability, relatively long reaction time (8–12 h) had to be used to achieve total conversion25 or to ensure effective reuse.26,27 There are no data for metal leaching in case of the polymer supported catalyst,25 but in the other case, an average of 2–4% loss of the original load of palladium was still observed in recycling studies.26,27
Di- and tricationic ionic liquids are known to have some advantageous properties compared to the monocationic versions, such as better thermal properties, lower miscibility with conventional organic solvents and more efficient stabilisation of transition metals.28 The introduction of imidazolium functionalities into polymeric supports, providing multiple ionic liquid-like structures, was found to enhance catalyst stability in Heck coupling.29 Supported palladium–carbene complexes obtained from dicationic ionic liquids also showed good performance in the same reaction.30 Dicationic triazolium salts anchored on halloysite was found to be useful for immobilisation of a palladium catalyst for a Suzuki coupling.31
At the same time, to the best of our knowledge, there is no example for the use of silica-grafted dicationic ionic liquids as supports in carbonylation reactions.
In the present paper we report the results of mono- and double carbonylation reactions of iodoarenes with amines as nucleophiles in the presence of the catalysts mentioned above.
Results and discussion
Preparation and characterisation of the catalysts
The dicationic ionic liquid phase 3 was prepared as shown in Scheme 1. Silica gel was treated with 3-aminopropyltriethoxysilane. The resulting aminopropyl silica (1) was reacted with glyoxal, formaldehyde and NH4Cl to construct the imidazole ring.32 Then, modified silica 2 was refluxed in the presence of 1-(2-bromoethyl)-3-methylimidazolium bromide33 in acetonitrile for 5 days. According to the weight increase in the last step, 55% of the imidazole groups were converted into the corresponding bis(imidazolium) bromide.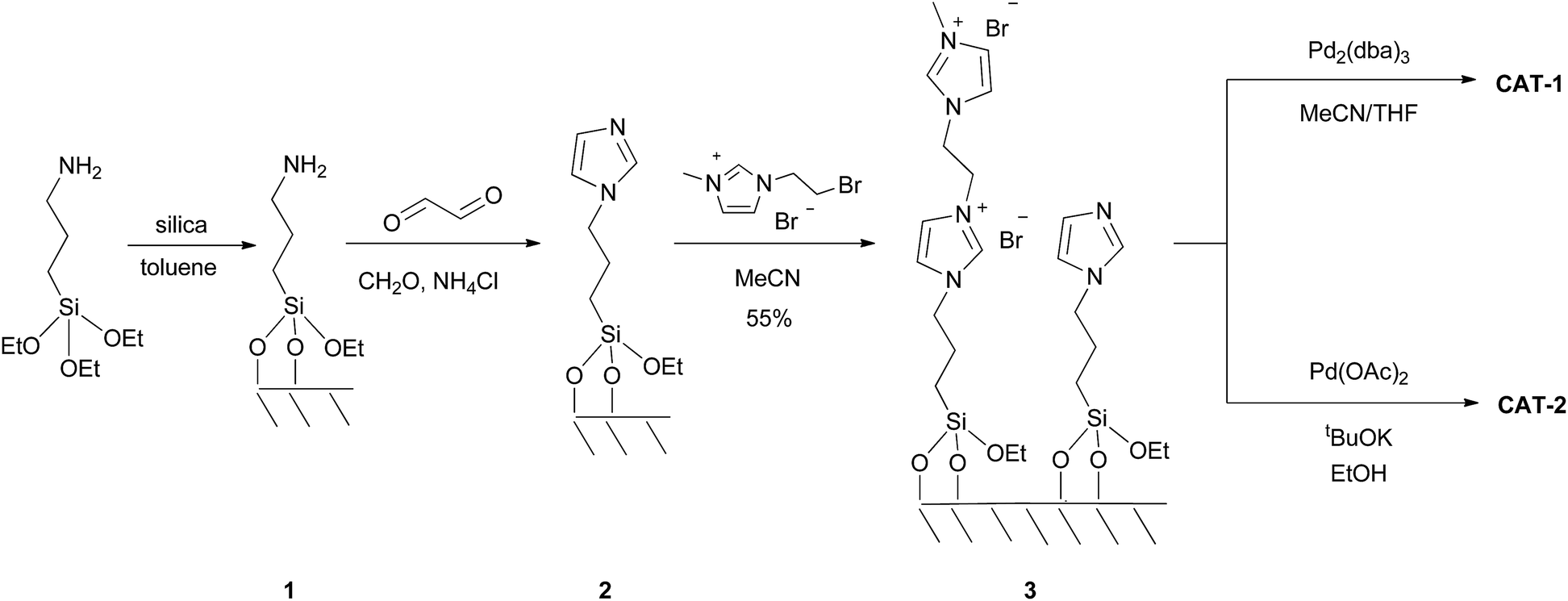 | ||
| Scheme 1 Preparation of SILP catalysts. | ||
The procedure was followed by FT-IR and solid phase NMR measurements. In the FT-IR spectrum of 1 the appearance of N–H stretching (3292 and 3362 cm−1) and N–H bending (1560 cm−1) vibrations34 showed the presence of the amine functionality on the surface of silica (Fig. 1). The disappearance of N–H stretching vibrations and the new bands at 1565, 1515, 1450 and 1410 cm−1,35 attributed to the imidazole ring, are in accordance with the formation of modified silica 2. The vibrations of the imidazole ring became stronger in the FT-IR spectrum of 3. 13C CP MAS NMR spectra of the solid materials also supported the formation of structures 2 and 3 (Fig. 2). The signals at 124 and 136 ppm in the spectrum of compound 2 proved the formation of the heterocycle. The new signals at 37 and 43 ppm were assigned to the methyl group and the methylene linkers between the two imidazole rings,36 respectively, in case of dicationic phase 3. In the 29Si CP MAS NMR spectrum of 3, the peaks around −60 ppm show the presence of organosiloxane moieties (Fig. S1†) that confirms the formation of covalent bonds between the ionic liquid and silica.37
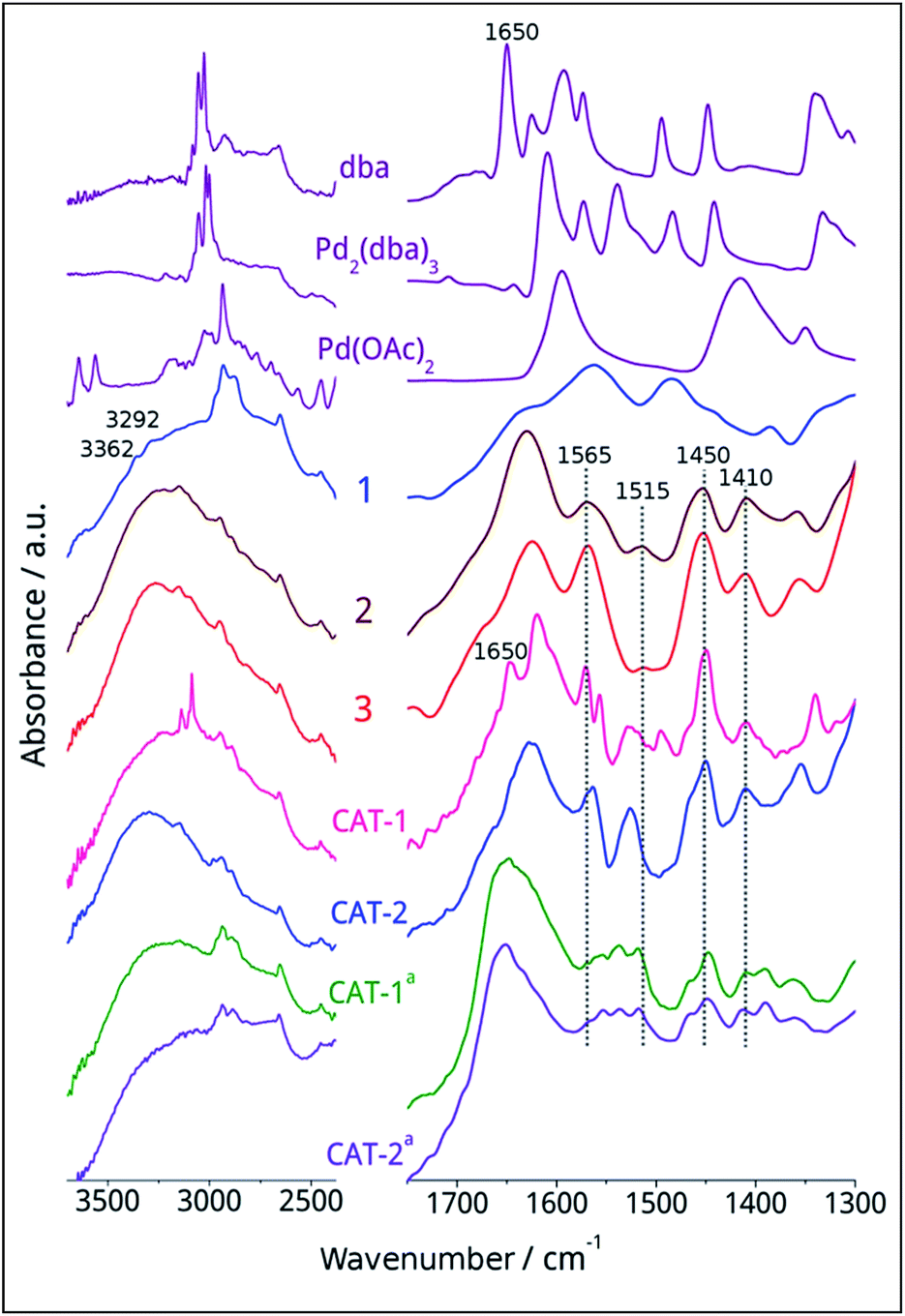 | ||
| Fig. 1 FT-IR spectra of ligand, Pd precursors, supported phases 1, 2 and 3, fresh and spent catalysts (aspent catalyst in the aminocarbonylation reaction of iodobenzene (4a) with morpholine (5a) in DMF). | ||
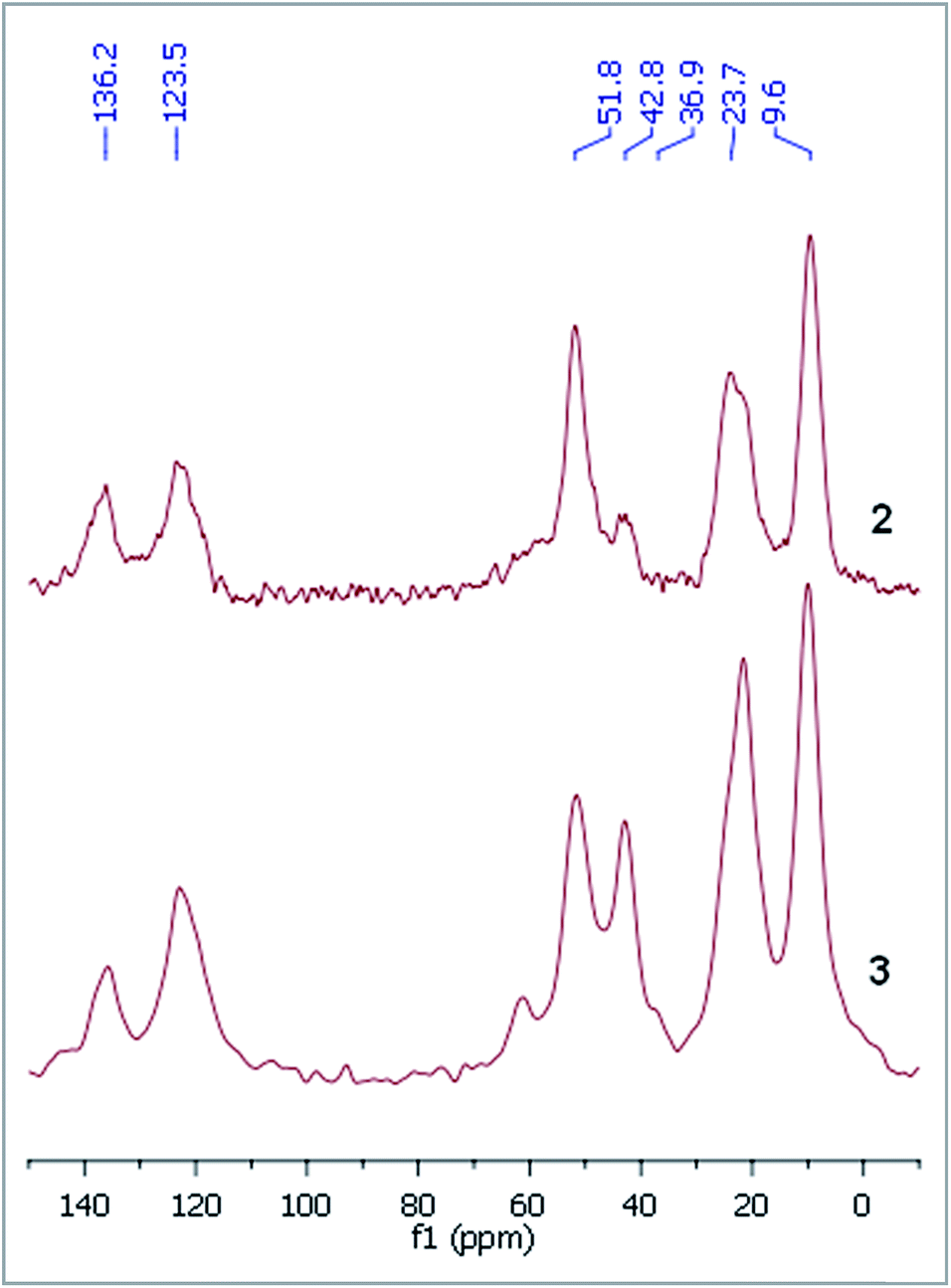 | ||
| Fig. 2 13C CP MAS NMR spectrum of 2 and 3. | ||
The catalysts were obtained from the SILP phase and Pd2(dba)3 (ref. 27) or Pd(OAc)2 precursors17 as reported previously. The palladium content of the catalysts was determined by ICP-AES (Table 1).
TEM measurements (Fig. S2†) proved the formation of palladium nanoparticles in both cases with an average diameter of 6.6 ± 0.9 nm (CAT-1) and 4.6 ± 0.8 nm (CAT-2).
The catalysts were investigated by X-ray photoelectron spectroscopy (XPS) analysis (Table 2). Bands at 335.0 eV and 340.3 eV were assigned to the Pd0 (3d5/2 and 3d3/2 respectively), and the ones at around 337.0 eV and 342.3 eV to Pd(II) (3d5/2 and 3d3/2 respectively). These results are typical for Pd(II) in similar conditions.17,26,38–40 The ratios for the Pd, Si and C are presented in the Table 2.
| Sample | Position of the peak Pd 3d5/2 | Ratio of the atomic concentrations | |||
|---|---|---|---|---|---|
| Pd0 (eV) | Pd(II) (eV) | Pd(II)/Pd0 | Pd/Si | Pd/C | |
| CAT-1 fresh | 335.0 | 336.9 | 5.72 × 10−1 | 5.79 × 10−2 | 3.87 × 10−2 |
| CAT-1 spent | 335.4 | 337.4 | 1.77 × 10−1 | 5.81 × 10−2 | 5.83 × 10−2 |
| CAT-2 fresh | 335.1 | 336.9 | 2.32 × 10−1 | 4.00 × 10−2 | 5.12 × 10−2 |
| CAT-2 spent | 335.3 | 337.1 | 4.60 × 10−1 | 5.21 × 10−2 | 5.70 × 10−2 |
Interestingly, Pd(II)/Pd0 ratio is a bit higher for CAT-1 that might be attributed to an oxidative addition of the surface organic moieties to Pd0 accompanied by deliberation of dba that can be observed also by FTIR measurements (Fig. 1). In case of CAT-2, the low Pd(II) content is probably a result of the reducing ability of the ethanol solvent used during immobilisation.
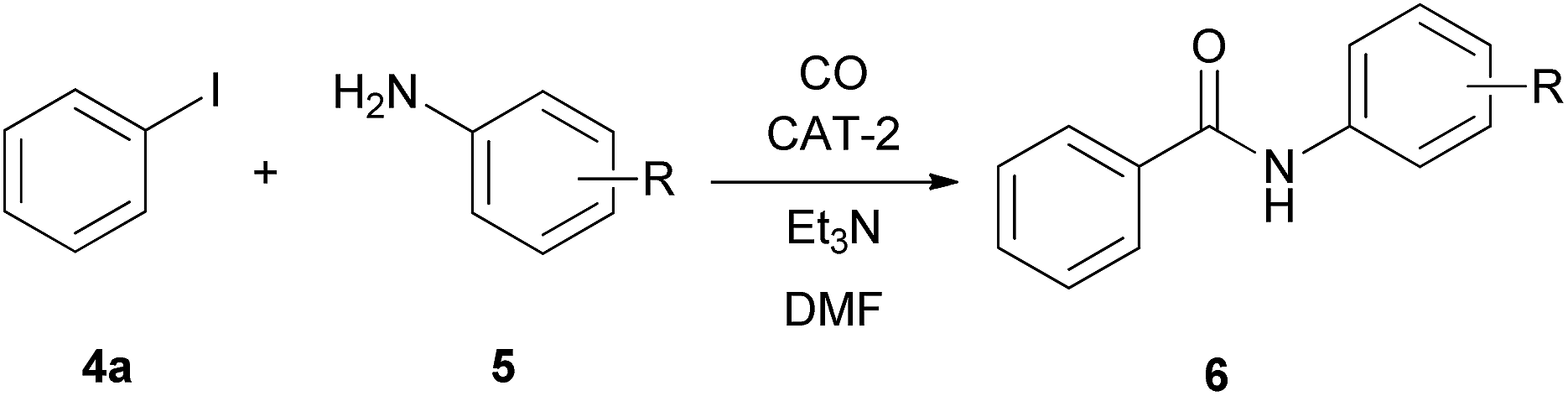
Aminocarbonylation of iodobenzene (4a) with morpholine (5a) in the presence of catalysts CAT-1 and CAT-2
The aminocarbonylation of iodobenzene (4a, Scheme 2) with morpholine (5a) as nucleophile was used as the model reaction. First, carbonylation was carried out at atmospheric pressure (Table 3, entries 1, 7). Full conversion of iodobenzene could not be achieved even after 8 hours and the selectivity for 7a was low. At the same time, under 30 bar pressure of CO iodobenzene was totally consumed after 3 hours, and α-ketoamide 7a was formed with excellent selectivity in the presence of both catalysts, CAT-1 and CAT-2 (Table 3, entries 2, 11). As a comparison, 88% conversion was observed after 3 hours in the presence of the monocationic SILP catalyst reported earlier.27 Aminocarbonylation in less polar solvents led to lower conversion of iodobenzene (4a) (Table 3, entries 3, 4). In toluene the monocarbonylated 6a was formed as the main product in the presence of triethylamine as the base. (Formation of the α-ketoamide is usually favoured in more polar solvents, as double carbonylation is thought to proceed via ionic intermediates. At the same time, the participation of neutral complexes is supposed in the catalytic cycle of monocarbonylation41) after changing the base to DBU, 7a was obtained selectively, and full conversion of 4a was achieved after 8 hours (Table 3, entries 5, 6). CAT-2 catalyst was tested under 5, 10 and 20 bar CO pressure and with the exception of the reaction at 5 bar, iodobenzene was totally consumed and good to excellent selectivity towards α-ketoamide 7a was observed (Table 3, entries 8–10).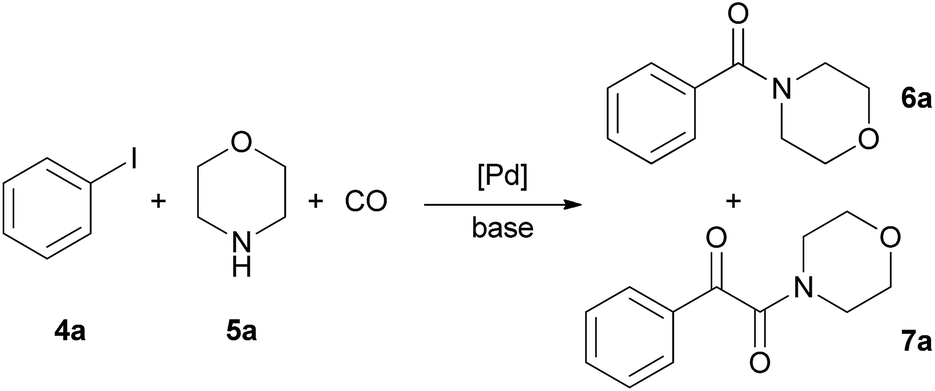 | ||
| Scheme 2 Aminocarbonylation of iodobenzene with morpholine. | ||
| Entry | Catalyst | CO pressure [bar] | R. time [h] | Solvent | Base | Conversionb [%] | Ratio of 6a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7ab :7ab |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 0.2 mmol 4a, 0.5 mmol 5a, 0.25 mmol base, 1.0 ml solvent, catalyst (2.8 μmol Pd, 1.4 mol%), 100 °C.b Determined by GC. | |||||||
| 1 | CAT-1 | 1 | 8 | DMF | Et3N | 74 | 76:24 |
| 2 | CAT-1 | 30 | 3 | DMF | Et3N | 100 | 5:95 |
| 3 | CAT-1 | 30 | 3 | Acetonitrile | Et3N | 88 | 5:95 |
| 4 | CAT-1 | 30 | 3 | Toluene | Et3N | 53 | 72:28 |
| 5 | CAT-1 | 30 | 3 | Toluene | DBU | 90 | 13:87 |
| 6 | CAT-1 | 30 | 8 | Toluene | DBU | 100 | 11:89 |
| 7 | CAT-2 | 1 | 8 | DMF | Et3N | 31 | 78:22 |
| 8 | CAT-2 | 5 | 3 | DMF | Et3N | 79 | 35:65 |
| 9 | CAT-2 | 10 | 3 | DMF | Et3N | 100 | 14:86 |
| 10 | CAT-2 | 20 | 3 | DMF | Et3N | 100 | 5:95 |
| 11 | CAT-2 | 30 | 3 | DMF | Et3N | 100 | 3:97 |
Recirculation experiments and palladium leaching
A main advantage of heterogeneous catalysts is the possibility of catalyst reuse. Accordingly, recycling experiments were carried out under different conditions with catalysts CAT-1 and CAT-2. Atmospheric carbonylation of 4a with morpholine reagent (5a) in the presence of CAT-1 led to 74%, 40% and 26% conversion in three successive runs. Then, the recyclability of this catalyst in different solvents was compared at 30 bar CO pressure (Fig. 3). In acetonitrile it could be reused with a small loss of activity, while in DMF (using triethylamine as base) and in toluene (using DBU as base) it could be recycled in 5 runs without significant change in the catalytic activity (Fig. 3, Table 4). The catalyst was also reused in further 5 runs and a drop in the activity was observed only after the 8th run in toluene (Table S1†).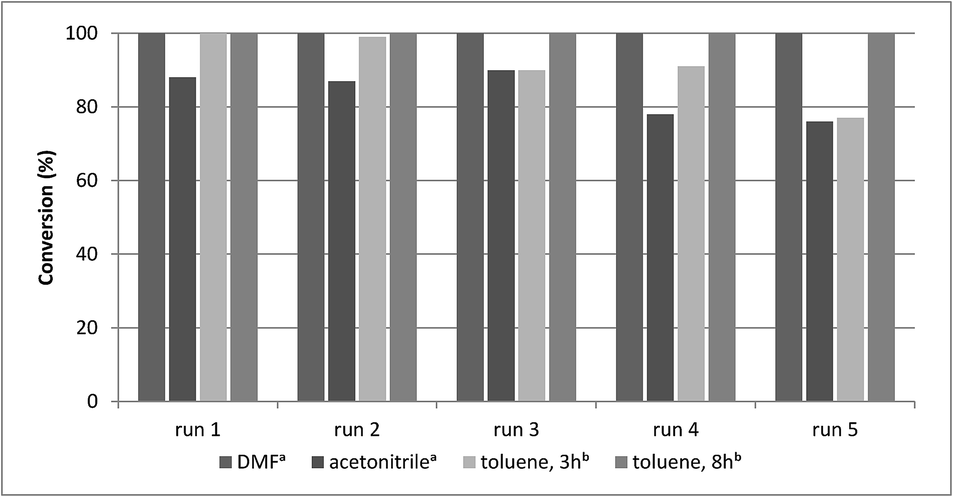 | ||
| Fig. 3 Recycling experiments with CAT-1 (0.2 mmol 4a, 0.5 mmol 5a, 0.25 mmol base, 1.0 ml solvent, CAT-1 (1.4 mol% Pd), 100 °C, conversion determined by GC. aWith Et3N. bWith DBU). | ||
| Entry | CO pressure [bar] | R. time [h] | Solvent | Base | Selectivity for 7ab | ||||
|---|---|---|---|---|---|---|---|---|---|
| Run 1 | Run 2 | Run 3 | Run 4 | Run 5 | |||||
| a Reaction conditions: 0.2 mmol 4a, 0.5 mmol 5a, 0.25 mmol base, 1.0 ml solvent, CAT-1 (2.8 μmol Pd, 1.4 mol%), 100 °C.b Determined by GC. | |||||||||
| 1 | 30 | 3 | DMF | Et3N | 95 | 94 | 97 | 98 | 96 |
| 2 | 1 | 8 | DMF | Et3N | 76 | 61 | 50 | n.d. | n.d. |
| 3 | 30 | 3 | Acetonitrile | Et3N | 95 | 94 | 95 | 96 | 98 |
| 4 | 30 | 3 | Toluene | Et3N | 28 | 29 | 26 | 27 | 30 |
| 5 | 30 | 3 | Toluene | DBU | 87 | 87 | 86 | 84 | 85 |
| 6 | 30 | 8 | Toluene | DBU | 89 | 91 | 88 | 89 | 87 |
In DMF, better results were obtained during the recycling experiments with CAT-2 catalyst (Fig. 4) than with CAT-1, surpassing even the efficiency of the CAT-1/toluene/DBU system (Fig. 3, Table S1,† entry 6). The effect of recycling on the catalytic features of the CAT-2 catalyst was studied under 5, 10, 20 and 30 bar CO pressure, and the pressure could be decreased to 20 bar without a significant loss in activity and selectivity (Fig. 4, Table S3†).
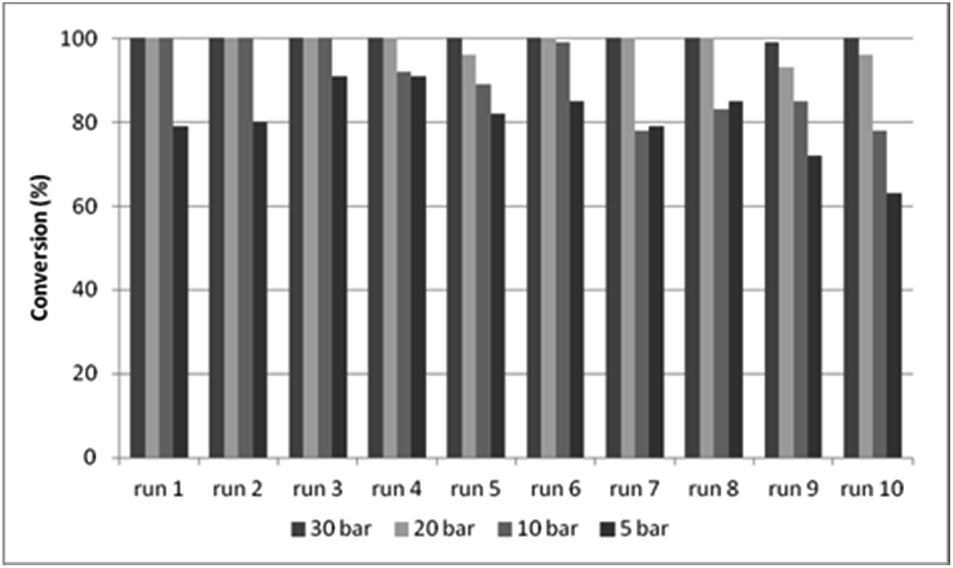 | ||
| Fig. 4 Recycling experiments with CAT-2 under different CO pressures (0.2 mmol 4a, 0.5 mmol 5a, 0.25 mmol Et3N, 1.0 ml DMF, CAT-2 (1.4 mol% Pd), 100 °C, 3 h, conversion determined by GC). | ||
The FT-IR spectra of fresh and spent catalysts were compared (Fig. 1). The FT-IR spectrum of fresh CAT-1 showed the immobilisation of Pd2(dba)3 on the surface and the presence of some free dba (1650 cm−1) could be observed. In case of CAT-2, no stretching vibrations of the carboxylate moiety of the AcO− ion could be detected even in the fresh catalyst. The IR spectra of spent CAT-1 and CAT-2 were very similar, preserving the bands attributed to the imidazolium cations at 1565, 1515, 1450 and 1410 cm−1. (In the case of spent catalysts, the DMF solvent could not be removed completely26 and the signal at 1660 cm−1 can be assigned to the amide band).
According to XPS measurements (Table 2), the Pd contents were similar for both of the samples before and after the catalytic cycle. At the same time, Pd(II)/Pd0 ratio slightly differs – in the case of CAT-1 Pd0 is enriched on the surface upon the catalytic reaction, while in CAT-2 the amount of Pd(II) increased. The catalytic cycle has no major effect on the Pd/Si and Pd/C ratios. Small amount of bromine can be detected on the “fresh” but not on the “spent” catalysts. However the lines appeared at ∼620.0 eV are assigned to I 3d and therefore to the presence of iodine (∼1 at%)42 after the catalytic cycle. This can be attributed to a Br → I exchange with the ammonium salt formed from Et3N base and deliberated HI. Nitrogen content was detected at peak energies that are typical for an organic environment. The peak at ∼400.0 eV is assigned to the N 1s in organic nitrogen species.42
TEM measurements (Fig. S2†) showed no aggregation of Pd nanoparticles after the reaction (average diameter 7.0 ± 1.3 nm (CAT-1) and 3.8 ± 0.4 nm (CAT-2)).
The amount of palladium loss during the aminocarbonylation reaction was determined by ICP measurements. In the reactions with CAT-1, a relatively high palladium leaching was detected in DMF, with a loss of 4.9% and 5.9% of the original load in the first two runs, respectively (Table 5, entry 1). At the same time, the use of toluene as solvent (entry 2) or CAT-2 as catalyst (entries 3, 4) led to a considerable decrease in palladium leaching that remained below the detection limit.
| Entry | Catalyst | CO pressure [bar] | R. time [h] | Solvent | Base | Pd leachingb [%] | |
|---|---|---|---|---|---|---|---|
| Run 1 | Run 2 | ||||||
| a Reaction conditions: 0.2 mmol 4a, 0.5 mmol 5a, 0.25 mmol base, 1.0 ml solvent, catalyst (2.8 μmol Pd, 1.4 mol%), 100 °C.b Determined by ICP.c Below the detection limit.d 120 °C. | |||||||
| 1 | CAT-1 | 30 | 3 | DMF | Et3N | 4.9 | 5.9 |
| 2 | CAT-1 | 30 | 8 | Toluene | DBU | <1.0c | <1.1c |
| 3 | CAT-2 | 30 | 3 | DMF | Et3N | <1.3c | <1.6c |
| 4 | CAT-2 | 20 | 3 | DMF | Et3N | <1.3c | <1.3c |
| 5d | CAT-2 | 5 | 3 | — | DABCO | <2.3c | <2.2c |
To get some information about the homogeneous or heterogeneous nature of the catalytic reaction, hot filtration and mercury poisoning tests were carried out with the catalysts (Table 6). In the first experiments (entries 1, 2) the reaction mixture was filtered after a half an hour. The filtrate was divided into 2 portions, (a small sample was analysed by GC) and one half of the mixture was heated for 3 hours at 20 or 30 bar. To the second portion a small amount of mercury was added and it was heated under similar conditions.
| Entry | Catalyst | Solvent | Pressure (bar) | Temp. (°C) | First step | Second step | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R. time | Conv. | Ratio of 6a:7ab |
Hg | R. time | Conv. | Ratio of 6a:7ab |
|||||
| a Reaction conditions: 0.2 mmol 4a, 0.5 mmol 5a, 0.25 mmol Et3N, 1.0 ml DMF, catalyst (2.8 μmol Pd, 1.4 mol%).b Determined by GC.c After filtration fresh reagents were added to the filtrate (0.2 mmol 4a, 0.5 mmol 5a, 0.25 mmol Et3N).d Solvent-free reaction (0.8 mmol 4a, 2.0 mmol 5a, 1.0 mmol DABCO, catalyst with 5.4 μmol Pd, 0.7 mol%).e Solvent-free reaction (0.4 mmol 4a, 1.0 mmol 5a, 0.5 mmol DABCO, catalyst with 2.7 μmol Pd, 0.7 mol%) it was extracted with toluene and the extract was heated in the second step.f After filtration fresh reagents were added to the toluene extract (0.4 mmol 4a, 1.0 mmol 5a, 0.5 mmol DABCO). | |||||||||||
| 1 | CAT-1 | DMF | 30 | 100 | 0.5 | 49 | 7:93 |
− | 3 | 80 | 6:94 |
| + | 3 | 55 | 7:93 |
||||||||
| 2 | CAT-2 | DMF | 20 | 100 | 0.5 | 38 | 3:97 |
− | 3 | 80 | 5:95 |
| + | 3 | 44 | 5:95 |
||||||||
| 3 | CAT-2 | DMF | 20 | 100 | 3 | 100 | 6:94 |
− | 3c | 50 | 6:94 |
| + | 3c | 50 | 6:94 |
||||||||
| 4 | CAT-2 | —d | 5 | 120 | 0.5 | 43 | 98:2 |
− | 3 | 99 | 85:15 |
| + | 3 | 75 | 84:16 |
||||||||
| 5 | CAT-2 | —e | 5 | 120 | 3 | 100 | 99:1 |
− | 3f | 49 | 99:1 |
| + | 3f | 49 | 99:1 |
||||||||
Some iodobenzene (4a) was consumed after the removal of the heterogeneous phases (Table 6, entries 1, 2) but the reaction was slower compared to the usual catalytic experiments (Table 3, entries 2, 10). Addition of mercury slowed down the reaction considerably, so it can be concluded that the catalytically active palladium species, leached into the mixture, are mainly nanoparticles. It should be mentioned that despite the relatively high conversion observed after the removal of the heterogeneous phase, the palladium leaching was below the detection limit (<30 ppm) after a 3 hour-long reaction in the presence of catalyst CAT-2 (Table 5, entry 3). It is supposed, that after the completion of the reaction the leached palladium species re-precipitate onto the solid phase.
To prove this, a modified hot filtration test was carried out (Table 6, entry 3). The reaction mixture was filtered after 3 hours (after 100% conversion) and fresh reagents were added to the filtrate. One half of the resulting mixture was heated under 20 bar CO pressure at 100 °C for 3 hours and to the second half, a small amount of mercury was added before heating (Table 6, entry 3). GC analysis of both of the reaction mixtures showed the presence of 50% iodobenzene and 50% of carbonylation products that means that no conversion of iodobenzene took place in the filtrate. After the removal of the solid phase, the reaction completely stopped proving the dissolution—re-precipitation theory.43
Double carbonylation with other nucleophiles/substrates
Based on the results discussed above, catalyst CAT-2 was chosen for aminocarbonylations carried out with other substrates. It was found effective in the double carbonylation of iodobenzene with other aliphatic amines (Table 7, entries 1–5) at 20 bar CO pressure in DMF. The catalyst was tested in two consecutive runs and the α-ketoamides were isolated in good to excellent yields.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | 5 | Run 1 | Run 2 | Yield of 7c [%] | ||
| Conv.b [%] | Ratio of 6:7b |
Conv.b [%] | Ratio of 6:7b |
||||
| a Reaction conditions: 0.2 mmol 4, 0.5 mmol 5, 0.25 mmol Et3N, 1.0 ml DMF, CAT-2 (2.8 μmol Pd, 1.4 mol%), 100 °C, 3 h.b Determined by GC.c Isolated yield from the combined reaction mixtures of the first two runs.d Side products: 8b, 9b and 10b formed with a total yield of 5% and 3% (GC) in the first and 2nd runs, respectively.e Side products: 8c, 9c and 10c formed with a total yield of 19% and 31% (GC) in the first and 2nd runs, respectively.f Side product: morpholino(4-aminophenyl)methanone.g Isolated yield for 6. | |||||||
| 1 | Ph | Morpholine | 100 | 5:95 |
100 | 7:93 |
90 |
| 2 | Ph | Piperidine | 100 | 4:96 |
100 | 3:97 |
91 |
| 3 | Ph | Pyrrolidine | 95 | 14:86 |
98 | 10:90 |
82 |
| 4 | Ph | Diethylamine | 100 | 17:83 |
100 | 24:76 |
71 |
| 5 | 4-CH3O–C6H4 | Morpholine | 100 | 7:93 |
100 | 8:92 |
86 |
| 6 | 3,4-(CH3)2–C6H3 | Morpholine | 100 | 4:96 |
100 | 4:96 |
86 |
| 7 | 1-Naphthyl | Morpholine | 100 | 40:60 |
100 | 34:66 |
48 |
| 8 | 3-F–C6H4 | Morpholine | 100 | 11:89 |
100 | 10:90 |
84 |
| 9 | 4-Cl–C6H4 | Morpholine | 100 | 11:89 |
100 | 7:93 |
85 |
| 10 | 3-Br–C6H4 | Morpholine | 100 | 4:91d |
100 | 2:95d |
50 |
| 11 | 4-Br–C6H4 | Morpholine | 100 | 3:78e |
100 | 3:66e |
61 |
| 12 | 4-NO2–C6H4 | Morpholine | 100 | 48:0f |
100 | 56:0f |
41g |
The double carbonylated products could be obtained in moderate to good yields in the reaction of other aryl iodides and morpholine as well (Table 7, entries 6–13). In the case of fluoro and chloro derivatives only the iodide functionality reacted (entries 9–10). When 3-bromoiodobenzene (entry 11) and 4-bromoiodobenzene (entry 12) were used as substrates, bis-carbonylated side products were formed (Scheme 3). However, no conversion of 1-bromo-4-chlorobenzene, 1-bromo-3-chlorobenzene, 1-bromo-4-fluorobenzene or bromobenzene was observed under identical conditions.
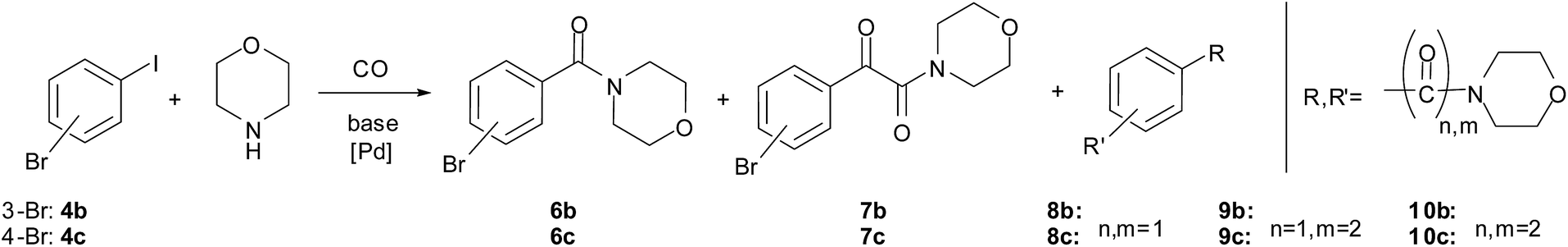 | ||
| Scheme 3 Aminocarbonylation of bromoiodobenzenes with morpholine. | ||
No formation of the corresponding α-ketoamide could be detected by GC-MS in the reaction of 1-iodo-4-nitrobenzene (Table 7, entry 12). Beside the expected amide product, the corresponding 4-amino-substituted amide, morpholino(4-aminophenyl)methanone was formed in a yield of 52% and 44% in the first and 2nd runs, respectively (determined by GC), via the reduction of the nitro group.
In the reaction of iodobenzene with aromatic amines no double carbonylation was observed, the amide product was formed selectively (Table 8). The catalyst was reused 4 times with full conversion of iodobenzene with the exception of p-anisidine (Table 8, entry 4). The corresponding substituted N-phenylbenzamides could be isolated in good to excellent yields.
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Conversion of 4ab [%] | Yield of 6c [%] | |||
| Run 1 | Run 2 | Run 3 | Run 4 | |||
| a Reaction conditions: 0.2 mmol 4, 0.5 mmol 5, 0.25 mmol Et3N, 1.0 ml DMF, CAT-2 (2.8 μmol Pd, 1.4 mol%), 100 °C, 3 h.b Determined by GC.c Isolated yield from the combined reaction mixtures of the first four runs. | ||||||
| 1 | H | 100 | 100 | 100 | 100 | 88 |
| 2 | 4-CH3 | 100 | 100 | 100 | 100 | 91 |
| 3 | 4-nBu | 100 | 100 | 100 | 100 | 92 |
| 4 | 4-CH3O | 100 | 97 | 98 | 100 | 92 |
| 5 | 4-NO2 | 100 | 100 | 100 | 100 | 85 |
Monocarbonylation of iodobenzene with morpholine
After the success of double carbonylations, reaction conditions were optimised to achieve selective formation of amide products even with aliphatic amines as nucleophiles. Reactions were carried out in toluene that usually favours formation of monocarbonylated products (Table 3, entry 4) or in the absence of a solvent. The latter condition had been found to lead to amides in good yields with a similar catalyst immobilised on a solid phase decorated with imidazolium ions.26 In toluene (Table 9, entry 1) the main product was indeed amide 6a, however, the conversion of iodobenzene was lower than in DMF even after a longer reaction (8 h instead of 3 h in DMF (Table 3, entry 11)). An increase in the amount of the base to 0.5 mmol lowered the activity of the catalyst (Table 9, entry 2). At 120 °C (entry 3) good selectivity for 6a and almost full conversion of iodobenzene was observed, but the latter decreased to 63% and 61% in the 2nd and 3rd runs, respectively, when the catalyst was recycled. The use of inorganic (Na2CO3, K2CO3, Cs2CO3) bases or DABCO did not lead to better results (Table 9, entries 4–7).| Entry | Base | Solvent | Pressure [bar] | Temp. [°C] | R. time [h] | Conversionb [%]/selectivity for 6ab [%] | ||
|---|---|---|---|---|---|---|---|---|
| Run 1 | Run 2 | Run 3 | ||||||
| a Reaction conditions: 0.2 mmol 4a, 0.5 mmol 5a, 0.25 mmol base, 1 ml solvent, CAT-2 (2.8 μmol Pd, 1.4 mol%).b Determined by GC.c 0.5 mmol Et3N.d Solvent-free reaction 0.4 mmol 4, 1.0 mmol 5, 0.5 mmol base, CAT-2 (2.8 μmol Pd, 0.7 mol%). | ||||||||
| 1 | Et3N | Toluene | 30 | 100 | 8 | 91/71 | 84/68 | 79/63 |
| 2 | Et3Nc | Toluene | 30 | 100 | 8 | 72/66 | 61/48 | n.d. |
| 3 | Et3N | Toluene | 30 | 120 | 8 | 99/89 | 63/85 | 61/86 |
| 4 | Na2CO3 | Toluene | 30 | 100 | 8 | 87/81 | 78/72 | n.d. |
| 5 | K2CO3 | Toluene | 30 | 100 | 8 | 73/75 | 37/65 | n.d. |
| 6 | Cs2CO3 | Toluene | 30 | 100 | 8 | 79/51 | 58/59 | n.d. |
| 7 | DABCO | Toluene | 30 | 100 | 8 | 88/70 | 78/70 | 80/71 |
| 8 | Et3N | —d | 1 | 100 | 8 | 59/100 | 32/100 | 14/100 |
| 9 | Na2CO3 | —d | 1 | 100 | 8 | 60/100 | 66/97 | 11/100 |
| 10 | DBU | —d | 1 | 100 | 8 | 90/97 | 77/100 | n.d. |
| 11 | DABCO | —d | 1 | 100 | 8 | 85/98 | 75/98 | 67/98 |
| 12 | DABCO | —d | 1 | 120 | 8 | 100/99 | 76/100 | 81/100 |
| 13 | Et3N | —d | 5 | 120 | 8 | 100/96 | 100/96 | 96/92 |
| 14 | DABCO | —d | 5 | 120 | 8 | 100/96 | 100/100 | 100/98 |
| 15 | DABCO | —d | 5 | 120 | 3 | 100/98 | 100/100 | 100/98 |
In the absence of a solvent, the monocarbonylated product 6a was formed in 59–87% yield in the presence of a variety of bases (Et3N, Na2CO3, DBU, DABCO) at atmospheric pressure of CO (Table 9, entries 8–11), but much lower conversions could be observed upon catalyst reuse. Under atmospheric conditions, the most efficient recycling could be achieved with DABCO as base at 120 °C (Table 9, entry 12).
By the increase of the CO pressure to 5 bar, better results were obtained (Table 9, entries 13–15). In the presence of DABCO as base, full conversion of iodobenzene was achieved even in 3 hour-long reactions. The catalyst could be reused efficiently leading to the selective formation of amide 6a and total conversion of iodobenzene (4a) in the first 9 cycles (Fig. 5).
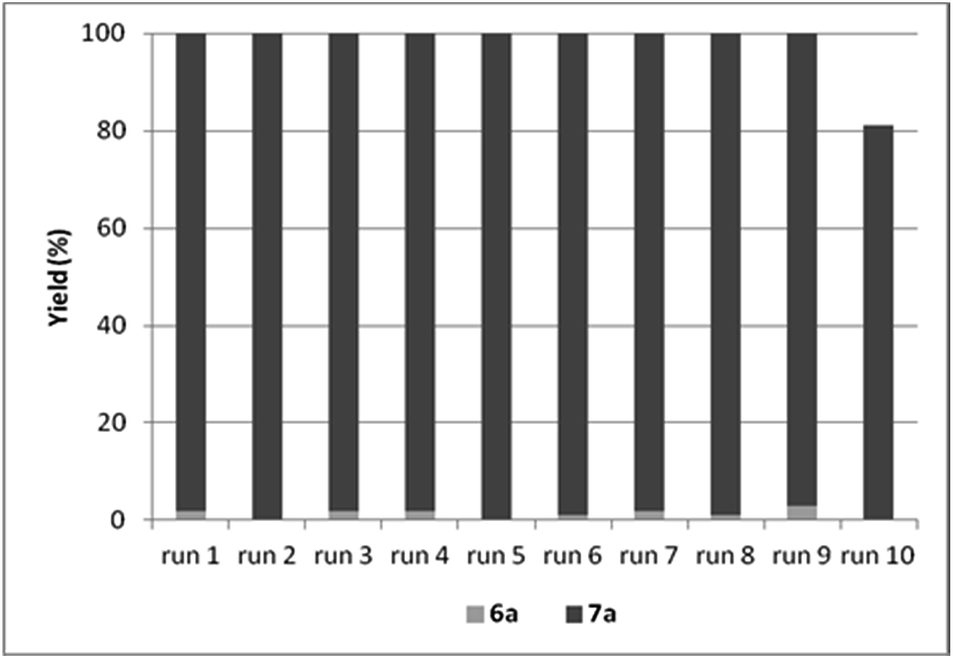 | ||
| Fig. 5 Recycling experiments with CAT-2 under solvent-free conditions (0.4 mmol 4a, 1.0 mmol 5a, 0.5 mmol DABCO, CAT-2 (2.8 μmol Pd, 0.7 mol%), 120 °C, 5 bar, 3 h, conversion determined by GC). | ||
Filtration and mercury poisoning tests were also carried out under solvent-free conditions (Table 6, entries 4, 5). The results were in good agreement with those in DMF. Catalytically active palladium species leached into the reaction mixture, and the presence of both nanoparticles and complexes could be detected (Table 6, entry 4). At the same time, when the mixture was extracted with toluene after completion of the reaction and fresh reagents were added to the extract, no conversion of iodobenzene could be observed (entry 5). This proves that similarly to the results obtained in DMF (entry 3), the leached palladium particles re-precipitated on the solid support at the end of the reaction.
Monocarbonylation with other nucleophiles/substrates
The CAT-2 catalyst was found to be efficient in the monocarbonylation of iodobenzene with other secondary amines as well (Table 10, entries 1–4). During carbonylations with primary aliphatic amines the amide products were isolated in acceptable yields (Table 10, entries 5–6). In the reaction of n-propylamine the corresponding imine (11d, Scheme 4) was obtained as side product instead of the expected α-ketoamide, via the condensation of the latter with the excess of the amine reagent. In the presence of aromatic amines lower conversion of iodobenzene was obtained (Table 10, entries 7, 9) under identical conditions. However, the N-arylbenzamides were isolated in good yields by the application of a longer reaction time (Table 10, entries 8, 10).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | 5 | Run 1 | Run 2 | Yield of 6c [%] | ||
| Conv.b [%] | Ratio of 6:7b |
Conv.b [%] | Ratio of 6:7b |
||||
| a Reaction conditions: 0.4 mmol 4, 1.0 mmol 5, 0.5 mmol DABCO, CAT-2 (2.8 μmol Pd, 0.7 mol%), 120 °C, 5 bar, 3 h.b Determined by GC.c Isolated yield from the combined reaction mixtures of the first two runs.d Side product: 11d (see Scheme 4) formed with a yield of 15% and 7% (GC) in the first and 2nd runs, respectively.e 8 h.f Side products: 8b, 9b and 10b (see Scheme 3) formed with a total yield of 58% and 48% (GC) in the first and 2nd runs, respectively.g Side products: 8b, 9b and 10b (see Scheme 3) formed with a total yield of 88% and 75% (GC) in the first and 2nd runs, respectively.h Isolated yield for 8b.i Side products: 8c, 9c and 10c (see Scheme 3) formed with a total yield of 25% and 51% (GC) in the first and 2nd runs, respectively.j Side products: 8c, 9c and 10c (see Scheme 3) formed with a total yield of 83% and 49% (GC) in the first and 2nd runs, respectively.k Isolated yield for 8c. | |||||||
| 1 | Ph | Morpholine | 100 | 98:2 |
100 | 100:0 |
91 |
| 2 | Ph | Piperidine | 100 | 95:5 |
100 | 97:3 |
93 |
| 3 | Ph | Pyrrolidine | 99 | 99:1 |
99 | 95:5 |
86 |
| 4 | Ph | Diethylamine | 100 | 74:26 |
94 | 78:22 |
72 |
| 5 | Ph | n-Propylamine | 100 | 85:0d |
100 | 93:0d |
75 |
| 6 | Ph | t-Butylamine | 89 | 75:25 |
84 | 84:16 |
63 |
| 7 | Ph | Aniline | 43 | 100:0 |
49 | 100:0 |
n.d. |
| 8e | Ph | Aniline | 98 | 100:0 |
100 | 100:0 |
89 |
| 9 | Ph | 4-Methylaniline | 83 | 100:0 |
32 | 100:0 |
n.d. |
| 10e | Ph | 4-Methylaniline | 100 | 100:0 |
98 | 100:0 |
90 |
| 11 | 4-CH3O–C6H4 | Morpholine | 100 | 100:0 |
100 | 97:0 |
87 |
| 12 | 3,4-(CH3)2–C6H3 | Morpholine | 100 | 100:0 |
92 | 100:0 |
93 |
| 13 | 1-Naphtyl | Morpholine | 100 | 97:3 |
100 | 97:3 |
95 |
| 14 | 3-F–C6H4 | Morpholine | 100 | 97:3 |
100 | 93:7 |
91 |
| 15 | 4-Cl–C6H4 | Morpholine | 100 | 100:0 |
100 | 100:0 |
96 |
| 16 | 3-Br–C6H4 | Morpholine | 94 | 37:1f |
79 | 39:0f |
31 |
| 17e | 3-Br–C6H4 | Morpholine | 99 | 11:0g |
93 | 19:0g |
73h |
| 18 | 4-Br–C6H4 | Morpholine | 94 | 72:1i |
82 | 37:1i |
48 |
| 19e | 4-Br–C6H4 | Morpholine | 100 | 17:0j |
97 | 50:0j |
58k |
| 20 | 4-Cl–C6H4 | 2-Morpholinoethylamine | 100 | 100:0 |
99 | 100:0 |
90 |

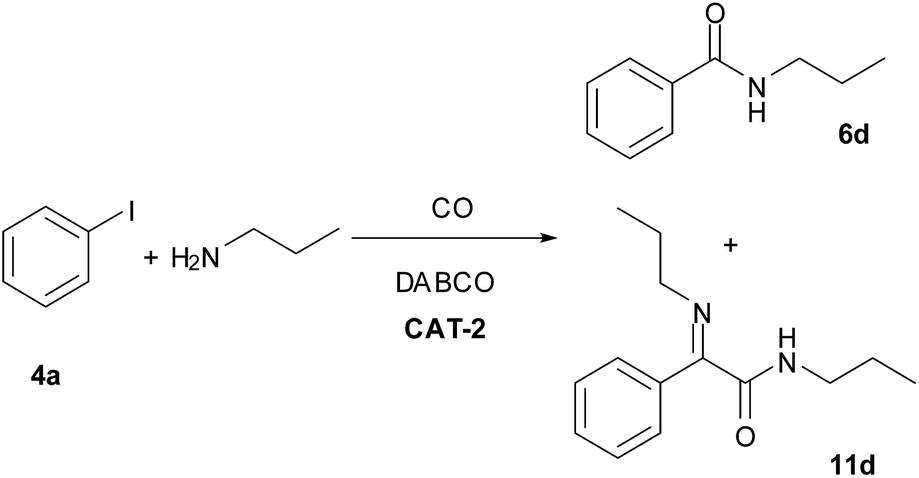 | ||
| Scheme 4 Aminocarbonylation of iodobenzene (4a) with n-propylamine (0.4 mmol 4a, 1.0 mmol n-propylamine, 0.5 mmol DABCO, CAT-2 (2.8 μmol Pd, 0.7 mol%), 120 °C, 5 bar, 3 h). | ||
In the reaction of iodobenzene derivatives, the monocarbonylated products were formed in good to excellent yield (Table 10, entries 11–15). In the case of 3-bromoiodobenzene and 4-bromoiodobenzene, the disubstituted products 8b and 8c were formed in considerable amounts (Table 10, entries 16, 18). When a longer reaction time was applied, the ratio of 8b and 8c increased in the reaction mixtures and they were isolated in 73% and 58% yield, respectively. Unfortunately, no carbonylation was observed using 1-bromo-4-chlorobenzene, 1-bromo-3-chlorobenzene, 1-bromo-4-fluorobenzene, 4-bromoacetophenone or bromobenzene as substrates, even in the presence of iodide ions added as KI or Bu4NI.
In the solvent-free aminocarbonylation reaction of 4-chloroiodobenzene and 4-(2-aminoethyl)morpholine, the antidepressant agent Moclobemide44 was synthesised with 90% isolated yield (Table 10, entry 20).
Conclusions
Modification of silica with dicationic organic moieties was shown to result in palladium catalysts with higher activity and better recyclability compared to similar derivatives immobilised on monocationic SILP phases. The use of Pd(OAc)2 as a precursor led to a more stable catalyst than that obtained with Pd2(dba)3·CHCl3 ensuring excellent recyclability as well as palladium leaching below the detection limit. The better performance can be attributed to the formation of smaller and more evenly distributed Pd-nanoparticles on the surface as shown by TEM measurements.N,N-Dialkyl-phenylglyoxylamide derivatives could be produced in excellent yield via double carbonylation reactions at 20 bar and 100 °C in DMF solvent with Et3N base. By a change in the reaction conditions (5 bar CO pressure, 120 °C in toluene, with DABCO as base) selective monocarbonylation took place resulting in the formation of N-alkyl- or N,N-dialkyl-benzamides. N-aryl benzamides were obtained as the only products in aminocarbonylation of iodobenzene with aniline derivatives even at higher pressure.
Dissolution — re-precipitation of active palladium species was proved by hot filtration tests.
Experimental
Preparation of 3-(2-bromoethyl)-1-methylimidazolium bromide
The 3-(2-bromoethyl)-1-methylimidazolium bromide was prepared according to a literature procedure.33 1,2-Dibromoethane (3.2 ml, 36.25 mmol) was added to a solution of 1-methylimidazole (500 μl, 6.13 mmol) in diethyl ether (3.5 ml) under Ar atmosphere, and the resulting mixture was stirred at room temperature for 4 days. During this time, a white precipitate was formed. It was collected by filtration and washed with diethyl ether (3 × 1 ml) and dried in vacuo. The filtrate was stirred at room temperature for further 24 hours to give a second crop of crystals. 3-(2-Bromoethyl)-1-methylimidazolium bromide was isolated as a hygroscopic white solid (0.90 g, 53%) and it was stored under argon. 1H NMR (400 MHz, DMSO-d6) δ 9.23 (s, 1H), 7.87–7.81 (m, 1H), 7.78–7.73 (m, 1H), 4.63 (t, J = 5.8 Hz, 2H), 3.95 (t, J = 5.8 Hz, 2H), 3.89 (s, 3H).Preparation of imidazole functionalised silica
Compound 2 was prepared by a modified method described in the literature.32 To a solution of (3-aminopropyl)triethoxysilane (2.3 ml, 10 mmol) in inert toluene (36 ml), 5.0 g silica (pre-treated by heating for 5 h at 250 °C) was added. The mixture was refluxed for 72 h. Then the white material was filtered and washed with toluene (3 × 5 ml) and diethyl ether (3 × 5 ml). The solid was dried to constant weight (8 h) in vacuo to produce 1. The aminopropyl group content of the silica was determined by measuring the weight increase of the material (0.73 mmol g−1 modified silica). 1.0 g of 1 was suspended in 10 ml methanol and 2.8 mmol glyoxal (40% solution in water) was added and was stirred overnight. 5.6 mmol NH4Cl and 5.6 mmol formaldehyde were added and it was diluted with 30 ml methanol. This was refluxed for an hour and H3PO4 (0.5 ml, 85%) was slowly added to the mixture before heating to reflux for 12 hours. The resulting material was filtered and washed with methanol (3 × 10 ml) and diethyl ether (3 × 10 ml). It was then dried in vacuo at 60 °C to constant weight (8 h) to give a yellowish solid product with 0.72 mmol g−1 imidazole functionality (determined by elemental analysis from the nitrogen content of the solid material: calculated for 0.72 mmol g−1 C8H14N2OSi: C: 6.93; H: 1.01; N: 2.02; found C: 6.34; H: 0.90; N: 2.02).Preparation of SILP phase 3
To a solution of 3-(2-bromoethyl)-1-methylimidazolium bromide (405 mg, 1.5 mmol) in 20 ml acetonitrile, 685 mg of 2 was added and the resulting mixture was refluxed for 5 days. Then the solid was filtered and washed with acetonitrile (3 × 5 ml), methanol (3 × 5 ml), acetone (3 × 5 ml) and diethyl ether (3 × 5 ml). The solid was dried to constant weight (8 h) in vacuo to produce 3, with 0.40 mmol g−1 dicationic moieties (determined by measuring the weight increase of the material). Elemental analysis: C: 9.28; H: 1.21; N: 3.11.Preparation of CAT-1
29.0 μmol (30.0 mg) Pd2(dba)3·CHCl3 was dissolved in a mixture of 2 ml acetonitrile and 2 ml THF. The mixture was stirred for 15 min at room temperature. Then 200 mg of 3 was added and the resulting mixture was stirred overnight. The solvents were removed in vacuo and the residue was dried in vacuo at 35 °C for 3 h and the catalyst was obtained as a dark grey solid. Palladium content of the catalyst: 2.64% (determined by ICP).Preparation of CAT-2
To a solution of Pd(OAc)2 (13.0 mg, 58.0 μmol) and potassium tert-butoxide (6.5 mg, 58.0 μmol) in 4 ml ethanol, 200 mg of 3 was added and the resulting mixture was stirred at room temperature overnight. It was filtered and washed with ethanol (3 × 2 ml) and diethyl ether (3 × 2 ml). The catalyst was dried in vacuo at 35 °C for 3 h and was obtained as a brown solid. Palladium content of the catalyst: 1.66% (determined by ICP).General procedure for aminocarbonylation reactions
General procedure for solvent-free reactions. The catalyst (with 2.8 μmol Pd content) was placed in a Schlenk tube. The atmosphere was changed to carbon monoxide. 0.4 mmol (45 μl) iodobenzene, 1.0 mmol (87 μl) morpholine and 0.5 mmol (70 μl) triethylamine was added via septum. The mixture was stirred in an oil bath for 8 hours at 100 °C. After cooling to room temperature, the products were extracted with 3 × 1 ml toluene. The reaction mixture was analysed by GC and the catalyst was reused.
General procedure for solvent-free reactions. In a typical experiment the catalyst (with 2.8 μmol Pd content) was placed in a stainless steel autoclave. 0.4 mmol (45 μl) iodobenzene, 1.0 mmol (87 μl) morpholine and 0.5 mmol (56.1 mg) DABCO was added. It was charged with carbon monoxide (5–30 bar) and was heated with stirring in an oil bath for 3 and 8 hours at 120 °C. After cooling to room temperature, the products were extracted with 3 × 1 ml toluene. The reaction mixture was analysed by GC and the catalyst was reused. The catalyst was washed with 2 × 1 ml methanol before recycling in every two runs to remove the ammonium salt formed during the reaction.
Analytical measurements
Reaction mixtures were analysed by gas chromatography (Hewlett Packard 5890) and GC-MS (Hewlett Packard 5971A GC-MSD, HP-1 column).The palladium-content of the catalysts and palladium leaching were determined by ICP.
FT-IR spectra were measured on a BRUKER Vertex 70 type spectrometer with a Bruker Platinum ATR adapter without sample preparation. The spectra were recorded at a resolution of 2 cm−1 with a room temperature DTGS detector (512 scans were co-added).
Surface compositions of CAT-1 and CAT-2 before (“fresh”) and after (“spent”) the catalytic test reactions were determined by X-ray photoelectron spectroscopy (XPS) performed by a KRATOS XSAM 800 XPS instrument. Al Kα characteristic X-ray line, 40 eV pass energy and FAT mode were applied for recording the XPS lines of Pd 3d, C 1s, O 1s, Br 3d, I 3d, N 1s and Si 2p. C 1s binding energy at 284.8 eV was used as reference for charge compensation. The surface concentrations of the elements were calculated from the integral intensities of the XPS lines using sensitivity factors given by the manufacturer.
Transmission Electron Microscope (TEM) investigations were carried out by a JEOL 3010 high resolution TEM operating at 300 kV, with a point resolution of 0.17 nm. The microscope was equipped with a GATAN Tridiem energy filter used for electron energy loss spectroscopy (EELS) elemental mapping. The samples were suspended in ethanol and drop-dried on carbon-coated microgrids for the measurements of the microstructure of the catalyst particles and their distribution over the support.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the GINOP-2.3.2-15-2016-00049 grant. The authors thank the National Research, Development and Innovation Office for the financial support (OTKA K105632).References
- R. Skoda-Földes, Top. Organomet. Chem., 2015, 51, 145–162 CrossRef.
- C. F. J. Barnard, Organometallics, 2008, 27, 5402–5422 CrossRef CAS.
- A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed.
- R. Grigg and S. P. Mutton, Tetrahedron, 2010, 66, 5515–5548 CrossRef CAS.
- D. Kumar, S. R. Vemula and G. R. Cook, ACS Catal., 2016, 6, 4920–4945 CrossRef CAS.
- S. Roy, S. Roy and G. W. Gribble, Tetrahedron, 2012, 68, 9867–9923 CrossRef CAS.
- C. De Risi, G. P. Pollini and V. Zanirato, Chem. Rev., 2016, 116, 3241–3305 CrossRef CAS PubMed.
- T. T. Dang, Y. Zhu, S. C. Ghosh, A. Chen, C. L. L. Chai and A. M. Seayad, Chem. Commun., 2012, 48, 1805–1807 RSC.
- T. T. Dang, Y. Zhu, J. S. Y. Ngiam, S. C. Ghosh, A. Chen and A. M. Seayad, ACS Catal., 2013, 3, 1406–1410 CrossRef CAS.
- T. T. Dang, A. Chen and A. M. Seayad, RSC Adv., 2014, 4, 30019–30027 RSC.
- A. O. Biying, K. T. Yuanting, N. S. Hosmane and Z. Yinghuai, J. Organomet. Chem., 2013, 747, 184–188 CrossRef CAS.
- F. Tinnis, O. Verho, K. P. J. Gustafson, C.-W. Tai, J.-E. Bäckvall and H. Adolfsson, Chem.–Eur. J., 2014, 20, 5885–5889 CrossRef CAS PubMed.
- R. S. Mane, T. Sasaki and B. M. Bhanage, RSC Adv., 2015, 5, 94776–94785 RSC.
- S. M. Islam, K. Ghosh, A. S. Roy and R. A. Molla, RSC Adv., 2014, 4, 38986–38999 RSC.
- M. B. Ibrahim, R. Suleiman, M. Fettouhi and B. E. Ali, RSC Adv., 2016, 6, 78826–78837 RSC.
- H. Mei, J. Hu, S. Xiao, Y. Lei and G. Li, Appl. Catal., A, 2014, 475, 40–47 CrossRef CAS.
- B. Urbán, M. Papp, D. Srankó and R. Skoda-Földes, J. Mol. Catal. A: Chem., 2015, 397, 150–157 CrossRef.
- M. V. Khedkar, T. Sasaki and B. M. Bhanage, ACS Catal., 2013, 3, 287–293 CrossRef CAS.
- Y. Yan, H. P. Zuo and Z. L. Jin, React. Funct. Polym., 1997, 32, 21–24 CrossRef CAS.
- J. Liu, S. Zheng, W. Su and C. Xia, Chin. J. Chem., 2009, 27, 623–627 CrossRef CAS.
- M. Genelot, N. Villandier, A. Bendjeriou, P. Jaithong, L. Djakovitch and V. Dufaudz, Catal. Sci. Technol., 2012, 2, 1886–1893 CAS.
- B. Chen, F. Li, Z. Huang, T. Lu and G. Yuan, Appl. Catal., A, 2014, 481, 54–63 CrossRef CAS.
- Z. Wang, C. Liu, Y. Huang, Y. Hu and B. Zhang, Chem. Commun., 2016, 52, 2960–2963 RSC.
- E. Takács, C. S. Varga, R. Skoda-Földes and L. Kollár, Tetrahedron Lett., 2007, 48, 2453–2456 CrossRef.
- R. A. Molla, M. A. Iqubal, K. Ghosh, A. S. Roy, Kamaluddin and S. M. Islam, RSC Adv., 2014, 4, 48177–48190 RSC.
- M. Papp, P. Szabó, D. Srankó and R. Skoda-Földes, RSC Adv., 2016, 6, 45349–45356 RSC.
- M. Papp and R. Skoda-Földes, J. Mol. Catal. A: Chem., 2013, 378, 193–199 CrossRef CAS.
- F. D'Anna and R. Noto, Eur. J. Org. Chem., 2014, 4201–4223 CrossRef.
- V. Sans, F. Gelat, M. I. Burguete, E. Garcia-Verdugo and S. V. Luis, Catal. Today, 2012, 196, 137–147 CrossRef CAS.
- J. Schwarz, V. P. Bohm, M. G. Gardiner, M. Grosche, W. A. Herrmann, W. Hieringer and G. Raudaschl-Sieber, Chem.–Eur. J., 2000, 6, 1773–1780 CrossRef CAS.
- M. Massaro, S. Riela, G. Cavallaro, C. G. Colletti, S. Milioto, R. Noto, F. Parisi and G. Lazzara, J. Mol. Catal. A: Chem., 2015, 408, 12–19 CrossRef CAS.
- A. Bayero, J. Pure Appl. Sci., 2009, 2, 61–69 Search PubMed.
- L. D. Field, B. A. Messerle, K. Q. Vuong and P. Turner, Organometallics, 2005, 24, 4241–4250 CrossRef CAS.
- Y. Jiang, Q. Gao, H. Yu, Y. Chen and F. Deng, Microporous Mesoporous Mater., 2007, 103, 316–324 CrossRef CAS.
- Y. Gushikem and J. C. Moreira, J. Colloid Interface Sci., 1985, 107, 70–74 CrossRef CAS.
- S. B. Aher and P. R. Bhagat, Res. Chem. Intermed., 2016, 42, 5587–5596 CrossRef CAS.
- L. Han, M. S. Park, S. J. Choi, Y. J. Kim, S. M. Lee and D. W. Park, Catal. Lett., 2012, 142, 259–266 CrossRef CAS.
- M. Brun, A. Berthet and J. C. Bertolini, J. Electron Spectrosc. Relat. Phenom., 1999, 104, 55–60 CrossRef CAS.
- C. Pavia, E. Ballerini, L. A. Bivona, F. Giacalone, C. Aprile, L. Vaccaro and M. Gruttadauria, Adv. Synth. Catal., 2013, 335, 2007–2018 CrossRef.
- B. Urbán, D. Srankó, G. Sáfrán, L. Ürge, F. Darvas, J. Bakos and R. Skoda-Földes, J. Mol. Catal. A: Chem., 2014, 395, 364–372 CrossRef.
- R. Skoda-Földes and L. Kollár, Curr. Org. Chem., 2002, 6, 1097–1119 CrossRef.
- C. D. Wagner, W. M. Riggs, L. E. David, J. F. Moulder and G. T. Muilenberg, Handbook of X-Ray Photoelectron Spectroscopy, Perkin-Elmer Corporation, Physical Electronics Division, Eden Prairie, Minn. 55344, 1979 Search PubMed.
- D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2–19 CrossRef CAS.
- A. Fitton, D. Faulds and K. L. Goa, Drugs, 1992, 43, 561–596 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation of the catalysts, general procedure for aminocarbonylation reactions, 29Si CP MAS NMR spectrum of 3, TEM images and XPS spectra of catalysts, data of recycling experiments with CAT-1 and CAT-2, characterisation of products, and 1H and 13C-NMR spectra of isolated products, references. See DOI: 10.1039/c7ra04680d |
| ‡ Present address: Department of Engineering Science and Mathematics, Luleå University of Technology, 97187 Luleå, Sweden. |
| This journal is © The Royal Society of Chemistry 2017 |