
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight on lithium polysulfide intermediates in a Li/S battery by density functional theory†
Qi Liua,
Daobin Mu *a,
Borong Wu*ab,
Lei Wanga,
Liang Gaia and
Feng Wuab
*a,
Borong Wu*ab,
Lei Wanga,
Liang Gaia and
Feng Wuab
aBeijing Key Laboratory of Environment Science and Engineering, School of Material Science and Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: mudb@bit.edu.cn
bCollaborative Innovation Center of Electric Vehicles in Beijing, Beijing, 100081, China. E-mail: wubr@bit.edu.cn
First published on 3rd July 2017
Abstract
Soluble lithium polysulfide intermediates dissolve and shuttle during the process of charge/discharge, leading to the rapid capacity decline of a Li–S battery. Density functional theory (DFT) computation is used to research the thermodynamic behavior of the polysulfides. The computation indicates that the stable molecular structures tend to be in a ring shape. This is helpful for cathode modification at a molecular level to fix the polysulfides.
With a growing demand from sophisticated functions in portable digital products to application in electric vehicles, a high energy storage system has become a necessity in our daily lives.1–4 Compared with conventional lithium-ion batteries, the Li–S battery exhibits a higher energy density of 2600 W h kg−1; the cathode of the Li–S battery enjoys a high theoretical specific capacity of 1672 mA h g−1,5,6 which is much higher that of the lithium ion battery, which is generally less than 300 mA h g−1. So, the Li–S battery has attracted more and more attention as a promising next generation energy storage system.7–13 In addition, the larger resource reserve of sulfur on the earth and the lower cost of sulfur provide strong market competitiveness. However, many challenges are still facing the Li–S battery technology. The first problem is the electrical insulation of sulfur;14 the second and the main problem for the Li–S battery cathode is the dissolution and shuttle behavior of polysulfide. Soluble lithium polysulfide intermediates (Li2Sx, 2 < x < 8) dissolve in organic electrolyte and shuttle from the cathode to the anode, leading to rapid capacity decline and low columbic efficiency.15,16 The poor electron conductivity of sulfur has been greatly improved by coupling high conductive materials such as multiwalled carbon nanotubes and graphene oxide. The dissolution and shuttle of polysulfides need to be studied further.
As for the dissolution of polysulfides in the Li–S battery, much work has been done to modify the (S) cathode.12,17–22 Zhang et al. have proposed a unique strategy by building a cooperative interface between the S cathode and a separator to suppress the polysulfide shuttle and enhance the reaction kinetics with LDH@NG separators.23 They also have designed a polar hybrid host material of TiC nanoparticles, which is grown within a porous graphene framework to localize the mobile polysulfide intermediates by chemisorption.24 In addition, chemical modification using N or O dopants has been proved to significantly enhance the interaction between the carbon hosts and the polysulfide guests, thereby effectively preventing the shuttle of polysulfides.25 Furthermore, research on electrolytes is also important to understand the mechanism of polysulfide dissolution and reaction.26 An in situ 7Li NMR technique has been used to probe the transient polysulfide redox reactions involving charged free radicals as intermediate species.27 A cation permselective membrane has been introduced to improve the stability and coulombic efficiency of lithium–sulfur batteries by confining the polysulfides on the cathode side.28 Different kinds of solvent structures have been investigated by changing the ratio of ether/alkyl moieties to vary the solubility of lithium polysulfide.29 The chemical reactivity of polysulfides with carbonate-based electrolytes has been investigated, and it is found that the polysulfides react with carbonate-based electrolytes via a nucleophilic addition or substitution reaction, leading to a sudden capacity fading of lithium–sulfur cells.30 First principles calculation has been carried out on the energetic and electronic properties of Li2S2, showing that Li2S2 is subject to spontaneous disproportionation.31 Synchrotron high-energy X-ray diffraction analysis and in situ transmission electron microscopy have been used to characterize dry powder deposits from lithium polysulfide solution and the lithium diffusion to sulfur.32 DFT calculations have revealed that Li2S8 accepts electrons more readily than S8 and Li2S6, so that it is thermodynamically and kinetically unstable.33 A solvent-in-salt electrolyte has been used in a Li–S cell, which can inhibit dissolution of polysulfide.9 The salt decomposition, solvation effects, interactions among intermediate products and other species have been investigated by density functional theory and ab initio molecular dynamics simulations.34 The theoretical predictions and experimental observations have also confirmed that DOL prefers to react with Li metal and release C2H4 gas, while the DME molecule has good stability.35 Although research on polysulfides from the point of view of the electrolyte has been given attention, the stable three-dimensional molecular structures of polysulfides in solution are rarely involved. In particular, the three-dimensional molecular structures of polysulfides could provide guidance for the modification of the cathode at a molecular level to fix polysulfides. In addition, the polysulfide degradation mechanism is still controversial, and this is a complex problem. Although some polysulfide mechanisms have been proposed,36–39 none of them could completely explain the reaction mechanism of the Li–S battery. The reaction contains complicated disproportionation. No single technique is capable of identifying and quantifying the polysulfide species.40 Fundamental thermodynamics steady state research could also analyze the polysulfide degradation mechanism, and the S–S bond breaking of polysulfides also needs to be further investigated to understand the distribution and transformation of polysulfides and to suppress the shuttle effect by researching the binding energies of S–S bonds. Oleg Borodin et al. have researched the solvation in carbonate esters by different kinds of quantum chemistry calculation and simulation.41–46 Lithium polysulfides in ether solvation should be further researched.
In this work, the DFT method is used to systematically research lithium polysulfide and its possible degradation in the Li–S battery. The stable three-dimensional molecular structure, solvation energy and the dissociation of polysulfides are studied in DOL and DME, respectively. The detailed calculations and analysis will be reported in this work.
Quantum chemical calculations were completed through the density functional theory (DFT) method with Becke's three parameters (B3) exchange functional along with the Lee–Yang–Parr (LYP) nonlocal correlation functional (B3LYP). All the structural optimizations were treated at B3LYP/6-31+G(d,p) level. In order to make zero-point energy (ZPE) corrections, frequency analyses are made with the corresponding basis sets. All the single point energy calculations were performed at B3LYP/6-311++G(3df,3dp) level for more accurate calculation. The outer solvation shells were also considered by using polarized continuum models (PCM). The outer shells solvent effects were completed by geometry optimization at C-PCM-B3LYP/6-31+G(d,p) in DOL solvent (dielectric constant of 7.1) and DME solvent (dielectric constant of 7.2), respectively. In addition, counterpoise corrections were employed to minimize the “Basis Set Superposition Error” (BSSE) when calculating binding energies. All of the DFT calculations were performed with the Gaussian 09 program package.47
Ionic association interactions are typically classified as either solvent-separated ion pair (SSIP), contact ion pair (CIP), or aggregate (AGG) coordination, depending upon whether the anions form coordinate bonds with zero, one, or more than one Li+ cations, respectively.41–44 In order to study the dissolution and dissociation of the lithium polysulfides, the CIP model is employed to optimize the stable three-dimensional molecular structures of lithium polysulfides in solution (Li2Sx, 1 ≤ x ≤ 8) by the DFT method. Fig. 1 shows the molecular structure of lithium polysulfides (Li2Sx, 1 ≤ x ≤ 8) in DOL. Six DOL molecules around one of the polysulfides are employed to simulate the solvation in all the quantum chemistry optimizations.
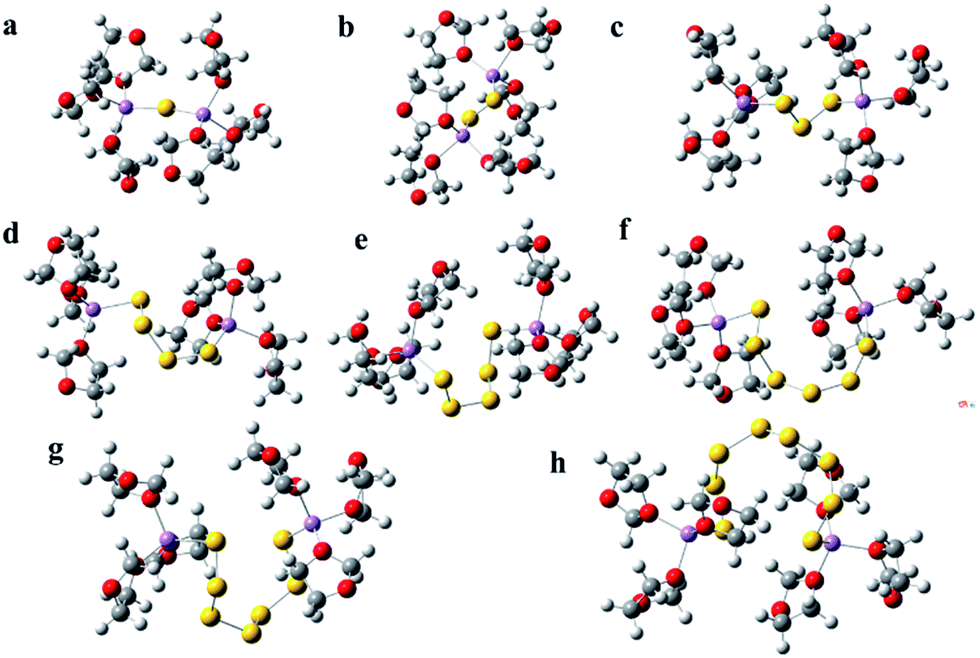 | ||
| Fig. 1 The solvation structure of lithium polysulfides (Li2Sx, 1 ≤ x ≤ 8, a–h) in DOL solvent; the lithium polysulfide geometries are surrounded by six DOL molecules. Dark gray, white, red, yellow and violet spheres denote C, H, O, S and Li atoms/ions, respectively. | ||
Fig. 1(a–f) show the solvated structures of Li2S, Li2S2, Li2S3, Li2S4, Li2S5, Li2S6, Li2S7 and Li2S8 in DOL, respectively. From the figure, it can clearly be seen that the short chain lithium polysulfides (Li2Sx, 1 ≤ x ≤ 3) have a linear sawtooth structure, but for the long chain lithium polysulfides (Li2Sx, 4 ≤ x ≤ 8), the solvated structures tend to be rings. In a previous study, many of the long chain polysulfides were regarded as linear chains. Our calculated results demonstrate that the stable long chain polysulfides have a cyclic structure, which is helpful for recognizing the polysulfides at a molecular level, which could better guide the modification of the cathode to suppress the shuttle of the polysulfides. Video 1† shows the process of structural optimization for Li2S6 which is surrounded by six DOL molecules (see ESI Video 1†). It can clearly be seen that at the beginning of the optimization the structure of Li2S6 is linear sawtooth, but as the optimization proceeds, the structure of Li2S6 gradually shrinks to a cyclic structure. In addition, the outer solvation shells of DOL are also considered by using PCM, and the optimized result also demonstrates that the stable structure of Li2S6 is cyclic under solvation conditions, and the implicit solvent has a negligible effect on the structure of lithium polysulfide (see Fig. S1†). LiTFSI is also introduced into the solvation structure, which indicates that the structure of polysulfide is also cyclic with the existence of LiTFSI (see Fig. S2†). The theoretical calculations demonstrate that a cyclic structure for long chain lithium polysulfides (Li2Sx, 4 ≤ x ≤ 8) is more stable in the DOL solvent. In addition, it can clearly be seen that there is no evident interaction between DOL and Sn groups; the DOL combines more easily with a Li atom. Therefore, it should be the case that the polar O atoms in the DOL combine with the Li atom in lithium polysulfides forming complexes in the dissolution process. And the Li–S bond is elongated, leading to the stronger negative electricity of the Sn groups, which accelerates the destruction of crystal structure for lithium polysulfides, owing to the two like electric charges of the Sn groups repelling each other. Therefore, the dissolution of lithium polysulfides should be ascribed to the solvation of the Li+ group when using a DOL solvent. It is also indicated that one Li+ combines with at most four DOL and three DME by ΔG calculation (see Fig. S3†). The dissolution of lithium polysulfides is in accord with the dissociation of lithium polysulfides.
Furthermore, the solvation of lithium polysulfides in DME was investigated. Four DME molecules are employed to surround the lithium polysulfides. This is similar to the solvation structures in DOL solvent; the short chains of lithium polysulfides (Li2Sx, 1 ≤ x ≤ 3) have a linear sawtooth structure, but the long chains of lithium polysulfides (Li2Sx, 4 ≤ x ≤ 8) have a cyclic structure in DME solvent (see Fig. 2). Video 2† also shows the optimization process of Li2S6 in DME solvent (see ESI Video 2†). From the video, it can be clearly seen that the linear sawtooth structure of Li2S6 gradually transforms to the ring structure in the optimization process. Fig. S4† also shows the optimized structure of Li2S6 in the outer solvation shells of DME by using PCM, and the cyclic structure is clearly shown. Therefore, the calculation results suggest that the cyclic structure for the long chain lithium polysulfides is more stable in both DOL and DME solvents. The difference for the solvation structure of lithium polysulfides in DME is that the lithium ions tend to locate on the middle of the Sn group at both ends, but for the DOL solvent, the lithium ions locate on the respective ends of the Sn group.
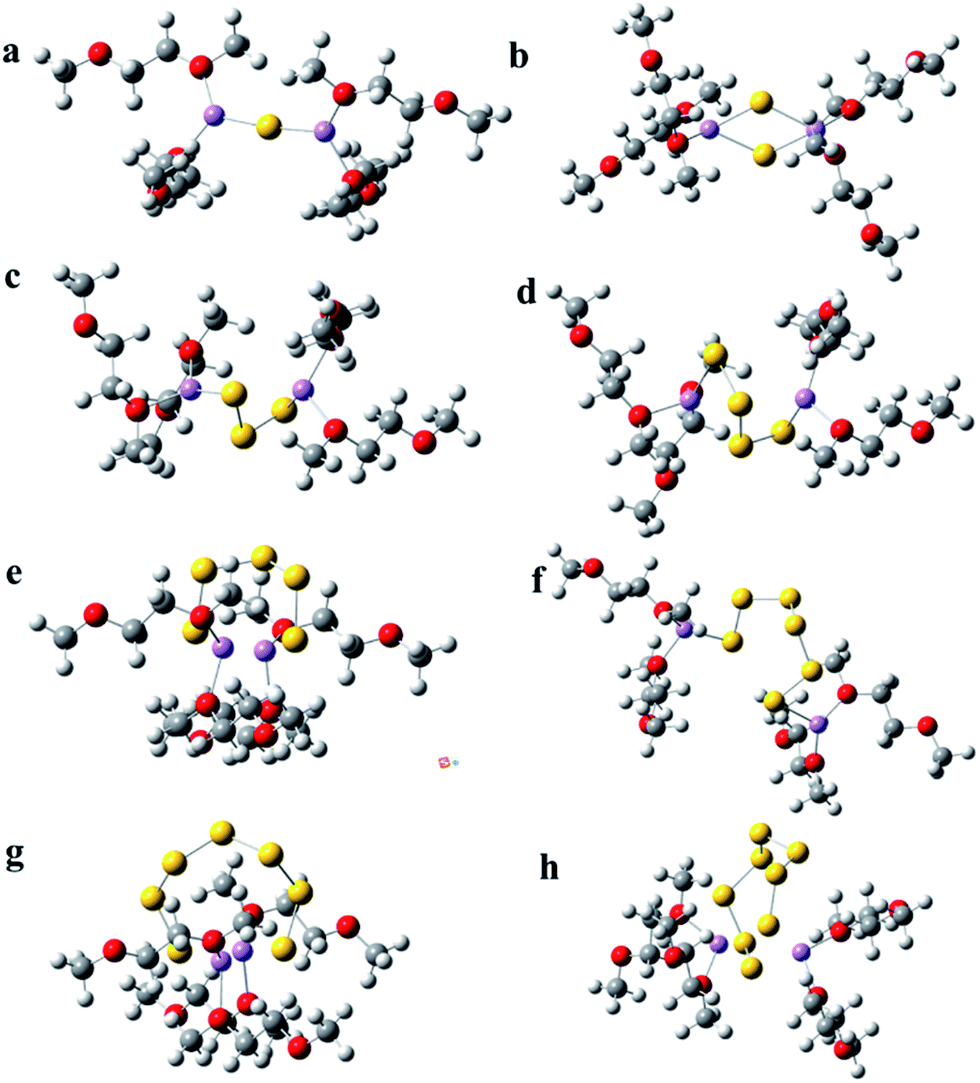 | ||
| Fig. 2 The solvation structure of polysulfide (Li2Sx, 1 ≤ x ≤ 8, a–h) in DME solvent; the polysulfide geometries are surrounded by four DME molecules. Dark grey, white, red, yellow and violet spheres denote C, H, O, S and Li atoms/ions, respectively. | ||
Moreover, the binding energies of all S–S bonds in lithium polysulfides in both DOL and DME solvent were analysed after optimization. The binding energies of S–S bonds are calculated by splitting all the lithium polysulfides at every S–S bond. Table 1 exhibits the S–S binding energies in DOL solvent. The binding energies of S1–S2, S2–S3 and S3–S4 for Li2S4 are 1.989, 1.465 and 1.986, respectively. The results suggest that the S2–S3 may be previously broken to form two S2. The binding energies of S1–S2, S2–S3, S3–S4 and S4–S5 for Li2S5 are 2.225, 1.472, 1.36 and 2.304 eV, respectively. This indicates that the S2–S3 or S3–S4 bonds may break to S2 and S3. Perla B. Balbuena48 et al.'s calculation also indicates that S5 later decomposes into S3 + S2 and S3 into S2 + S. In addition, the binding energies of S1–S2, S2–S3, S3–S4, S4–S5 and S5–S6 bonds for Li2S6 are 2.344, 1.487, 1.242, 1.647 and 2.238 eV, indicating that S3–S4 might be broken into two S3. Similarly, the S3–S4 bond may undergo bond rupture and form S3 and S4 radicals in Li2S7. For the Li2S8, the S3–S4 or S5–S6 bond may be broken into S3 and S5 radicals.
| DOL | S1–S2 | S2–S3 | S3–S4 | S4–S5 | S5–S6 | S6–S7 | S7–S8 |
|---|---|---|---|---|---|---|---|
| Li2S2 | 3.319 | — | — | — | — | — | — |
| Li2S3 | 2.018 | 2.127 | — | — | — | — | — |
| Li2S4 | 1.989 | 1.465 | 1.986 | — | — | — | — |
| Li2S5 | 2.225 | 1.472 | 1.36 | 2.304 | — | — | — |
| Li2S6 | 2.344 | 1.487 | 1.242 | 1.647 | 2.238 | — | — |
| Li2S7 | 2.321 | 1.663 | 1.407 | 1.527 | 1.62 | 2.309 | — |
| Li2S8 | 2.37 | 1.689 | 1.262 | 1.829 | 1.319 | 1.769 | 2.303 |
Table 2 shows the binding energies of S–S bonds in lithium polysulfides after optimization in DME solvent. The binding energy of the S2–S3 for the Li2S4 is 2.109 eV, which is less than that of the S1–S2 (2.435 eV) and S3–S4 (2.435 eV), indicating that the two S2 radicals may be formed more easily. For the Li2S5, S3 and S2 formation may occur. The binding energy of the S3–S4 bond (3.151 eV) is the minimum for Li2S6, indicating that two S3 may be formed by breaking the S3–S4 bond. For the Li2S7, S3 and S4 may be obtained easily. Similarly, for the Li2S8, S3–S4 (3.441 eV) or S5–S6 (3.44 eV) may be broken to S3 and S5. Ab initio molecular dynamics simulation also suggests that the PS chain of Li2S8 reacts with two Li atoms and, in most cases, it was observed to break into Li2S3 and Li2S5.49
| DME | S1–S2 | S2–S3 | S3–S4 | S4–S5 | S5–S6 | S6–S7 | S7–S8 |
|---|---|---|---|---|---|---|---|
| Li2S2 | 3.561 | — | — | — | — | — | — |
| Li2S3 | 2.861 | 2.745 | — | — | — | — | — |
| Li2S4 | 2.435 | 2.109 | 2.435 | — | — | — | — |
| Li2S5 | 3.914 | 3.348 | 2.865 | 4.103 | — | — | — |
| Li2S6 | 4.079 | 3.457 | 3.151 | 3.457 | 4.079 | — | — |
| Li2S7 | 3.985 | 3.609 | 3.162 | 3.468 | 3.415 | 4.148 | — |
| Li2S8 | 4.135 | 3.676 | 3.441 | 3.817 | 3.44 | 3.677 | 4.135 |
The Raman spectra of lithium polysulfides in a DOL solvation structure were also calculated. Fig. S5† shows the Raman spectra of Li2Sn (2 ≤ n ≤ 8), which clearly show that polysulfide species generally stay in the wavenumber region of 150–550 cm−1, which is in accord with previous studies.50–54 In addition, characteristic polysulfide Raman position lines can be found in the area between 400 and 500 cm−1 (S–S stretch vibration) and below 250 cm−1 (bending and torsional modes).51
Conclusions
The lithium polysulfide reaction mechanism is crucial for the high energy density Li–S battery. DFT calculations suggest that the stable molecular structure for short chain lithium polysulfides (Li2Sx, 1 ≤ x ≤ 3) is linear sawtooth, but the stable molecule structure for long chain lithium polysulfides (Li2Sx, 4 ≤ x ≤ 8) tends to a ring in both DOL and DME solvents, which is important for cathode modification at a molecular level to fix the polysulfides. In addition, S2 and S, two S2, S3 and S2, two S3, S3 and S4, and S3 and S5 may be formed by the decomposition of Li2S3, Li2S4, Li2S5, Li2S6, Li2S7, and Li2S8, respectively in both DOL and DME solvation. Furthermore, the mechanism of lithium polysulfide decomposition is complicated, and more combinational work of calculation and experiment is needed to clarify it completely. In addition, further investigations with SSIP and AGG models are needed to understand the solvation of lithium polysulfides.Acknowledgements
This project was financially supported by NSAF (Grant No. U1530155); the the International Science & Technology Cooperation Program of China under contract No. 2016YFE0102200; the National Key Basic Research Program of China (Contract No. 2015CB251100); Industry University Research Project in Shenzhen (Contract No. 2012CXHZ008).Notes and references
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS PubMed.
- B. Kang and G. Ceder, Nature, 2009, 458, 190 CrossRef CAS PubMed.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2013, 46, 1135 CrossRef CAS PubMed.
- R. Demir-Cakan, M. Morcrette, F. Nouar, C. Davoisne, T. Devic and D. Gonbeau, et al., J. Am. Chem. Soc., 2011, 133, 16154 CrossRef CAS PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19 CrossRef CAS PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500 CrossRef CAS PubMed.
- L. Suo, Y.-S. Hu and H. Li, et al., Nat. Commun., 2013, 4 CAS.
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125 CrossRef CAS PubMed.
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751 CrossRef CAS PubMed.
- Z. Li and J. Zhang, et al., Angew. Chem., Int. Ed., 2015, 53, 12886 CrossRef PubMed.
- X. Gu, S. Zhang and Y. Hou, Chin. J. Chem., 2016, 34, 13 CrossRef CAS.
- X.-B. Cheng, H.-J. Peng, J.-Q. Huang, R. Zhang, C.-Z. Zhao and Q. Zhang, ACS Nano, 2015, 9, 6373 CrossRef CAS PubMed.
- M.-K. Song, Y. Zhang and E. J. Cairns, Nano Lett., 2013, 13, 5891 CrossRef CAS PubMed.
- X. Liang, Z. Wen, Y. Liu, H. Zhang, J. Jin, M. Wu and X. Wu, J. Power Sources, 2012, 206, 409 CrossRef CAS.
- R. Sahore, L. P. Estevez, A. Ramanujapuram, F. J. DiSalvo and E. P. Giannelis, J. Power Sources, 2015, 297, 188–194 CrossRef CAS.
- J. Zhang, Y. Cai and Q. Zhong, et al., Nanoscale, 2015, 7, 17791 RSC.
- L. Qie and A. Manthiram, Adv. Mater., 2015, 27, 1694 CrossRef CAS PubMed.
- R. Fang, S. Zhao, P. Hou, M. Cheng, S. Wang, H. M. Cheng, C. Liu and F. Li, Adv. Mater., 2016, 28, 3374 CrossRef CAS PubMed.
- M. R. Kaiser, X. Liang, H. K. Liu, S. X. Dou and J. Z. Wang, Carbon, 2016, 103, 163 CrossRef CAS.
- X. Chen, H. Peng, R. Zhang, T. Hou, J. Huang, B. Li and Q. Zhang, ACS Energy Lett., 2017, 2, 795 CrossRef CAS.
- H. Peng, Z. Zhang, J. Huang, G. Zhang, J. Xie, W. Xu, J. Shi, X. Chen, X. Cheng and Q. Zhang, Adv. Mater., 2016, 28, 9551 CrossRef CAS PubMed.
- H. Peng, G. Zhang, X. Chen, Z. Zhang, W. Xu, J. Huang and Q. Zhang, Angew. Chem., 2016, 128, 13184 CrossRef.
- T. Hou, X. Chen, H. Peng, J. Huang, B. Li, Q. Zhang and B. Li, Small, 2016, 12, 3283 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503 CrossRef CAS PubMed.
- J. Xiao, J. Z. Hu, H. Chen, M. Vijayakumar, J. Zheng, H. Pan and E. D. Walter, et al., Nano Lett., 2015, 15, 3309 CrossRef CAS PubMed.
- J.-Q. Huang, Q. Zhang, H.-J. Peng, X.-Y. Liu, W.-Z. Qian and F. Wei, Energy Environ. Sci., 2014, 7, 347 CAS.
- C. Barchasz, J.-C. Lepretre, S. Patoux and F. Alloin, Electrochim. Acta, 2013, 89, 737 CrossRef CAS.
- T. Yim, M.-S. Park, J.-S. Yu, K. J. Kim, K. Y. Im, J.-H. Kim, G. Jeong and Y. N. Jo, et al., Electrochim. Acta, 2013, 107, 454 CrossRef CAS.
- Z. Feng, C. Kim, A. Vijh, M. Armand, K. H. Bevan and K. Zaghib, J. Power Sources, 2014, 272, 518 CrossRef CAS.
- R. Xu, I. Belharouak, X. Zhang, R. Chamoun, C. Yu, Y. Ren, A. Nie, R. Shahbazian-Yassar and J. Lu, et al., ACS Appl. Mater. Interfaces, 2014, 6, 21938 CAS.
- J.-J. Chen, R.-M. Yuan, J.-M. Feng, Q. Zhang, J.-X. Huang, G. Fu and M.-S. Zheng, et al., Chem. Mater., 2015, 27, 2048 CrossRef CAS.
- L. E. Camacho-Forero, T. W. Smith and P. B. Balbuena, J. Phys. Chem. C, 2017, 121, 182–194 CAS.
- X. Chen, T. Hou, B. Li, C. Yan, L. Zhu, C. Guan, X. Cheng, H. Peng, J. Huang, Q. Zhang, DOI:10.1016/j.ensm.2017.01.003.
- N. A. Canas, S. Wolf, N. Wagner and K. Friedrich, J. Power Sources, 2013, 226, 313 CrossRef CAS.
- M. Cuisinier, P.-E. Cabelguen, S. Evers, G. He, M. Kolbeck, A. Garsuch and T. Bolin, et al., J. Phys. Chem. Lett., 2013, 4, 3227 CrossRef CAS.
- A. Kawase, S. Shirai, Y. Yamoto, R. Arakawa and T. Takata, Phys. Chem. Chem. Phys., 2014, 16, 9344 RSC.
- Q. Wang, J. Zheng, E. Walter, H. Pan, D. Lv, P. Zuo, H. Chen and Z. D. Deng, et al., J. Electrochem. Soc., 2015, 162, A474 CrossRef CAS.
- M. Wild, L. O'Neill, T. Zhang, R. Purkayastha, G. Minton, M. Marinescu and G. J. Offer, Energy Environ. Sci., 2015, 8, 3477 CAS.
- S. Han, S. Yun, O. Borodin, D. M. Seo, R. D. Sommer, V. G. Young Jr and W. A. Henderson, J. Phys. Chem. C, 2015, 119, 8492 CAS.
- D. M. Seo, O. Borodin, S. Han, Q. Ly, P. D. Boyle and W. A. Henderson, J. Electrochem. Soc., 2012, 159, A553 CrossRef CAS.
- D. M. Seo, O. Borodin, S. Han, P. D. Boyle and W. A. Henderson, J. Electrochem. Soc., 2012, 159, A1489 CrossRef CAS.
- D. M. Seo, O. Borodin, D. Balogh, M. O'Connell, Q. Ly, S. Han, S. Passerini and W. A. Henderson, J. Electrochem. Soc., 2013, 160, A1061 CrossRef CAS.
- J. W. Smith, R. K. Lam, A. T. Sheardy, O. Shih, A. M. Rizzuto, O. Borodin, S. J. Harris, D. Prendergaste and R. J. Saykally, Phys. Chem. Chem. Phys., 2014, 16, 23568 RSC.
- O. Borodin and G. D. Smith, J. Phys. Chem. B, 2009, 113, 1763 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- L. E. Camacho-Forero, T. W. Smith, S. Bertolini and P. B. Balbuena, J. Phys. Chem. C, 2015, 119, 26828 CAS.
- Z. Liu, S. Bertolini, P. B. Balbuena and P. P. Mukherjee, ACS Appl. Mater. Interfaces, 2016, 8, 4700 CAS.
- J. Yeon, J. Jang, J. Han, J. Cho, K. Lee and N. Choi, J. Electrochem. Soc., 2012, 159, A1308 CrossRef CAS.
- M. Hagen, P. Schiffels, M. Hammer, S. Dorfler, J. Tubke, M. J. Hoffmann, H. Althues and S. Kaskel, J. Electrochem. Soc., 2013, 160, A1205 CrossRef CAS.
- W. Zhu, A. Paolella, C.-S. Kim, D. Liu, Z. Feng, C. Gagnon, J. Trottier, A. Vijh, A. Guerfi, A. Mauger, C. M. Julien, M. Armandd and K. Zaghib, Sustainable Energy Fuels, 2017, 1, 737 Search PubMed.
- J. Sun, Y. Sun, M. Pasta, G. Zhou, Y. Li, W. Liu, F. Xiong and Y. Cui, Adv. Mater., 2016, 28, 9797 CrossRef CAS PubMed.
- H. Yao, K. Yan, W. Li, G. Zheng, D. Kong, Z. Seh, V. Narasimhan, Z. Liang and Y. Cui, Energy Environ. Sci., 2014, 7, 3381 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Distances of all S–S bonds in lithium polysulfides after optimization. The distances and binding energy between polysulfide ions and lithium ions in DOL and DME solvation. Two videos for the process of structure optimization for Li2S6 in DOL and DME. The solvation structure of Li2S6 in the polarized continuum model of DOL and DME respectively. The curve of Gibbs free energy of Li+ solvation. The calculated Raman spectroscopy of lithium polysulfides. See DOI: 10.1039/c7ra04673a |
| This journal is © The Royal Society of Chemistry 2017 |