
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bisphenol A exposure triggers apoptosis via three signaling pathways in Caenorhabditis elegans
Yun Wang *ab,
Lianfeng Zhangc,
Xun Luo*a,
Shunchang Wanga and
Yuanyuan Wanga
*ab,
Lianfeng Zhangc,
Xun Luo*a,
Shunchang Wanga and
Yuanyuan Wanga
aSchool of Bioengineering, Huainan Normal University, Huainan, Anhui 232001, China. E-mail: wy2015@ustc.edu.cn; luoxun72@163.com; scwangl@hotmail.com; 1290821431@qq.com; Fax: +86-551-63601443
bSchool of Life Sciences, CAS Center for Excellence in Molecular Cell Science, University of Science and Technology of China, Hefei, Anhui 230027, China
cSchool of Chemistry and Material Engineering, Huainan Normal University, Huainan, Anhui 232001, China. E-mail: zhanglianfenghfgd@163.com
First published on 27th June 2017
Abstract
Bisphenol A (BPA) is a well-known xenoestrogen that is responsible for multiple adverse effects including reproductive toxicity and apoptosis. However, the mechanism of apoptosis remains elusive. Our present study investigated the mode of BPA's action by evaluating its effect on germline apoptosis in C. elegans. The DNA damage response (DDR) pathway, mitogen-activated protein kinase (MAPK) signaling cascades and insulin-like growth factor-1 (IGF-1) networks were selected as candidates in our model, which may be responsible for BPA-induced apoptosis. The wild type and related mutants were treated with BPA in young adult C. elegans. The results indicate that: (1) in the DDR pathway, both caspase protein CED-3 and Apaf-1 protein CED-4 were required for BPA-induced apoptosis. The apoptosis was blocked by ced-9, and was activated by egl-1. Checkpoint protein HUS-1 exerted proapoptotic effects, and cep-1, the homologue of p53, also participated in BPA-induced apoptosis. (2) As for MAPK signaling cascades, MAPKKK Raf LIN-45, MAPKK MEK-2/MEK-1/JKK-1 and MAPK JNK-1 may play important roles in the BPA-induced apoptotic process. (3) The results also indicated that IGF-1 network and its target protein DAF-16/FOXO, played important roles in BPA-induced apoptosis. DAF-2/IGF-1R and AKT-1/PKB negatively regulated the apoptosis via DAF-16 induction. Our present studies uncover a signaling network of BPA-induced apoptotic process.
1. Introduction
Bisphenol A (BPA), as an organic chemical raw material, has been frequently used in a wide range of consumer products, such as drinking bottles, food containers and toys, and has become ubiquitous in the environment.1 It is implied that humans might be potentially exposed to BPA via the food chain and occupational routes. BPA can be detected in up to 92.6% human bodies and there is an increasing concern about its higher bioaccumulation in developing organisms.2,3 BPA can produce detrimental effects on the body. Di Donato et al. found that BPA interfered with normal hormone signal pathway transduction, resulting in prolonged exposure of receptors to stimuli or interference with cellular hormone signaling in target cells.4 Meanwhile, more and more studies have provided evidences for the adverse effects of BPA (especially at the high dosages) in different systems both in vivo and in vitro.5–8There have been growing concerns about the reproductive toxicity of bisphenol A to various species in recent years.9 Exposure to BPA significantly reduced the number of germ cells and caused embryo abnormalities in the testis of mammals, and induced spermatocytes apoptosis in rare minnow testis, which was probably under regulation of intrinsic mitochondrial apoptotic pathway.10 It could cause endocrine disruption and oxidative stress in male offspring, leading to inhibition of spermatogenesis in SD rats.11 The exposure of BPA to pregnant rats could increase fetal resorption, induce gross fetal anomalies, and decrease spermatogenesis in offspring.12 It should be noted that the reproductive toxicity of BPA has been variable in different experiments, animal species, doses, duration of exposure, and the biological end points. Thus, comprehensive study is an urgent demand to illustrate the reproductive dysfunction of BPA in intact organisms.
Apoptosis is a programmed event during animal normal development, which plays an important role in removing defective germ cells, maintaining suitable germ cell to Sertoli cell ratio, and controlling the sperm production.13 BPA has been reported to induce autophagy and apoptosis in Sertoli and Leyding cells of SD rats testis and cell death in Sertoli-germ cells in male rats.14 It was reported that BPA can induce the activation of protein kinase A (PKA) and Akt in testicular Leydig cells.15 In human granulosa cells, BPA was shown to induce peroxisome proliferator-activated receptor gamma (PPARc) and to downregulate FSH-stimulated steroidogenesis and proliferation via the activation of GATA sequence-binding transcription factors.16 Furthermore, the Pten/Akt signaling pathway was shown to participate in BPA-induced apoptosis of rat Sertoli cells.17 Exposure to BPA might induce dysfunction of insulin secretion and increase apoptosis in rat insulinoma cells through mitochondrial damage.18 In summary, BPA has been shown to be involved in cell proliferation, apoptosis, and steroidogenesis through various receptors and/or signaling pathways in various cell models. However, little is known about the underlying mechanism of germ cell apoptosis induced by BPA, especially the involvement of apoptosis signal pathways.
The nematode Caenorhabditis elegans is a powerful genetic model to explore environmental toxicants in vivo, especially for dissecting key pathways that are essential for stress response.19 The transparent cuticle allows observing and distinguishing germ cells at the whole organism level. The C. elegans germline apoptosis during oogenesis is a fundamentally important reproductive process, which is evolutionarily conserved from worms to humans. The validity to study genes and pathways related to human health and disease has been well established in C. elegans by dissecting the pathway controlling apoptosis.20
In C. elegans, the evolutionarily conserved DDR signaling is composed of HUS-1, CEP-1, EGL-1, CED-9, CED-4, and CED-3. Conventional MAPKs include ERK MAPK, JNK MAPK, and p38 MAPK. IGF-1 signaling pathway is composed of DAF-2/IGF-1R, AGE-1/PI3K and the subsequent PDK-1, AKT-1/2 and SGK-1.21 In this study, using signal pathways that mentioned above, we found a signaling network of BPA-induced apoptotic process.
2. Materials and methods
2.1. Strains and reagents
The strains used in this study were as follows: wild type Bristol N2, ced-3(n717), ced-4(n1162), egl-1(n1084n3082), ced-9(n1950), cep-1(gk138), hus-1(op241), lin-45(n2520), mek-2(n1989), mpk-1(ku1), nsy-1(ag3), mek-1(ks54); jkk-1(km1); mkk-4(ju91), jnk-1(gk7), sek-1(ag1), pmk-1(km25), daf-2(e1370), age-1(mg109), pdk-1(sa680), akt-1(ok525), daf-16(mu86) and TJ356 which were provided by the Caenorhabditis Genetics Center (Minneapolis, MN, USA). All strains were maintained on nematode growth medium (NGM) plates that were seeded with Escherichia coli OP50 as a food source by standard procedures.22 Gravid hermaphrodites were lysed in an alkaline hypochlorite solution to obtain age-synchronized worms, and the young adult worms were subjected to BPA treatment at 20 °C. BPA (Sigma, St. Louis, USA) was dissolved in 100% DMSO and diluted with K-medium at the final DMSO concentration of 0.5% for all doses.All the experimental animals used were treated in accordance with the recommendations in the Guide for the Regulation for the Administration of Affairs Concerning Experimental Animals of P. R. China, and approved by Anhui Administration Committee of Laboratory Animals.
2.2. Sample preparation
Animal handling and chemical exposure were performed as described previously.23 Apoptotic germ cells and gene expression were assessed in the synchronized young adult worms that were exposed to the indicated BPA concentrations. For each concentration and control, three independent assays were conducted for all of the tests.2.3. Apoptosis assay
Apoptotic germ cells were measured by acridine orange (AO) staining using a procedure as described previously.21 Apoptotic cells were examined under a fluorescent microscopy. At least 30 worms were measured for each treatment. The apoptotic cells appeared yellow or yellow-orange, which represented increased DNA fragmentation, whereas intact cells were uniformly green. Worms were then picked and mounted onto agar pads on microscope slides in 60 μg mL−1 levamisole, after which they were examined with an Olympus IX71 inverted microscope (Olympus, Japan). Pictures were taken using a Zeiss Imager A2 (Jena, Germany). Only the gonad arm in the posterior part of the body was scored.2.4. DAF-16 translocation determination
The TJ356 strain was used to determine the cellular distribution of DAF-16 in the living worm. In this strain, GFP was fused to the daf-16 gene.24 To determine whether BPA exposure affected the DAF-16 translocation from cytoplasm to nucleus in intestine cells, the percentages of translocation were measured using a procedure as described previously.252.5. Real-time PCR
Synchronized wild type young adult hermaphrodites were grown at 20 °C and exposed to vehicle DMSO or 50 μM BPA for 6 h. Worms were then collected and frozen in liquid nitrogen. cDNAs were synthesized with random hexamers by using the PrimeScript RT Reagent Kit (TaKaRa Bio, Dalian, Liaoning, China) according to the manufacturer's protocol. act-1 was used as the internal control, and the RNA levels of the genes of interest were normalized to the act-1 level for comparison. mRNA expression was assessed by quantitative real-time PCR on an ABI 7500 FAST Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using SuperReal PreMix (SYBR Green) in a 96-well plate. The gene expression data were analyzed using the comparative 2−ΔΔCt method.26 Triplicates for each sample were included for one single reaction.2.6. Statistical tools
All values were expressed as the mean ± standard error of the mean (SEM). One-way analysis of variance (ANOVA) followed by Duncan's test (SPSS 22.0) was performed to assess the statistical significance. Differences were considered significant at P < 0.05.3. Results
3.1. BPA induces germ cell death of wild type worms
C. elegans germ cell corpses were vitally stained by AO after 6 h of BPA exposure, and apoptotic cells were scored (Fig. 1A). The average number of spontaneous germ cell corpses in wild type worms was 3.45 ± 0.20. As shown in Fig. 1B, there was a dose-dependent increase of germ cell death in C. elegans exposed to BPA. A significant increase of germ cell death was observed in worms exposed to BPA at concentrations of 1, 10, 50 and/or 200 μM for 6 h, respectively. The germ cell corpses in worms exposed to 50 μM BPA was 7.85 ± 0.32, which was 2.28-fold changes compared with the control. However, as the BPA concentration increased to 200 μM, the level of apoptotic cells did not raise significantly compared with 50 μM group. Thus, 6 h exposure of 50 μM BPA was used in the following experiments.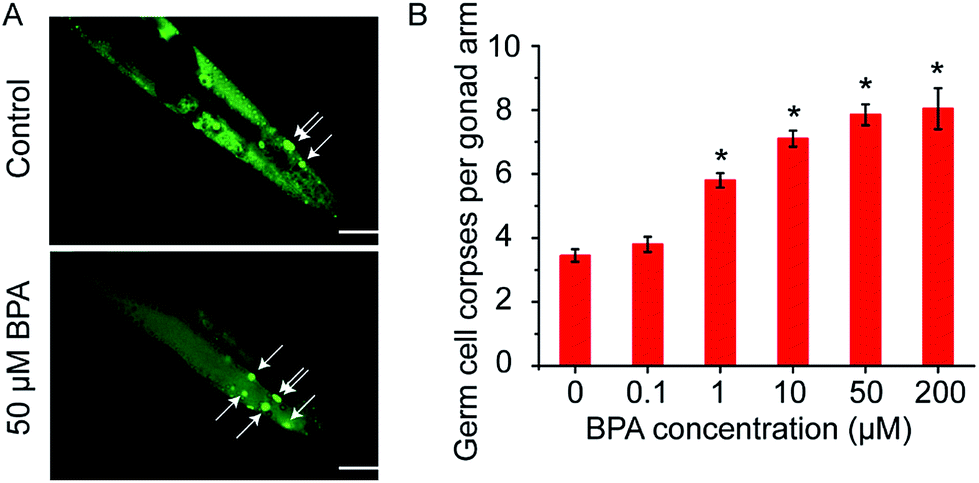 | ||
| Fig. 1 BPA induces C. elegans germline apoptosis. (A) Representative pictures of germline apoptosis of untreated control and 50 μM of BPA treated for 6 h. Germline apoptosis was assayed by AO staining. Germ cell corpses are indicated by white arrows. The scale bar represents 20 μm. (B) Wild type young adult hermaphrodites were treated with indicated concentrations of BPA for 6 h, and germ cell corpses per gonad arm were determined. All values are represented as the mean ± SEM, n = 30; * represents P < 0.05, BPA treatment vs. control. | ||
3.2. DDR signaling pathway is essential for BPA-induced germ cell death
In C. elegans apoptotic machinery, the caspase protein CED-3 and CED-4 are required for the killing process. CED-9 inhibits programmed cell death by sequestering CED-3 and CED-4. To confirm the germ cell corpses induced by BPA under the core apoptosis machinery, animals with loss-of-function (lf) mutantions ced-3(n717), ced-4(n1162), hus-1(op241), cep-1(gk138), egl-1(n1084n3082) and gain-of-function (gf) mutantion ced-9(n1950) were exposed to BPA respectively. Few germline cell corpses were observed in the ced-3(n717) and ced-4(n1162) under BPA exposure (Fig. 2A), suggesting the essential roles of ced-3 and ced-4 in mediating BPA-induced germline apoptosis.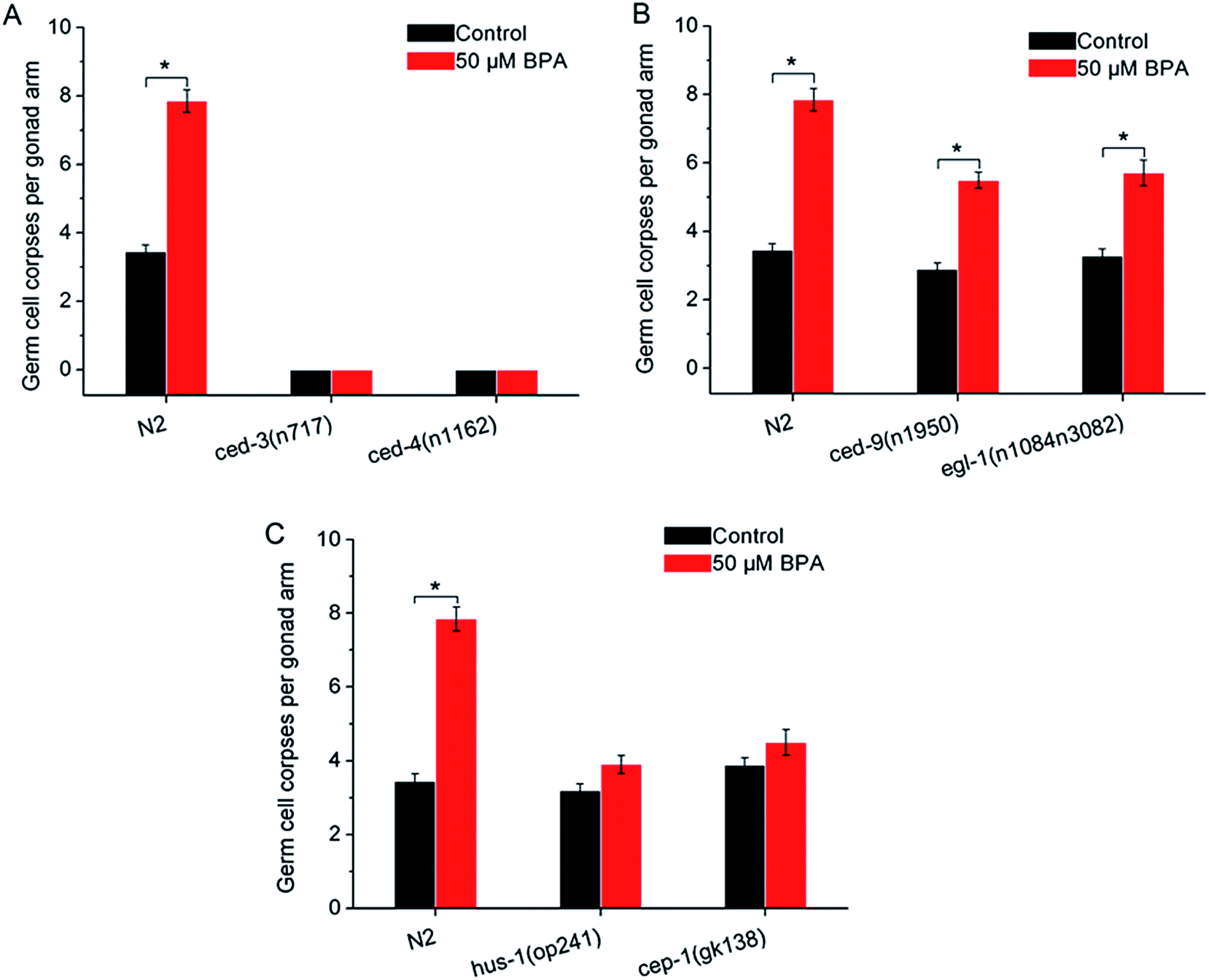 | ||
| Fig. 2 Core apoptotic machinery is required for BPA-induced germline apoptosis. (A) Germline apoptosis induced by BPA in ced-3(n717), ced-4(n1162) strains, and (B) in ced-9(n1950) and egl-1(n1084n3082) strains. (C) The checkpoint gene hus-1 played proapoptotic role in BPA-induced apoptosis, and CEP-1 promoted DNA damage-induced apoptosis. For comparison, germline apoptosis data for the wild type in all figures herein are presented in the same manner as shown in Fig. 1. All values are represented as the mean ± SEM; * represents P < 0.05, BPA treatment vs. control. | ||
The C. elegans ced-9 encodes a protein homologous to mammalian BCL-2. Under BPA stress, germline apoptosis in gf strain ced-9(n1950) was much lower than that of wild type, indicating that ced-9 exerted anti-apoptotic effects under BPA stress (Fig. 2B). egl-1 encodes a protein that contains a region similar to the BH3 (Bcl-2 homology region 3). Using lf strain egl-1(n1084n3082), the result showed that egl-1 was required for BPA-induced germline apoptosis (Fig. 2B).
In mammalian cells, checkpoint proteins, such as HUS-1, transduce DNA damage signals and mediate apoptosis.27 Tumor suppressor protein p53 was proved to be necessary for genotoxic agents-induced apoptosis. To confirm whether these checkpoint proteins are involved in BPA-induced apoptosis, worms carrying mutated alleles of hus-1 and cep-1 were treated with BPA. Germ cell corpses in both hus-1(op241) and cep-1(gk138) strains remained unchanged under BPA stress compared with the corresponding control (Fig. 2C), indicating that HUS-1 was indispensable and CEP-1 promoted DNA damage-induced apoptosis under BPA stress in worms.
3.3. MAPK signaling pathways are involved in BPA-induced germ cell death
In C. elegans, there are some core components in MAPK signaling pathways, such as ERK MAPK signaling pathway including lin-45, mek-2 and mpk-1, JNK MAPK signaling pathway including nsy-1, mek-1, jkk-1, mkk-4 and jnk-1, p38 MAPK signaling pathway including nsy-1, sek-1 and pmk-1. To confirm the role of ERK MAPK signaling pathway in BPA-induced apoptosis, lf mutants including lin-45(n2520), mek-2(n1989) and mpk-1(ku1) were exposed to BPA. The mpk-1(ku1) mutant showed a much higher induction of cell death like wild type worm, whereas germline apoptosis in lin-45 or mek-2 mutant was significantly suppressed compared with wild type worms (Fig. 3A). In other word, germ cell corpses in both lin-45(n2520) and mek-2(n1989) strains were significantly alleviated compared with the wild type under BPA stress (P < 0.05), while, without BPA exposure, the levels of apoptotic cells of these two mutants did not change significantly compared with wild type. The present results suggested that lin-45 and mek-2 were required for BPA-induced apoptosis.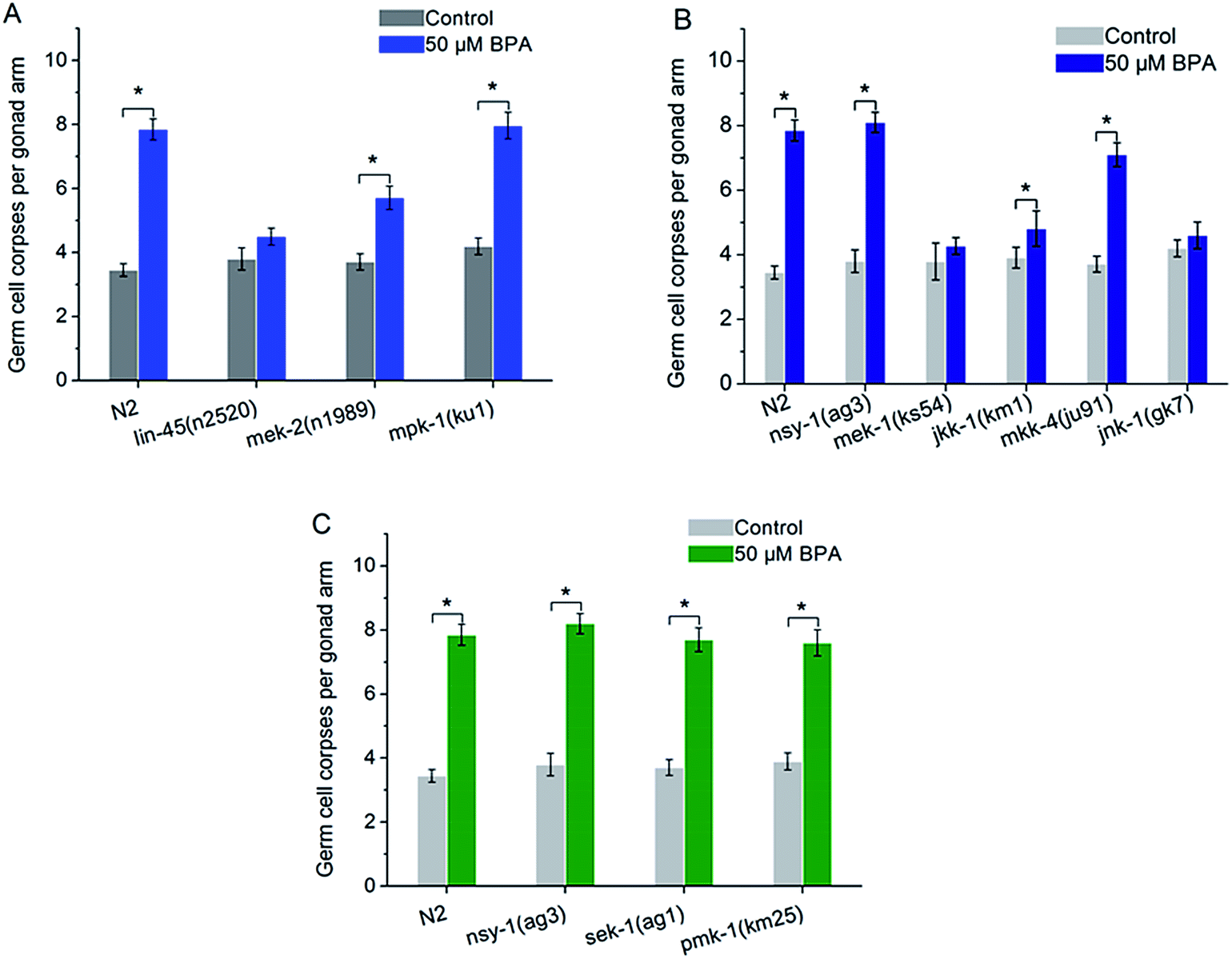 | ||
| Fig. 3 ERK MAPK, JNK MAPK, but not p38 MAPK cascades are required for BPA-induced germline apoptosis. Role of (A) ERK MAPK cascade, (B) JNK MAPK cascade, and (C) p38 MAPK cascade on BPA-induced germline apoptosis. All values are represented as the mean ± SEM; * represents P < 0.05, BPA treatment vs. control. | ||
nsy-1 is a MAPKKK family member that activates JNK-1 and p38 MAPK cascades through its substrate MAPKKs. The mek-1 and jkk-1 genes encode MKK7-like MAPKKs. mkk-4 encodes a protein that is homologous to the MKK4 family of MAPKK. NSY-1 and MKK-4 are activators of the C. elegans JNK homologue JNK-1.28 In the present study, germline apoptosis induced by BPA remained unchanged in the mek-1(ks54) or jnk-1(gk7) strain compared to that of the corresponding control (Fig. 3B). Meanwhile, we found that BPA-induced germline apoptosis in jkk-1(km1) strain was significantly lower than that of wild type (P < 0.05) (Fig. 3B). In contrast, BPA-induced germline apoptosis increased significantly in the nsy-1(ag3), mkk-4(ju91) mutants like wild type (Fig. 3B). These results indicated that MEK-1, JKK-1 and JNK-1 were indispensible to BPA-induced germline apoptosis, whilst the function of NSY-1, MKK-4 may be not or only partially involved in the apoptosis induction.
The C. elegans NSY-1-SEK-1-PMK-1 signaling cascade is a conserved p38 MAPK pathway. PMK-1 can be activated by MKK3/6-type MAPKK SEK-1, which is in turn activated by MAPKKK NSY-1.29 As shown in Fig. 3C, BPA-induced apoptosis in the mutated strain of nsy-1(ag3), sek-1(ag1) or pmk-1(km25) displayed a similar pattern to wild type strain (Fig. 3C). The present results suggested that p38 MAPK pathway was not required for BPA-induced apoptosis.
3.4. IGF-1 signaling pathway is required for BPA-induced germ cell death
To investigate whether IGF-1 signaling pathway was involved in BPA-induced germline apoptosis, daf-2 and its downstream genes lf mutants were exposed to 50 μM BPA (Fig. 4A). The C. elegans daf-2 encodes an insulin-like growth factor-1 receptor (IGF-1R). Compared to wild type worms, daf-2(e1370) strain produced more germ cell corpses. The mammalian PI3K homolog age-1 of C. elegans lies downstream of daf-2. In this study, germline apoptosis in age-1(mg109) showed no difference compared to wild type. The C. elegans pdk-1 lies downstream of age-1 and encodes a protein homologous to mammalian 3-phosphoinositide-dependent kinase-1 (PDK1), and germline apoptosis in pdk-1(sa680) strain showed a minor increase compared to that of wild type. The C. elegans AKT-1 is a candidate to transduce IGF-1 signals. In our study, dysfunction of akt-1 led to a major induction of germline apoptosis compared with wild type. C. elegans DAF-16, the only forkhead box class O (FOXO) homologue, notably, antagonized IGF-1 signals in signaling the BPA-induced apoptosis. As shown in Fig. 4A, germline apoptosis in daf-16(mu86) showed no difference compared to wild type. Further analysis indicated that DAF-16::GFP nuclear translocation occurred in a dose-dependent manner under BPA for 6 h (Fig. 4B and C). It is suggested that oxidative stress might be one of the early DNA damage events for apoptosis progression.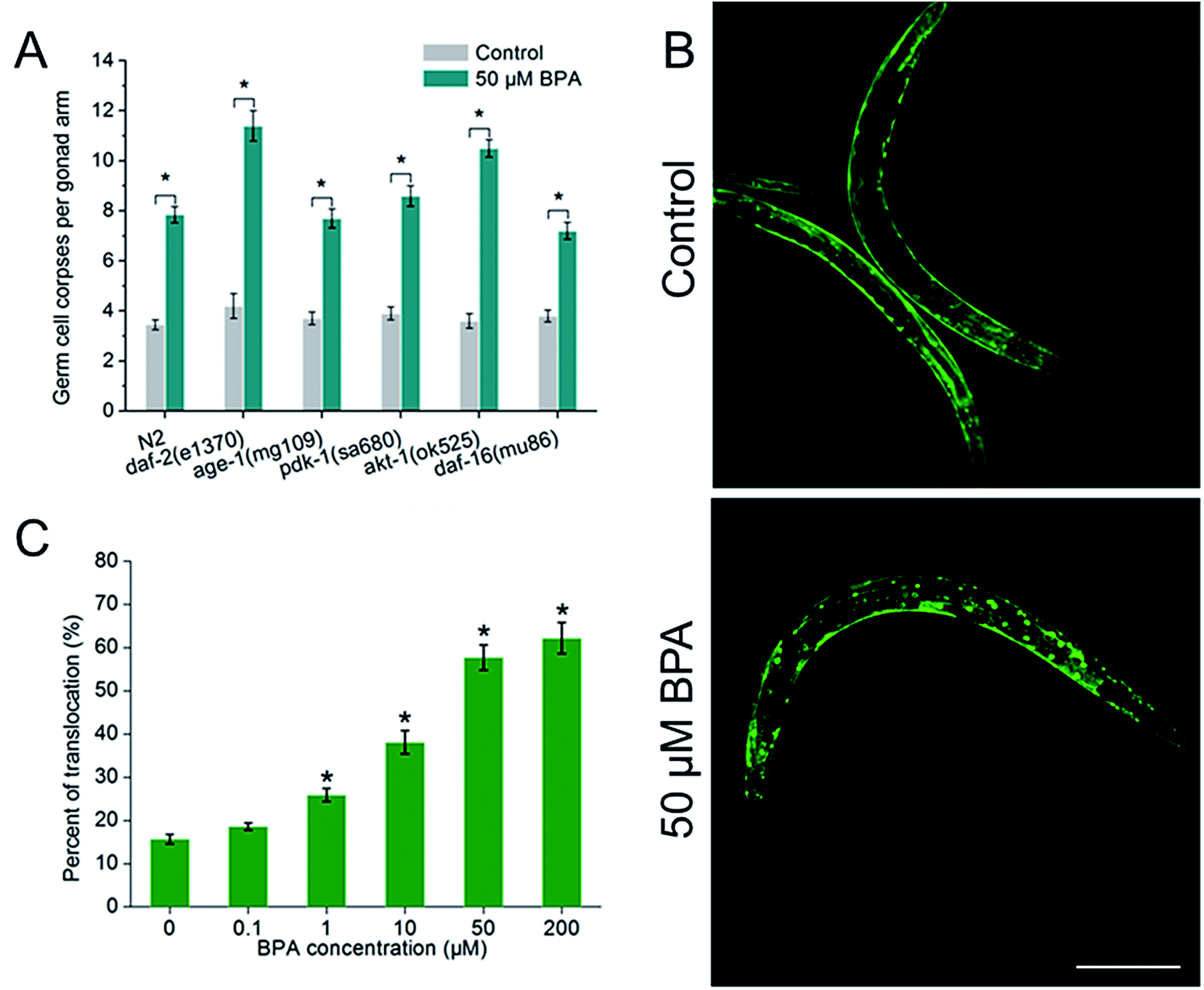 | ||
| Fig. 4 IGF-1 network and DAF-16/FOXO, play important roles in BPA-induced apoptosis. (A) daf-2 and akt-1 negatively regulated the BPA-induced apoptosis. daf-16 antagonized IGF-1 signals in signaling the BPA-induced apoptosis. (B) Representative pictures of DAF-16 translocation of control and 50 μM of BPA treated for 6 h. The scale bar represents 50 μm. (C) Quantification of DAF-16 translocation of TJ356 hermaphrodites which were treated with indicated concentrations of BPA for 6 h. All values are represented as the mean ± SEM; * represents P < 0.05, BPA treatment vs. control. | ||
To further understand the molecular mechanisms of the stress response in vivo, the mRNA levels of all of the potential apoptosis-related genes mentioned above were measured. Synchronized wild type young adult were exposed to vehicle DMSO or 50 μM BPA for 6 h, and the gene whose mRNA expression level significantly changed were displayed (Fig. 5). It is obvious that BPA exposure could induce alterations in most of the involved genes in wild type worms.
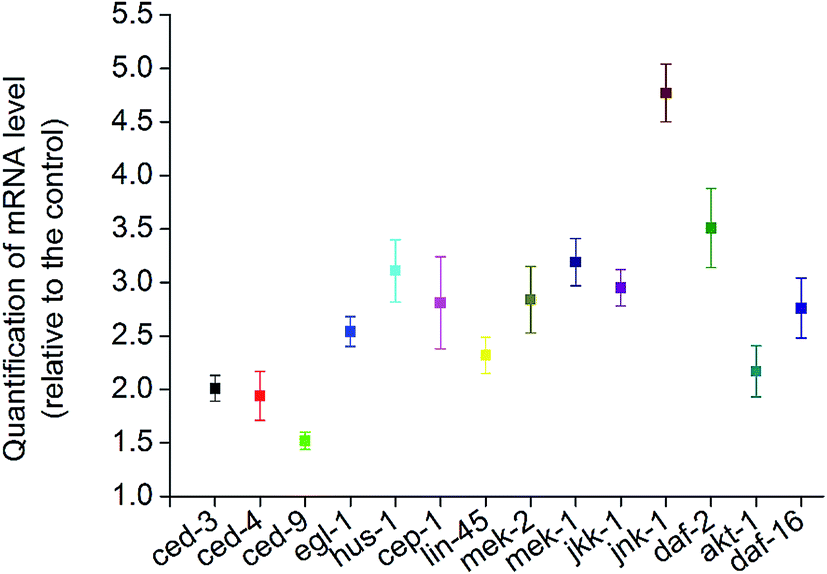 | ||
| Fig. 5 Quantitative summary of stress-related gene expression profiles tested in wild type exposed to BPA for 6 h. Values of gene expression were normalized using actin mRNA and represent means (n = 3) relative to the untreated control. | ||
4. Discussion
BPA is a well-known environmental estrogenic disruptor that causes a series of adverse effects. Previous research suggested that BPA exposure could cause an accelerated aging process in C. elegans mediated by the induction of oxidative stress.30 In this study, we firstly confirmed that BPA exposure could cause germline apoptosis in a dose-dependent manner. Then, we wanted to address the mechanism of the apoptotic process. Therefore, we studied BPA-induced apoptosis in C. elegans strains carrying mutated genes encoding key regulators of apoptosis.Our work demonstrates that there are three distinct pathways of BPA-induced apoptosis (Fig. 6). (1) DDR signaling pathway is essential for BPA-induced germ cell death. (2) ERK or JNK MAPK signaling pathways are involved in BPA-induced apoptosis. (3) IGF-1 signaling pathway is required for BPA-induced germ cell death.
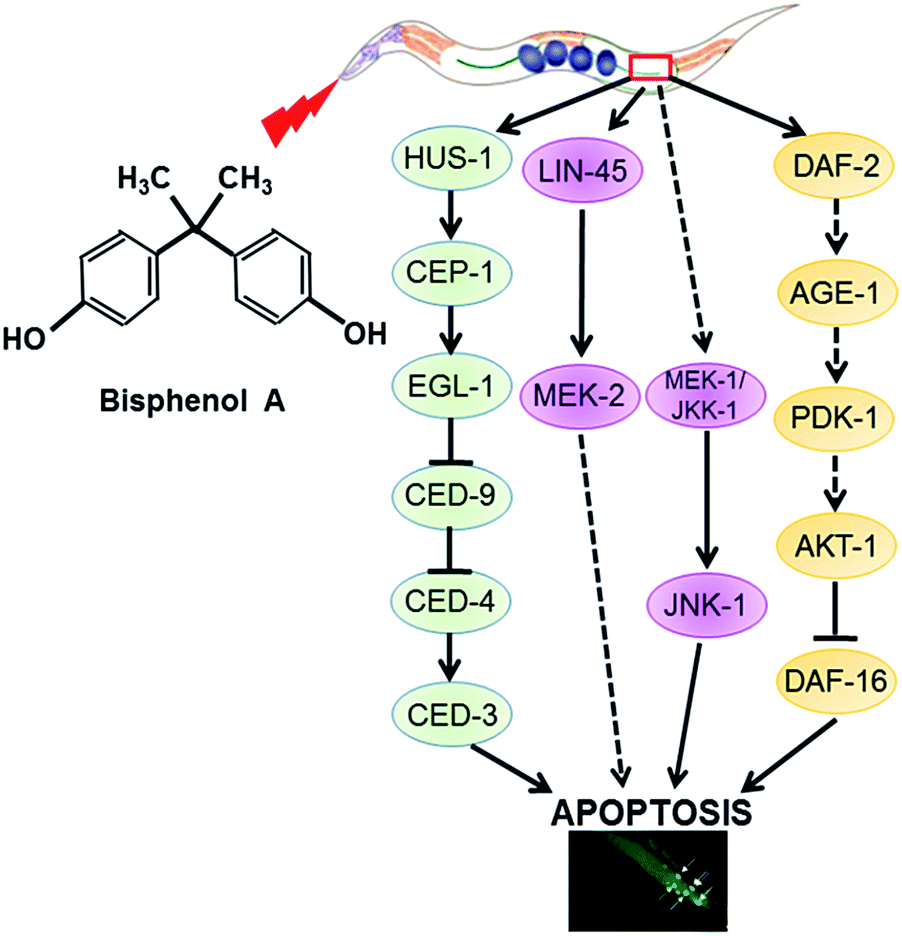 | ||
| Fig. 6 Proposed working model for BPA-induced apoptotic signaling pathways in C. elegans. | ||
Due to the absence of germline apoptosis physiologically or under BPA exposure in ced-3 and ced-4 mutants, CED-3 and CED-4 play pivotal roles on BPA-induced apoptosis. CED-3 is activated by CED-4, which is in turn activated by EGL-1 through binding to CED-9. Take into consideration of the transcription levels of ced-3, ced-4, egl-1 and ced-9, the present study confirmed that the core apoptotic machinery proteins CED-3, CED-4 and EGL-1 were required for BPA-induced apoptosis, and CED-9 acted as an antiapoptotic effector. Previous study demonstrated that ROS production and DNA damage are two important events under BPA exposure in C. elegans.30,31 DNA damage-induced apoptosis required checkpoint gene such as hus-1. In C. elegans, cep-1 is the only p53-like gene, which has been shown to be essential for apoptosis induced by DNA-damage agents.32 The present results suggested that hus-1 and cep-1 were both required for BPA-induced apoptosis, which were strengthened by the elevated expression of them (Fig. 5).
MAPKs phosphorylate specific serines and threonines of target protein substrates and regulate many diverse physiological processes, including stress responses and apoptosis. Recent data have implicated role ERK MAPK pathway in apoptosis promotion in several cell types under genotoxic or nongenotoxic stresses.33 Our study showed that the members of ERK signaling behaved differently under BPA exposure. MAPKKK Raf LIN-45 and MAPKK MEK-2 played pivotal roles in mediating BPA-induced germline apoptosis, while MPK-1 was not essential. The C. elegans nsy-1 gene encodes a mammalian homolog of ASK1, while mkk-4 and jnk-1 encode the homologs of mammalian MKK4 and JNK, respectively. The nsy-1 lf mutant conferred enhanced susceptibility to killing by pathogens,34 while the mutants of mkk-4 and jnk-1 exhibited hypersensitivity to persistent toxic pollutants.35 The roles of JNK in BPA-induced apoptosis are controversial: JNK-dependent apoptosis was observed in mouse hippocampal HT-22 cells and 15P-1 Sertoli cells,36 but it seemed unnecessary in HT4 neuronal cells.34 In our cases, the results indicated that BPA-induced apoptosis was independent of NSY-1 and MKK-4, but dependent of MEK-1, JKK-1 and JNK-1. Notably, jnk-1 mRNA accumulated nearly 5-fold (Fig. 5). In general, p38 MAPK pathway is associated with diverse cellular processes including apoptosis and stress responses.37,38 Unexpectedly, the present results demonstrated that p38 MAPK pathway was not required for BPA-induced apoptosis. In summary, these data indicate that the intracellular signaling pathways of BPA-induced germline apoptosis is dependent on ERK and JNK MAPK cascades.
Recent studies reported that several members of IGF-1 signaling were involved in apoptosis regulation in response to BPA.39 Our results indicated that C. elegans DAF-2/IGF-1R and AKT-1 acted as anti-apoptotic effectors in response to BPA exposure, however, age-1 and pdk-1 may not be important in response to BPA exposure (Fig. 4A and 5). DAF-16, the only C. elegans FOXO homologue, was reported to play anti-apoptotic roles under genotoxic stress.40 DAF-16 is negatively regulated by DAF-2 through AGE-1, PDK-1 and AKT-1 cascades.41 In this study, there may be some antagonistic or cooperative effects between DAF-2 and DAF-16, because BPA-induced apoptosis promoted by AKT-1 or DAF-2 dysfunctions were attenuated by DAF-16 depletion. Based on the above observations, we speculate that DAF-16 play apoptotic role under BPA stress through the canonical insulin/IGF-1 pathway.42 On the other hand, BPA-induced apoptosis accompanied by induction of daf-16 transcription (Fig. 5). Thus, signals from DAF-2 to DAF-16 might bypass AGE-1/PDK-1/AKT-1 cascades under BPA exposure (Fig. 6), which were consistent with previous findings.43 In C. elegans, mutations in daf-16 increased sensitivities to oxidative stress, whereas mutation in daf-2 caused resistance effects.44 On the other hand, DAF-16::GFP nuclear translocation in soma under BPA exposure was ascribed to their hypersensitive to oxidative stress.
Furthermore, clearly more research is necessary to systematically analyze the pathways of BPA-induced apoptosis. There may be crosstalk among the three signaling pathways mentioned above, as it is reported that insulin/IGF-1 signaling can selectively engage AKT-2/DAF-16 to promote DNA damage-induced germ cell apoptosis downstream of CEP-1 through the Ras pathway.45 In addition, if the inhibitors or RNA interference can be used to determine genes involved in BPA-induced apoptosis, the conclusions must be more solid.
5. Conclusion
In summary, using C. elegans as an in vivo model to investigate mechanisms on BPA-induced apoptosis, we conclude that DDR signaling components HUS-1, CEP-1, EGL-1, CED-9, CED-4 and CED-3 acted as core apoptotic machinery; ERK and JNK MAPK signaling components LIN-45, MEK-2, MEK-1, JKK-1 and JNK-1 are essential; and IGF-1 signaling components DAF-2 and AKT-1 negatively regulated the apoptosis. DAF-16/FOXO exerted apoptotic role on IGF-1 signals in BPA-induced apoptosis. Our data establish that DNA damage response pathway plays essential roles in the BPA-induced apoptosis. MAPK cascades and IGF-1 signaling pathway also partially participates in the process. These findings suggest a potential disease mechanism involving genotoxic and/or non-genotoxic effects of BPA that warrants future investigation.Author contributions
Yun Wang, Xun Luo and Shunchang Wang conceived and designed the experiments; Yun Wang, Lianfeng Zhang and Yuanyuan Wang performed the experiments; Yun Wang analyzed the data and wrote the paper.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful to the Caenorhabditis Genetics Center (CGC), the International C. elegans Gene Knockout Consortium for providing the strains. We gratefully acknowledged financial support from the National Natural Science Foundation of China (20907016), the Natural Science Foundation of Anhui Province (1608085MC68), China Postdoctoral Science Foundation (2015M582006).References
- P. Fenichel, N. Chevalier and F. Brucker-Davis, Ann. Endocrinol., 2013, 74, 211–220 CrossRef CAS PubMed.
- A. M. Calafat, X. Ye, L. Y. Wong, J. A. Reidy and L. L. Needham, Environ. Health Perspect., 2008, 116, 39–44 CrossRef CAS PubMed.
- E. Kitraki, I. Nalvarte, A. Alavianghavanini and J. Rüegg, Mol. Cell. Endocrinol., 2015, 417, 191–199 CrossRef CAS PubMed.
- M. Di Donato, G. Cernera, P. Giovannelli, G. Galasso, A. Bilancio, A. Migliaccio and G. Castoria, Mol. Cell. Endocrinol., 2017 DOI:10.1016/j.mce.2017.02.045.
- M. A. Ahbab, N. Barlas and G. Karabulut, Toxicol. Ind. Health, 2015, 30, 63–69 Search PubMed.
- C. Fang, B. Ning, A. B. Waqar, M. Niimi, S. Li, K. Satoh, M. Shiomi, T. Ye, S. Dong and J. Fan, J. Appl. Toxicol., 2015, 35, 1058–1070 CrossRef CAS PubMed.
- S. Khan, S. Beigh, B. P. Chaudhari, S. Sharma, A. H. A. Sayed, S. Ahmad, F. Ahmad, S. Parvez and S. Raisuddin, Environ. Toxicol., 2015, 31, 1922–1934 CrossRef PubMed.
- X. Pan, X. Wang, Y. Sun, Z. Dou and Z. Li, Int. J. Clin. Exp. Med., 2015, 8, 8720–8729 CAS.
- A. Zivgal and J. A. Flaws, Fertil. Steril., 2016, 106, 827–856 CrossRef CAS PubMed.
- Y. Zhang, M. Cheng, L. Wu, G. Zhang and Z. Wang, Aquat. Toxicol., 2016, 179, 18–26 CrossRef CAS PubMed.
- C. Quan, C. Wang, P. Duan, W. Huang, W. Chen, S. Tang and K. Yang, Environ. Toxicol., 2016 DOI:10.1002/tox.22339.
- S. Medwid, H. Guan and K. Yang, Environ. Toxicol. Pharmacol., 2016, 43, 203–208 CrossRef CAS PubMed.
- K. K. Shukla, A. A. Mahdi and S. Rajender, Front. Biosci., 2012, 4, 746–754 CrossRef.
- C. Quan, C. Wang, P. Duan, W. Huang and K. D. Yang, Environ. Toxicol., 2017 DOI:10.1002/tox.22300.
- J. Y. Kim, E. H. Han, H. G. Kim, K. N. Oh, S. K. Kim, K. Y. Lee and H. G. Jeong, Toxicol. Lett., 2010, 193, 200–208 CrossRef CAS PubMed.
- J. Kwintkiewicz, Y. Nishi, T. Yanase and L. C. Giudice, Environ. Health Perspect., 2010, 118, 400–406 CrossRef CAS PubMed.
- C. Wang, W. Fu, C. Quan, M. Yan, C. Liu, S. Qi and K. Yang, Environ. Toxicol., 2014, 30, 793–802 CrossRef PubMed.
- Y. Lin, X. Sun, L. Qiu, J. Wei, Q. Huang, C. Fang, T. Ye, M. Kang, H. Shen and S. Dong, Cell Death Dis., 2013, 4, e460 CrossRef CAS PubMed.
- M. C. K. Leung, P. L. Williams, A. Benedetto, C. Au, K. J. Helmcke, M. Aschner and J. N. Meyer, Toxicol. Sci., 2008, 106, 5–28 CrossRef CAS PubMed.
- P. Laun, S. Büttner, M. Rinnerthaler, W. C. Burhans and M. Breitenbach, Yeast Aging and Apoptosis, Springer, Netherlands, 2012 Search PubMed.
- S. Wang, X. Teng, Y. Wang, H. Q. Yu, X. Luo, A. Xu and L. Wu, Chemosphere, 2014, 112, 248–255 CrossRef CAS PubMed.
- S. Brenner, Genetics, 1974, 77, 71–94 CAS.
- P. L. Williams and D. B. Dusenbery, Environ. Toxicol. Chem., 1990, 9, 1285–1290 CrossRef CAS.
- R. Y. Lee, J. Hench and G. Ruvkun, Curr. Biol., 2002, 11, 1950–1957 CrossRef.
- Y. Wang, F. Jian, J. Wu and S. Wang, Bull. Environ. Contam. Toxicol., 2012, 89, 704–711 CrossRef CAS PubMed.
- Q. Q. Li, J. Skinner and J. E. Bennett, BMC Mol. Biol., 2012, 13, 22 CrossRef CAS PubMed.
- E. R. Hofmann, S. Milstein, S. J. Boulton, M. Ye, J. J. Hofmann, L. Stergiou, A. Gartner, M. Vidal and M. O. Hengartner, Curr. Biol., 2002, 12, 1908–1918 CrossRef CAS PubMed.
- D. H. Kim, R. Feinbaum, G. Alloing, F. E. Emerson, D. A. Garsin, H. Inoue, M. Tanaka-Hino, N. Hisamoto, K. Matsumoto, M. W. Tan and F. M. Ausubel, Science, 2002, 297, 623–626 CrossRef CAS PubMed.
- K. Berman, J. McKay, L. Avery and M. Cobb, Mol. Cell Biol. Res. Commun., 2001, 4, 337–344 CrossRef CAS PubMed.
- L. Tan, S. Wang, Y. Wang, M. He and D. Liu, Toxicol. Lett., 2015, 235, 75–83 CrossRef CAS PubMed.
- Y. Chen, L. Shu, Z. Qiu, D. Y. Lee, S. J. Settle, S. Que Hee, D. Telesca, X. Yang and P. Allard, PLoS Genet., 2016, 12, e1006223 Search PubMed.
- W. B. Derry, A. P. Putzke and J. H. Rothman, Science, 2001, 294, 591–595 CrossRef CAS PubMed.
- S. Cagnol and J. C. Chambard, FEBS J., 2010, 277, 2–21 CrossRef CAS PubMed.
- D. H. Kim, N. T. Liberati, T. Mizuno, H. Inoue, N. Hisamoto, K. Matsumoto and F. M. Ausubel, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10990–10994 CrossRef CAS PubMed.
- H. Du, M. Wang, L. Wang, H. Dai, M. Wang, W. Hong, X. Nie, L. Wu and A. Xu, Toxicol. Sci., 2015, 145, 118–127 CrossRef CAS PubMed.
- S. Lee, K. Suk, I. K. Kim, I. S. Jang, J. W. Park, V. J. Johnson, T. K. Kwon, B. J. Choi and S. H. Kim, J. Neurosci. Res., 2008, 86, 2932–2942 CrossRef CAS PubMed.
- G. L. Johnson and R. Lapadat, Science, 2002, 298, 1911–1912 CrossRef CAS PubMed.
- Y. Wang, S. Wang, X. Luo, Y. Yang, F. Jian, X. Wang and L. Xie, Chemosphere, 2014, 108, 231–238 CrossRef CAS PubMed.
- F. Ariemma, V. D'Esposito, D. Liguoro, F. Oriente, S. Cabaro, A. Liotti, I. Cimmino, M. Longo, F. Beguinot and P. Formisano, PLoS One, 2016, 11, e0150762 Search PubMed.
- C. Quevedo, D. R. Kaplan and W. B. Derry, Curr. Biol., 2007, 17, 286–292 CrossRef CAS PubMed.
- A. Mukhopadhyay, S. W. Oh and H. A. Tissenbaum, Exp. Gerontol., 2006, 41, 928–934 CrossRef CAS PubMed.
- D. van Heemst, Aging Dis., 2010, 1, 147–157 Search PubMed.
- M. Hertweck, C. Göbel and R. Baumeister, Dev. Cell, 2004, 6, 577–588 CrossRef CAS PubMed.
- S. Heniskorenblit, P. Zhang, M. Hansen, M. Mccormick, S. J. Lee, M. Cary and C. Kenyon, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9730–9735 CrossRef CAS PubMed.
- A. J. Perrin, M. Gunda, B. Yu, K. Yen, S. Ito, S. Forster, H. A. Tissenbaum and W. B. Derry, Cell Death Differ., 2013, 20, 97–107 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |