
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoparticles co-delivering pVSVMP and pIL12 for synergistic gene therapy of colon cancer
Yuanyuan Xiao†
 a,
Yuping Yang†a,
Yujiao Wu†a,
Chunmei Wanga,
Hao Chenga,
Wei Zhaoab,
Yang Lia,
Beibei Liua,
Jianlin Longacd,
Wenhao Guoae,
Guangping Gaoa and
Maling Gou*a
a,
Yuping Yang†a,
Yujiao Wu†a,
Chunmei Wanga,
Hao Chenga,
Wei Zhaoab,
Yang Lia,
Beibei Liua,
Jianlin Longacd,
Wenhao Guoae,
Guangping Gaoa and
Maling Gou*a
aState Key Laboratory of Biotherapy, Cancer Center, West China Hospital, Sichuan University, Collaborative Innovation Center for Biotherapy, Chengdu 610041, P. R. China. E-mail: goumaling@scu.edu.cn; Fax: +86-028-85164060; Tel: +86-028-85164061
bDepartment of Thoracic Oncology, Cancer Center, West China Hospital, Medical School, Sichuan University, Chengdu 610041, P. R. China
cLung Cancer Center, Cancer Center, West China Hospital, Medical School, Sichuan University, Chengdu 610041, P. R. China
dDepartment of Oncology, West China Guang'an Hospital, Sichuan University, Guang'an 638000, P. R. China
eDepartment of Abdominal Oncology, Cancer Center, West China Hospital, Medical School, Sichuan University, Chengdu 610041, P. R. China
First published on 28th June 2017
Abstract
Nanoparticles delivering therapeutic genes have promising applications in cancer treatments. Here, we used biodegradable heparin–polyethyleneimine (HPEI) nanoparticles to co-deliver two genes (vesicular stomatitis virus matrix protein, VSVMP; interleukin-12, IL12) for cancer therapy. These HPEI nanoparticles can bind the plasmid expressing VSVMP (pVSVMP) and the plasmid expressing IL12 (pIL12) through electrostatic interactions, and mediate the simultaneous expression of VSVMP and IL12 in colon cancer cells in vitro and in vivo. After intraperitoneal administration, the HPEI/pIL12 + pVSVMP complexes efficiently inhibit the growth of intraperitoneal metastasis of C-26 colon cancer, showing synergistic anticancer efficacy, without obvious systemic adverse effects. While VSVMP induces apoptosis, VSVMP and IL12 synergistically generate an anticancer immune response, which renders the potent anticancer effect of HPEI/pIL12 + pVSVMP. Our data suggest that VSVMP and IL12 could synergistically inhibit cancer survival in vivo, and the HPEI nanoparticle co-delivered VSVMP and IL12 might have potential application in treating intraperitoneal metastasis of colon cancer.
Introduction
Colorectal cancer has a high incidence and mortality rate, making it a great challenge to human health.1,2 During the past 15 years, several traditional therapeutic approaches (i.e. surgery, radiofrequency ablation, chemotherapy, cryosurgery and radiation therapy) have been used for the treatment of colon cancer.3,4 However, there are still some problems on clinical application, such as local recurrences, distant metastases, and side effects.5–7 Therefore, there is an urgent need to develop new therapeutics for the treatment of colon cancer.Gene therapy is a promising technology which is involved in the delivery of DNA, RNA or antisense oligonucleotides to treat genetic diseases, and has been adapted for cancer therapy.8–12 For recent years, researchers have made great efforts to create potent anticancer therapies. Approximately two-thirds of the clinical trials worldwide in gene therapy have been aimed at the treatment of various types of solid or hematological malignancies.13 The vesicular stomatitis virus matrix protein (VSVMP) from vesicular stomatitis virus (VSV) causes apoptosis of cells by inhibiting host gene expression and disorganizing cytoskeletal elements.14–16 Previous studies in our laboratory have demonstrated that gene therapy with VSVMP alone or combined with chemotherapy efficiently inhibit the growth of solid tumor and significantly prolong the survival time. VSVMP could directly induce tumor cell apoptosis, its antitumor effect was also accompanied by cytotoxic T lymphocytes (CTLs) and NK cells activation.17–23 During the past two decades, a great number of researches have demonstrated that gene therapy with IL12 has a potent anticancer activity in various tumor models.24–26 IL12, as a heterodimeric pro-inflammatory cytokine, could activate both innate and adaptive immunities and induce the production of interferon gamma (IFN-γ).24,27,28 In this regard, we wonder whether gene therapy with IL12, which is capable of activating immune effector cells, could enhance the antitumor effects of VSVMP.
Recently, both viral vectors and non-viral vectors have been investigated as delivery vehicles in cancer gene therapy. Compared with viral vectors, non-viral vectors are demonstrated to show several important safety advantages, such as reduced pathogenicity, absence of risk of transgenes insertional mutagenesis, low immune-toxicity, handy chemical modification, low cost and simply production.29 Non-viral vectors, such as polyethyleneimine (PEI), due to its strong condensation of DNA, which further facilitates endocytosis and prevents the DNA degradation from the endosomal disruption, has been widely investigated and applied as a gene delivery vehicle.30,31 However, PEI still have some disadvantages, such as cytotoxicity and non-biodegradability,32,33 and several strategies have been investigated to modify PEI to address these issues.34–36 Our previous studies have demonstrated that heparin–polyethyleneimine (HPEI) nanoparticles, which were prepared by covalently conjugating low molecular weight PEI2K with heparin, could efficiently and safely deliver gene into cells, overcoming the limitations of PEI such as the high cytotoxicity and non-biodegradability.23,37,38 Simultaneous delivery of multiple therapeutic candidates via nanoparticles is a promising choice for the treatment of cancer.39–43 Here we try to use the biodegradable HPEI nanoparticles to co-deliver pVSVMP and pIL12. Our hypothesis is that, co-delivery of VSVMP and IL12 by HPEI nanoparticles could show the synergistic effect and perform excellent inhibition of colon cancer.
In the present study, HPEI nanoparticles as synergistic combinational delivery vehicles were prepared and then electrostatically complexed with pVSVMP and pIL12 to form the HPEI/pIL12 + pVSVMP complexes. The physicochemical properties of the complexes (i.e. especially size, zeta-potential, gel retardation and morphology) were characterized. This HPEI nanoparticle could efficiently bind pVSVMP and pIL12 through electrostatic interaction, and mediate the simultaneous expression of VSVMP and IL12 in colon cancer cells in vitro and in vivo. To further study the combinatory therapy effects of HPEI/pIL12 + pVSVMP complexes, the anticancer activities of HPEI/pIL12 + pVSVMP complexes were investigated. The synergistic mechanisms were studied by using TUNEL assay, Ki67 staining and flow cytometry assay. Our results suggested that the synergistic antitumor effect of HPEI/pIL12 + pVSVMP complexes may result from induction of cancer cell apoptosis, inhibition of tumor cell proliferation and activation of immune effector cells (Fig. 1).
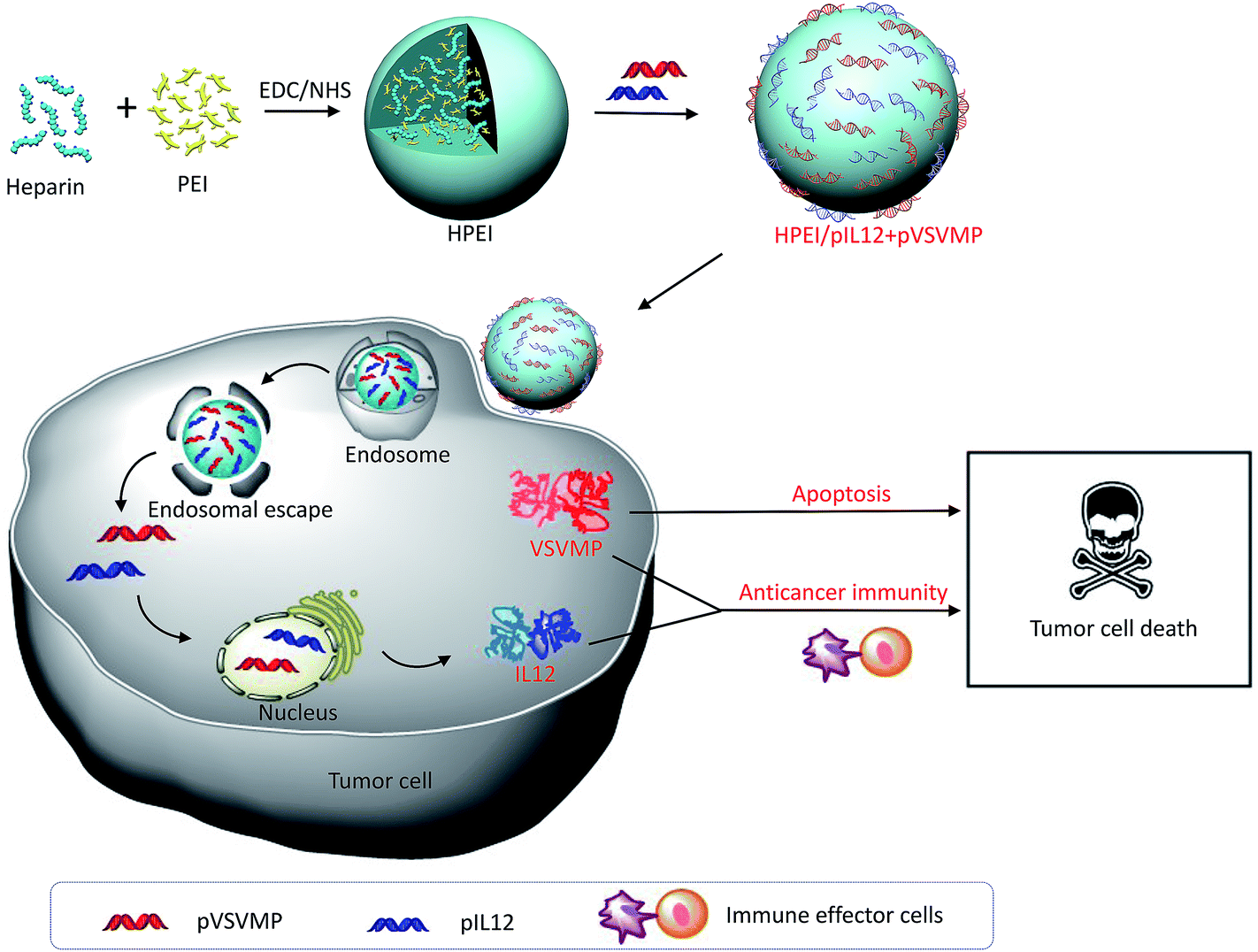 | ||
| Fig. 1 Schematic illustration of co-delivered pIL12 and pVSVMP by HPEI nanoparticle for improving anticancer efficacy by synergistic therapeutic effects. | ||
Materials and methods
Materials
Poly-ethyleneimine (molecular weight 2000 [PEI2K]), 2-(N-morpholino)ethane-sulfonic acid (MES), 1-ethyl-3-(3-dimethyl-aminopropyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma (St Louis, MO, USA). Dulbecco's Modified Eagle's Medium (DMEM), fetal bovine serum (FBS), penicillin, and streptomycin were purchased from HyClone™ (GE Healthcare UK Ltd, Little Chalfont, UK). DNA ladder was provided by Fermentas (Thermo Fisher Scientific Inc., Waltham, MA, USA). Balb/c mice (6–8 weeks old) were obtained from the Beijing HFK Bioscience (Beijing, China). All animal procedures were performed according to the guidelines of Sichuan University and approved by the Animal Care Committee of Sichuan University (Chengdu, China).Construction and purification of plasmid-based expression vectors: pVSVMP and pIL12
VSVMP was inserted into the pVAX plasmid at a BamHI site and a EcoRI site forming the expression plasmid (pVSVMP). The plasmid (pIL12) encoding p35 and p40 subunits of murine IL12 was constructed at the BamHI site and the XhoI site of the pVAX vector (Invitrogen, San Diego, CA, U.S.). The empty plasmid pVAX was used as negative control (pEP). The recombinant plasmid and negative control plasmid were prepared using the Endofree Plasmid kit (Omega Bio-Tek, Norcross, GA, USA) in accordance with the manufacturer's instructions. Escherichia coli colonies were cultured in Luria-Bertani broth with kanamycin (50 μg mL−1).Preparation and characterization of HPEI nanoparticles
HPEI nanoparticles were prepared as described previously.23,44 Briefly, 50 mg of heparin was dissolved in a MES buffer solution (100 mL, 0.05 M); then, 30 mg of NHS and 20 mg of EDC were added into the aforementioned solution to activate the carboxylic acid groups of heparin. After 2 h of reaction, this solution was dropped into 20 mL of PEI2K solution (7.5 mg mL−1) while stirring consistently at room temperature overnight. Later the obtained HPEI nanoparticles were dialyzed (molecular weight cut off = 8000–14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) in distilled water for 3 days. Then, the HPEI nanoparticles were filtered by using a syringe filter (pore size = 220 nm) (Millex-LG; EMD Millipore, Billerica, MA, USA). The HPEI nanoparticles were adjusted to 1 mg mL−1 and stored at 4 °C. The morphology of the HPEI nanoparticles was observed under a transmission electron microscope (TEM) (H-6009IV; Hitachi Ltd., Tokyo, Japan). The HPEI nanoparticles were diluted with distilled water and placed on a copper grid covered with nitrocellulose. The size and zeta potential of the HPEI nanoparticles or HPEI/pIL12 + pVSVMP complexes were determined by dynamic light scattering (Nano-ZS; Malvern Instruments, Malvern, UK) at 25 °C. All results were obtained as the mean of three test runs.
000) in distilled water for 3 days. Then, the HPEI nanoparticles were filtered by using a syringe filter (pore size = 220 nm) (Millex-LG; EMD Millipore, Billerica, MA, USA). The HPEI nanoparticles were adjusted to 1 mg mL−1 and stored at 4 °C. The morphology of the HPEI nanoparticles was observed under a transmission electron microscope (TEM) (H-6009IV; Hitachi Ltd., Tokyo, Japan). The HPEI nanoparticles were diluted with distilled water and placed on a copper grid covered with nitrocellulose. The size and zeta potential of the HPEI nanoparticles or HPEI/pIL12 + pVSVMP complexes were determined by dynamic light scattering (Nano-ZS; Malvern Instruments, Malvern, UK) at 25 °C. All results were obtained as the mean of three test runs.
Gel retardation assay
The HPEI/DNA complexes were incubated at room temperature for 30 min and then electrophoresed on 1% (w/v) agarose gel (Thermo Fisher Scientific) for 30 min at 100 V. The agarose gel was dissolved in TAE buffer (40 mM Tris/HCl, 1% acetic acid, 1 mM ethylene diamine tetra-acetic acid, pH 7.4) and stained with Golden View™. The electrophoresis gels were illuminated by using a gel documentation system (Gel Doc 1000; Bio-Rad Laboratories Inc., Hercules, CA, USA).In vitro gene transfection
C-26 cells were seeded into 6-well plates (Corning Incorporated, Corning, NY, USA) at a density of 2 × 105 cells per well in 2 mL of complete DMEM medium. After 24 h incubation, the medium was replaced with 800 μL serum-free DMEM medium per well. pGFP as a report gene was added at 2 μg per well, while the mass ratio of HPEI/pGFP and PEI25K/pGFP was 5/1, 1/1 respectively. After 6 h incubation, the medium was replaced by complete DMEM medium. 24 h later, the expression of green fluorescent protein (GFP) was observed by an Olympus IX 71 inverted fluorescence microscope (Olympus Corporation, Tokyo, Japan), and the transfection efficiency was recorded by flow cytometry (Epics Elite ESP, USA).Cytotoxicity assay
The cytotoxicity of HPEI nanoparticles on the C-26 cells was measured by using MTT assay. In those tests, C-26 cells were plated at a density of 5 × 103 cells per well in 100 μL of DMEM medium in 96-well plates. 24 h later, the cells were treated with different concentrations of HPEI nanoparticles or PEI25K for 48 h. Subsequently, 10 μL of 5 mg mL−1 concentration of MTT reagent was added to each well and incubated at 37 °C for 3 h in the dark until the purple formazan crystal was visible. Dimethyl sulfoxide (DMSO, 150 μL) was add to each well and cells were gently shaken for about 30 min in order to dissolve the formazan product. A microplate reader was used to measure the absorbance value of each well at a wavelength of 540 nm. The relative cell viability was expressed as a percentage relative to the untreated control wells. Experiments were performed three times independently.Anticancer activity of HPEI/pIL12 + pVSVMP complexes on C-26 cells in vitro
C-26 cells were plated in 96-well plates at a density of 5 × 103 cells per well in 100 μL of DMEM medium and incubated for 24 h. Normal saline (NS) and HPEI nanoparticles were used as controls. HPEI/pEP complexes, HPEI/pIL12 complexes, HPEI/pVSVMP complexes, or HPEI/pIL12 + pVSVMP complexes (5 μg HPEI/1 μg DNA) were prepared in DMEM medium without serum. The cells were then treated with those samples separately for 48 h. The viability of cells was evaluated by MTT assays.Furthermore, the apoptosis induction by HPEI/pIL12 + pVSVMP complexes were investigated in vitro. C-26 cells were plated in 6-well plates at a density of 2 × 105 cells per well in 2 mL of complete DMEM medium and incubated with NS, HPEI nanoparticles (10 μg HPEI), HPEI/pEP complexes (10 μg HPEI/2 μg DNA), HPEI/pIL12 complexes (10 μg HPEI/2 μg DNA), HPEI/pVSVMP complexes (10 μg HPEI/2 μg DNA) or HPEI/pIL12 + pVSVMP complexes (10 μg HPEI/1 μg + 1 μg DNA) separately. After 6 h incubation, the medium was replaced by completed medium. 48 h later, apoptosis was examined by flow cytometric analysis using propidium iodide and annexin-V staining method.
Murine colon cancer model
Tumor models were established by intraperitoneal (i.p.) injection of C-26 cells (about 5 × 105 cells/200 μL serum-free DMEM) into Balb/c mice on day 0. Three days after inoculation, mice were randomly divided into five groups, and were then intraperitoneally injected with 7 dosages of NS, HPEI/pEP complexes (25 μg/5 μg), HPEI/pIL12 complexes (25 μg/5 μg), HPEI/pVSVMP complexes (25 μg/5 μg), HPEI/pIL12 + pVSVMP complexes (25 μg/2.5 μg + 2.5 μg) once every two days. The weights of mice were recorded every day. All mice were sacrificed by cervical vertebra dislocation on day 17, tumor weight, ascites volume and number of nodules were recorded and analyzed. Vital organs and tumors were obtained and fixed in 4% paraformaldehyde or frozen in liquid nitrogen immediately. The ascites was collected for the ELISA analysis. The anticancer effects were determined by tumor weight, ascites volume and tumor nodule numbers.Enzyme linked immunosorbent assay (ELISA)
About 48 h after C-26 cells transfection with NS, HPEI nanoparticles, HPEI/pEP complexes, HPEI/pIL12 complexes, HPEI/pVSVMP complexes or HPEI/pIL12 + pVSVMP complexes, the cell culture supernatants of each group were collected for IL12p70 ELISA analysis (eBioscience, Inc., San Diego, CA, U.S.). The expression of IL12 in tumors from mouse models were detected by IL12p70 ELISA analysis. The level of secreted IFN-γ in tumors and ascites were also determined using commercially IFN-γ ELISA kits (eBioscience, Inc., San Diego, CA, U.S.). For tumor samples, the tumors were first removed and weighted, and then were cut into pieces and homogenized. The whole process was performed on ice. The homogenized supernatants were subsequently collected by centrifuging for 10 min at 10000 rpm at 4 °C. All the ELISA assays were performed in accordance with the manufacturer's instructions.
Real-time PCR
Total RNA was isolated from cells and tumor tissues by using an RNA simple Total RNA Kit (TIANGEN, Beijing, China), and was then reverse-transcribed by using a PrimeScript™ RT reagent Kit with gDNA Eraser (TaKaRa, Liaoning, China). The amount of cDNA used as templates was normalized to β-actin. The following sets of primers were used: VSVMP: forward, 5′-CGC GGA TCC ATC ATG AGT TCC TTA AAG AAG-3′, reverse, 5′-CGG AAT TCT CAT TTG AAG TGG CTG ATA GAA TCC-3′; IL12p40: forward, 5′-AAC CTC ACC TGT ACA CGC C-3′, reverse, 5′-CAA GTC CAT GTT TCT TTG CAC G-3′; IFN-γ: forward, 5′-TCA GCA ACA GCA AGG CGA AA-3′, reverse, 5′-CCG CTT CCT GAG GCT GGA T-3′; β-actin, forward, 5′-AGA GCT ACG AGC TGC CTG AC-3′, reverse, 5′-AGC ACT GTG TTG GCG TAC AG-3′. The RT-PCR conditions comprised a 30 second step at 95 °C, followed by 95 °C for 5 seconds and 56 °C for 20 seconds for 39 cycles with melting curve analysis. Relative quantification of each gene was calculated after normalization to β-actin.Western blot analysis
Protein concentrations of tumor lysates and the transfected cells were quantified with bicinchoninic acid assay protein assay kit (Bio-Rad Laboratories Inc.). Proteins from each sample were loaded into 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis and transferred to Millipore PVDF membranes. The membranes were blocked with 5% nonfat dried milk and incubated with anti-VSVMP (Chengdu Zen Bioscience Co., Ltd., Chengdu, Sichuan, China) or anti-β-actin (Santa Cruz Biotechnology Inc., Dallas, TX, USA) primary antibody at 4 °C overnight. The membranes were further incubated with corresponding secondary antibody conjugated with horseradish peroxidase (HRP) and developed with an enhanced chemiluminescence detection reagents (EMD Millipore, Billerica, MA, USA).Flow cytometric analysis
For the analysis of lymphocytes infiltration in tumor tissues. Specimens of about 1 mg of tumor tissue were taken from the NS, HPEI/pEP complexes, HPEI/pIL12 complexes, HPEI/pVSVMP complexes or and HPEI/pIL12 + pVSVMP complexes groups and digested into single-cell suspensions, then stained with FITC-CD3 and PE-CD8 antibodies for 30 min on ice. Mouse spleens from each group were harvested and filtrated with cell strainer (74 μm) to form a single cell suspension. The single cells of each group were stained with PE-CD107a (10 μL mL−1) in the presence of interleukin-2 (100 U mL−1) and monensin for 4 h at 37 °C. Phorbol-12-myristate-13-acetate (PMA, 2.5 μg mL−1; Sigma Chem. Co., St. Louis, MO, US) and ionophore (Ionomycin, 0.5 μg mL−1; Sigma Chem. Co., St. Louis, MO, US) were used as positive controls. Cells were then stained with FITC-CD49b for 30 min at 4 °C. All the antibodies were purchased from BD biosciences. The analysis was performed on a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, U.S.) and data were analyzed by FlowJo software.TUNEL assay, immunohistochemistry and H&E staining
Tissues were fixed in 4% paraformaldehyde solution for more than 24 h, then hydrated and embedded in paraffin blocks. The embedded tissues were sliced (3–4 μm) and stained with hematoxylin and eosin. Immunohistochemical analysis of Ki67 antigen was carried out with rabbit anti-human Ki67 antibody (Novus Biologicals, Littleton, CO, US) using the labeled streptavidin-biotin method as previously described.45 Ki67 expression was quantified by calculating the number of positive cells/total number of cells in five randomly selected areas at a 200× magnification. A commercially available terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) immunofluorescence kit (Promega, Madison, WI, U.S.) was used to examine the incidence of apoptosis in each group following the manufacturer's instructions. TUNEL positive cells were assessed by counting the apoptotic cells under a fluorescence microscope (Leica Microsystems CMS GmbH, Wetzlar, Germany) at a 200× magnification in randomly selected areas.Safety evaluation
To evaluate the potential administration side effects and toxicity, relevant indices such as weight, diarrhea, appearance, skin ulcerations and toxic deaths were observed. Vital organs (heart, liver, spleen, lung, and kidney) were harvested and stained with H&E using standard methods.Results and discussion
Preparation and characterization of HPEI/pIL12 + pVSVMP complexes
To efficiently deliver gene into tumor cell, the biodegradable cationic HPEI nanoparticles were prepared through amide bond heparin conjugated PEI molecules. The schematic representation of the preparation of HPEI/pIL12 + pVSVMP complexes was exhibited in Fig. 1. The particle size distribution spectrum of HPEI/pIL12 + pVSVMP complexes was presented in Fig. 2A, it was indicated that the complexes had a mean particle size of 101 nm. As shown in Fig. 2B, the HPEI/pIL12 + pVSVMP complexes had a zeta potential of 35 mV. In addition, the transmission electron microscopy (TEM) image of HPEI/pIL12 + pVSVMP complexes indicated that those complexes were monodisperse and had the mean particle size of 53 nm (shown in Fig. 2C).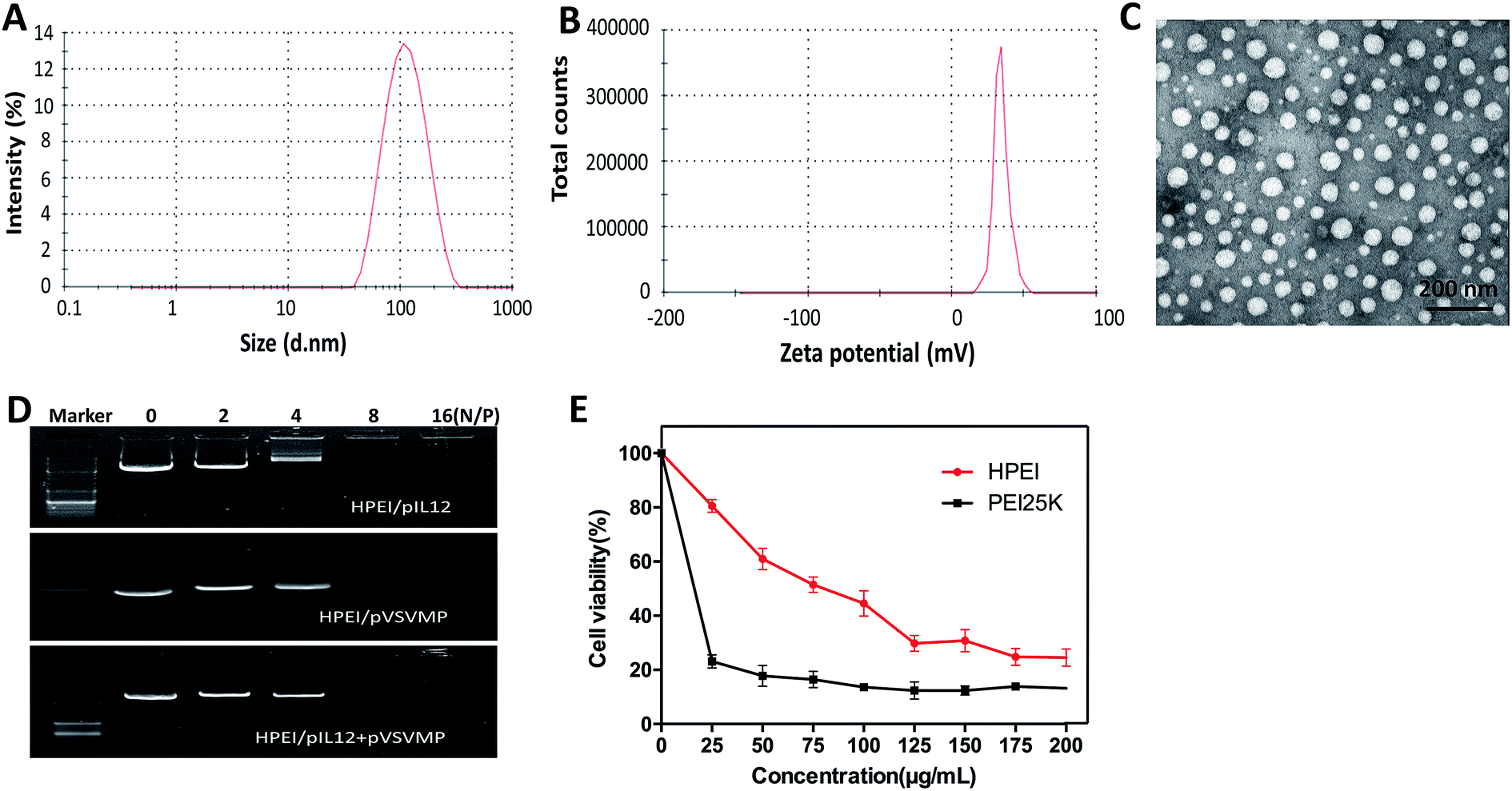 | ||
| Fig. 2 Characterization of HPEI/pIL12 + pVSVMP complexes. (A) Size distribution spectrum of HPEI/pIL12 + pVSVMP complexes. (B) Zeta potential spectrum of HPEI/pIL12 + pVSVMP complexes. (C) Transmission electron microscopic image of HPEI/pIL12 + pVSVMP complexes. (D) The ability of HPEI nanoparticles binding plasmids determined by gel retardation assay. (E) C-26 cells were treated with PEI 25 K or HPEI in various concentrations, cell viability was measured by MTT assay. | ||
Gel retardation assays were used to evaluate the ability of the HPEI nanoparticles that bound to pIL12 and pVSVMP. As shown in Fig. 2D, the HPEI nanoparticles with positive charges could efficiently bind to negative plasmids. When the N/P ratio of HPEI to DNA was ≥8, a complete retardation of DNA was achieved, no other band of free DNA was observed on the gel. This suggested that the anionic pVSVMP and pIL12 were completely absorbed on the surface of HPEI nanoparticles due to electrostatic interaction, forming HPEI/pIL12 + pVSVMP complexes.
Then, we compared the cytotoxicity and transfection efficiency of HPEI nanoparticles with PEI25K (as a standard transfection agent) in vitro. MTT assay revealed that, at a concentration of 25 μg mL−1, PEI25K could significantly inhibit the growth of C-26 cells, showing an obvious toxicity, while HPEI nanoparticles had much less toxicity, the results were presented in Fig. 2E. The transfection efficiency of HPEI nanoparticles and PEI25K in C-26 cells were determined by flow cytometry, the results showed that the transfection efficiency of HPEI nanoparticles was 35.7 ± 2.3%, while the transfection efficiency of PEI25K was 34.1 ± 1.9% (Fig. 3A–C). The transfection efficiency of HPEI nanoparticles on the C-26 cells is comparable to that of PEI25K (p = 0.29) and with less cytotoxicity than PEI25K. Those results are consistent with previous reports that HPEI could efficiently carry DNA with low cytotoxicity,23,44 showing great promising as gene co-delivery vehicles.
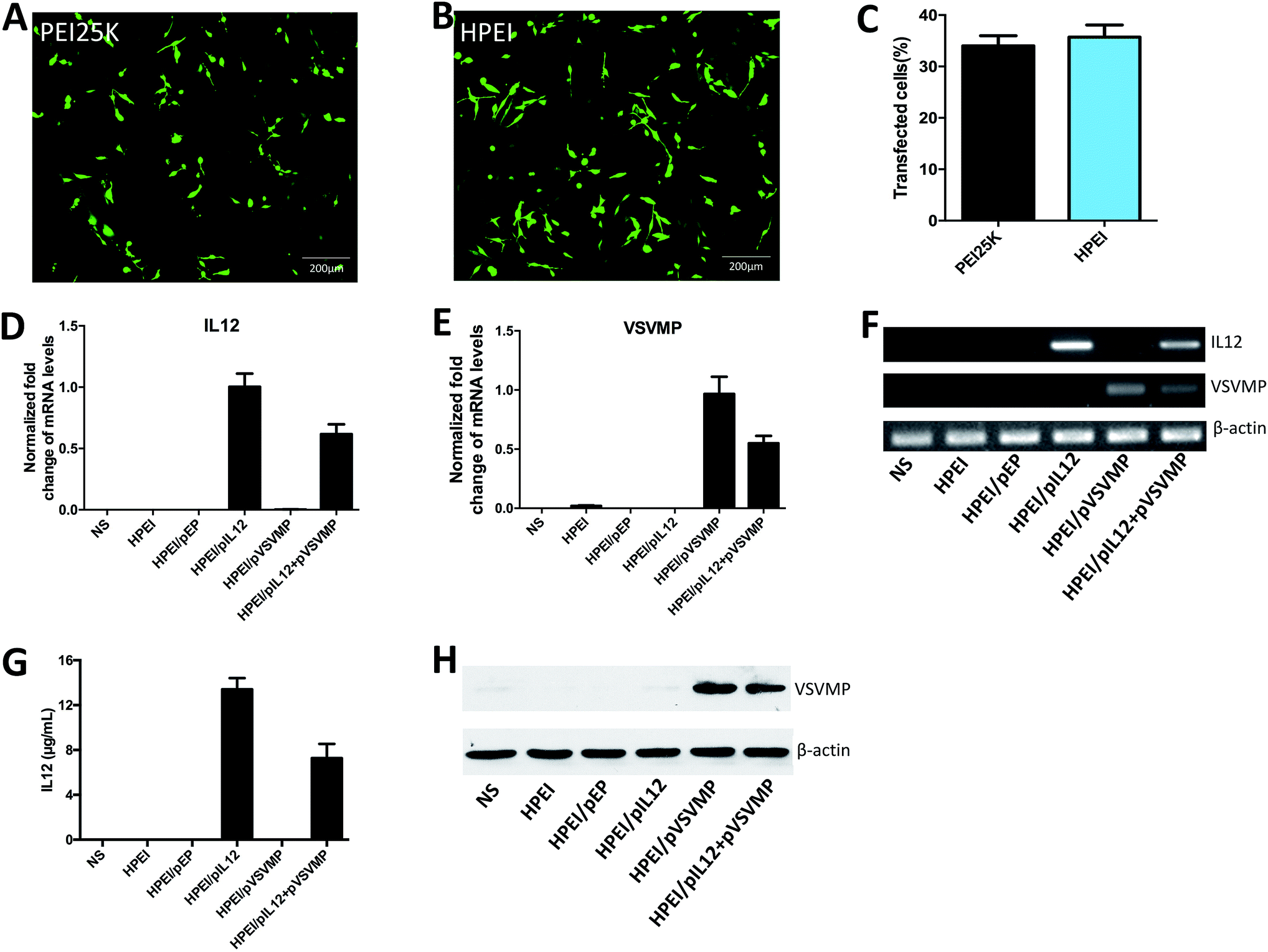 | ||
| Fig. 3 Gene expression mediated by HPEI nanoparticles in vitro. (A) Fluorescent image of C-26 cells transfected by PEI25K/pGFP. (B) Fluorescent image of C-26 cells transfected by HPEI/pGFP. (C) The transfection efficiency of HPEI and PEI25K were determined by flow cytometry. (D, E and F) 48 h after C-26 cells transfected with HPEI/pIL12 + pVSVMP, the expression of IL12 and VSVMP were determined by RT-PCR. (G) Expression of IL12 examined by ELISA assay. (H) Expression of VSVMP at the protein expression level was determined by western blot analysis. | ||
Anticancer activity of HPEI/pIL12 + pVSVMP complexes in vitro
To evaluate the anticancer activity of HPEI/pIL12 + pVSVMP complexes on the C-26 cell line. We firstly measured the expression of DNA plasmids in C-26 cell line by RT-PCR, ELISA and western blot analysis. 48 h after transfection, the expression of IL12 could be detected in HPEI/pIL12 complexes group and HPEI/pIL12 + pVSVMP complexes group (Fig. 3D, F and G). While the expression of VSVMP could be detected in HPEI/pVSVMP complexes group and HPEI/pIL12 + pVSVMP complexes group (as shown in Fig. 3E, F and H). Then the viability of C-26 cells was determined by MTT assay and the results were presented in Fig. 4A. Co-delivery of pVSVMP and pIL12 by HPEI nanoparticles efficiently inhibited the viability of C-26 cells, while treatment with NS, HPEI complexes, HPEI/pEP complexes or HPEI/pIL12 complexes did not significantly reduce cell viability. Those results implied that co-delivered VSVMP and IL12 by HPEI nanoparticles could efficiently inhibit the survival of C-26 colon cancer cells in vitro.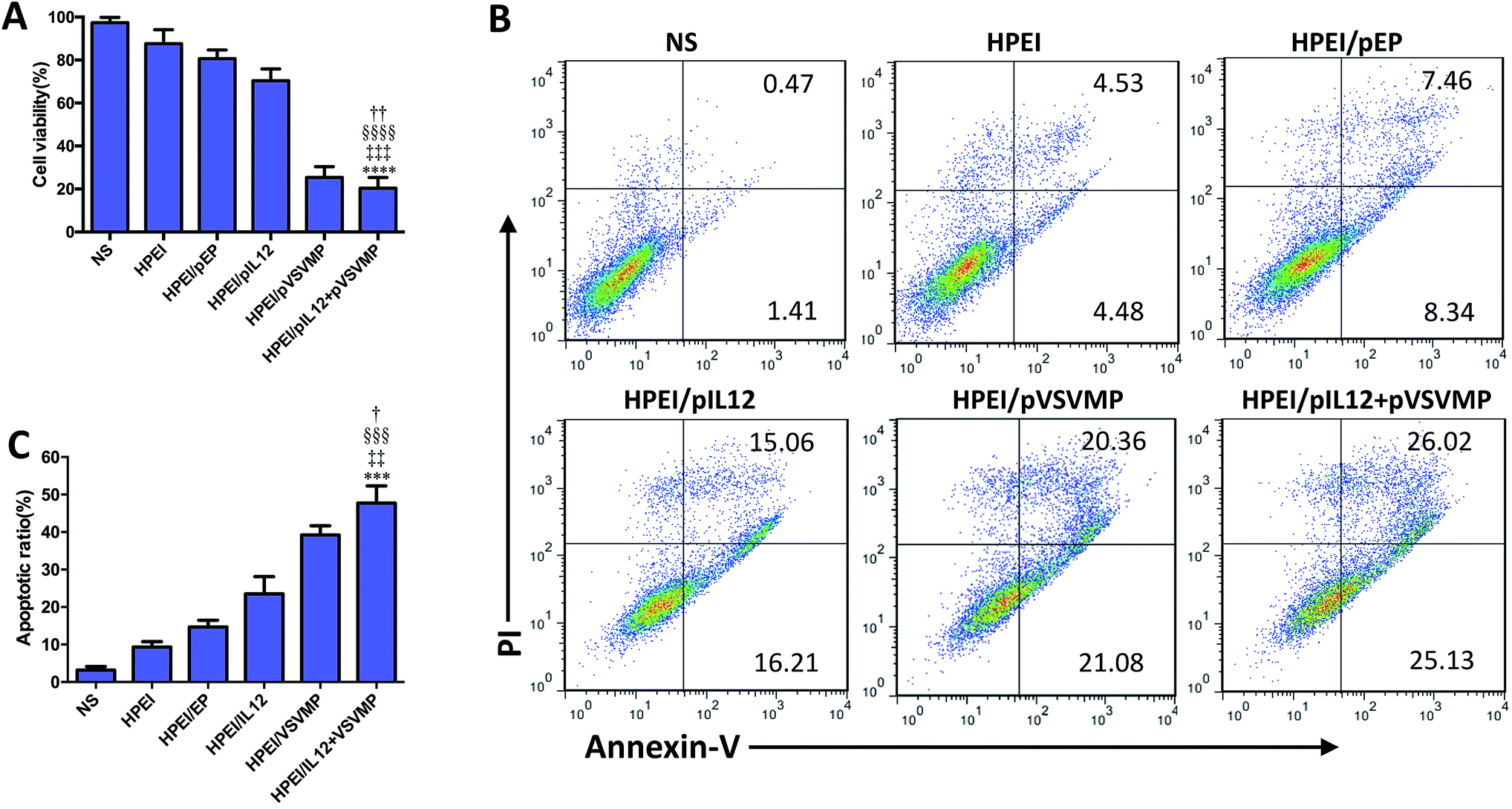 | ||
| Fig. 4 Apoptosis induced by HPEI/pIL12 + pVSVMP complexes on C-26 cells in vitro. (A) Cell viability determined by MTT assay. (B and C) C-26 cells were treated with NS, HPEI alone, HPEI/EP, HPEI/pIL12, HPEI/pVSVMP or HPEI/pIL12 + pVSVMP complexes for 48 h. Cells were then stained with propidium iodide and annexin-V to evaluate apoptotic ratio by flow cytometric analysis. All data were representative of three independent experiments. Data are presented as the mean ± SD by t-test (***p < 0.001, ****p < 0.0001 vs. NS; ‡‡p < 0.01, ‡‡‡p < 0.001 vs. HPEI; §§§p < 0.001, §§§§p < 0.0001 vs. HPEI/pEP; †p < 0.05, ††p < 0.01 vs. HPEI/pIL12). | ||
Furthermore, propidium iodide and annexin-V staining method was performed to reveal whether apoptosis was one of the main factors that contributed to the inhibition of C-26 colon cancer cells. As shown in Fig. 4B and C, 47.7 ± 7.5% of apoptotic cells were observed in response to the treatment with HPEI/pIL12 + pVSVMP complexes (10 μg HPEI/1 μg pVSVMP +1 μg pIL12), 39.2 ± 4.2% in HPEI/pVSVMP group (10 μg HPEI/2 μg pVSVMP), 23.5 ± 7.9% in HPEI/pIL12 group (10 μg HPEI/2 μg pIL12), while 15.3 ± 4.1% in HPEI/EP group (10 μg HPEI/2 μg pEP), and only 9.3 ± 2.4% were observed in the HPEI group (10 μg HPEI). These results suggested that the induction of apoptosis was associated with the anticancer effects of HPEI/pIL12 + pVSVMP complexes mediated C-26 colon cancer gene therapy. However, the data in MTT-based cytotoxicity assay and PI/annexin-V based flow cytometric apoptosis assay suggested that there was no significant difference between HPEI/pVSVMP complexes and HPEI/pIL12 + pVSVMP complexes. The reason maybe in part due to the IL12 show strong antitumor effect by sensing innate and adaptive lymphocytes,46,47 rather than directly perform effective anti-cancer activity in vitro.
Anticancer effect of HPEI/pIL12 + pVSVMP complexes in vivo
To study the anticancer activity of HPEI/pIL12 + pVSVMP complexes on the abdominal cavity metastases of C-26 colon cancer, tumor-bearing mice were divided into five groups and treated with NS, HPEI/pEP complexes, HPEI/pIL12 complexes, HPEI/pVSVMP complexes and HPEI/pIL12 + pVSVMP complexes by intraperitoneal injection every two days. The metastatic tumor tissues in each group were harvested and weighed at day 17. As shown in Fig. 5A and B, the tumor progress was significantly inhibited in mice treated with HPEI/pIL12 + pVSVMP complexes compared with those treated with NS, HPEI/pEP complexes, HPEI/pIL12 complexes or HPEI/pVSVMP complexes. The average tumor weight in HPEI/pIL12 + pVSVMP complexes group was (0.66 ± 0.48) g, while it was (4.57 ± 0.57) g in the NS group, (2.75 ± 0.82) g in the HPEI/pEP complexes group, (1.54 ± 0.59) g in the HPEI/pIL12 complexes group, and (1.52 ± 0.62) g in the HPEI/pVSVMP complexes group, respectively. It was obvious that the mice treated with HPEI/pIL12 + pVSVMP complexes showed a statistically significant reduction in tumor weight than other groups (Fig. 5C). Moreover, as shown in Fig. 5D, there was a statistically significant decrease in the ascites volume of the mice treated with HPEI/pIL12 + pVSVMP complexes (0.21 ± 0.18) mL, compared with (4.14 ± 1.35) mL in the mice in the NS group, (2.6 ± 0.79) mL in the mice treated with HPEI/pEP complexes. In addition, compared with treatment with NS, HPEI/pEP complexes, HPEI/pIL12 complexes or HPEI/pVSVMP complexes, treatment with HPEI/pIL12 + pVSVMP complexes also showed more efficiently inhibition in abdominal cavity metastases of C-26 colon carcinomas by reducing the number of tumor nodules from 125 to 28 (Fig. 5E). All these results suggested that HPEI/pIL12 + pVSVMP complexes exerted potent synergistic antitumor effects and significantly inhibited the growth of colon cancer cells in vivo.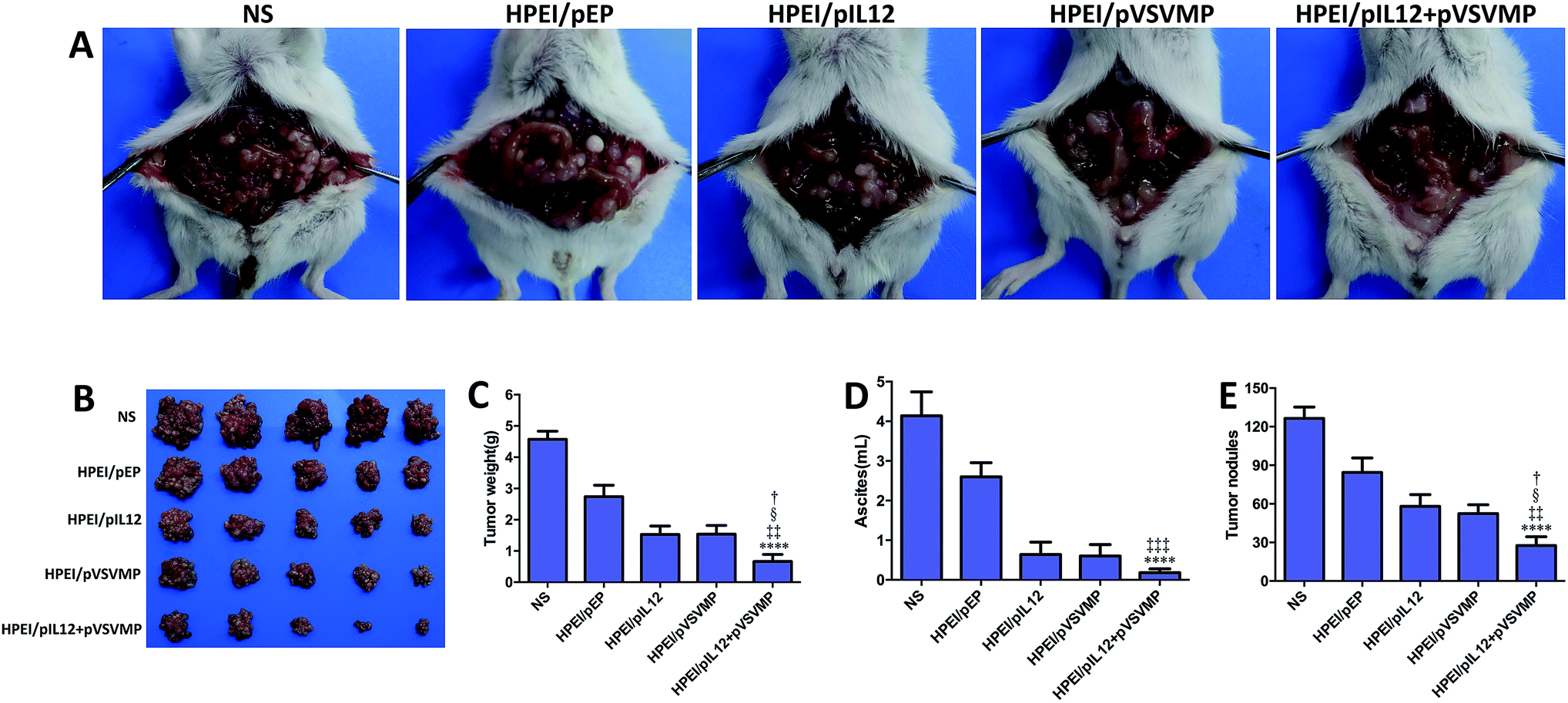 | ||
| Fig. 5 HPEI/pIL12 + pVSVMP complexes inhibited the growth of abdominal metastases of C-26 colon cancer model in vivo. (A) Representative photographs of colon cancer in NS, HPEI/pEP, HPEI/pIL12, HPEI/pVSVMP or HPEI/pIL12 + pVSVMP complexes group, respectively. (B) Tumor images of each group. (C) The mean tumor weight of abdominal colon cancer metastasis. (D) Ascites volume. (E) Tumor nodules. All data were representative of three independent experiments. Data are presented as the mean ± SD by t-test (****p < 0.0001 vs. NS; ‡‡p < 0.01, ‡‡‡p < 0.001 vs. HPEI/pEP; §p < 0.05 vs. HPEI/pIL12; †p < 0.05 vs. HPEI/pVSVMP). | ||
Furthermore, we confirmed the expression of IL12 in tumor tissues by RT-PCR and ELISA. As shown in Fig. 6A, D and E. IL12 could be detected in mice treated with HPEI/pIL12 complexes or HPEI/pIL12 + pVSVMP complexes. We evaluated the expression of VSVMP by RT-PCR and western blot analysis, the results indicated that both HPEI/pVSVMP complexes and HPEI/pIL12 + pVSVMP complexes group had the expression of VSVMP (Fig. 6B and H). Previous studies have demonstrated that IL12 stimulation could induce the secretion of IFN-γ,47,48 we further determined IFN-γ level in tumor and ascites by RT-PCR and ELISA. As shown in Fig. 6C, D, F and G, the expression of IFN-γ in tumor and ascites was significantly increased in mice treated with HPEI/pIL12 + pVSVMP complexes, compared with NS, HPEI/pEP complexes, HPEI/pIL12 complexes, or HPEI/pVSVMP complexes.
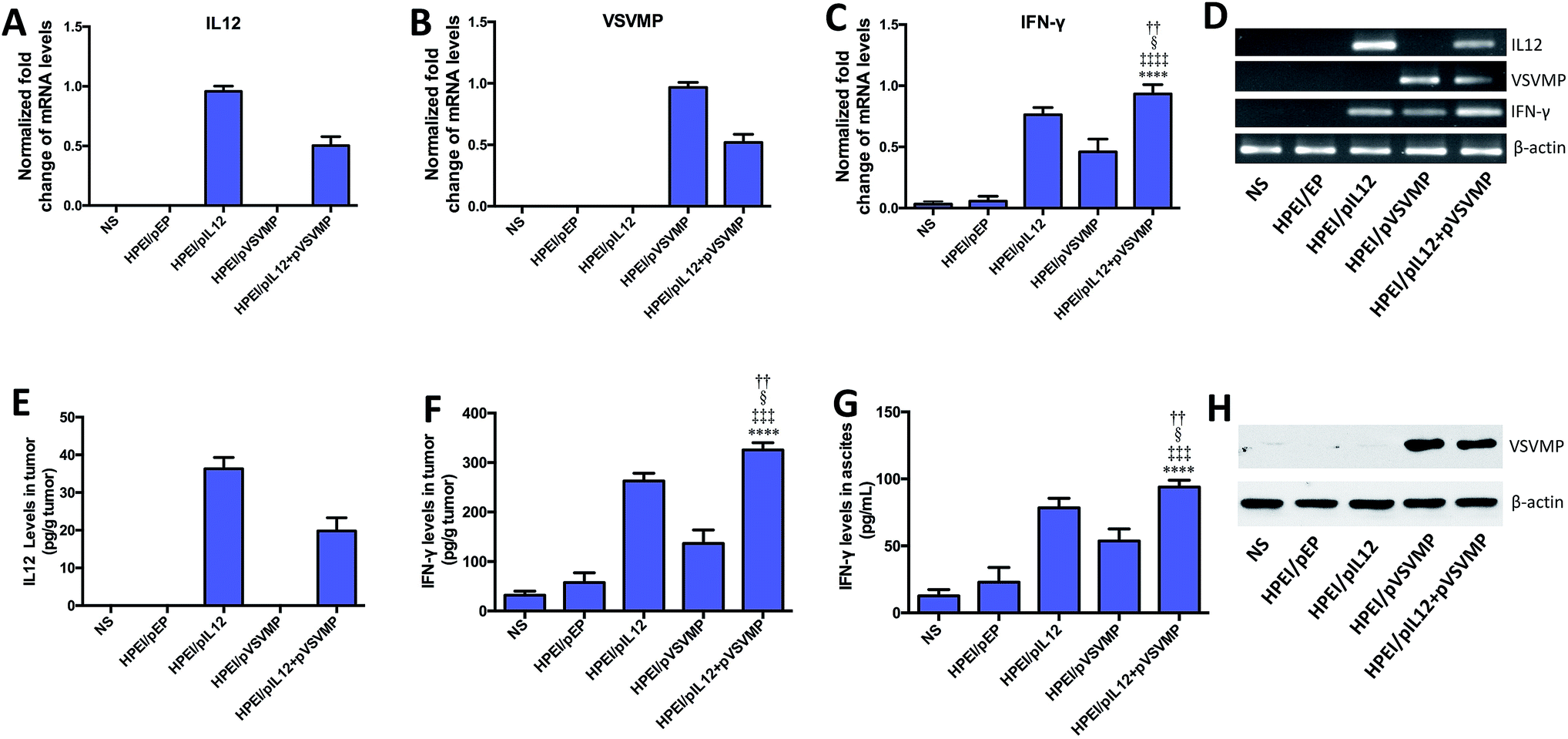 | ||
| Fig. 6 Gene expression mediated by HPEI/pIL12 + pVSVMP complexes on C-26 colon cancer mice model. (A–D) Relative expression of IL12, VSVMP and IFN-γ in each group were detected by real-time RT-PCR. (E) Expression of IL12 in tumor tissues was detected by ELISA assay. (F and G) Expression of IFN-γ in tumor tissues and ascites were examined by ELISA assay. (H) Expression of VSVMP at the protein expression level were determined by western blot analysis. Data are presented as the mean ± SD by t-test (****p < 0.0001 vs. NS; ‡‡‡p < 0.001, ‡‡‡‡p < 0.0001 vs. HPEI/pEP; §p < 0.05 vs. HPEI/pIL12; ††p < 0.01 vs. HPEI/pVSVMP). | ||
Mechanisms underlying the anticancer effects of HPEI/pIL12 + pVSVMP complexes
To explore the mechanisms involved in the anticancer activity of HPEI/pIL12 + pVSVMP complexes, tumor cell apoptosis and proliferation were examined. As shown in Fig. 7A and B, the TUNEL assay results revealed that significant increase of apoptosis positive cells was induced in tumor tissues treated with HPEI/pIL12 + pVSVMP complexes (131.7 ± 6.3) which was more than in those treated with NS (1.5 ± 1.2), HPEI/pEP complexes (4.0 ± 1.0), HPEI/pIL12 complexes (63.6 ± 6.5) or HPEI/pVSVMP complexes (81.3 ± 7.1). Moreover, the proliferation of tumor cells was analyzed by immune-histochemical staining for Ki67, a proliferation marker (Fig. 7A and C). Compared with NS (61.3 ± 3.2), HPEI/pEP complexes (54.1 ± 3.9), HPEI/pIL12 complexes (24.3 ± 3.1) or HPEI/pVSVMP complexes (21.2 ± 2.2), the percentage of Ki67 positive cells in the HPEI/pIL12 + pVSVMP complexes group (12.5 ± 1.9) was significant lower. H&E staining of tumor sections indicated that there were more survival cancer cells in tumors treated with NS or HPEI/pEP. In contrast, tumor treated with HPEI/pIL12 complexes, HPEI/pVSVMP complexes or HPEI/pIL12 + pVSVMP complexes showed more dead cancer cells (Fig. 7A). Thus, the results suggested that the superior anticancer effects of HPEI/pIL12 + pVSVMP complexes may partly result from the induction of the tumor cells apoptosis and inhibition of tumor cells proliferation.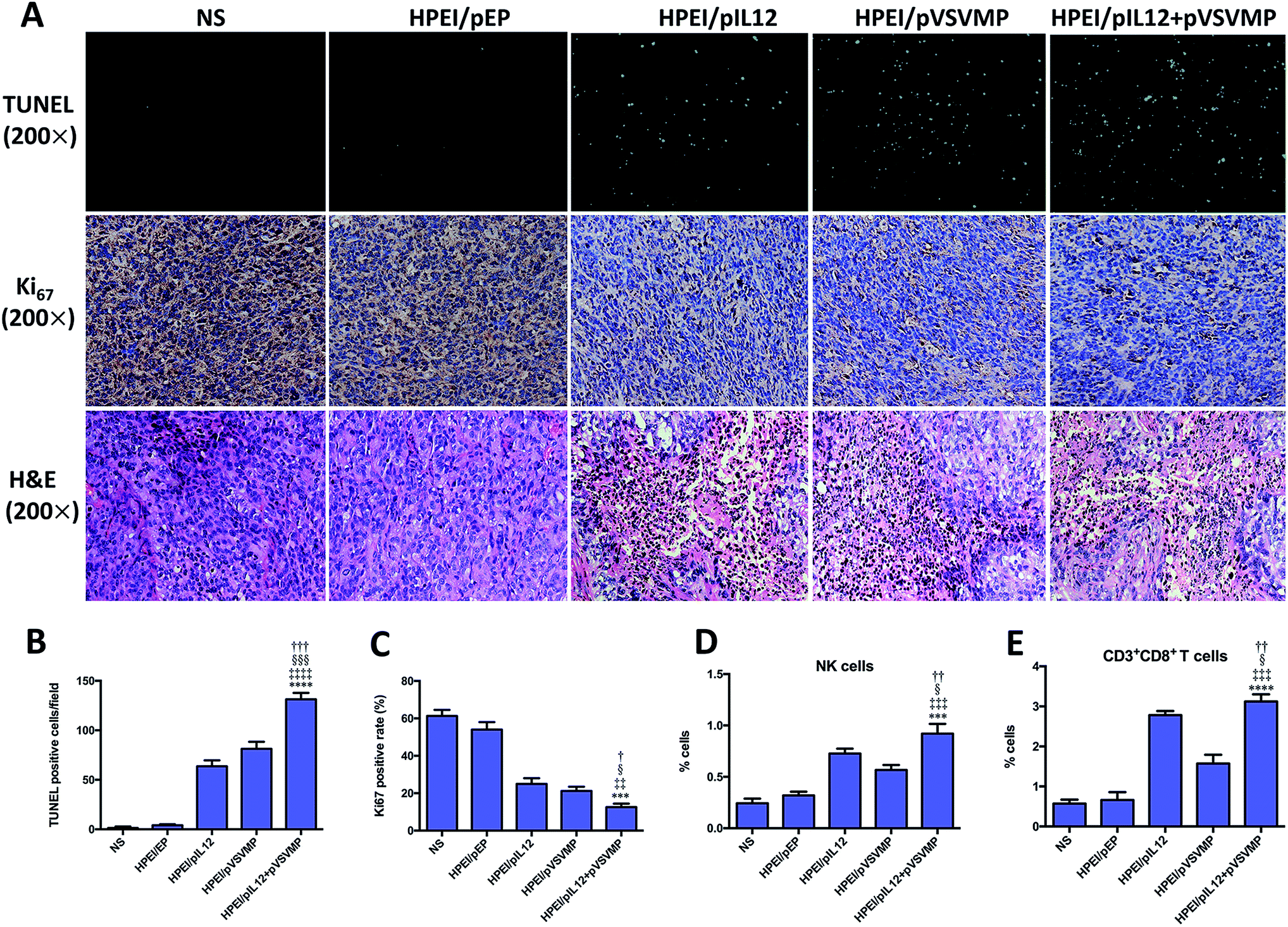 | ||
| Fig. 7 Antitumor mechanisms of HPEI/pIL12 + pVSVMP complexes. (A) TUNEL assay, Ki67 staining and H&E staining of the tumor tissues in each treatment group. (B and C) Tumor cell apoptosis and proliferation were assessed by counting the number of TUNEL-positive cells per field and the Ki67-positive rate (three high power fields per slide). (D) Splenic single cells were prepared and analyzed by flow cytometry for detecting activated NK cells. (E) Single-cell suspensions isolated from tumors were analyzed by flow cytometry for the presence of CD8+ T cells. Data are presented as the mean ± SD by t-test (***p < 0.001, ****p < 0.0001 vs. NS; ‡‡p < 0.01, ‡‡‡p < 0.001, ‡‡‡‡p < 0.0001 vs. HPEI/pEP; §p < 0.05, §§§p < 0.001 vs. HPEI/pIL12; ††p < 0.05, ††p < 0.01, †††p < 0.01 vs. HPEI/pVSVMP). | ||
Moreover, quantifications of lymphocytes infiltration in spleen and tumor were performed by flow cytometry analysis. Two types of potential effector populations were stained with antibodies: PE-CD107a and FITC-CD49b (NK cells), FITC-CD3 and PE-CD8 (CD8+ T cells). As shown in Fig. 7D, A higher ratio of activated NK cells from total spleen cells were detected in the HPEI/pIL12 + pVSVMP complexes group (0.92 ± 0.09%) than in the NS group (0.24 ± 0.05%), HPEI/pEP complexes group (0.32 ± 0.04%), HPEI/pIL12 complexes group (0.72 ± 0.05%) or HPEI/pVSVMP complexes group (0.57 ± 0.06%). In addition, as shown in Fig. 7E, the percentage of CD8+ T cells in tumor was also dramatically increased in HPEI/pIL12 + pVSVMP complexes group (3.05 ± 0.23%), which was higher than in NS group (0.57 ± 0.10%), HPEI/pEP group (0.66 ± 0.19%), HPEI/pIL12 group (3.03 ± 0.13%) or HPEI/pVSVMP group (1.57 ± 0.22%). These results are in line with that of other studies,19,20,46,47 suggesting that HPEI/pIL12 + pVSVMP complexes treatment could enhance the activation of NK cells and CD8+ T cells compared with either treatment alone in vivo.
IL12 could activate both innate NK and CD8+ T cells,27,47,48 while VSVMP is capable of directly inducing tumor cell death.14,15 Our data indicated that gene therapy with IL12 could enhance the antitumor effects of VSVMP. This synergistic antitumor effect of co-delivered IL12 and VSVMP by HPEI nanoparticles may result from induction of tumor cell apoptosis, inhibition of tumor cell proliferation, and activation of immune effector cells.
HPEI/pIL12 + pVSVMP safety and toxicity assessment in mice
To examine potential side effects of HPEI/pIL12 + pVSVMP complexes in mice. Some key mouse organs, including the heart, liver, spleen, lung and kidney, were harvested and performed H&E staining for histopathological analysis. As shown in Fig. 8, no significant pathological changes were observed. Those results indicated that HPEI/pIL12 + pVSVMP complexes had no obvious toxic and side effects.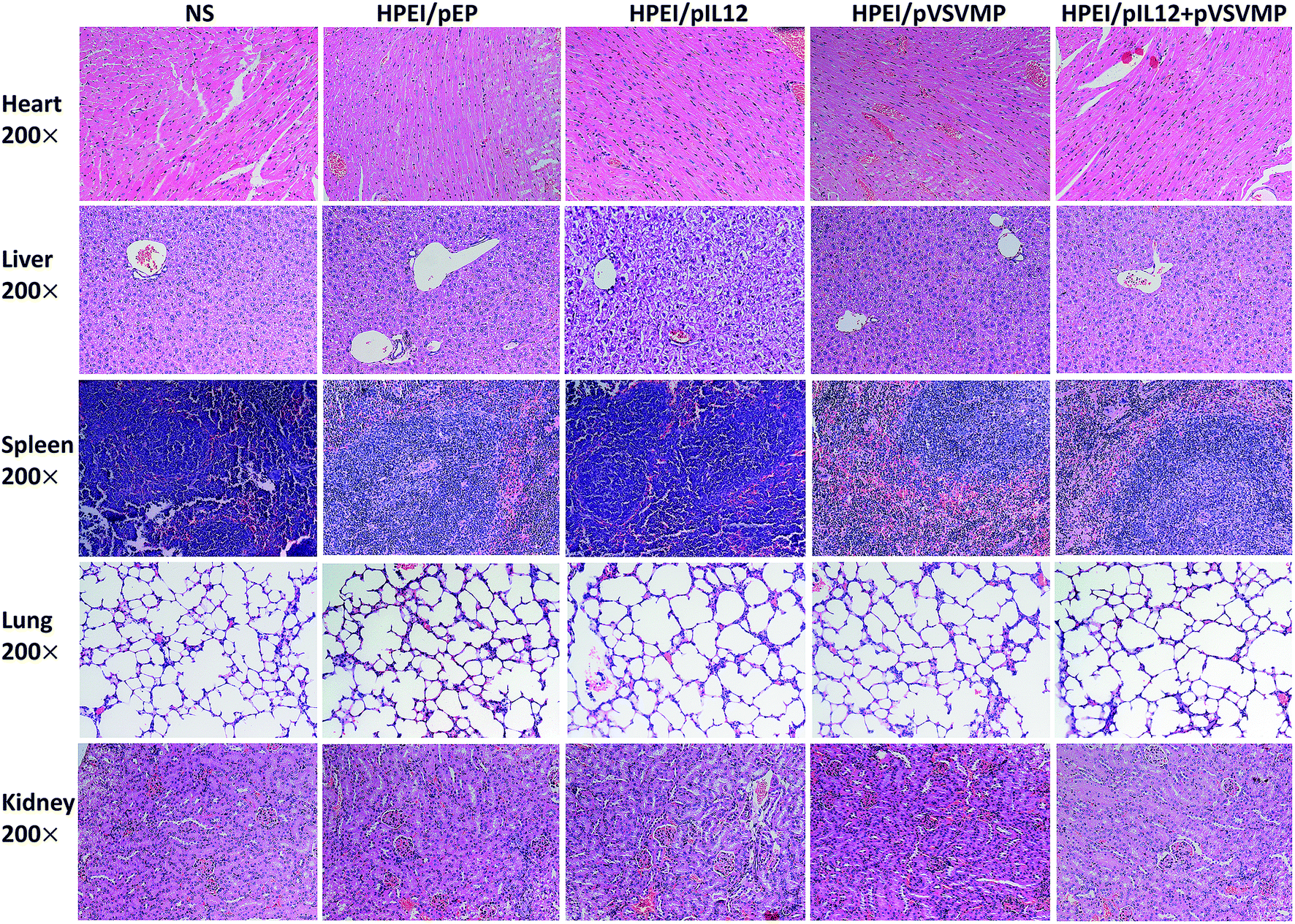 | ||
| Fig. 8 Histological examinations of H&E-stained vital organ sections. The heart, liver, spleen, lung, and kidney in NS, HPEI/pEP, HPEI/pIL12, HPEI/pVSVMP or HPEI/pIL12 + pVSVMP complexes group were collected and conducted with HE staining, respectively. No significant pathological changes were detected. | ||
Conclusions
In summary, HPEI nanoparticles could co-bind pVSVMP and pIL12 through electrostatic interactions, and mediate the simultaneous expression of VSVMP and IL12 in colon cancer cells in vitro and in vivo. Intraperitoneal administration of HPEI/pIL12 + pVSVMP complexes could efficiently inhibit the growth of intraperitoneal metastasis of C-26 colon cancer, showing synergistic anticancer effects, without obvious systemic adverse effects. This study suggested that VSVMP and IL12 could synergistically inhibit colon cancer, and the HPEI nanoparticle co-delivered VSVMP and IL12 might have potential application in treating intraperitoneal metastasis of colon cancer.Authors' contributions
M. L. G. and Y. Y. X. conceived and designed the experiments. Y. Y. X., Y. P. Y., Y. J. W. analyzed the data. Y. Y. X., Y. P. Y., Y. J. W., C. M. W., H. C., W. Z., Y. L., J. L. L., W. H. G. performed the experiments. G. P. G. supervised the experiments. M. L. G. and Y. ·Y. X. wrote the main manuscript text.Conflict of interest
No potential conflicts of interest were disclosed.Acknowledgements
This work was supported by the National Natural Science Foundation (81422025, 81572990), the Foundation for Distinguished Young Scientists of Sichuan Province (2016JQ0020), Sichuan province science and technology support plan (2015FZ0040, 2015SZ0049), and the Outstanding Young Scholars Research Fund of Sichuan University (2014SCU04A12).References
- C. E. DeSantis, R. L. Siegel, A. G. Sauer, K. D. Miller, S. A. Fedewa, K. I. Alcaraz and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 290–308 CrossRef PubMed.
- R. L. Siegel, K. D. Miller, S. A. Fedewa, D. J. Ahnen, R. G. S. Meester, A. Barzi and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 177–193 CrossRef PubMed.
- F. Ciardiello, Lancet Oncol., 2015, 16, 1444–1445 CrossRef PubMed.
- A. R. Marley and H. Nan, Int. J. Mol. Epidemiol. Genet., 2016, 7, 105–114 Search PubMed.
- D. C. Damin and A. R. Lazzaron, World J. Gastroenterol., 2014, 20, 877–887 CrossRef PubMed.
- U. T. Sankpal, G. P. Nagaraju, S. R. Gottipolu, M. Hurtado, C. G. Jordan, J. W. Simecka, M. Shoji, B. El-Rayes and R. Basha, Oncotarget, 2016, 7, 3186–3200 Search PubMed.
- J. Prados, C. Melguizo, H. Roldan, P. J. Alvarez, R. Ortiz, J. L. Arias and A. Aranega, BioDrugs, 2013, 27, 317–327 CrossRef CAS PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- D. Ibraheem, A. Elaissari and H. Fessi, Int. J. Pharm., 2014, 459, 70–83 CrossRef CAS PubMed.
- I. M. Verma and N. Somia, Nature, 1997, 389, 239–242 CrossRef CAS PubMed.
- K. J. Kauffman, M. J. Webber and D. G. Anderson, J. Controlled Release, 2016, 240, 227–234 CrossRef CAS PubMed.
- M. Foldvari, D. W. Chen, N. Nafissi, D. Calderon, L. Narsineni and A. Rafiee, J. Controlled Release, 2016, 240, 165–190 CrossRef CAS PubMed.
- S. L. Ginn, I. E. Alexander, M. L. Edelstein, M. R. Abedi and J. Wixon, J. Gene Med., 2013, 15, 65–77 CrossRef CAS PubMed.
- S. A. Kopecky and D. S. Lyles, J. Virol., 2003, 77, 5524–5528 CrossRef CAS PubMed.
- D. F. Gaddy and D. S. Lyles, J. Virol., 2005, 79, 4170–4179 CrossRef CAS PubMed.
- S. A. Kopecky and D. S. Lyles, J. Virol., 2003, 77, 4658–4669 CrossRef CAS PubMed.
- X. M. Jing, Y. J. Wen, W. Shi, Q. Q. Tang, J. Li and X. C. Chen, Cancer Gene Ther., 2012, 19, 101–109 CrossRef CAS PubMed.
- Q. Zhong, Y. J. Wen, H. S. Yang, H. Luo, A. F. Fu, F. Yang, L. J. Chen, X. Chen, X. R. Qi, H. G. Lin, Y. Wan, X. C. Chen, Y. Q. Wei and X. Zhao, Ann. Oncol., 2008, 19, 1584–1591 CrossRef CAS PubMed.
- J. M. Zhao, Y. J. Wen, Q. Li, Y. S. Wang, H. B. Wu, J. R. Xu, X. C. Chen, Y. Wu, L. Y. Fan, H. S. Yang, T. Liu, Z. Y. Ding, X. B. Du, P. Diao, J. Li, H. B. Wu, B. Kan, S. Lei, H. X. Deng, Y. Q. Mao, X. Zhao and Y. Q. Wei, FASEB J., 2008, 22, 4272–4280 CrossRef CAS PubMed.
- X. Lin, X. Chen, Y. Wei, J. Zhao, L. Fan, Y. Wen, H. Wu and X. Zhao, Gynecol. Oncol., 2007, 104, 540–546 CrossRef CAS PubMed.
- Z. Y. He, F. Deng, X. W. Wei, C. C. Ma, M. Luo, P. Zhang, Y. X. Sang, X. Liang, L. Liu, H. X. Qin, Y. L. Shen, T. Liu, Y. T. Liu, W. Wang, Y. J. Wen, X. Zhao, X. N. Zhang, Z. Y. Qian and Y. Q. Wei, Sci. Rep., 2016, 6, 23764 CrossRef CAS PubMed.
- S. Luo, P. Chen, Z. C. Luo, P. Zhang, P. Sun, W. Shi, Z. Y. Li, X. L. Zhang, L. Q. Wang, X. Chen, Y. Q. Wei and Y. J. Wen, Cancer Sci., 2010, 101, 1219–1225 CrossRef CAS PubMed.
- M. Gou, K. Men, J. Zhang, Y. Li, J. Song, S. Luo, H. Shi, Y. Wen, G. Guo, M. Huang, X. Zhao, Z. Qian and Y. Wei, ACS Nano, 2010, 4, 5573–5584 CrossRef CAS PubMed.
- W. Lasek, R. Zagozdzon and M. Jakobisiak, Cancer Immunol. Immunother., 2014, 63, 419–435 CrossRef CAS PubMed.
- M. L. Lucas, L. Heller, D. Coppola and R. Heller, Mol. Ther., 2002, 5, 668–675 CrossRef CAS PubMed.
- S. Li, X. Zhang and X. Xia, J. Natl. Cancer Inst., 2002, 94, 762–768 CrossRef CAS PubMed.
- G. Trinchieri, Nat. Rev. Immunol., 2003, 3, 133–146 CrossRef CAS PubMed.
- T. Yue, X. Zheng, Y. Dou, X. Zheng, R. Sun, Z. Tian and H. Wei, BMC Cancer, 2016, 16, 665 CrossRef PubMed.
- M. Ramamoorth and A. Narvekar, J. Clin. Diagn. Res., 2015, 9, GE01–GE06 Search PubMed.
- M. Neu, D. Fischer and T. Kissel, J. Gene Med., 2005, 7, 992–1009 CrossRef CAS PubMed.
- U. Lungwitz, M. Breunig, T. Blunk and A. Gopferich, Eur. J. Pharm. Biopharm., 2005, 60, 247–266 CrossRef CAS PubMed.
- G. Liang, Y. Li, W. Feng, X. Wang, A. Jing, J. Li and K. Ma, Int. J. Nanomed., 2016, 11, 6079–6088 CrossRef PubMed.
- H. K. Nguyen, P. Lemieux, S. V. Vinogradov, C. L. Gebhart, N. Guerin, G. Paradis, T. K. Bronich, V. Y. Alakhov and A. V. Kabanov, Gene Ther., 2000, 7, 126–138 CrossRef CAS PubMed.
- S. Gao, H. Tian, Z. Xing, D. Zhang, Y. Guo, Z. Guo, X. Zhu and X. Chen, J. Controlled Release, 2016, 243, 357–369 CrossRef CAS PubMed.
- P. Davoodi, M. P. Srinivasan and C. H. Wang, Acta Biomater., 2016, 39, 79–93 CrossRef CAS PubMed.
- M. A. Sheikh, Y. S. Malik, Z. Xing, Z. Guo, H. Tian, X. Zhu and X. Chen, Acta Biomater., 2017, 54, 58–68 CrossRef CAS PubMed.
- P. Liu, M. Gou, T. Yi, C. Xie, X. Qi, S. Zhou, H. Deng, Y. Wei and X. Zhao, Oncol. Rep., 2012, 27, 363–370 CAS.
- F. Yang, M. Gou, H. Deng, T. Yi, Q. Zhong, Y. Wei and X. Zhao, Oncol. Rep., 2012, 28, 668–676 CAS.
- Y. Xiong, Y. Zhao, L. Miao, C. M. Lin and L. Huang, J. Controlled Release, 2016, 244, 63–73 CrossRef CAS PubMed.
- X. Liang, B. Shi, K. Wang, M. Fan, D. Jiao, J. Ao, N. Song, C. Wang, J. Gu and Z. Li, Biomaterials, 2016, 82, 194–207 CrossRef CAS PubMed.
- P. A. Ascierto and F. M. Marincola, J. Transl. Med., 2011, 9, 115 CrossRef PubMed.
- Y. Guo, W. He, S. Yang, D. Zhao, Z. Li and Y. Luan, Colloids Surf., B, 2016, 151, 119–127 CrossRef PubMed.
- Y. Cui, M. Zhang, F. Zeng, H. Jin, Q. Xu and Y. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 32159–32169 CAS.
- L. Luo, T. Du, J. Zhang, W. Zhao, H. Cheng, Y. Yang, Y. Wu, C. Wang, K. Men and M. Gou, Int. J. Nanomed., 2016, 11, 501–512 CrossRef CAS PubMed.
- A. Kawabata, A. Baoum, N. Ohta, S. Jacquez, G. M. Seo, C. Berkland and M. Tamura, Cancer Res., 2012, 72, 2057–2067 CrossRef CAS PubMed.
- R. Mortarini, A. Borri, G. Tragni, I. Bersani, C. Vegetti, E. Bajetta, S. Pilotti, V. Cerundolo and A. Anichini, Cancer Res., 2000, 60, 3559–3568 CAS.
- K. S. Wang, D. A. Frank and J. Ritz, Blood, 2000, 95, 3183–3190 CAS.
- S. X. Yang, W. S. Wei, Q. W. Ouyan, Q. H. Jiang, Y. F. Zou, W. Qu, J. H. Tu, Z. B. Zhou, H. L. Ding, C. W. Xie, Q. M. Lei and C. R. Zhong, Biomed. Pharmacother., 2016, 84, 1466–1471 CrossRef CAS PubMed.
Footnote |
| † Those authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |