
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of mesoporous NiAl2O4/MOx (M = La, Ce, Ca, Mg)–γ-Al2O3 nanocomposites for dry reforming of methane†
Li Zhanga,
Xueguang Wang *ab,
Chenju Chena,
Xiujing Zoub,
Xingfu Shangb,
Weizhong Dingab and
Xionggang Lu*a
*ab,
Chenju Chena,
Xiujing Zoub,
Xingfu Shangb,
Weizhong Dingab and
Xionggang Lu*a
aState Key Laboratory of Advanced Special Steel, Shanghai University, Shanghai 200072, China. E-mail: wxg228@shu.edu.cn; luxg@shu.edu.cn
bShanghai Key Laboratory of Advanced Ferrometallurgy, Shanghai University, China
First published on 30th June 2017
Abstract
One-pot synthesized mesoporous NiAl2O4/γ-Al2O3 and NiAl2O4/MOx (M = La, Ce, Ca, Mg)–γ-Al2O3 nanocomposites with excellent textural properties were employed for the dry reforming of methane (DRM). NiAl2O4/La2O3/γ-Al2O3-imp prepared via a traditional impregnation method was used for comparison. The promotion effect of modifiers on the physicochemical properties and catalytic performance of the catalysts was systematically investigated. Characterization and evaluation results indicated that the modified catalysts showed higher activities and better coking-resistance than Ni/γ-Al2O3, and Ni/La2O3–γ-Al2O3 was found to be the most effective one. All the catalysts with or without modifiers presented similar Ni particle sizes due to the enhanced metal–support interaction derived from the reduction of the NiAl2O4 precursor. However, more medium-strength basic sites on the catalyst surface were obtained by adding promoters, which could facilitate the adsorption/activation of CO2 and the gasification of amorphous carbon, improving the catalytic properties and accelerating the coke elimination rate. Additionally, the incorporation of promoters also prevented the phase transformation of γ-alumina.
1. Introduction
Recently, a major scientific challenge has been to effectively utilize greenhouse gases (CO2, CH4) to produce value-added commodity chemicals or fuels. In this regard, compared with the partial oxidation of methane and steam reforming of methane, dry reforming of methane (DRM) can be considered as a reliable option.1–3
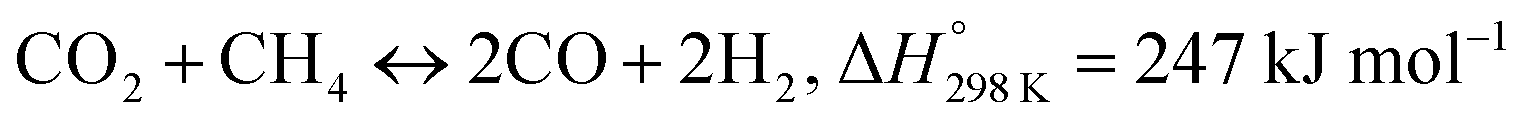 | (1) |
Eqn (1) involves the reforming of methane in the presence of CO2 to produce syngas (CO + H2), which also can effectively convert the cheap energy source of natural gas into syngas.4,5 Syngas with a low H2/CO molar ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) is suitable to serve as a feedstock for further synthesis of oxygenated compounds and liquid hydrocarbons.6–8 Transition metals such as Ni, Co, Pt, and Ru are widely used as the main active components for their high activity in breaking C–H bonds, but attention has been preferentially focused on the development of Ni-based catalysts, due to their extensive availability, high initial catalytic activity and low-cost.9,10 However, Ni-based catalysts are inclined to sinter and be subjected to heavy carbon under high reaction temperatures, which are the major drawbacks resulting in their rapid deactivation.11–13 Therefore, developing new catalyst systems with enhanced anti-sintering and -coking capacities remains the bottleneck restricting the industrialization of DRM process.
:1) is suitable to serve as a feedstock for further synthesis of oxygenated compounds and liquid hydrocarbons.6–8 Transition metals such as Ni, Co, Pt, and Ru are widely used as the main active components for their high activity in breaking C–H bonds, but attention has been preferentially focused on the development of Ni-based catalysts, due to their extensive availability, high initial catalytic activity and low-cost.9,10 However, Ni-based catalysts are inclined to sinter and be subjected to heavy carbon under high reaction temperatures, which are the major drawbacks resulting in their rapid deactivation.11–13 Therefore, developing new catalyst systems with enhanced anti-sintering and -coking capacities remains the bottleneck restricting the industrialization of DRM process.
It has been reported that the support materials can greatly affect the performance of Ni-based catalysts.14–16 Alumina supported Ni oxides have been the commonly investigated system for DRM reaction due to the outstanding thermal stability and catalytic activity.17–21 Numerous approaches have been conducted to develop highly efficient Ni-based catalysts for the reforming reactions, such as improving the Lewis basic properties of the supports,22,23 developing novel preparation methods,24–27 designing bimetallic or trimetallic catalysts.28,29 For example, immobilizing the Ni nanoparticles inside the pore channels of order mesoporous alumina matrix by the confinement effect of the mesopores can prevent the sintering of Ni particles during the DRM reaction.30,31 Yu et al.6 have reported that the Ni–Co bimetallic catalysts show high catalytic activities and stabilities due to the enhanced metal dispersion and small metal size. However, among them, the addition of modifiers, including alkali, alkaline earth, and rare-earth metal oxides was widely reported to be an effective strategy which can provide more basic sites on the surface of the catalysts, favour the adsorption/activation of CO2, enhance the coke-resistant ability and prevent the sintering of active sites and supports.32–37 Therefore, in pioneer literatures, lots of oxides, especially MgO,7 CaO,30 La2O3,38 and CeO2,39 have been used as promoters to improve the catalytic properties and reduce coke deposition. Recently, many new methods were developed to add the promoters. Ma et al.38 prepared the La-modified ordered mesoporous Ni-based catalysts via a one-pot evaporation induced self-assembly method. Ni/SiO2 doped with La2O3 catalysts were synthesized through an in situ self-assembled core–shell precursor route and used for dry reforming of methane.40 Catalyst-supports, 10 mol% X–ZrO2 (X = La, Ce, Pr, Nd, Y) were prepared using a complex polymerized method, and catalyst of 1 wt% Ni/10 mol% La–ZrO2 showed very high DRM activity.41
In our previous study, mesoporous crystalline γ-aluminas with excellent textural properties and thermal and hydrothermal stability were synthesized via the one-pot partial hydrolysis of Al(NO3)3 aqueous solution with (NH4)2CO3,42 and based on this result, mesoporous NiAl2O4/γ-Al2O3 nanocomposites with high specific surface areas, large pore volumes and narrow pore size distributions were also successfully prepared and showed excellent catalytic performance for DRM after H2 reduction.43 The mesoporous spinel structure could effectively stabilize the metallic Ni during the process of in situ reduction and DRM reaction at high temperatures. Therefore, in order to develop more efficient Ni-based catalysts, we intend to systematically investigate the influence of different chemical environments and electronic effects caused by doping La, Ce, Ca, and Mg promoters on the catalytic performance of the catalysts for DRM reaction.
Herein, mesoporous NiAl2O4/γ-Al2O3 and NiAl2O4/MOx (M = La, Ce, Ca, Mg)–γ-Al2O3 materials were successfully synthesized through the facile one-pot partial hydrolysis of metal nitrates aqueous solution with (NH4)2CO3. Without any surfactants that makes the prepared procedure to be cost-effective and environmental-friendly. The physicochemical properties and catalytic performance of the La, Ce, Ca, and Mg elements modified catalysts were systematically investigated, and also compared with that of the catalyst prepared through the traditional impregnation method. All of La2O3, CeO2, CaO, and MgO could improve the performance of the Ni/γ-Al2O3 catalyst and reduce the amount of inert carbon species, especially Ni/La2O3–γ-Al2O3.
2. Experimental
2.1 Chemicals
Aluminum nitrate nonahydrate (Al(NO3)3·9H2O), nickel nitrate hexahydrate (Ni(NO3)2·6H2O), lanthanum nitrate hexahydrate (La(NO3)2·6H2O), cerium nitrate hexahydrate (Ce(NO3)2·6H2O), calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), magnesium nitrate hexahydrate (Mg(NO3)2·6H2O), and ammonium carbonate ((NH4)2CO3) were all analytical reagent and purchased from Sinopharm Chemical Reagent Co., Ltd., using as received without any purification.2.2 Catalyst preparation
The mesoporous NiAl2O4/γ-Al2O3 and NiAl2O4/MOx (M = La, Ce, Ca, Mg)–γ-Al2O3 materials were prepared by our previously reported one-pot synthesis route.42,43 Based on the preliminary investigations, the contents of Ni and M were fixed at 13 wt% and 3 wt%, respectively, in all the materials investigated in this paper. Typically, 0.1 mol of Al(NO3)3·9H2O and certain amount of metal nitrates were together dissolved in 50 mL of deionized water at room temperature. After heating the mixed solution to 70 °C, (NH4)2CO3 aqueous solution (1 mol L−1) was added dropwise into the system under vigorous stirring until the pellucid gel was formed. Then, the gel was aged at room temperature for 48 h, subsequently, dispersed in a glass dish to dry at 100 °C for 24 h. Finally, the solid at heating speed of 1 °C min−1 was calcined in air at 200 °C and 800 °C for 10 h, respectively.For comparison, NiAl2O4/La2O3/γ-Al2O3-imp sample was prepared by the wet impregnation method. The prepared γ-Al2O3 by the one-pot rout was impregnated with the aqueous solution containing required amount of Ni(NO3)2·6H2O and La(NO3)2·6H2O, followed by stirring for 5 h and drying in water-bath at 80 °C. Then, the sample was ground and calcined at 800 °C in air for 10 h.
2.3 Catalyst characterization
Chemical composition analysis was obtained by Perkin Elmer 7300DV inductively coupled plasma atomic emission spectrometer (ICP-AES). X-ray diffraction (XRD) experiments were performed using a Rigaku D/MAX-2200 diffractometer with Cu Kα radiation (λ = 0.15418 nm). The average crystallite sizes of the metal particles were calculated using the Scherrer equation. UV-vis absorption spectra were recorded with a UV-2501 spectrophotometer at ambient temperature, with barium sulfate as a standard for background correction. The specific surface area (SBET), pore size distribution (PSD), and pore volume (Vp) were evaluated from N2 sorption isotherms measured by Micromeritics ASAP 2020 instrument at −196 °C. Before that, the samples were first degassed at 250 °C for 8 h. SEM images were measured with a Hitachi S-4800 electron microscope. TEM images were obtained on a JEOL JEM-2010F electron microscope, and the Ni particle size distributions were obtained by counting ca. 200–250 particles in the TEM images through the Nano Measurer software. H2 temperature-programmed reduction (H2-TPR) experiments were operated on a fixed bed reactor equipped with a thermal conductivity detector (TCD). 100 mg of sample was first pretreated at 200 °C for 1 h, and then reduced under the 5 vol% H2/Ar stream from 200 to 1000 °C at a heating rate of 10 °C min−1. X-ray photoelectron spectra (XPS) were recorded on an ESCALAB 250Xi spectrometer equipped with an Al Kα radiation. Charging effects were calibrated by using the containment carbon (C1s = 284.8 eV). NH3 temperature-programmed desorption (NH3-TPD) analyses were conducted on Micromeritics ASAP 2920. The sample (100 mg) was first in situ reduced at 800 °C for 2 h in 20 vol% H2/He stream (50 mL min−1). After cooling to 40 °C, 10 vol% NH3/He (30 mL min−1) was introduced for 1 h. Then, the sample was purged in flowing He until the baseline was smooth. Finally, NH3-TPD was performed with a heating rate of 10 °C min−1 to 800 °C under He stream. Coke formation was quantitatively analyzed by thermogravimetric (TG) analyzer (Netzsch STA 4449 F3). The used catalysts were heated from 50 °C to 900 °C at a heating speed of 10 °C min−1 in flowing air (30 mL min−1). Raman spectra were collected on a RENISHAW INVIA Raman microscope equipped with a 532 nm laser beam under room temperature.2.4 Catalyst evaluation
The dry reforming of methane was carried out in a quartz tube fixed-bed reactor (i.d. 8 mm) at temperatures from 550 to 800 °C under atmospheric pressure. Prior to each reaction, 100 mg of catalyst (40–60 mesh) diluted with 900 mg of quartz particles (40–60 mesh) was in situ reduced at 800 °C for 2 h in a 20 vol% H2/N2 flow (50 mL min−1). The reaction gas consisted of 50 vol% CH4 and 50 vol% CO2 without dilution gas with the gas hourly space velocity (GHSV) of 1.8 × 105 mL gcat−1 h−1 was fed into the reactor via mass flow controller. Product gases were analyzed by online GC-TCD gas chromatograph with a TDX-01 packed column. An ice-water trap was placed before analyzing to remove the water. In each test, the carbon mass balance was more than 97% on the basis of carbon in the starting reactants. The conversions of CO2 and CH4, and H2/CO molar ratios were calculated based on the following equations:
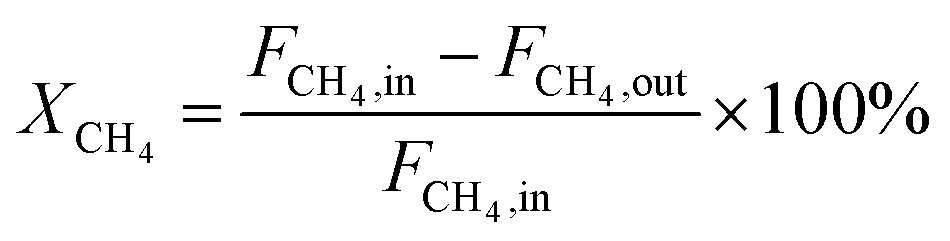 | (2) |
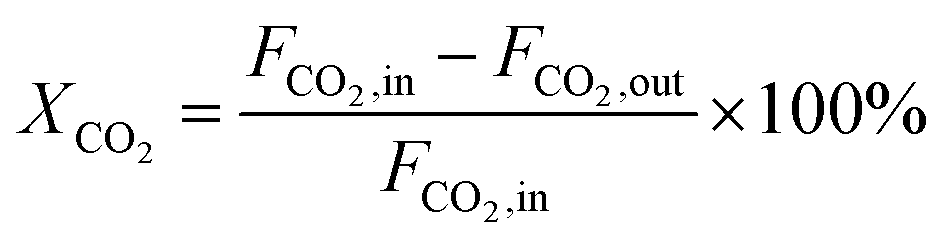 | (3) |
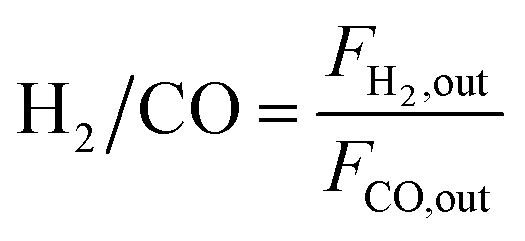 | (4) |
100 mg of catalyst (40–60 mesh) diluted with 900 mg of quartz particles (40–60 mesh) was in situ reduced at 800 °C for 2 h in a 20 vol% H2/N2 flow (50 mL min−1), then the long-term stability tests were conducted under the reaction conditions: GHSV = 1.8 × 105 mL gcat−1 h−1, CO2/CH4 = 1, 750 °C, 1 atm. The catalysts suffered from t h DRM reaction were denoted as Ni/(MOx)–γ-Al2O3-St. The estimation of the turnover frequency (TOF) was described in ESI.†
3. Results and discussion
3.1 Catalytic performance
Blank experiments were first carried out using quartz particles, pure γ-Al2O3 and MOx (M = La, Ce, Ca, Mg)–γ-Al2O3 as the bed materials, and the conversions of CH4 and CO2 could be negligible. Additionally, preliminary experiments showed that the catalytic activities of all the Ni/(MOx)–γ-Al2O3 catalysts would gradually decrease in the initial ca. 80–100 h reaction under the given conditions, followed by almost constant conversions of CO2 and CH4. Therefore, Ni/(MOx)–γ-Al2O3 were first stabilized for 100 h DRM reaction under the conditions: GHSV = 1.8 × 105 mL gcat−1 h−1, CO2/CH4 = 1, 750 °C, 1 atm, and then used to investigate the influences of reaction temperatures on the catalytic properties in the range of 550–800 °C. As illustrated in Fig. 1, the catalytic properties of Ni/(MOx)–γ-Al2O3 for DRM were strongly dependent on the reaction temperature, that is, the CO2 and CH4 conversions increased with the rising of reaction temperature, implying the endothermic character of dry reforming of methane. However, the conversions obtained were far away from the thermodynamic equilibrium values (Table S1†). For all the catalysts, the CO2 conversion was always higher than that of CH4, and the H2/CO ratio was also less than 1:1 in the studied temperature range. According to previous reports,6,14,44 this was mainly due to the existence of reverse water gas shift reaction (RWGS), which could consume part of CO2 and H2 in the reaction system. In addition, the H2/CO ratios gradually closed to unity with elevating temperature due to the suppression of RWGS reaction and enhancement of methane cracking at high temperature.
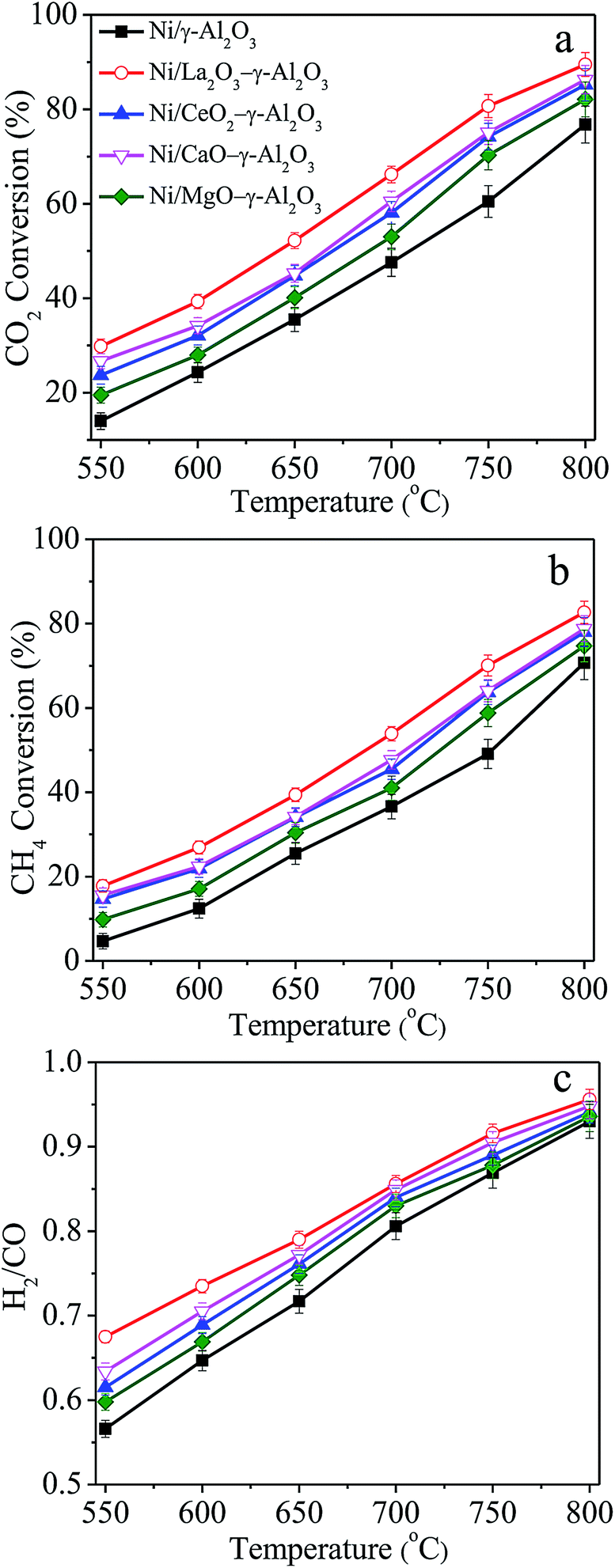 | ||
| Fig. 1 (a) CO2 conversion, (b) CH4 conversion, and (c) H2/CO ratio as functions of reaction temperature over Ni/(MOx)–γ-Al2O3 catalysts stabilized after 100 h DRM reaction. Reaction conditions: GHSV = 1.8 × 105 mL gcat−1 h−1, CO2/CH4 = 1, 1 atm. | ||
Moreover, the promoted catalysts presented higher CO2 and CH4 conversions in comparison with the Ni/γ-Al2O3 catalyst. This result indicated that the presence of modifiers in the support played a positive role in improving the catalytic performance of the catalysts. La-modified Ni/γ-Al2O3 gave the highest activity, and the CO2 and CH4 conversions over Ni/γ-Al2O3 increased by ca. 20% after adding La element. Additionally, it also could be found that the conversions of CO2 and CH4 over Ni/CeO2–γ-Al2O3 were almost identical to those over Ni/CaO–γ-Al2O3, and Mg modifier showed the least improvement on the catalytic performance. The change sequence of H2/CO molar ratios was quite similar to that of CO2 and CH4 conversions. Typically, the higher H2/CO molar ratio corresponded to higher CO2 and CH4 conversions. This result could be explained by the variation in CO2 concentration in the product gas, affecting the RWGS reaction. For different catalyst systems, these modifiers customarily acted a complicated role in affecting the catalytic activity for DRM. For example, La and Ca had been reported to be very beneficial to enhance the catalytic activities over the Ni-based catalysts.30,45 However, Xu46 and Alipour7 declared that the incorporation of La and Ca would cause a decrease in the catalytic activities over Ni/Al2O3 catalyst.
Because the methane dissociation was considered as the rate-determining step for the DRM reaction,14,47 the CH4 reaction rates were normalized to the surface exposed Ni atoms to gain insight into the intrinsic activities of Ni/γ-Al2O3 and Ni/MOx (M = La, Ce, Ca, Mg)–γ-Al2O3. Fig. S1† showed that the CH4 conversion of Ni/La2O3–γ-Al2O3 would become constant when the GHSV > 1.5 × 106 mL gcat−1 h−1, at 750 °C. Therefore, the steady-state TOFCH4 was measured over the catalysts stabilized after 100 h DRM reaction under the given conditions (ESI†). As listed in Table 1, the TOFCH4 values over La, Ce, Ca, and Mg modified catalysts were 5.64 s−1, 4.84 s−1, 5.01 s−1, and 4.08 s−1, respectively. They were higher than that of 3.31 s−1 over Ni/γ-Al2O3. This result confirmed that the intrinsic activity of Ni/γ-Al2O3 for DRM could be effectively enhanced by doping moderate amount of promoters. These TOFCH4 values were also higher than those over various Ni-based catalysts for DRM reaction reported in previous studies. Wang et al.31 reported the relatively low TOFCH4 of 0.7–1.2 s−1 over ordered mesoporous Ni–Ce–Al catalysts. Using the rare-earth oxides modified NiMgAl catalysts, the TOFCH4 values between 2024 and 2349 h−1 were presented.8 For 5% Ni/Ce0.6Zr0.4O2 (1.25) and Ni/α-Al2O3–ZrO2, the TOFCH4 values were 2.5 s−1 and 3.0 s−1, respectively.48,49
| Sample | CH4 conversion (%) | TOFCH4 (s−1) |
|---|---|---|
| a TOFCH4 is defined as the number of converted CH4 molecules per surface Ni atom and second. Reaction conditions: GHSV = 1.8 × 106 mL gcat−1 h−1 (STP), CO2/CH4 = 1.0, 750 °C, 1 atm. | ||
| NiAl2O4/γ-Al2O3 | 10.2 | 3.31 |
| NiAl2O4/La2O3–γ-Al2O3 | 17.1 | 5.64 |
| NiAl2O4/CeO2–γ-Al2O3 | 13.8 | 4.84 |
| NiAl2O4/CaO–γ-Al2O3 | 14.5 | 5.01 |
| NiAl2O4/MgO–γ-Al2O3 | 12.6 | 4.08 |
Fig. 2 illustrated the dependences of the catalytic activities and H2/CO molar ratios on the reaction time over Ni/(MOx)–γ-Al2O3 and Ni/La2O3/γ-Al2O3-imp for the DRM to investigate the catalytic stabilities of the various catalysts. It was observed that all the catalysts prepared following the one-pot method showed a stabilization period, that is, the catalytic activities and H2/CO molar ratios over Ni/γ-Al2O3 and Ni/MOx–γ-Al2O3 gradually decreased in the initial ca. 60–100 h under the given conditions, and kept unchanged during the following period, showing very high catalytic stability. Taking Ni/La2O3–γ-Al2O3 as an example, the conversions of CO2, CH4 and H2/CO molar ratio gently decreased from ca. 89.5%, ca. 81.9% and 0.95 to ca. 81.0%, ca. 70.0% and ca. 0.90, respectively, after the initial 100 h reaction, followed by almost constant catalytic properties. After 100 h time-on-stream, the sequence of the steady-state catalytic performance was as follows: Ni/La2O3–γ-Al2O3 > Ni/CaO–γ-Al2O3 ≈ Ni/CeO2–γ-Al2O3 > Ni/MgO–γ-Al2O3 > Ni/γ-Al2O3. However, the conventional Ni/La2O3/γ-Al2O3-imp catalyst showed a rapid drop in the catalytic activity at the initial stage, and then gradually deactivated to lower conversions with the reaction kept going. At the same time, a decrease in H2/CO molar ratio was also observed for the Ni-impregnated samples due to the enhancement of RWGS by the presence of more unreacted CO2. It has been reported that carbon deposition and sintering of Ni nanoparticles which would reduce the number of the active sites were mainly responsible for the deactivation of traditional Ni-impregnated catalysts during the stability test.31,39,50 This further demonstrated that the present catalysts synthesized by the one-pot hydrolysis method were the extremely suitable and promising candidates to realize the industrialization of DRM reaction.
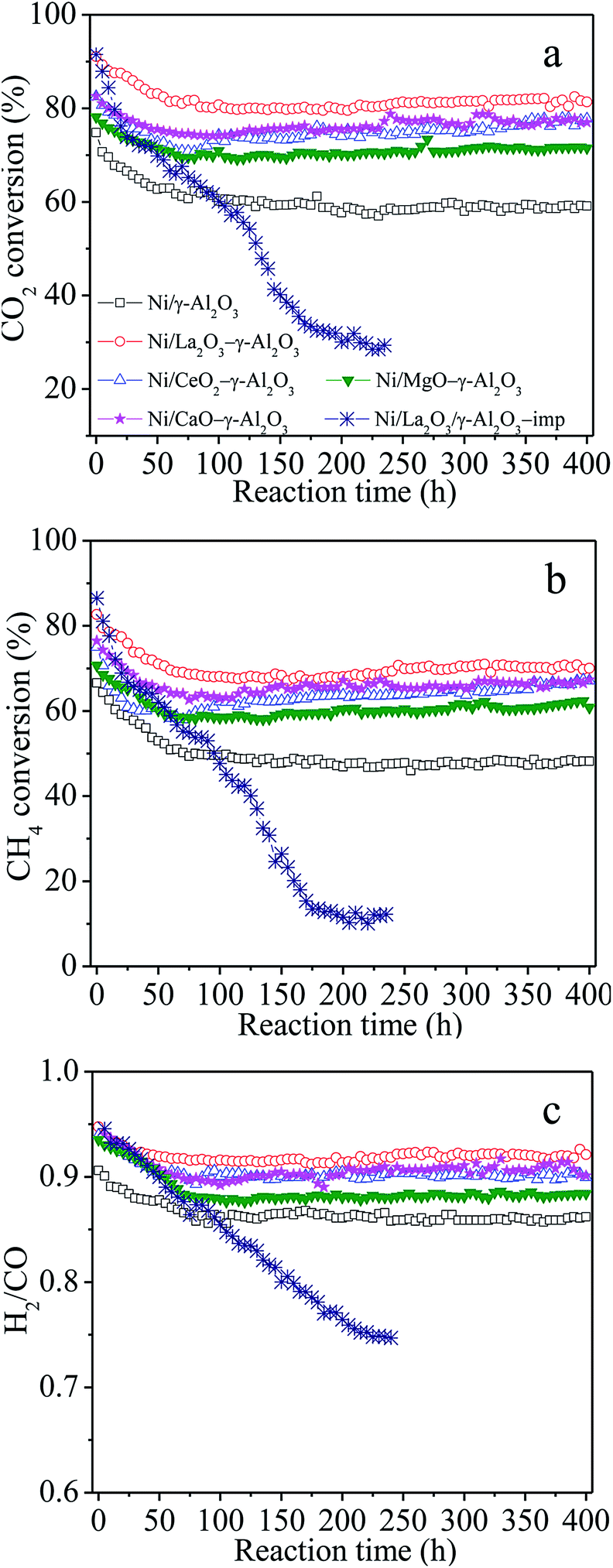 | ||
| Fig. 2 Catalytic properties as a function of reaction time for the DRM over the Ni/(MOx)–γ-Al2O3 and Ni/La2O3/γ-Al2O3-imp catalysts. Reaction conditions: GHSV = 1.8 × 105 mL gcat−1 h−1 (STP), CO2/CH4 = 1, 750 °C, 1 atm. | ||
3.2 Characterization of the various samples
Table 2 showed the amount of Ni and M (M = La, Ce, Ca, Mg) determined by ICP-AES, expressed as weight percentage. It was found that the actual chemical compositions were closed to the nominal ones, which illustrated that the metals had been successfully dispersed into the frameworks. As shown in Fig. S2,† the HAADF-STEM images and EDX spectrum of the representative NiAl2O4/La2O3–γ-Al2O3 sample confirmed that the Ni and promoter elements were uniformly distributed throughout the γ-Al2O3 matrices due to the advantage of one-pot method.| Sample | Actual amount (wt%) by ICP | BE of Ni 2p3/2 (eV) | Ni/Al atomic ratiob | |
|---|---|---|---|---|
| Ni | Ma | |||
| a M = La, Ce, Ca, and Mg.b The values in parentheses are the nominal surface Ni/Al molar ratios. | ||||
| NiAl2O4/γ-Al2O3 | 11.70 | — | 856.2 | 0.082(0.135) |
| NiAl2O4/La2O3–γ-Al2O3 | 11.59 | 2.71 | 856.1 | 0.098(0.141) |
| NiAl2O4/CeO2–γ-Al2O3 | 11.74 | 2.89 | 856.1 | 0.085(0.141) |
| NiAl2O4/CaO–γ-Al2O3 | 11.68 | 2.91 | 856.3 | 0.092(0.142) |
| NiAl2O4/MgO–γ-Al2O3 | 11.62 | 2.85 | 856.2 | 0.093(0.144) |
The NiAl2O4/γ-Al2O3 solid solution was successfully formed in the materials after 800 °C calcination, illustrating that addition of La, Ce, Ca, and Mg would not change the crystalline phases, and the NiAl2O4 precursor could be reduced to metallic Ni catalyst through H2 reduction (Fig. S3†). Fig. 3(1) and (2) showed the XRD patterns of the spent Ni/γ-Al2O3 and Ni/MOx (M = La, Ce, Ca, Mg)–γ-Al2O3 catalysts after 100 h and 400 h reaction, respectively. The metallic Ni phase was also detected in all the used catalysts. As depicted in Fig. 3(2), a group of new reflections corresponding to α-Al2O3 (JCPDS card 11-0661) was observed in Ni/γ-Al2O3-S400, illustrating the phase transformation from γ-Al2O3 to α-Al2O3, which was one of the major reasons for the loss of specific surface area and severe coke deposition over Ni/γ-Al2O3 during the DRM process. However, the crystal structures and crystalline phases of the Ni/MOx–γ-Al2O3-S400 catalysts had no obvious change compared with those of the reduced catalysts (Fig. S3†). This suggested that the addition of promoters played an important role in stabilizing the catalyst structures during long-term DRM reaction. In addition, Ni/γ-Al2O3-S100 and Ni/γ-Al2O3-S400 showed the strongest graphite diffraction peaks at a 2θ value of ca. 26°, respectively, which was indicative of larger amount of carbon deposition.
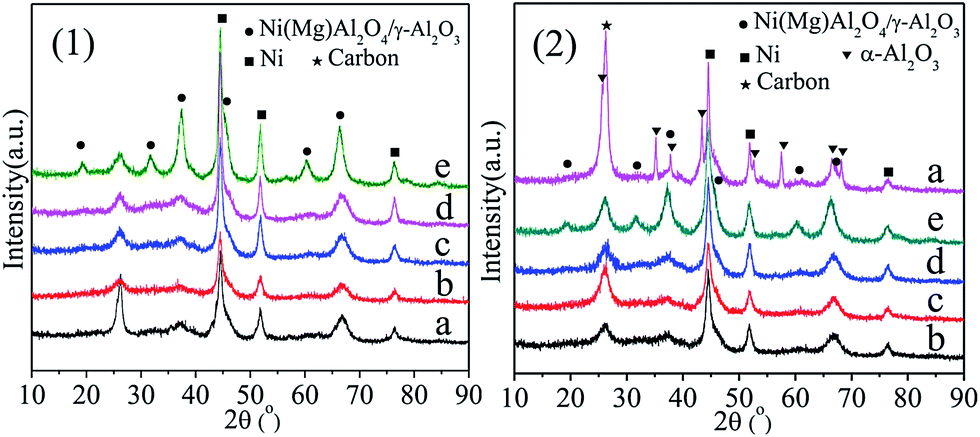 | ||
| Fig. 3 XRD patterns of the spent catalysts (1) after 100 h reaction, and (2) after 400 h reaction. (a) Ni/γ-Al2O3, (b) Ni/La2O3–γ-Al2O3, (c) Ni/CeO2–γ-Al2O3, (d) Ni/CaO–γ-Al2O3, and (e) Ni/MgO–γ-Al2O3. Reaction conditions: GHSV = 1.8 × 105 mL gcat−1 h−1 (STP), CO2/CH4 = 1, 750 °C, 1 atm. | ||
The average crystallite sizes of the metallic Ni over Ni/(MOx)–γ-Al2O3 presented a similar value of ca. 16 nm (Table S2†). After 100 h reaction, the sizes of Ni particles decreased from ca. 16 nm to ca. 9 nm, however those still kept ca. 9 nm after 400 h reaction (Table 4), which powerfully confirmed the excellent sintering resistance of Ni particles. But, the mechanism of the decrease in Ni particle sizes in the initial stabilization period was still incompletely identified. Montero et al.51 also found a decrease in the Ni crystallite sizes over the used catalysts from 10.0 to 5.1 nm, and they attributed this to the formation of NiC. Slagtern et al.52 speculated that the apparent decrease in Ni particle sizes was probably due to a continued reduction of the catalyst during the reforming reaction. We thought that the reaction atmospheres would affect the interaction between Ni crystallites and support, resulting in re-construction and/or re-distribution of Ni species on catalyst surface, which led to the decrease in the Ni crystallite sizes.53
For NiAl2O4/La2O3/γ-Al2O3-imp, shown in Fig. S3,† NiAl2O4 spinel and NiO phase were simultaneously observed, and the mean size of Ni particles over Ni/La2O3/γ-Al2O3-imp was ca. 18.9 nm (Table S2†), slightly higher than that over Ni/La2O3–γ-Al2O3. After 235 h DRM reaction, the metallic Ni particles were aggregated to bigger ones of ca. 28.9 nm (Table 4). A strong diffraction peak at 26° ascribed to coke deposition was also observed in Ni/La2O3/γ-Al2O3-imp-S235, which demonstrated that severe carbon deposition occurred. Therefore, the sintering of Ni particles and surface coke deposition were mainly responsible for the deactivation of Ni/La2O3/γ-Al2O3-imp.
The UV-vis absorption spectra of the calcined samples were presented in Fig. 4. It was previously reported that the bands located at 550–645 nm corresponded to the tetrahedral Ni2+ species in the NiAl2O4 lattice, whereas, those around 720 nm and a shoulder around 650 nm were due to the d–d transition of octahedral Ni2+ of Ni oxides.17,54,55 The presence of the Ni2+ in octahedral environment could evidence the formation of NiAl2O4 structure in the inverse coordination.56 The Ni2+ ions in the tetrahedral sites corresponds to “readily reduced” nickel and that in the octahedral sites corresponds to “hard to reduce” nickel.57,58 Therefore, the main two bands at 590 and 630 nm clearly found in the visible domain in Fig. 4 were ascribed to tetrahedral Ni2+ ions for normal NiAl2O4, avoiding the formation of hardly reduced inverse NiAl2O4 in the present materials. This could improve the reducibility of the NiAl2O4/(MOx)–γ-Al2O3 materials.
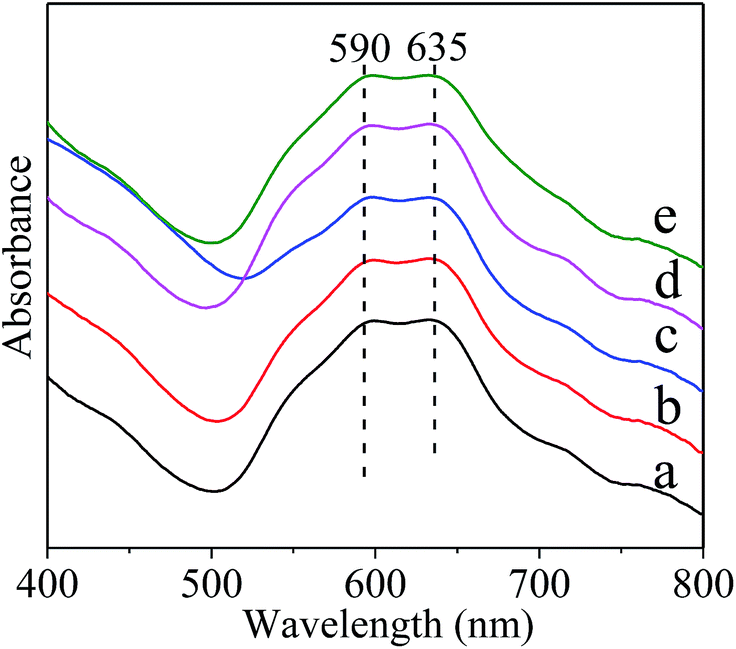 | ||
| Fig. 4 Normalized UV-vis absorption spectra of the fresh materials. (a) NiAl2O4/γ-Al2O3, (b) NiAl2O4/La2O3–γ-Al2O3, (c) NiAl2O4/CeO2–γ-Al2O3, (d) NiAl2O4/CaO–γ-Al2O3, and (e) NiAl2O4/MgO–γ-Al2O3. | ||
H2-TPR was believed to reflect the energy bonds between metal species and their environment,59 and the H2-TPR profiles were displayed in Fig. 5. All NiAl2O4/(MOx)–γ-Al2O3 samples showed similar profiles of H2 consumption with only a strong and relatively symmetric peak located in the range of 809–841 °C, indicating the reduction of Ni2+ into Ni.8 No reduction peaks in the lower temperature region less than 750 °C were observed, confirming the formation of single-phase NiAl2O4/γ-Al2O3 solid solutions without formation of any NiO species as well as the stronger interaction between metal Ni and support, which was in favour of improving the dispersion of Ni and suppressing the sintering of Ni particles in the reduction and reaction process.30,60 Additionally, the prepared materials displayed a shift for the reduction temperatures. For instance, NiAl2O4/MgO–γ-Al2O3 showed the highest reduction temperature at 841 °C, and that of NiAl2O4/La2O3–γ-Al2O3 decreased to 809 °C. We thought that the variations in the degree of crystallinity and crystallite sizes of NiAl2O4/γ-Al2O3 solid solutions influenced the reduction temperatures of Ni2+ ions in the matrices.
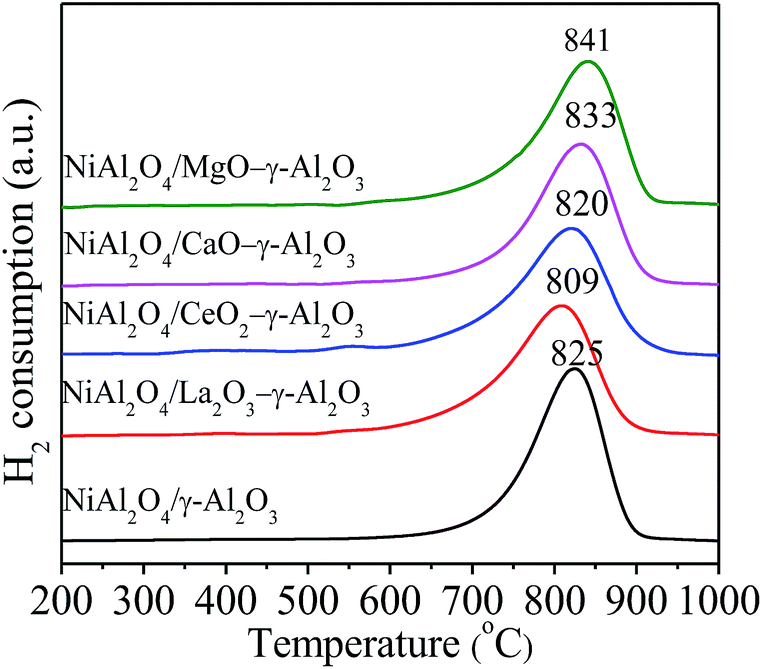 | ||
| Fig. 5 H2-TPR profiles of the NiAl2O4/(MOx)–γ-Al2O3 samples. | ||
Fig. 6 and Table 2 presented the Ni 2p XPS spectra, the related binding energies (BEs) of the Ni 2p3/2 levels, respectively, to determine the chemical environment and oxidation state of the surface Ni species in NiAl2O4/γ-Al2O3 and NiAl2O4/MOx (M = La, Ce, Ca, Mg)–γ-Al2O3 materials. For all the samples, similar intensities of the peaks for Ni 2p were observed due to the same Ni content. In addition, all the Ni 2p3/2 peaks located in a BE range of 856.0–856.5 eV accompanied by a satellite peak at 862.4–862.8 eV and a spin–orbit coupling energy gas of ca. 17.6 eV. The reported binding energy of Ni 2p3/2 for NiAl2O4 spinel was ca. 856.0 eV, which was much higher than ca. 854.0 eV ascribed to the BE of Ni 2p3/2 in pure NiO.43,46 This forcefully verified that the Ni2+ ions were reacted with γ-Al2O3 to form NiAl2O4 spinel in NiAl2O4/(MOx)–γ-Al2O3 materials, in combination with the XRD and H2-TPR measurement. Table 2 also listed the surface atomic composition of Ni relative to Al. The value of Ni/Al over each sample was smaller than the corresponding nominal one. This phenomenon could be attributed to the diffusion of Ni2+ into γ-Al2O3 lattice to form NiAl2O4.
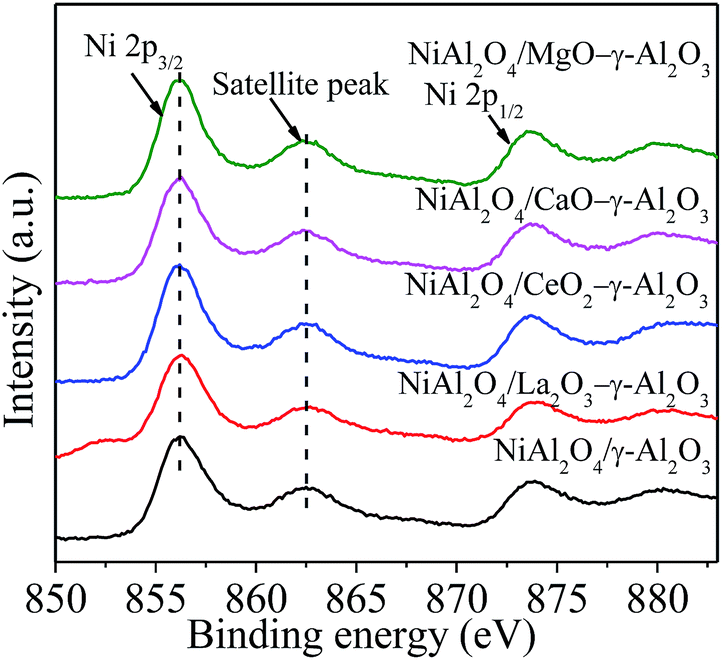 | ||
| Fig. 6 Ni 2p XPS spectra of the NiAl2O4/(MOx)–γ-Al2O3 materials. | ||
N2 adsorption–desorption isotherms and pore size distributions (Fig. S5†) and TEM images (Fig. S6†) verified that all the reduced samples possessed the uniform mesopores according to the IUPA classification.6,8 The stabilization effect of modifiers on the mesostructure of Ni/γ-Al2O3 was also verified powerfully by N2 sorption isotherms of the spent catalysts after 400 h reaction in Fig. 7(a) and (b), where deposited carbon was removed. Except Ni/γ-Al2O3-S400, each of the spent catalysts exhibited similar N2 sorption isotherms and narrow pore size distributions located in the range of 4.0–8.0 nm to the reduced counterpart (Fig. S5†). This was an indication that the addition of modifiers was helpful for the maintaining of mesoporous frameworks of the Ni/γ-Al2O3 during the stability test. It was noted that the SBET and Vp of Ni/MOx–γ-Al2O3-S400 after removing coke in Table 4 showed slight decreases compared with those of the reduced catalysts in Table 3, likely due to structural shrinkage and surface re-construction in the reaction process. However, the Ni/γ-Al2O3-S400 after removing coke exhibited sharp decreases in the SBET and Vp from 151 m2 g−1 and 0.24 cm3 g−1 to 55 m2 g−1 and 0.17 cm3 g−1, respectively.
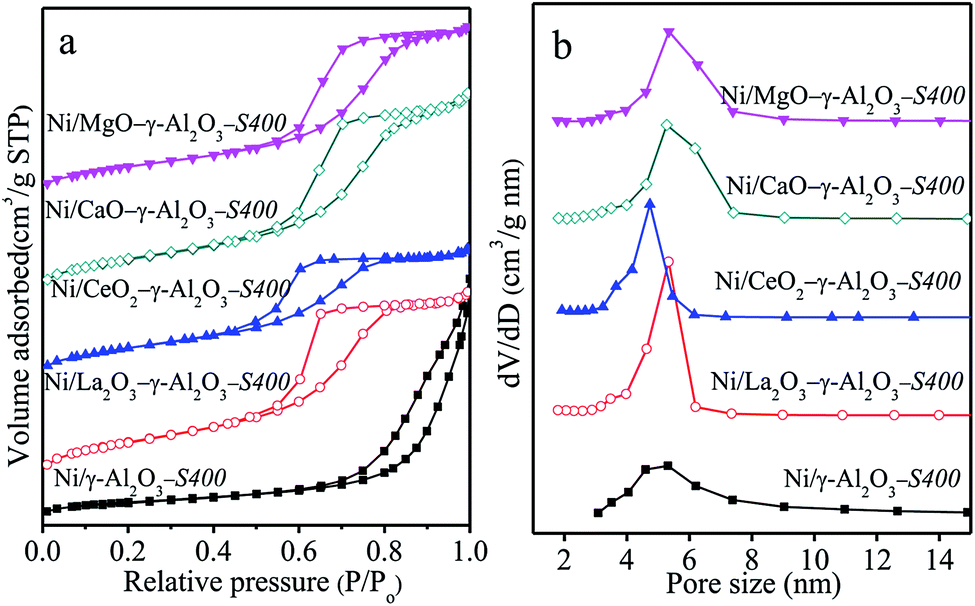 | ||
| Fig. 7 (a) Nitrogen adsorption–desorption isotherms, and (b) BJH pore size distributions of the spent Ni/(MOx)–γ-Al2O3-S400 catalysts after being calcined in air at 600 °C for 2 h to remove the carbon deposited. | ||
| Sample | SBET (m2 g−1) | Vp (cm3 g−1) | Relative amount of NH3 desorptiona |
|---|---|---|---|
| a Defined as the total amount of NH3 desorbed per gram of each catalyst divided by the total amount of NH3 desorbed (100 as the reference) per gram of Ni/γ-Al2O3 by the integration of TPD curves. | |||
| Ni/γ-Al2O3 | 151 | 0.24 | 100 |
| Ni/La2O3–γ-Al2O3 | 152 | 0.24 | 70 |
| Ni/CeO2–γ-Al2O3 | 147 | 0.23 | 82 |
| Ni/CaO–γ-Al2O3 | 142 | 0.22 | 80 |
| Ni/MgO–γ-Al2O3 | 114 | 0.19 | 85 |
The NH3-TPD analysis was used to examine the acid and base properties on the surfaces of the reduced catalysts, and the acquired profiles were displayed in Fig. 8. All the profiles mainly including two clear bands distributed in the desorption regions of 50–200 °C and 200–500 °C, respectively, corresponding to two types of acidic sites. According to the pioneer literatures, the desorption regions located in 50–200 °C were attributed to the weak acidic sites and physically adsorbed NH3, and the desorption regions in 200–500 °C were associated with the NH3 desorption from medium-strength acidic sites.57 All the TPD profiles showed the similar shape indicated that these acidic sites had similar coordination environments in the tested materials. The positions of the desorption peaks had no apparent variations with addition of promoters, but the intensities were different with each other. This implied that the doping of promoter elements mainly influenced the quantity of the medium-strength acidic sites on the catalyst surface. Table 1 gave the relative amount of NH3 desorption on the catalyst surfaces to facilitate the above comparison. The sequence of the relative amount of desorbed NH3 from the reduced catalysts could be summarized as following: Ni/γ-Al2O3 > Ni/MgO–γ-Al2O3 > Ni/CeO2–γ-Al2O3 ≈ Ni/CaO–γ-Al2O3 > Ni/La2O3–γ-Al2O3. The more amount of desorbed NH3 meant the more amount of acidic sites and less amount of basic sites. Therefore, the order of the Lewis basicity of the catalysts was Ni/La2O3–γ-Al2O3 > Ni/CaO–γ-Al2O3 ≈ Ni/CeO2–γ-Al2O3 > Ni/MgO–γ-Al2O3 > Ni/γ-Al2O3. From the above analyses, it could be kwon that all the catalysts presented the similar mesoporous spinel structures and almost the identical Ni particle sizes. Thus, it could be judged that there was a close relationship between the activities and the Lewis basicity of the catalysts. The incorporation of promoters could favour the Lewis basicity of the support, and mainly increase the amount of medium-strength basic sites, which could simultaneously enhance the chemisorption and activation of CO2 on the catalyst surfaces. This would increase the CO2 concentration on the surfaces of the catalysts, leading to higher catalytic activities. Hence, different promoters improved the catalytic activity in a different degree. Additionally, the enhancement of the basic sites on the catalyst surface could also effectively hinder the formation of carbon deposition, which would be investigated in the following section in detailed.
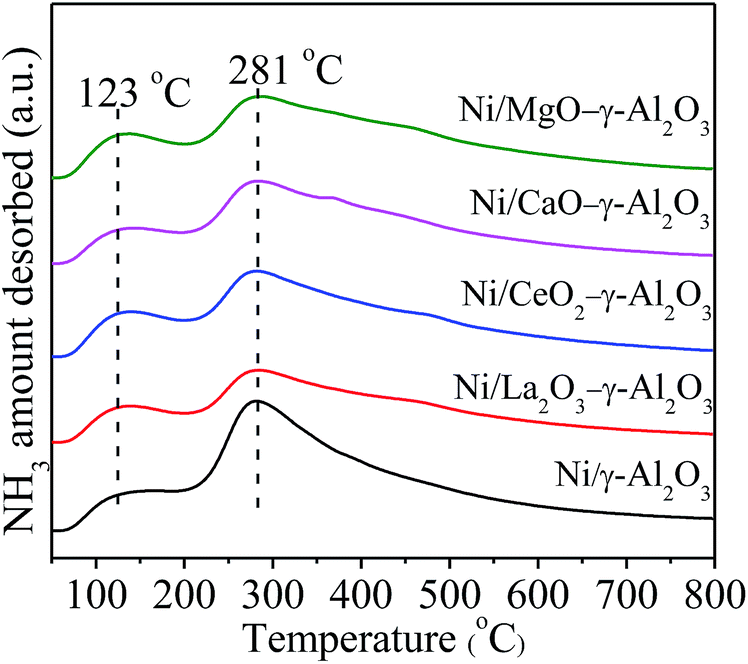 | ||
| Fig. 8 NH3-TPD profiles for the reduced Ni/(MOx)–γ-Al2O3 materials. | ||
3.3 Deactivation analysis of spent catalysts
It is well known that the carbon deposition is more severe in DRM process than that in the steam reforming reaction for Ni-based catalysts. Therefore, it is very crucial to investigate the coke-resistant capacities of Ni/(MOx)–γ-Al2O3 via various techniques of TEM, SEM, TG, and Raman to realize its industrialized application in DRM, and the acquired results were analyzed in the following section.As summarized in Table 4, the mean sizes of Ni particles after stability test estimated from the TEM images (Fig. S7†) were similar to those calculated by the XRD patterns. It could be found that the change in Ni particle sizes took place mainly in the initial stabilization period, and the maintenance of the small Ni particles in the next reaction pledged the excellent stabilities of Ni/(MOx)–γ-Al2O3. TEM images (Fig. S7†) showed that severe agglomeration of Ni crystallites occurred over Ni/La2O3/γ-Al2O3-imp-S235, which was consistent with the XRD results (Fig. S4†). The carbon species, including whiskers or filaments carbon and nano-onion carbon, were determined by the strength of the metal–support interaction and the balance between carbon deposition and carbon diffusion.61 It had been reported that the active sites could not be encapsulated by the filamentous carbon, whereas, the nano-onion carbon which acted like a shell would lead to a significant lose of active sites and catalyst deactivation.46,61,62 The accumulation of carbon deposition was also depicted in the TEM and SEM images. It was obvious that the carbon was existed mainly in the dominant form of carbon filaments. Only a few of short and gracile filamentous carbon covered on Ni/La2O3–γ-Al2O3-S400, but a substantial amount of long and thick filamentous carbon was clearly observed depositing on Ni/γ-Al2O3-S400 and Ni/La2O3/γ-Al2O3-imp-S235, indicating the excellent coking-resistance of the promoted catalysts prepared by the one-pot method. Moreover, we noted that carbon nano-onions with embedded big sintered Ni particles were formed over Ni/La2O3/γ-Al2O3-imp-S235 (Fig. 9), which would aggravate the deactivation of the Ni-impregnated catalyst.
| Sample | Ni particle size (nm) by | Carbon amount by TG (mg gcat−1) | SBETb (m2 g−1) | Vpb (cm3 g−1) | |
|---|---|---|---|---|---|
| XRD | TEM | ||||
| a Reaction conditions: GHSV = 1.8 × 105 mL gcat−1 h−1, CO2/CH4 = 1, 750 °C, 1 atm, and the catalysts suffered from t h DRM reaction were denoted as Ni/(MOx)–γ-Al2O3-St.b SBET and Vp after removing carbon in air at 600 °C for 2 h. | |||||
| Ni/γ-Al2O3-S100 | 8.7 | 9.1 ± 1.4 | 709 | — | — |
| Ni/La2O3–γ-Al2O3-S100 | 9.1 | 9.1 ± 1.7 | 307 | — | — |
| Ni/CeO2–γ-Al2O3-S100 | 9.5 | 9.9 ± 1.9 | 395 | — | — |
| Ni/CaO–γ-Al2O3-S100 | 9.3 | 9.8 ± 1.6 | 336 | — | — |
| Ni/MgO–γ-Al2O3-S100 | 8.1 | 9.1 ± 1.4 | 408 | — | — |
| Ni/γ-Al2O3-S400 | 8.8 | 9.2 ± 1.8 | 1641 | 55 | 0.17 |
| Ni/La2O3–γ-Al2O3-S400 | 8.9 | 9.0 ± 2.0 | 373 | 134 | 0.21 |
| Ni/CeO2–γ-Al2O3-S400 | 8.2 | 9.1 ± 2.2 | 567 | 117 | 0.19 |
| Ni/CaO–γ-Al2O3-S400 | 9.0 | 9.7 ± 2.1 | 428 | 123 | 0.22 |
| Ni/MgO–γ-Al2O3-S400 | 8.6 | 9.3 ± 2.0 | 538 | 105 | 0.19 |
| Ni/La2O3/γ-Al2O3-imp-S235 | 26.2 | 28.9 ± 3.0 | 1512 | — | — |
 | ||
| Fig. 9 TEM and SEM images of the various spent catalysts. Reaction conditions: GHSV = 1.8 × 105 mL gcat−1 h−1 (STP), CO2/CH4 = 1, 750 °C, 1 atm. | ||
The TG-DSC measurement was conducted in air to quantify the carbon deposition based on the weight loss due to the removal of carbon, and identify the types of the coke over the spent samples. All the TG profiles in Fig. 10 showed a sharply downward trend in the range of 500–800 °C, which was derived from the oxidation of deposited carbon. The weight loss over Ni/γ-Al2O3-S400 was more severe than that over Ni/MOx–γ-Al2O3-S400. Although the Ni/La2O3/γ-Al2O3-imp just used in DRM for 235 h, it still appeared a large weight loss. Table 4 gave the quantity of the deposited carbon calculated from the corresponding weight loss in TG profiles. It could be seen that the amounts of carbon deposited over La, Ce, Ca, and Mg modified catalysts were smaller than that deposited on the Ni/γ-Al2O3, among them, the Ni/La2O3–γ-Al2O3 showed the least amount of carbon deposition. For example, after 400 h reaction, La, Ce, Ca, and Mg modified catalysts showed the amounts of carbon were 373 mg gcat−1, 567 mg gcat−1, 428 mg gcat−1, and 538 mg gcat−1, respectively, whereas, that over Ni/γ-Al2O3 was significantly increased to 1641 mg gcat−1. It was obvious that the amounts of the carbon deposition matched well with the trend of the basicity of the catalysts. Compared the amounts of carbon over Ni/La2O3–γ-Al2O3-S100 and Ni/La2O3–γ-Al2O3-S400, it was interesting to find that the carbon deposition mainly took place in the initial stabilization stage, and during the following 300 h period, the amounts of carbon were very small. This result was consistent with the variation with the Ni particle sizes during the DRM reaction. Although the Ni particle sizes of Ni/MOx–γ-Al2O3-S400 were almost identical (Table 4), the quantities of the carbon deposition were totally different (Table 4). This observation demonstrated that the physicochemical environment of metal Ni on the catalyst surfaces affected the formation of coke. Combined the results of NH3-TPD and TG, it was reasonable to conclude that the surface acid–base property of the catalyst was the main influence factor. The improvement of the Lewis basicities of the catalysts, especially the increase in the number of medium-strength basic sites on the catalyst surfaces by doping La, Ce, Ca, and Mg elements, assisted in the adsorption/activation of CO2 and oxidation of surface carbon, lowering the rate of coke deposition. Additionally, the carbon deposition over Ni/La2O3/γ-Al2O3-imp-S400 was up to 1512 mg gcat−1, revealing that the sever coke deposition could be effectively inhibited by employing the one-pot synthesis method.
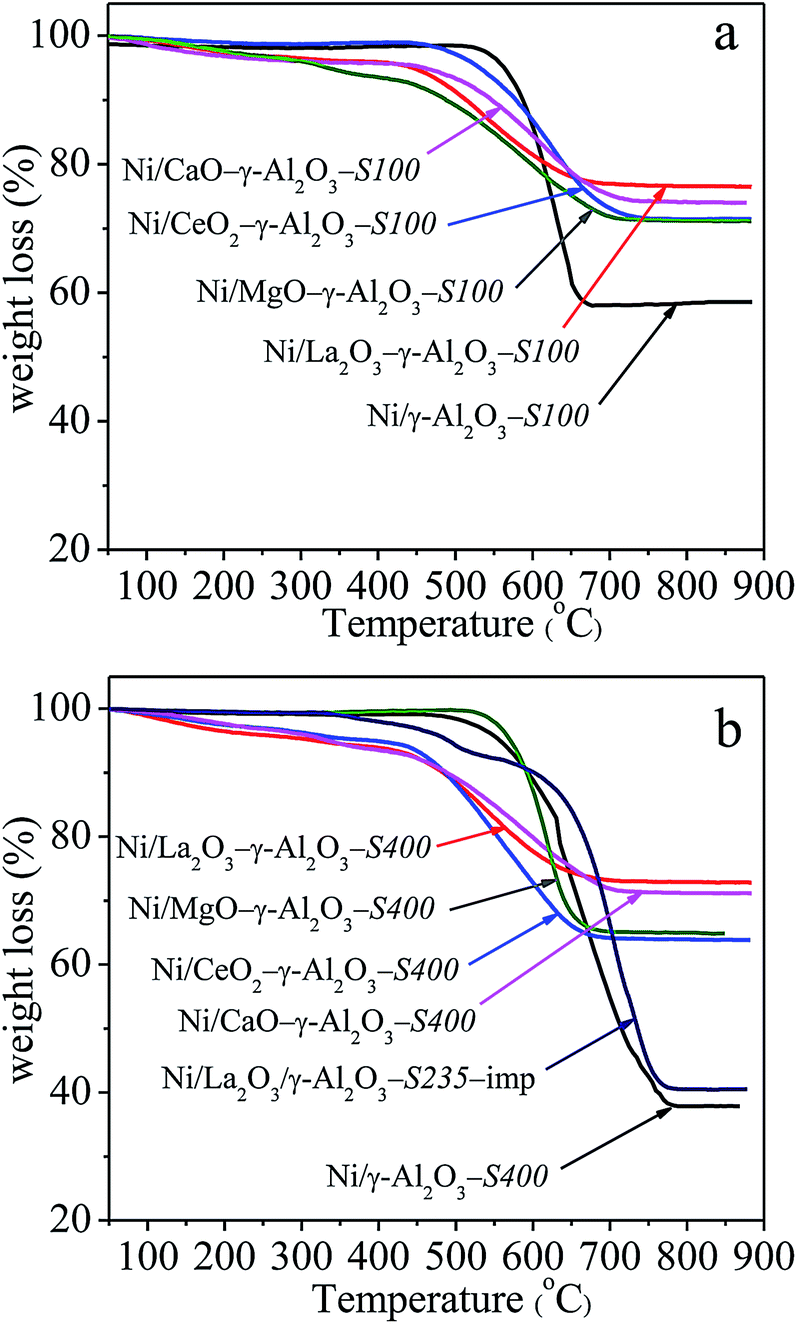 | ||
| Fig. 10 TG profiles of the spent catalysts. Reaction conditions: GHSV = 1.8 × 105 mL gcat−1 h−1 (STP), CO2/CH4 = 1, 750 °C, 1 atm. | ||
The oxidation of carbon deposition was an exothermic process, therefore, the types of the carbon species could be distinguished on the basis of temperatures of the exothermic peaks in DSC profiles. It could be seen in Fig. 11 that there were three types of coke with different reactivities over the spent catalysts. The peaks at low temperature around 325 °C were assigned to the oxidation of amorphous carbon or active carbon species (Cα), which was usually formed via the cracking of CH4 during the initial reaction stage and accounted for the formation of CO. The peaks around 500 °C could be attributed to less-active carbon species (Cβ) derived from further dehydrogenation of amorphous carbon, whereas, the high-temperature peaks above 590 °C corresponded to the inert carbonaceous species (Cγ) with different degrees of graphitization.8,31,61,63 It was observed that the spent catalysts performed quite different exothermic peaks. All DSC curves for Ni/(MOx)–γ-Al2O3-S400 showed a weak exothermic peak around 320 °C corresponded to Cα, which could be easily eliminated by oxidation. The carbon species formed on Ni/γ-Al2O3-S400 were mainly Cγ identified from the intense exothermic peak at 655 °C. Ni/La2O3–γ-Al2O3-S400 showed a big exothermic peak at 510 °C and a small shoulder peak at 470 °C corresponded to Cβ. The main peak of Ni/CeO–γ-Al2O3-S400 appeared at 525 °C was attributed to Cβ, meanwhile, a strong shoulder peak at 600 °C (Cγ) was also observed.
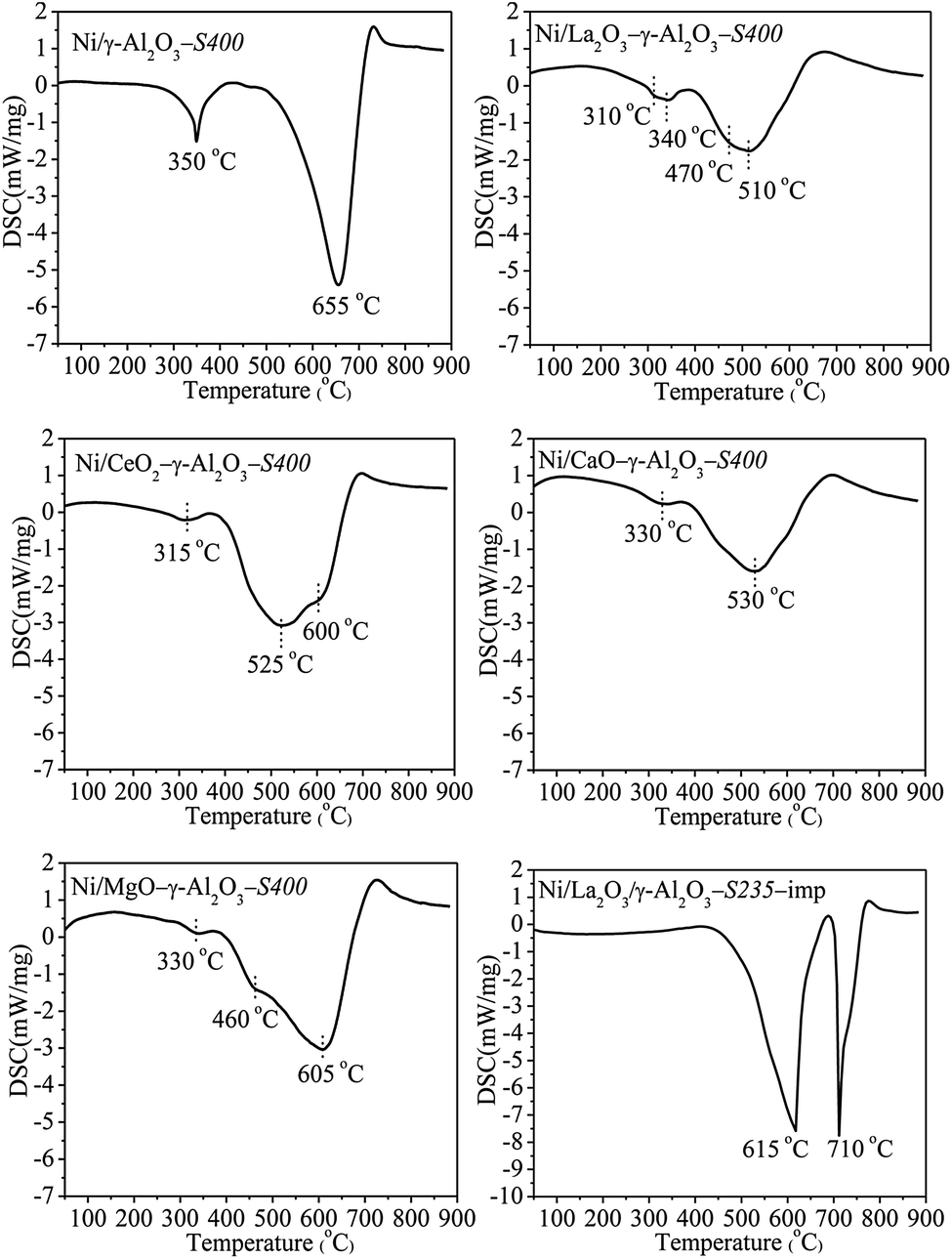 | ||
| Fig. 11 DSC profiles of the spent catalysts. Reaction conditions: GHSV = 1.8 × 105 mL gcat−1 h−1 (STP), CO2/CH4 = 1, 750 °C, 1 atm. | ||
However, only one peak at 530 °C for Cβ was found over Ni/CaO–γ-Al2O3-S400. Ni/MgO–γ-Al2O3-S400 presented an exothermic peak at 605 °C (Cγ) accompanied by a shoulder peak at 460 °C (Cβ). These results revealed that the promoters significantly influenced the distribution of the carbon species deposited on the catalyst surfaces, and the reactivities of the carbon species increased with the increase in the basicities of the promoters. In addition, besides the exothermic peaks at 615 °C and 710 °C, no Cα and Cβ were found over Ni/La2O3/γ-Al2O3-imp-S235 indicated that both of them transformed into Cγ with the highest degree of graphitization. This result once again proved that the catalysts prepared via the one-pot method possessed more excellent coke resistance than the catalysts prepared through the traditional impregnation method.
Raman spectroscopy of the used catalysts was provided to further study the structure and properties of carbonaceous species, shown in Fig. 12. The Raman spectra displayed two obvious peaks located around 1340 cm−1 and 1576 cm−1, which corresponded to D band and G band, respectively. The disorders and defects caused by structural imperfections induced the D band, whereas, the G band produced through C–C stretching vibration of all pairs of sp2 C atoms in carboatomic ring or long-chain.10,15,64 Generally, the intensity ratio of D band to G band (ID/IG) was used to measure the graphitic degree of carbon deposition. The ID/IG ratios of the spent catalysts obviously decreased in the following sequence: Ni/La2O3–γ-Al2O3-S400 (1.94) > Ni/CaO–γ-Al2O3-S400 (1.62) > Ni/CeO2–γ-Al2O3-S400 (1.59) > Ni/MgO–γ-Al2O3-S400 (1.48) > Ni/γ-Al2O3-S400 (0.76), illustrating that more disordered carbonaceous species with lower degree of graphitization deposited over catalysts with stronger Lewis basicity, which was matched well with the DSC results.
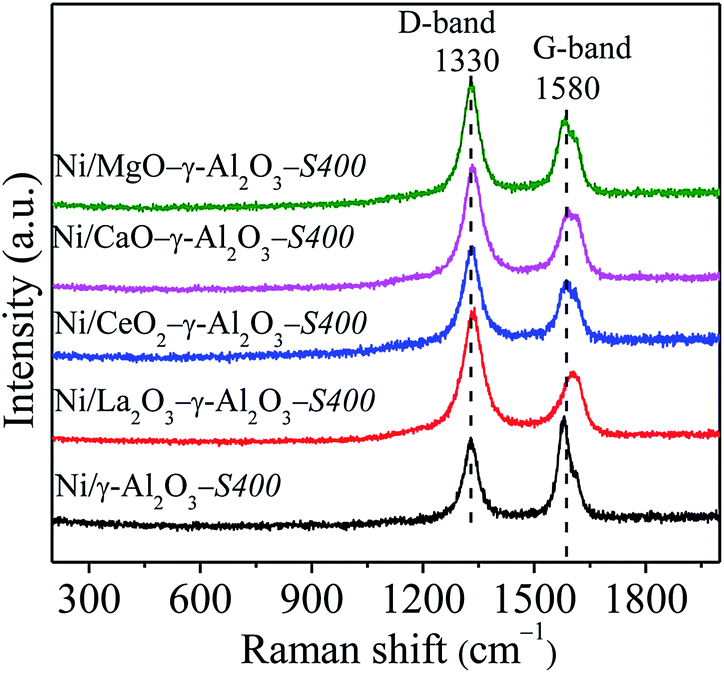 | ||
| Fig. 12 Raman spectra of the spent catalysts. Reaction conditions: GHSV = 1.8 × 105 mL gcat−1 h−1 (STP), CO2/CH4 = 1, 750 °C, 1 atm. | ||
The two side reactions CH4 decomposition (CH4 → C + 2H2) and CO disproportionation (2CO → C + CO2) in DRM reforming procedure have been considered to lead to the coke formation.65 However, under high temperatures, the carbon resource mainly came from CH4 cracking on the Ni active sites. In the early stage of the DRM reaction, CH4 decomposition would create a large amount of amorphous carbon, namely the easily oxidized intermediate CHx species. With the reforming reaction to continue, they gradually transformed into inert carbon species through further dehydrogenation, polymerization, and rearrangement.30 The addition of La could effectively enhance the surface basicity of the catalyst, increasing the rates of absorption/activation of CO2, accelerating the gasification of intermediate CHx species and preventing the formation of inactive carbon during the dry reforming of methane.25 Although the promoting mechanism of Ca and Mg in suppressing coke was similar to that of La, they showed relatively poor effect. As for the Ce modified catalyst, it possessed enhanced redox property and plentiful oxygen vacancies. During the reforming reaction, the cyclic transformation between Ce3+ and Ce4+ according to the following reaction: Ce2O3 + CO2 → 2CeO2 + CO and 2CeO2 + C → Ce2O3 + CO promoted the elimination of coke deposition.8,10,46
4. Conclusions
In summary, mesoporous NiAl2O4/γ-Al2O3 and NiAl2O4/MOx (M = La, Ce, Ca, Mg)–γ-Al2O3 were prepared through the one-pot hydrolysis of mixed metal nitrates with (NH4)2CO3, and investigated for the DRM reaction. After in situ reduction at 800 °C, these composite oxides showed high catalytic activities and long-term stabilities under GHSV = 1.8 × 105 mL gcat−1 h−1, especially the La-modified Ni/γ-Al2O3 showed the best catalytic performance. The results of the various characterization measurements indicated that normal NiAl2O4 spinel precursor was successfully formed after 800 °C calcination, which was beneficial to improve the dispersion of Ni particles and strengthen the metal–support interaction. The addition of La, Ce, Ca, and Mg had little effect on the textural properties and crystalline phases of the prepared materials and Ni particle sizes, but they could enhance the strength and increase the number of medium-strength basic sites on the catalyst surfaces, and the addition of promoter oxides inhibited phase transformation of γ-Al2O3. Mesoporous Ni/MOx–γ-Al2O3 also showed high resistance to coke deposition in the presence of promoters, due to the promotion of the chemisorptions and dissociation of CO2 originated from the enhancement of surface basicity of the catalysts. Additionally, Ni/La2O3–γ-Al2O3 presented the least amount of carbon and the most active carbon species. The comparative investigation for DRM over Ni/La2O3/γ-Al2O3-imp illustrated that the severe sintering of Ni particles and surface coke deposition were mainly responsible for the deactivation of Ni/La2O3/γ-Al2O3-imp. This result revealed that the one-pot strategy was a more suitable and promising method to obtain ideal candidates for the industrialized application of dry reforming of methane.Acknowledgements
This research was supported by Open Project of State Key Laboratory of Advanced Special Steel of Shanghai University, the Major State Basic Research Development Program of China (No. 2014CB643403), National Science Fund for Distinguished Young Scholars (No. 51225401, 51574164).References
- M. M. Naira and S. Kaliaguine, New J. Chem., 2016, 40, 4049–4060 RSC
.
- Q. L. Zhang, T. F. Zhang, Y. Z. Shi, B. Zhao, M. Z. Wang, Q. X. Liu, J. Wang, K. X. Long, Y. K. Duan and P. Ning, J. CO2 Util., 2017, 17, 10–19 CrossRef CAS
- X. Liao, R. Gerdts, S. F. Parker, L. N. Chi, Y. X. Zhao, M. Hill, J. Q. Guo, M. O. Jones and Z. Jiang, Phys. Chem. Chem. Phys., 2016, 18, 17311–17319 RSC
- J. M. Lavoie, Front. Chem., 2014, 2, 1–17 CAS
- C. Z. Wang, N. N. Sun, W. Wei and Y. X. Zhao, Int. J. Hydrogen Energy, 2016, 41, 19014–19024 CrossRef CAS
- X. P. Yu, F. B. Zhang and W. Chu, RSC Adv., 2016, 6, 70537–70546 RSC
- Z. Alipour, M. Rezaei and F. Meshkani, J. Ind. Eng. Chem., 2014, 20, 2858–2863 CrossRef CAS
- Y. Cao, H. R. Li, J. P. Zhang, L. Y. Shi and D. S. Zhang, RSC Adv., 2016, 6, 112215–112225 RSC
- M. G. Jeong, S. Y. Kim, D. H. Kim, S. W. Han, I. H. Kim, M. Lee, Y. K. Hwang and Y. D. Kim, Appl. Catal., A, 2016, 515, 45–50 CrossRef CAS
- X. Y. Zhao, H. R. Li, J. P. Zhang, L. Y. Shi and D. S. Zhang, Int. J. Hydrogen Energy, 2016, 41, 2447–2456 CrossRef CAS
- L. Karam, S. Casale, H. E. Zakhem and N. E. Hassan, J. CO2 Util., 2017, 17, 119–124 CrossRef CAS
- Z. Y. Liu, D. C. Grinter, P. G. Lustemberg, T. D. Nguyen-Phan, Y. H. Zhou, S. Luo, I. Waluyo, E. J. Crumlin, D. J. Stacchiola, J. Zhou, J. Carrasco, H. F. Busnengo, M. V. Ganduglia-Pirovano, S. D. Senanayake and J. A. Rodriguez, Angew. Chem., Int. Ed., 2016, 55, 7455–7459 CrossRef CAS PubMed
- H. S. Roh and K. W. Jun, Catal. Surv. Asia, 2008, 12, 239–252 CrossRef CAS
- X. Y. Li, D. Li, H. Tian, L. Zeng, Z. J. Zhao and J. L. Gong, Appl. Catal., B, 2017, 202, 683–694 CrossRef CAS
- D. L. Li, R. L. Li, M. M. Lu, X. Y. Lin, Y. Y. Zhan and L. L. Jiang, Appl. Catal., B, 2017, 200, 566–577 CrossRef CAS
- H. R. Liu, L. Yao, H. B. H. Taief, M. Benzina, P. D. Costa and M. E. Gálvez, Catal. Today DOI:10.1016/j.cattod.2016.12.017
- S. Sepehri, M. Rezaei, G. Garbarino and G. Busca, Int. J. Hydrogen Energy, 2016, 41, 8855–8862 CrossRef CAS
- X. J. Du, D. S. Zhang, R. H. Gao, L. Huang, L. Y. Shi and J. P. Zhang, Chem. Commun., 2013, 49, 6770–6772 RSC
- H. R. Liu, D. Wierzbicki, R. Debek, M. Motak, T. Grzybek, P. D. Costa and M. Elena Gálvez, Fuel, 2016, 182, 8–16 CrossRef CAS
- K. Jabbour, P. Massiani, A. Davidson, S. Casale and N. E. Hassan, Appl. Catal., B, 2017, 201, 527–542 CrossRef CAS
- C. J. Liu, J. Y. Ye, J. J. Jiang and Y. X. Pan, ChemCatChem, 2011, 3, 529–541 CrossRef CAS
- D. Horiuchi, K. Sakuma, T. Fukui, Y. Kubo, T. Osaki and T. Mori, Appl. Catal., A, 1996, 144, 111–120 CrossRef
- Y. Z. Chen, B. J. Liaw and W. H. Lai, Appl. Catal., A, 2002, 230, 73–83 CrossRef CAS
- L. Zhang, Q. Zhang, Y. Liu and Y. Zhang, Appl. Surf. Sci., 2016, 389, 25–33 CrossRef CAS
- F. Guo, J. Q. Xu and W. Chu, Catal. Today, 2015, 256, 124–129 CrossRef CAS
- E. Baktash, P. Littlewood, R. Schomäcker, A. Thomas and P. C. Stair, Appl. Catal., B, 2015, 179, 122–127 CrossRef CAS
- J. T. Feng, Y. J. Lin, X. Duan and D. Q. Li, J. Catal., 2009, 266, 351–358 CrossRef CAS
- A. Tsoukalou, Q. Imtiaz, S. M. Kim, P. M. Abdala, S. Yoon and C. R. Müller, J. Catal., 2016, 343, 208–214 CrossRef CAS
- L. L. Xu, F. G. Wang, M. D. Chen, J. Zhang, K. D. Yuan, L. J. Wang, K. Wu, G. Q. Xu and W. Chen, ChemCatChem, 2016, 8, 2536–2548 CrossRef CAS
- L. L. Xu, H. L. Song and L. J. Chou, ACS Catal., 2012, 2, 1331–1342 CrossRef CAS
- N. Wang, K. Shen, L. H. Huang, X. P. Yu, W. Z. Qian and W. Chu, ACS Catal., 2013, 3, 1638–1651 CrossRef CAS
- M. H. Amin, J. Tardio and S. K. Bhargava, Int. J. Hydrogen Energy, 2013, 38, 14223–14231 CrossRef CAS
- M. Yu, K. Zhu, Z. C. Liu, H. P. Xiao, W. Deng and X. G. Zhou, Appl. Catal., B, 2014, 148–149, 177–190 CrossRef CAS
- J. Ma, N. N. Sun, X. L. Zhang, N. Zhao, F. K. Xiao, W. Wei and Y. H. Sun, Catal. Today, 2009, 148, 221–231 CrossRef CAS
- P. Osorio-Vargas, N. A. Flores-González, R. M. Navarro, J. L. G. Fierro, C. H. Campos and P. Reyes, Catal. Today, 2015, 259, 27–38 CrossRef
- X. Yang, J. W. Da, H. T. Yu and H. Wang, Fuel, 2016, 179, 353–361 CrossRef CAS
- M. W. Tan, X. G. Wang, X. F. Shang, X. J. Zou, X. G. Lu and W. Z. Ding, J. Catal., 2014, 314, 117–131 CrossRef CAS
- H. Y. Ma, L. Zeng, H. Tian, D. Li, X. Wang, X. Y. Li and J. L. Gong, Appl. Catal., B, 2016, 181, 321–331 CrossRef CAS
- S. R. Li and J. L. Gong, Chem. Soc. Rev., 2014, 43, 7245–7256 RSC
- L. Mo, K. K. M. Leong and S. Kawi, Catal. Sci. Technol., 2014, 4, 2107–2114 CAS
- T. Yabe, K. Mitarai, K. Oshima, S. Ogo and Y. Sekine, Fuel Process. Technol., 2017, 158, 96–103 CrossRef CAS
- X. F. Shang, X. G. Wang, W. X. Nie, X. F. Guo, X. F. Zou, W. Z. Ding and X. G. Lu, J. Mater. Chem., 2012, 22, 23806–23814 RSC
- L. Zhang, X. G. Wang, X. F. Shang, M. W. Tan, W. Z. Ding and X. G. Lu, J. Energy Chem., 2017, 26, 93–100 CrossRef
- X. Z. Fang, C. Peng, H. G. Peng, W. M. Liu, X. L. Xu, X. Wang, C. Q. Li and W. F. Zhou, ChemCatChem, 2015, 7, 3753–3762 CrossRef CAS
- J. Q. Zhu, X. X. Peng, L. Yao, J. Shen, D. M. Tong and C. W. Hu, Int. J. Hydrogen Energy, 2011, 36, 7094–7104 CrossRef CAS
- L. L. Xu, Z. C. Miao, H. L. Song and L. J. Chou, Int. J. Hydrogen Energy, 2014, 39, 3253–3268 CrossRef CAS
- J. Munera, S. Irusta, L. Cornaglia, E. Lombardo, D. Vargascesar and M. Schmal, J. Catal., 2007, 245, 25–34 CrossRef CAS
- T. Sukonket, A. Khan, B. Saha, H. Ibrahim, S. Tantayanon, P. Kumar and R. Idem, Energy Fuels, 2011, 25, 864–877 CrossRef CAS
- F. Pompeo, N. N. Nichio, M. M. V. M. Souza, D. V. Cesar, O. A. Ferretti and M. Schmal, Appl. Catal., A, 2007, 316, 175–183 CrossRef CAS
- J. Liu, H. Peng, W. Liu, X. Xu, X. Wang, C. Li, W. Zhou, P. Yuan, X. Chen, W. Zhang and H. Zhan, ChemCatChem, 2014, 6, 2095–2104 CrossRef CAS
- C. Montero, A. Ochoa, P. Castaño, J. Bilbao and A. G. Gayubo, J. Catal., 2015, 331, 181–192 CrossRef CAS
- Å. Slagtern, U. Olsbye, R. Blom, I. M. Dahl and H. Fjellvåg, Appl. Catal., A, 1997, 165, 379–390 CrossRef
- K. Takehira, T. Shishido, P. Wang, T. Kosaka and K. Takaki, J. Catal., 2004, 221, 43–54 CrossRef CAS
- C. J. González, Z. Boukha, B. D. Rivas, J. J. Delgado, M. Á. Cauqui, J. R. G. Velasco, J. I. G. Ortiz and R. L. Fonseca, Appl. Catal., A, 2013, 466, 9–20 CrossRef
- Y. Kathiraser, W. Thitsartarn, K. Sutthiumporn and S. Kawi, J. Phys. Chem. C, 2013, 117, 8120–8130 CAS
- M. Wu and D. M. Hercules, J. Phys. Chem., 1979, 83, 2003–2008 CrossRef CAS
- M. C. Sanchez-Sanchez, R. M. Navarro and J. L. G. Fierro, Int. J. Hydrogen Energy, 2007, 32, 1462–1471 CrossRef CAS
- A. Cimino, M. Lo jacono and M. Schiavello, J. Phys. Chem., 1975, 79, 243–249 CrossRef CAS
- Q. Zhang, T. Wu, P. Zhang, R. J. Qi, R. Huang, X. F. Song and L. Gao, RSC Adv., 2014, 4, 51184–51193 RSC
- Z. Y Shang, S. G. Li, L. Li, G. Z Liu and X. H. Liang, Appl. Catal., A, 1997, 165, 379–390 CrossRef
- L. Zhou, L. D. Li, N. N. Wei, J. Li and J. M. Basset, ChemCatChem, 2015, 7, 2508–2516 CrossRef CAS
- R. Q. Yang, C. Xing, C. X. Lv, L. Shi and N. Tsubaki, Appl. Catal., A, 2010, 385, 92–100 CrossRef CAS
- W. W. Yang, H. M. Liu, Y. M. Li, H. Wu and D. H. He, Int. J. Hydrogen Energy, 2016, 41, 1513–1523 CrossRef CAS
- X. Y. Quek, D. Liu, W. N. E. Cheo, H. Wang, Y. Chen and Y. Yang, Appl. Catal., B, 2010, 95, 374–382 CrossRef CAS
- D. P. Liu, X. Y. Quek, W. N. E. Cheo, R. Lau, A. Borgna and Y. H. Yang, J. Catal., 2009, 266, 380–390 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04497f |
| This journal is © The Royal Society of Chemistry 2017 |