
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst-principles study on structural, electronic, vibrational and thermodynamic properties of Sr10(PO4)6X2 (X = F, Cl, Br)
Zhihong Yuan a,
Tao Gao*ab,
Yuanlei Zhenga,
Shenggui Maa,
Mingli Yangab and
Piheng Chen*c
a,
Tao Gao*ab,
Yuanlei Zhenga,
Shenggui Maa,
Mingli Yangab and
Piheng Chen*c
aInstitute of Atomic and Molecular Physics, Sichuan University, Chengdu, 610065, China. E-mail: gaotao@scu.edu.cn
bKey Laboratory of High Energy Density Physics and Technology of Ministry of Education, Sichuan University, Chengdu, 610064, China
cScience and Technology on Surface Physics and Chemistry Laboratory, Mianyang, 621907, China. E-mail: chenph@live.cn
First published on 12th June 2017
Abstract
A theoretical investigation on the structural stability, electronic, vibrational, and thermodynamic properties of the strontium apatites Sr10(PO4)6X2 (X = F, Cl, Br) is systematically conducted by the first-principles calculations. Results of cohesive energies and formation enthalpies suggest that the thermal stability of strontium apatites decreases from Sr10(PO4)6F2 (Sr-FAP) to Sr10(PO4)6Cl2 (Sr-ClAP) and further to Sr10(PO4)6Br2 (Sr-BrAP); such a tendency is also be observed with regard to the band gaps. Using linear-response approach, the detailed vibrational properties of Sr10(PO4)6X2 (X = F, Cl, Br) are obtained. According to the calculated phonon dispersions, it is concluded that strontium apatites Sr10(PO4)6X2 (X = F, Cl, Br) are dynamically stable, and the phonon behaviors are generally similar to these apatites, but most of the vibrational frequencies decrease from Sr-FAP, Sr-ClAP to Sr-BrAP. The assignment of the vibrational modes at the gamma point demonstrate that all the silent mode Bg, Bu and E2u are affected and the only optically active mode involved is the Raman active mode E2g with the replacement of larger Cl− and Br− for F−. The results calculated with the quasi-harmonic approximation (QHA) show that Sr10(PO4)6X2 (X = F, Cl, Br) exhibits similar but slightly different behaviors in terms of its thermodynamic properties, which is expected because the halogen atoms F, Cl and Br are in the same VIIA group. Significantly, all the present calculation results are satisfactory compared to the existing experimental and theoretical results.
1. Introduction
Phosphate-bearing apatites with the general formula M10(PO4)6X2 (M = Ca, Sr, Ba, Pb, etc.; X = F, Cl, Br, OH, etc.) have been attracting considerable attention from scientists due to their significance in many fields, which vary from geochronology to environment remediation and optoelectronic as well as biomaterials and medical sciences.1–7 Calcium apatites which are the typical representatives of the apatite family have been the most extensively investigated, due to their proverbial industrial and biological applications.8–14 Furthermore, researchers have been constantly discussing the various ionic substitutions in calcium apatites15–20 for the sake of better applications of apatites.Strontium (Sr) is one of the most common substituents in the apatite structure. The presence and behavior of Sr in apatite compounds are of great significance in geology, biology and materials science, and some studies have paid more attention to the Ca10−xSrx(PO4)6X2 (X = F, Cl) apatites.21–23 In this paper, we focus on the Sr-richest apatites Sr10(PO4)6X2 (X = F, Cl, Br) which as the Sr end-member apatites have many practical applications. Firstly, Sr10(PO4)6X2 (X = F, Cl, Br) are well-known hosts for functional rare earth ions with subsequent applications as fluorescent and laser materials.24–30 Secondly, Sr10(PO4)6F2 (Sr-FAP) is of interest in biological fields because of the ready entry of Sr into the food chain and its subsequent incorporation in bone.31–34 Thirdly, as the member of apatite family, Sr10(PO4)6X2 (X = F, Cl, Br) are considered to be suitable for nuclear waste storage, such as the glass encapsulated Sr10(PO4)6Cl2 (Sr-ClAP) can immobilize most of the radwaste elements in the composite matrix.35
There are some investigations have been carried on the crystal Sr10(PO4)6X2 (X = F, Cl, Br). The crystal structures of Sr10(PO4)6X2 (X = F, Cl, Br) were determined with X-ray diffraction.36–38 Several groups15,39–42 have studied the infrared (IR) and Raman (R) spectra of Sr10(PO4)6X2 (X = F, Cl, Br). The phase transitions and thermal expansion of Sr-FAP and Sr-ClAP were studied by high temperature X-ray diffraction and differential thermal analysis.43,44 The results indicate Sr-FAP and Sr-ClAP have large thermal expansion coefficient and there are not any phase transformations for Sr-FAP and Sr-ClAP in temperature interval 298–1723 K. The formation enthalpies of Sr-FAP and Sr-ClAP were given at the standard of 298 K and 1 bar.45–47 Specially, the heat capacity and enthalpy increments of Sr-ClAP were measured by Hrudananda Jena et al.35,48,49 Furthermore, the thermodynamics functions of Sr-ClAP were computed by least-squares fitting method from the measured enthalpy increments and compared with the Ca10(PO4)6Cl2 and Ba10(PO4)6Cl2, which indicate that the specific heat capacity of the alkaline earth chloroapatites increases in the order barium < strontium < calcium and the alkaline earth chloroapatites can withstand decay heat of the radio nuclides without decomposition or degradation.49 Theoretically, the electronic properties and elastic constants of Sr10(PO4)6X2 (X = F, Cl, Br) were calculated using density functional theory (DFT) with the generalized gradient approximation (GGA) functional.50 Besides, the elastic constants of Sr-FAP and Sr-ClAP also were predicted using the program GULP.51
As mentioned above, there is presently a dearth of vibrational and thermodynamic information for Sr10(PO4)6X2 (X = F, Cl, Br). However, the thermodynamic functions such as enthalpy, heat capacity and entropy for Sr10(PO4)6X2 (X = F, Cl, Br) are very important as the apatites (especially the Sr10(PO4)6Cl2) becoming more and more important for radioactive waste immobilization. Computational prediction for these properties is of interest and motivates us to acquire further knowledge to define better applications of these apatites. The aim of this paper is to provide the first systematic study of the structural, electronic, vibrational and thermodynamic properties of the strontium apatites Sr10(PO4)6X2 (X = F, Cl, Br) from first-principles calculations.
The remainder of this paper is structured as follows: in Section 2, the Computational details are briefly described. Section 3 is devoted to Results and discussions, we present the structural, electronic, vibrational and thermodynamic properties of Sr10(PO4)6X2 (X = F, Cl, Br). Finally, the Conclusions of the present work are given in Section 4.
2. Computational details
In the present work, the first-principles calculations based on the DFT52 are performed by the Vienna Ab initio Simulation Package (VASP) code.53 The local density approximation (LDA) with Ceperley–Alder (CA) functional54 is applied to describe the exchange correlation energy, the valence electron configurations for these elements are Sr 4s24p65s2, P 3s23p3, O 2s22p4, F 2s22p5, Cl 3s23p5 and Br 4s24p5, respectively. The primitive cells of Sr10(PO4)6X2 (X = F, Cl, Br), which contain 42 atoms are chosen in all our calculations in order to reduce the computational cost. The energy cutoff of 520 eV is chosen to determine the number of plane waves in expansion, the k-point mesh is set at 2π × 0.04 Å−1 for samplings in the irreducible edge of Brillouin zone (BZ)55 during our calculations. Both the kinetic-energy cutoff and k-point sampling have been tested to be sufficient for convergence. Besides, the total energy is converged to 10−7 eV and the force acting on each atom is converged to less than 0.01 eV Å−1 throughout the present calculations. The equilibrium structures of Sr10(PO4)6X2 (X = F, Cl, Br) are obtained by minimizing the total energy with respect to the fully optimized of cell volume, cell shape and atomic positions in primitive cells.The phonon calculations are performed by the linear response approach as implemented in the PHONOPY code,56 with VASP code used as the computational engine. The vibrational properties including the phonon dispersion curves and phonon density of states, Born effective charge tensor and associated longitudinal optical and transverse optical (LO–TO) splitting of optical modes, as well as the phonon frequencies at the Brillouin zone center are obtained in the framework of the density functional perturbation theory (DFPT). Thereafter, some phonon related thermodynamic properties such as Helmholtz free energy F, internal energy E, entropy S and specific heat CV (CP) are predicted within the QHA.
3. Results and discussions
3.1. Structural properties and phase stability
The apatites Sr10(PO4)6X2 (X = F, Cl, Br) have the fairly complex hexagonal crystal structures with the space group P63/m (no. 176). The models are illustrated in Fig. 1, which are composed of six PO4 groups surrounded by ten Sr atoms with two X ions located along the c axis. Of which ten Sr atoms occupy two different crystallographic symmetry sites 4f and 6h. The PO4 groups occupy 6h sites, while the oxygen atoms have three unique environments. The very difference for these apatites is the positions of halogen atoms. The F atoms lie at the z = 0.25 and z = 0.75, occupy the 2a positions in Sr-FAP, whereas the Cl and Br atoms lie at z = 0 and z = 1/2, occupy the 2b positions in Sr-ClAP and Sr-BrAP. | ||
| Fig. 1 The crystal structures of Sr10(PO4)6X2 (X = F, Cl, Br), the atomic species are labeled on the balls. And the yellow balls represent the F atoms, blue balls represent the Cl or Br atoms (the figures are generated by the VESTA code57). | ||
In the first step, the crystal structures of Sr10(PO4)6X2 (X = F, Cl, Br) are fully optimized by minimizing the total energy. The obtained results after relaxation are summarized in Table 1 and in comparison with the experimental36–38 and other theoretical values.50 Obviously, the optimized structure parameters in our calculations with LDA underestimate by 1–2% with respect to the experiment values, while the theoretical values calculated with GGA overestimate within 1–2%, which due to the general feature of DFT that LDA underestimate and GGA overestimate the structure parameters. Therefore, the optimized structures are sufficient to allow us to make a further study of the electronic, vibrational and thermodynamic properties of these apatites. After analysis, we note the replacement of F by Cl and Br causes the cell volumes of the apatites to expand, but leading to a decreased c/a ratio of the unit cell, which can be explained by the fact that the halogen ionic radius increased from F− to Br−.
| Apatite | Lattice parameters (Å) | V (Å3) | c/a | Ref. | |
|---|---|---|---|---|---|
| a | c | ||||
| a Ref. 36.b Ref. 37.c Ref. 38.d Ref. 50. | |||||
| Sr-FAP | 9.568 | 7.183 | 569.47 | 0.751 | This work |
| 9.678 | 7.275 | 590.11 | 0.752 | Expta | |
| 9.841 | 7.365 | 617.80 | 0.748 | Cal.d | |
| Sr-ClAP | 9.711 | 7.085 | 578.63 | 0.730 | This work |
| 9.859 | 7.205 | 606.58 | 0.731 | Exptb | |
| 10.015 | 7.258 | 630.45 | 0.725 | Cal.d | |
| Sr-BrAP | 9.796 | 7.101 | 590.15 | 0.725 | This work |
| 9.972 | 7.214 | 621.26 | 0.723 | Exptc | |
| 10.108 | 7.272 | 643.36 | 0.719 | Cal.d | |
The stability for apatites is very important to control sintering or thermal processing conditions for the design and preparation of apatite ceramics. In order to determine the relative stability of Sr10(PO4)6X2 (X = F, Cl, Br) apatites, the cohesive energy and formation enthalpy of Sr10(PO4)6X2 (X = F, Cl, Br) are calculated by the following definitions:
| Ec(Sr10(PO4)6X2) = E(Sr10(PO4)6X2) − 10Eiso(Sr) − 6Eiso(P) − 24Eiso(O) − 2Eiso(X) |
| ΔH(Sr10(PO4)6X2) = E(Sr10(PO4)6X2) − 10E(Sr) − 6E(P) − 12E(O2) − E(X2) |
The calculated cohesive energies and formation enthalpies of these apatites are tabulated in Table 2, along with the experimental data and other published. Apparently, our calculated formation enthalpies of these apatites are agree well with the experimental values.45–47 The negative cohesive energies and formation enthalpies show these strontium apatites are stable. Compared with the results given by Michael Kocher,50 we find that GGA could obtain more negative cohesive energy and formation enthalpy, but the LDA opposite. Further analysis, the stability of these apatites is related to the halogen anion radius and decreases in the following order Sr-FAP > Sr-ClAP > Sr-BrAP, which is reflected by the more negative cohesive energy and formation enthalpy of Sr-FAP than that of Sr-ClAP and Sr-BrAP. Such a tendency has been observed in recent publications of calcium apatites12 and barium apatites.16
3.2. Electronic structure
Fig. 2 shows the calculated energy band structure of each apatite along the same high symmetry reciprocal space path. The Fermi level (Ef) is chosen to align to zero and expressed by a dash line. The main features of the band structures are common to each apatite. All these apatites are insulator materials and characterized by an indirect band gap. Table 3 provides our calculated band gaps and other theoretical values for each apatite, it is observed whether our results or the values given by Michael Kocher,50 the band gap values decrease from Sr-FAP to Sr-ClAP to Sr-BrAP. As far as we know, for some have been studied apatites,9,12,16,20 (such as calcium apatites, barium apatites and cadmium apatites) the band gaps decrease with the reduction of X ion electronegativity. In addition, as already noted that DFT predictions always underestimate the true band gap so the real values may be somewhat higher than calculated here.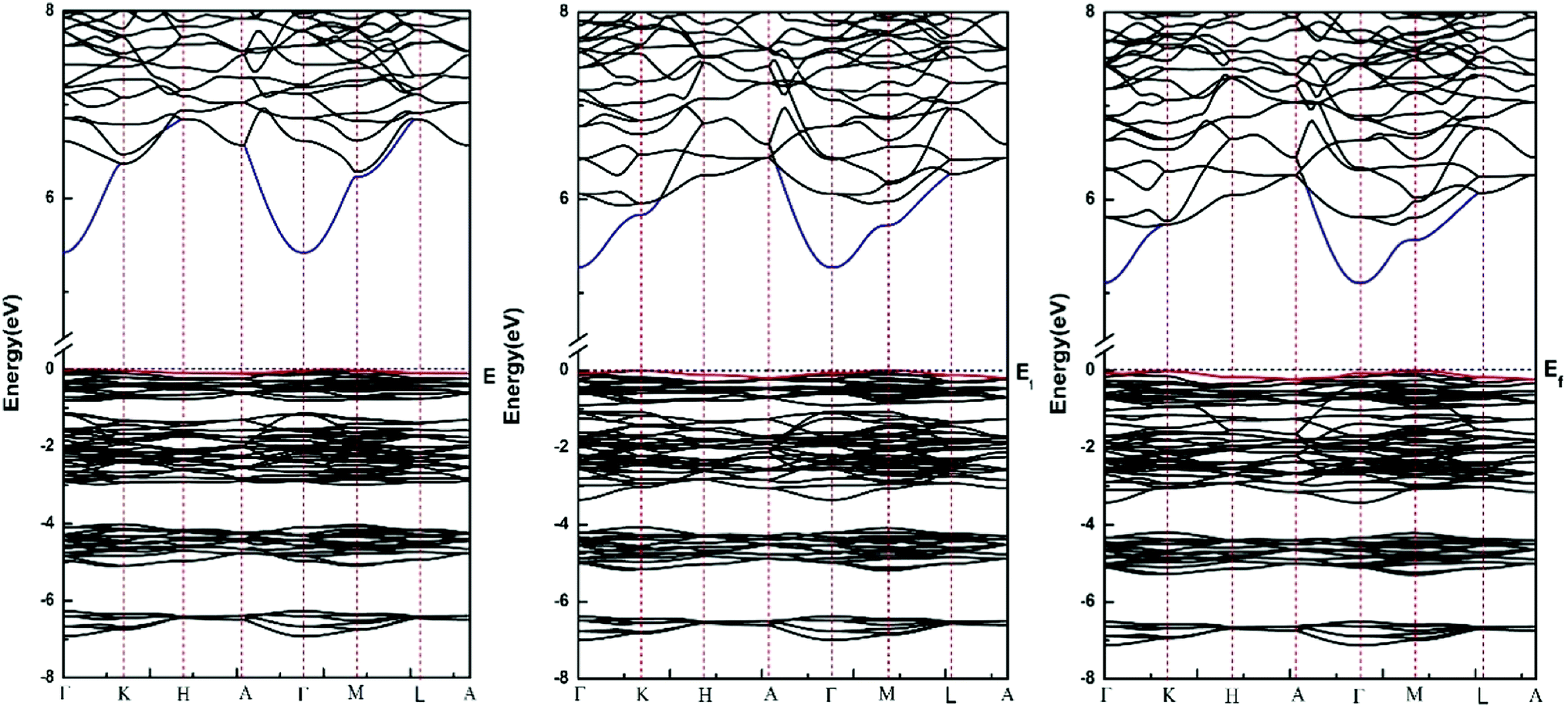 | ||
| Fig. 2 Calculated band structures for Sr-FAP, Sr-ClAP and Sr-BrAP apatites. | ||
Fig. 3 plots the total density of states (TDOS) and the partial densities of states for each apatite. The DOSs of these apatites are very similar along with slight differences as with the band structures. The DOSs below Fermi energy in the energy range of −10 eV to 0 eV are described by four main peaks (A–D). Peak A mostly derived from O 2p and P 3s states with some O 2s contributions, while peak B is dominated by O 2p and P 3p states. As for peaks C and D are mainly composed of O 2p states and the peak C has receive approximately 10% of their value from the X p states. The conduction band is chiefly contributed from Sr 4d states with some mixing of O 2s, O 2p and F p states. There are also three main peaks (1–3) below −10 eV for Sr10(PO4)6X2 (X = F, Cl, Br). Of these peaks, the peak 1 is primarily from O 2s and P 3s states, while the peak 2 is mainly from the O 2s states and P 3p states. Another significant peak 3 for these apatites principally made up by Sr 4p with the minor contribution of O 2s and O 2p states, while for Sr-ClAP and Sr-BrAP the Cl 3s and Br 4s states also contribute to the peak 3. Furthermore, from the X PDOS in Fig. 3, we note about −15 eV there is a peak 3′ which also comes from Cl 3s (Br 4s) states, and Sr-FAP has a small peak 1′ at about −21 eV from its F s states. The similar phenomena are also reported in the recent publications of calcium apatites12 and barium apatites16 calculated with the DFT.
 | ||
| Fig. 3 Total density of states (TDOS) and partial density of states (PDOS) for Sr-FAP, Sr-ClAP and Sr-BrAP apatites. | ||
3.3. Vibrational properties
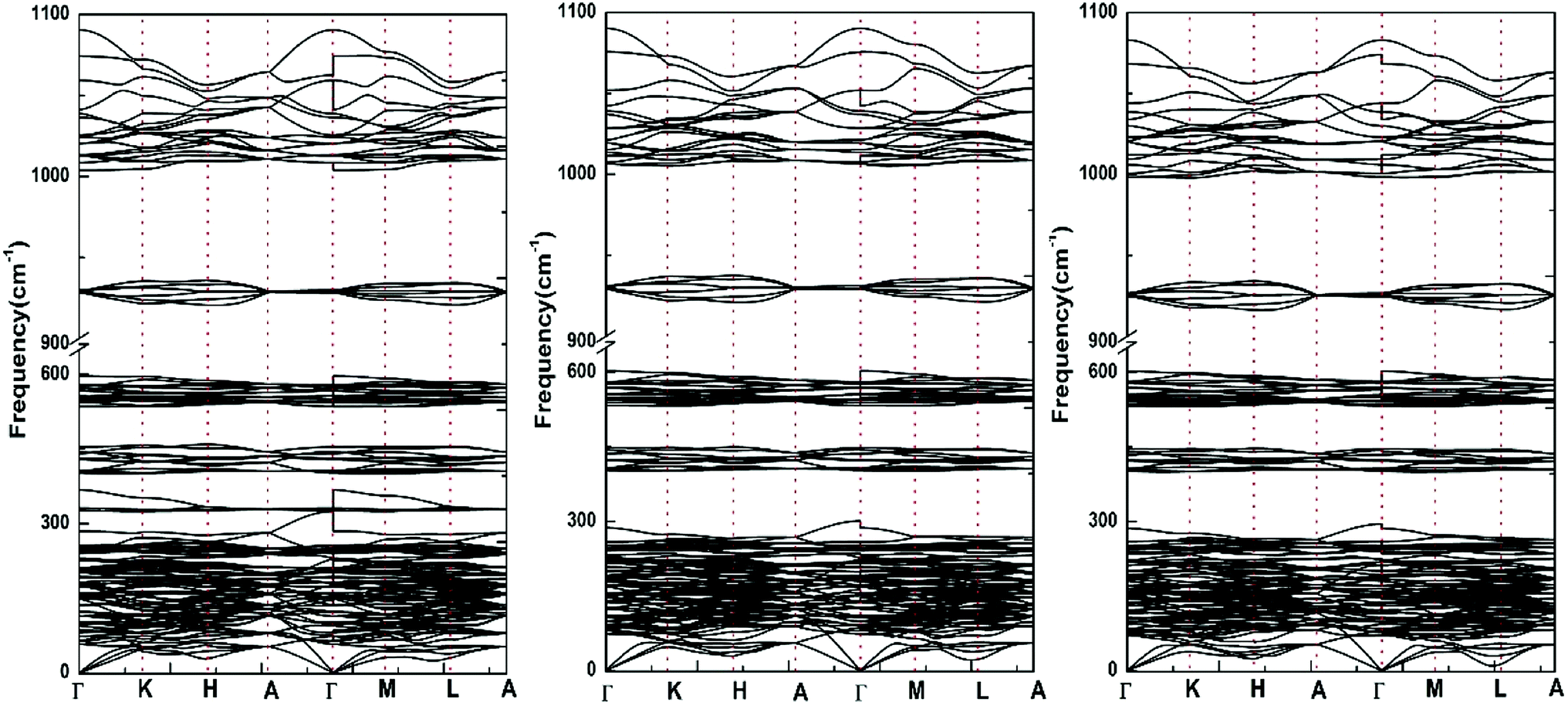 | ||
| Fig. 4 Calculated phonon dispersion curves (including LO–TO splitting) for Sr-FAP, Sr-ClAP and Sr-BrAP apatites. | ||
Since all the primitive cells of hexagonal Sr-FAP, Sr-ClAP and Sr-BrAP contain 42 atoms, the complete phonon spectrum for each apatite consists 126 dispersion curves, of which 3 are acoustical modes and the remaining 123 are optical modes. According to the standard group-theoretical analysis based on the P63/m space group, the Sr-FAP yields the optical phonon modes at the Γ point, as following:
| Γopt = 12Ag + 8E1g + 13E2g + 8Au + 12E1u + 12Bu + 9Bg + 8E2u |
The Sr-ClAP and Sr-BrAP yield the optical phonon modes at the Γ point, as following:
| Γopt = 12Ag + 8E1g + 12E2g + 8Au + 12E1u + 13Bu + 8Bg + 9E2u |
Of these modes, Ag, E1g and E2g are corresponding to Raman active modes, whereas the Au and E1u are infrared active modes, Bg, Bu and E2u are silent modes. The A and E modes are singly and doubly degenerate modes, respectively.
Interestingly, with the replacement of larger Cl− and Br− for F−, all the silent modes Bg, Bu and E2u are affected and the only optically active modes involved is the Raman active mode E2g, which can be explained by the different Wyckoff sites of X (X = F, Cl, Br) ions in theirs apatite structures.
Fig. 5 illustrates the phonon total density of state and the phonon partial density of states for each apatite. It is distinctly for these apatites that the modes beyond 350 cm−1 up to 1100 cm−1 are contributed from PO4 group vibrations. And since these selected apatites differ only in X atoms, their high-frequency spectra are naturally very similar. Some characteristic differences among these apatites can be found at the low frequencies (<350 cm−1). The massive modes of these apatites below 300 cm−1 involve the vibrations of all bonds are the major contributions to thermodynamics. But owing to the fact that atomic mass of F is lighter than that of Cl and Br, the frequencies in the 300–350 cm−1 region are associated with the Sr–F bond only. In addition, we find the low frequencies increased with the increase in the atomic mass (F < Cl < Br).
 | ||
| Fig. 5 Phonon total density of states (PTDOS) and phonon partial density of states (PPDOS) for Sr-FAP, Sr-ClAP and Sr-BrAP apatites. | ||
 ,
, 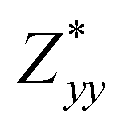 and
and 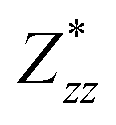 for each atom possess two independent components
for each atom possess two independent components 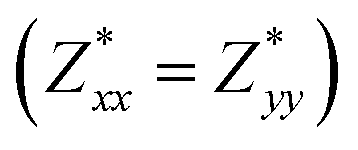 . Clearly, the Z* diagonal components are close to the nominal ionic values of Sr (+2), O (−2), P (+3), F (−1), Cl (−1), Br (−1), which confirm the ionic character of these apatites from a chemical point of view. Table 5 presents the calculated diagonal elements of ε. Due to the symmetry of the crystal, three diagonal elements εxx, εyy, εzz also exhibit two independent components (εxx = εyy). We note the ε increase with replacement of F by Cl and Br. And the εxx (εyy) is larger than the εzz for Sr-FAP, while for Sr-ClAP and Sr-BrAP, the εxx (εyy) is smaller than the εzz in our calculations.
. Clearly, the Z* diagonal components are close to the nominal ionic values of Sr (+2), O (−2), P (+3), F (−1), Cl (−1), Br (−1), which confirm the ionic character of these apatites from a chemical point of view. Table 5 presents the calculated diagonal elements of ε. Due to the symmetry of the crystal, three diagonal elements εxx, εyy, εzz also exhibit two independent components (εxx = εyy). We note the ε increase with replacement of F by Cl and Br. And the εxx (εyy) is larger than the εzz for Sr-FAP, while for Sr-ClAP and Sr-BrAP, the εxx (εyy) is smaller than the εzz in our calculations.
| Site | Atom | Sr-FAP | Sr-ClAP | Sr-BrAP | |||
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
||
| 4f | Sr1 | 2.408 | 2.630 | 2.485 | 2.555 | 2.505 | 2.544 |
| 6h | Sr2 | 2.562 | 2.494 | 2.500 | 2.702 | 2.495 | 2.692 |
| 6h | P | 3.310 | 3.109 | 3.386 | 3.181 | 3.410 | 3.217 |
| 6h | O1 | −2.010 | −1.423 | −2.017 | −1.413 | −2.025 | −1.406 |
| 6h | O2 | −1.935 | −1.334 | −2.045 | −1.321 | −2.087 | −1.325 |
| 12i | O3 | −1.498 | −2.181 | −1.563 | −2.187 | −1.578 | −2.197 |
| 2a | F | −1.609 | −0.714 | ||||
| 2b | Cl | −1.068 | −1.440 | ||||
| 2b | Br | −0.932 | −1.447 | ||||
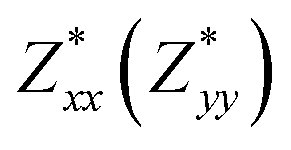
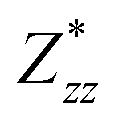
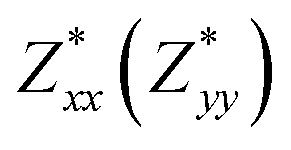
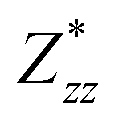
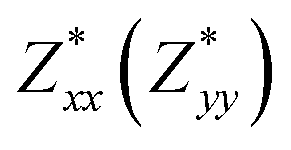

| Apatite | Sr-FAP | Sr-ClAP | Sr-BrAP |
|---|---|---|---|
| εxx(εxx) | 2.92 | 3.09 | 3.15 |
| εzz | 2.91 | 3.11 | 3.19 |
Taking the calculated macroscopic static dielectric tensor and the Born effective charge tensor into consideration, the long-range coulomb (dipole–dipole) interaction causes the splitting of optical modes at Γ point. The splitting of the LO and TO modes at the Γ point is evident from the phonon dispersion curves that displayed in Fig. 4. The IR vibration frequencies have LO–TO splitting at various frequencies and the LO frequency is larger than the TO frequency. Tables 6 and 7 provide the phonon frequencies of Raman and infrared (including LO–TO splitting) modes, respectively. The comparison between the calculated and the measured frequencies15,42 gives the maximum deviations of 5.6%, and the frequencies from other groups39–41 also show good consistency with our calculation. The IR spectra of Ca10(PO4)6F2 has been obtained by Etienne Balan et al.11 with DFT, the calculated frequencies underestimate about 10% with respect to the measured IR frequencies. Therefore, the agreement is generally excellent between our calculated and the experimental values. Further analysis, there is a sole Raman active mode E2g which is mainly associated with the atomic vibrations from F atoms according to our calculated phonon partial density of states. In addition, we find the mode Au (5) which include the vibrations of PO4 group and Sr, and the mode Au (8) which correspond to the vibrations of P–O bond exhibit the very large LO–TO splitting (>45 cm−1), indicating these modes are sensitive to the long-range Coulomb interaction.
| Modes | Sr-FAP | Sr-ClAP | Sr-BrAP | |
|---|---|---|---|---|
| This work | Expta | This work | This work | |
| a Ref. 42. | ||||
| E1g | 58.0 | 73.6 | 76.7 | |
| 97.1 | 98.9 | 98.2 | ||
| 152.2 | 150.6 | 151.3 | ||
| 204.8 | 198.8 | 192.4 | ||
| 256.0 | 252.1 | 247.1 | ||
| 405.3 | 421 | 405.3 | 403.5 | |
| 556.4 | 580 | 556.5 | 555.7 | |
| 1012.7 | 1027 | 1019.5 | 1020.2 | |
| E2g | 73.4 | 86.0 | 84.4 | |
| 101.4 | 109.1 | 105.4 | ||
| 149.7 | 152.4 | 148.0 | ||
| 176.2 | 172.6 | 165.4 | ||
| 202.2 | 191.5 | 191.0 | ||
| 241.8 | 247.5 | 250.6 | ||
| 326.5 | ||||
| 428.9 | 442 | 422.8 | 421.8 | |
| 546.6 | 572 | 545.5 | 543.6 | |
| 579.5 | 601 | 579.2 | 579.0 | |
| 929.8 | 951 | 931.2 | 926.8 | |
| 1013.3 | 1028 | 1006.3 | 998.4 | |
| 1025.5 | 1037 | 1028.5 | 1022.9 | |
| Ag | 121.5 | 107.9 | 99.5 | |
| 150.4 | 139.4 | 136.1 | ||
| 181.8 | 169.0 | 159.2 | ||
| 191.5 | 179.6 | 180.4 | ||
| 210.7 | 219.5 | 218.8 | ||
| 248.6 | 244.8 | 246.2 | ||
| 431.1 | 444 | 424.1 | 423.8 | |
| 557.1 | 580 | 554.5 | 551.0 | |
| 571.5 | 592 | 576.5 | 578.6 | |
| 928.3 | 951 | 929.2 | 925.3 | |
| 1024.3 | 1041 | 1013.6 | 1006.1 | |
| 1038.9 | 1054 | 1037.1 | 1030.1 | |
| Modes | Sr-FAP | Sr-ClAP | Sr-BrAP | ||||||
|---|---|---|---|---|---|---|---|---|---|
| This work | Expta | This work | Expta | This work | Expta | ||||
| LO | TO | LO | TO | LO | TO | ||||
| a Ref. 15. | |||||||||
| E1u(1) | 86.4 | 80.9 | 83.1 | 79.9 | 72.9 | 71.1 | |||
| E1u(2) | 157.7 | 157.2 | 145.2 | 137.1 | 136.3 | 122.5 | |||
| E1u(3) | 199.7 | 172.7 | 183.4 | 168.3 | 160.1 | 158.3 | |||
| E1u(4) | 221.0 | 203.6 | 199.1 | 187.2 | 183.2 | 170.6 | |||
| E1u(5) | 274.7 | 249.6 | 231.2 | 208.2 | 218.9 | 188.1 | |||
| E1u(6) | 356.7 | 331.3 | 291.9 | 258.4 | 292.2 | 259.7 | |||
| E1u(7) | 441.2 | 441.1 | 458 | 436.9 | 436.3 | 459 | 436.3 | 434.4 | 458 |
| E1u(8) | 548.7 | 543.8 | 570 | 544.9 | 539.1 | 565 | 542.6 | 536.1 | 562 |
| E1u(9) | 584.4 | 567.0 | 590 | 584.0 | 569.2 | 591 | 584.2 | 569.7 | 593 |
| E1u(10) | 929.0 | 928.2 | 949 | 930.4 | 928.9 | 949 | 926.2 | 924.4 | 945 |
| E1u(11) | 1050.0 | 1008.5 | 1026 | 1044.4 | 1007.4 | 1028 | 1035.0 | 1000.6 | 1024 |
| E1u(12) | 1094.3 | 1059.3 | 1075 | 1090.7 | 1052.0 | 1069 | 1083.2 | 1044.0 | 1061 |
| Au(1) | 73.7 | 65.3 | 91.6 | 91.4 | 77.6 | 77.5 | |||
| Au(2) | 123.8 | 123.5 | 124.2 | 123.7 | 120.2 | 118.5 | |||
| Au(3) | 142.1 | 137.0 | 160.9 | 150.6 | 153.8 | 144.3 | |||
| Au(4) | 250.6 | 226.3 | 238.4 | 225.4 | 227.4 | 220.4 | |||
| Au(5) | 299.6 | 254.5 | 298.4 | 243.1 | 293.0 | 235.8 | |||
| Au(6) | 444.8 | 443.6 | 458 | 445.4 | 443.6 | 459 | 443.8 | 441.4 | 458 |
| Au(7) | 561.8 | 535.2 | 570 | 557.4 | 533.2 | 565 | 555.0 | 533.2 | 562 |
| Au(8) | 1080.9 | 1003.7 | 1026 | 1083.4 | 1011.4 | 1028 | 1081.0 | 1012.1 | 1024 |
3.4. Thermodynamic properties
To our knowledge there are some publications focused on the thermodynamic properties of Sr-ClAP, but no experimental or theoretical works exploring the thermodynamic properties of Sr-FAP and Sr-BrAP. As we know that the first-principles for phonon calculations are limited to T = 0 K yet the detailed thermodynamic properties of the crystals could be derived by phonons based on the QHA. Neglecting the thermal electronic contributions and focus only the contribution of atomic vibrations, the Helmholtz free energy F of a system at temperature T can be expressed as:| F(VP,T) = Ec(V) + Fvib(V,T) |
where kB is the Boltzman constant, ω is the phonon frequencies, g(ω) is the phonon density of states with
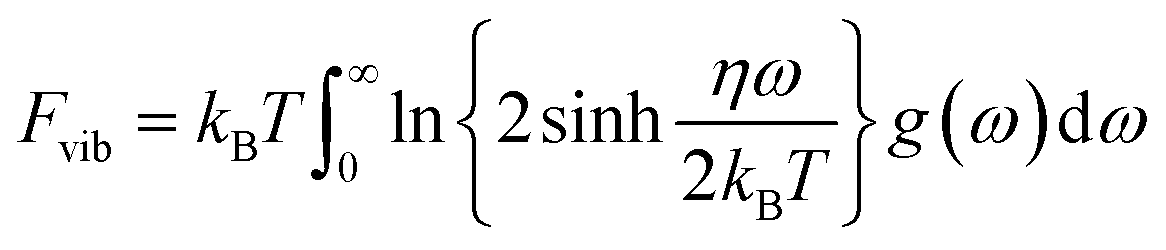
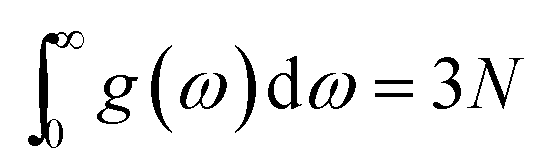 .
.
When the Helmholtz free energy F obtained, the entropy S and internal energy E can be derived by thermodynamic relations, S = −(∂F/∂T)V and E = F + TS respectively. And the heat capacity at constant volume CV can be estimated directly from Helmholtz free energy by CV = −T(∂2F/∂T2)V. Moreover, the heat capacity at constant pressure CP which is generally measured in experiments can be calculated using thermodynamic relation: CP = CV + α2VBT, where B is the bulk modulus, V is the volume and α is the volume thermal expansion coefficient. The volume thermal expansion coefficient α is calculated according to the formula 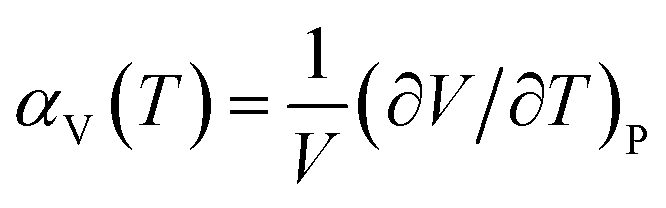 , which is shown in Fig. 6. It is found that our calculated results are smaller than experiment values.43 One reason is from the fact that the synthetic apatites are polycrystalline and the measurements are conducted at very high temperatures in the experiment, but our calculations are based on ideal single crystal under the 0 K. Another important reason is the neglect anharmonicity and thermal electronic contributions may be the important contributions for these materials.
, which is shown in Fig. 6. It is found that our calculated results are smaller than experiment values.43 One reason is from the fact that the synthetic apatites are polycrystalline and the measurements are conducted at very high temperatures in the experiment, but our calculations are based on ideal single crystal under the 0 K. Another important reason is the neglect anharmonicity and thermal electronic contributions may be the important contributions for these materials.
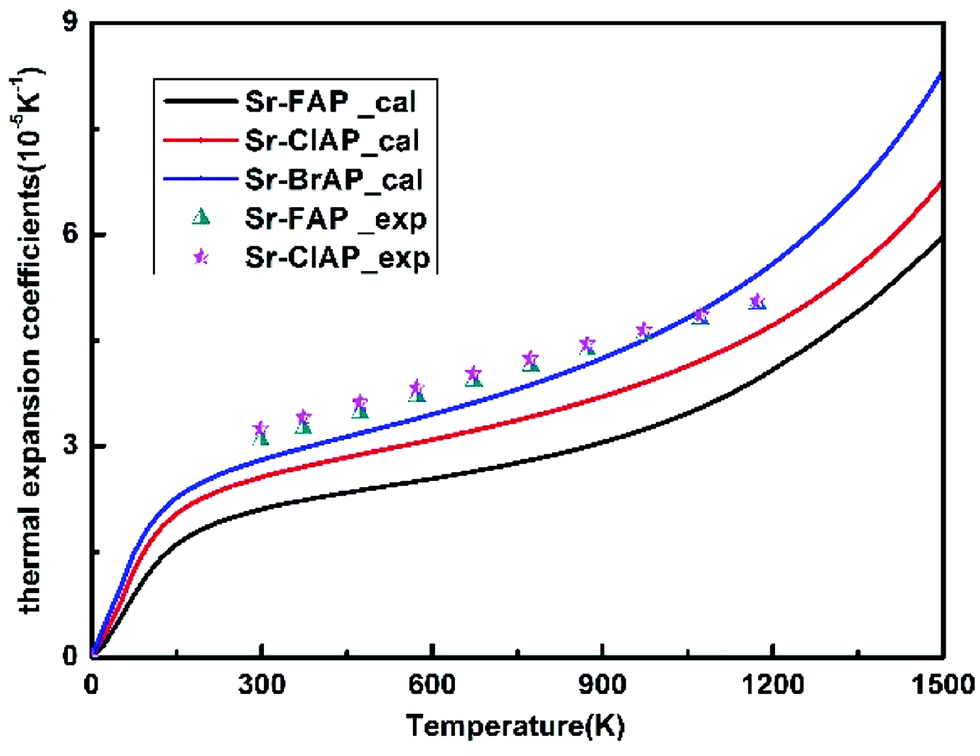 | ||
| Fig. 6 Thermal expansion coefficient αV of Sr-FAP, Sr-ClAP and Sr-BrAP apatites as a function of temperature at 0 GPa. Experimental data from ref. 43. | ||
The predicted thermodynamic properties under different temperature T for Sr10(PO4)6X2 (X = F, Cl, Br) are depicted in Fig. 7. The difference of thermodynamic properties for Sr-FAP, Sr-ClAP and Sr-BrAP is small, which is consistent with their similar phonon dispersion curves. From Fig. 7a, it is found the temperature-dependent F shows a decreasing trend from X = F, Cl, to Br. The differences of the temperature-dependent E between these apatites are too small to be observed in Fig. 7b, though a decreasing trend of E is shown from X = F, Cl, to Br. In Fig. 7c, S shows a relatively evident increasing trend from X = F, Cl, to Br, which can be explained by the increasing trend of phonon DOSs at the low phonon frequency. Fig. 7d gives the relationships between the specific heat CV and CP versus temperature that both CV and CP exhibit an increasing trend with increasing atom mass (F < Cl < Br) at the given temperature. And with increasing temperature, the both CV and CP grow rapidly up to 700 K, then above 700 K, CP keeps positive slope but CV tends to be a constant value of 1033 J K−1 mol−1 that is conform to the well-known Dulong–Petit limit of CV = 3nNkB = 1047 J K−1 mol−1.
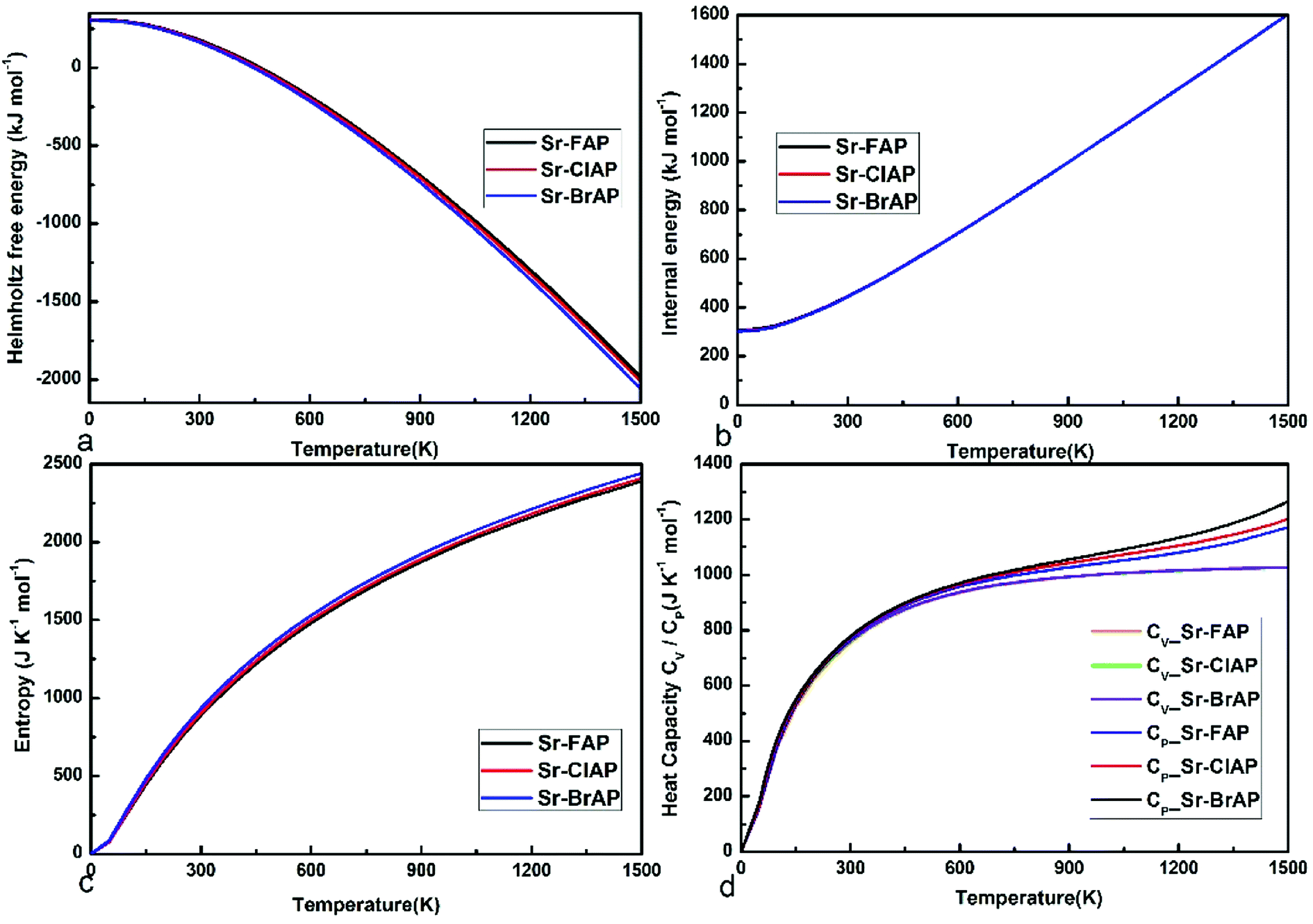 | ||
| Fig. 7 The thermodynamic properties of Sr-FAP, Sr-ClAP and Sr-BrAP apatites. (a) Helmholtz free energy F, (b) internal energy E, (c) entropy S, (d) specific heat CV (CP). | ||
In order to compare our results with the experimental data, Table 8 lists the S, H − H298 and CP at selected temperatures from 298 K to 1500 K for Sr-ClAP. For studies at constant pressure, the appropriate thermodynamic function of enthalpy is H = E + PV, where E is the internal energy of the system, P is the pressure, and V is the volume. All our studies are carried out at P = 0 GPa and hence PV = 0, as a result the H = E. It is found the calculated S, H − H298 and CP of Sr-ClAP exhibit reasonable agreement with the experimental values.35,48,49 So our predicted thermodynamic properties for Sr10(PO4)6X2 (X = F, Cl, Br) are reliable. There are no experimental or theoretical works exploring the thermodynamic properties of crystal Sr-FAP and Sr-BrAP for comparison with the present data, we hope our predicted results can give a reference for the future study.
| T(K) | S (J K−1 mol−1) | H − H298 (kJ mol−1) | CP (J K−1 mol−1) | ||||
|---|---|---|---|---|---|---|---|
| This work | Expta | This work | Expta | This work | Exptb | Expta | |
| a Ref. 49.b Ref. 48. | |||||||
| 0 | 901 | 904 | 0 | 0 | 0 | — | — |
| 300 | 906 | 909 | 1.52 | 1.41 | 773 | 844 | 764 |
| 400 | 1138 | 1153 | 82.1 | 86.8 | 861 | 995 | 921 |
| 500 | 1333 | 1367 | 169.6 | 181.8 | 922 | 1070 | 991 |
| 600 | 1500 | 1551 | 261.5 | 283.8 | 964 | 1115 | 1027 |
| 700 | 1647 | 1711 | 356.3 | 387.6 | 996 | 1146 | 1047 |
| 800 | 1776 | 1852 | 453.7 | 492.9 | 1021 | 1169 | 1058 |
| 900 | 1892 | 1977 | 552.3 | 599.0 | 1042 | 1064 | |
| 1000 | 1997 | 2089 | 652.1 | 705.6 | 1063 | 1067 | |
| 1100 | 2093 | 2191 | 752.7 | 812.4 | 1083 | 1068 | |
| 1200 | 2182 | 2284 | 854.0 | 919.2 | 1105 | 1068 | |
| 1300 | 2263 | 2369 | 955.8 | 1025.9 | 1130 | 1066 | |
| 1400 | 2339 | 2448 | 1058.0 | 1132.5 | 1161 | 1064 | |
| 1500 | 2409 | 2522 | 1160.5 | 1238.8 | 1201 | 1062 | |
4. Conclusions
In summary, we have done a comprehensive investigations on the structural stability, electronic, vibrational, and thermodynamic properties of the strontium apatites Sr10(PO4)6X2 (X = F, Cl, Br) within DFT and DFPT in combination with QHA.It was observed that with the increase in the size of the c-axis ion (F− < Cl− < Br−), the cell volumes of Sr10(PO4)6X2 (X = F, Cl, Br) compounds increased, and the optimized structure parameters of Sr10(PO4)6X2 (X = F, Cl, Br) are in good agreement with the experimental values. The results of cohesive energies and formation enthalpies show the stability of strontium apatites is related to the anion radius and decreases in the following sequence: Sr-FAP > Sr-ClAP > Sr-BrAP. The electronic structures indicate that strontium apatites Sr10(PO4)6X2 (X = F, Cl, Br) are insulator materials with the indirect band gaps, and the band gap values decrease from Sr-FAP, Sr-ClAP, to Sr-BrAP. The detailed vibrational properties are obtained using DFPT. The calculated phonon frequencies are in good agreement with the reported experimental infrared and Raman data in the literatures. According to the calculated phonon dispersions, it is concluded that strontium apatites Sr10(PO4)6X2 (X = F, Cl, Br) are dynamically stable, and the phonon behaviors are generally similar for these apatites, but most of the vibrational frequencies decrease from Sr-FAP, Sr-ClAP to Sr-BrAP, which is a consequence of the atomic mass increase (F− < Cl− < Br−). The vibrational modes at the gamma point are analyzed from group theory, it is demonstrated that with the replacement of larger Cl− and Br− for F−, all the silent modes Bg, Bu and E2u are affected and the only optically active modes involved is the Raman active mode E2g, which can be explained by the different Wyckoff sites of X (X = F, Cl, Br) ions in theirs apatite structures. From phonon density states, the high-frequency spectra for these selected apatites are naturally very similar, the low frequencies increased with the increase in the atomic mass (F < Cl < Br), and the frequencies in the 300–350 cm−1 region are associated with the Sr–F bond only. Some phonon related thermodynamic properties of Sr10(PO4)6X2 (X = F, Cl, Br) are predicted from the temperature of 0 to 1500 K and discussed. When the mass of the halogen atoms increase, the entropy S and specific heat CV and CP both increase, while free energy F decreases. On the whole, these considered apatites exhibit similar but slightly different behaviors for the thermodynamic properties, which is expected because the halogen atoms F, Cl, Br are in the same VIIA group. Our calculated thermodynamic properties of Sr-ClAP are in reasonable agreement with the experimental values.
We hope that our work could provide a useful guidance for future experimental and theoretical works.
Acknowledgements
This work was financially supported by the National High Technology Research and Development Program of China (2AA034202) and the National Natural Science Foundation of China (No. 11305147).References
- K. A. Gross and C. C. Berndt, Rev. Mineral. Geochem., 2002, 48, 631–672 CrossRef CAS.
- J. D. Pasteris, Am. Mineral., 2016, 101, 2594–2610 CrossRef.
- J. F. Rakovan and J. D. Pasteris, Elements, 2015, 11, 195–200 CrossRef CAS.
- M. Mathew and S. Takagi, J. Res. Natl. Inst. Stand. Technol., 2001, 106, 1035 CrossRef CAS PubMed.
- P. H. Mercier, Y. Le Page, P. S. Whitfield, L. D. Mitchell, I. J. Davidson and T. White, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 635–655 Search PubMed.
- C. Combes, S. Cazalbou and C. Rey, Minerals, 2016, 6, 34 CrossRef.
- Y. Pan and M. Fleet, Rev. Mineral. Geochem., 2002, 48, 13–50 CrossRef CAS.
- J. M. Hughes, M. Cameron and K. D. Crowley, Am. Mineral., 1989, 74, 870–876 CAS.
- P. Rulis, L. Ouyang and W. Ching, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 155104 CrossRef.
- J. C. Elliott, Rev. Mineral. Geochem., 2002, 48, 427–453 CrossRef CAS.
- E. Balan, S. Delattre, D. Roche, L. Segalen, G. Morin, M. Guillaumet, M. Blanchard, M. Lazzeri, C. Brouder and E. K. Salje, Phys. Chem. Miner., 2011, 38, 111–122 CrossRef CAS.
- C.-X. Li, Y.-H. Duan and W.-C. Hu, J. Alloys Compd., 2015, 619, 66–77 CrossRef CAS.
- A. Slepko and A. A. Demkov, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 134108 CrossRef.
- S. S. Bhat, U. V. Waghmare and U. Ramamurty, Cryst. Growth Des., 2014, 14, 3131–3141 CAS.
- W. Klee and G. Engel, J. Inorg. Nucl. Chem., 1970, 32, 1837–1843 CrossRef CAS.
- Z. Junhui, D. Yonghua, M. Lishi and L. Runyue, J. Alloys Compd., 2016, 680, 121–128 CrossRef.
- S. Aryal, K. Matsunaga and W.-Y. Ching, J. Mech. Behav. Biomed. Mater., 2015, 47, 135–146 CrossRef CAS PubMed.
- Z.-S. Tao, W.-S. Zhou, X.-W. He, W. Liu, B.-L. Bai, Q. Zhou, Z.-L. Huang, K.-k. Tu, H. Li and T. Sun, Mater. Sci. Eng., C, 2016, 62, 226–232 CrossRef CAS PubMed.
- C. Drouet, J. Chem. Thermodyn., 2015, 81, 143–159 CrossRef CAS.
- A. P. Soroka, V. L. Karbivskyy and V. H. Kasiyanenko, Funct. Mater., 2015, 22, 79–92 CrossRef CAS.
- K. Sudarsanan and R. Young, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 1525–1530 CrossRef.
- E. M. Michie, R. W. Grimes, S. K. Fong and B. L. Metcalfe, J. Solid State Chem., 2008, 181, 3287–3293 CrossRef CAS.
- A. M. Goryaeva, V. S. Urusov and N. N. Eremin, Eur. J. Mineral., 2013, 25, 947–955 CrossRef CAS.
- C. Guo, L. Luan, X. Ding, F. Zhang, F. G. Shi, F. Gao and L. Liang, Appl. Phys. B: Lasers Opt., 2009, 95, 779–785 CrossRef CAS.
- M. Kottaisamy, R. Jagannathan, P. Jeyagopal, R. Rao and R. Narayanan, J. Phys. D: Appl. Phys., 2001, 27, 2210 CrossRef.
- S. A. Payne, L. D. DeLoach, L. K. Smith, W. L. Kway, J. B. Tassano, W. F. Krupke, B. H. Chai and G. Loutts, J. Appl. Phys., 1994, 76, 497–503 CrossRef CAS.
- Q. Wang, S. Zhao, X. Zhang, L. Sun and S. Zhang, Opt. Commun., 1996, 128, 73–75 CrossRef CAS.
- C. Wu, J. Zhang, P. Feng, Y. Duan, Z. Zhang and Y. Wang, J. Lumin., 2014, 147, 229–234 CrossRef CAS.
- Q. Zeng, H. Liang, G. Zhang, M. D. Birowosuto, Z. Tian, H. Lin, Y. Fu, P. Dorenbos and Q. Su, J. Phys.: Condens. Matter, 2006, 18, 9549 CrossRef CAS.
- J. Liu, Z.-M. Zhang, Z.-C. Wu, F.-F. Wang and Z.-J. Li, Mater. Sci. Eng., B, 2017, 221, 10–16 CrossRef CAS.
- P. Marie, Bone, 2006, 38, 10–14 CrossRef PubMed.
- J. F. Rakovan and J. M. Hughes, Can. Mineral., 2000, 38, 839–845 CrossRef CAS.
- C. Wu, Y. Ramaswamy, D. Kwik and H. Zreiqat, Biomaterials, 2007, 28, 3171–3181 CrossRef CAS PubMed.
- W. Zhang, Y. Shen, H. Pan, K. Lin, X. Liu, B. W. Darvell, W. W. Lu, J. Chang, L. Deng and D. Wang, Acta Biomater., 2011, 7, 800–808 CrossRef CAS PubMed.
- B. K. Maji, H. Jena, M. Krishnaiah and R. Asuvathraman, J. Therm. Anal. Calorim., 2016, 124, 857–863 CrossRef CAS.
- S. H. Swafford and E. M. Holt, Solid State Sci., 2002, 4, 807–812 CrossRef CAS.
- K. Sudarsanan and R. Young, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1381–1386 CrossRef.
- P. Alberius-Henning, C. Mattsson and S. Lidin, Z. Kristallogr. - New Cryst. Struct., 2000, 215, 345–346 CAS.
- A. Aissa, B. Badraoui, R. Thouvenot and M. Debbabi, Eur. J. Inorg. Chem., 2004, 2004, 3828–3836 CrossRef.
- S. Zhai, S. R. Shieh, W. Xue and T. Xie, Phys. Chem. Miner., 2015, 42, 579–585 CrossRef CAS.
- C. E. Bonner, C. C. Chess, C. Meegoda, S. Stefanos and G. B. Loutts, Opt. Mater., 2004, 26, 17–22 CrossRef CAS.
- A. Schulte, S. C. Buchter and B. H. Chai, International Society for Optics and Photonics, 1995, 2380, 34–42 CAS.
- N. Chernorukov, A. Knyazev and E. Bulanov, Inorg. Mater., 2011, 47, 172–177 CrossRef CAS.
- A. V. Knyazev, N. G. Chernorukov and E. N. Bulanov, Mater. Chem. Phys., 2012, 132, 773–781 CrossRef CAS.
- M. Jemal, A. B. Cherifa, I. Khattech and I. Ntahomvukiye, Thermochim. Acta, 1995, 259, 13–21 CrossRef CAS.
- I. Khattech and M. Jemal, Thermochim. Acta, 1997, 298, 23–30 CrossRef CAS.
- I. Khattech and M. Jemal, Thermochim. Acta, 1997, 298, 17–21 CrossRef CAS.
- R. V. Krishnan, H. Jena, K. G. Kutty and K. Nagarajan, Thermochim. Acta, 2008, 478, 13–16 CrossRef CAS.
- R. Babu, H. Jena, K. G. Kutty and K. Nagarajan, Thermochim. Acta, 2011, 526, 78–82 CrossRef CAS.
- M. Kocher, A. Jain, S. P. Ong and G. Hautier, Materials Project, https://www.materialsproject.org/materials/mp-6669, mp-23121, mp-23141.
- J. A. L. Rabone and N. De Leeuw, J. Comput. Chem., 2006, 27, 253–266 CrossRef CAS PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- D. M. Ceperley and B. Alder, Phys. Rev. Lett., 1980, 45, 566 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- P. V. Balachandran, K. Rajan and J. M. Rondinelli, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 612–615 CAS.
- Y. Wang, S.-L. Shang, H. Fang, Z.-K. Liu and L.-Q. Chen, npj Comput. Mater., 2016, 2, 16006 CrossRef.
- S. Baroni, S. De Gironcoli, A. Dal Corso and P. Giannozzi, Rev. Mod. Phys., 2001, 73, 515 CrossRef CAS.
- X. Gonze and C. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10355 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |