
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA single fluorescent chemosensor for multiple targets of Cu2+, Fe2+/3+ and Al3+ in living cells and a near-perfect aqueous solution†
Tae Geun Joa,
Jae Min Jung*a,
Jiyeon Hanb,
Mi Hee Lim b and
Cheal Kim*a
b and
Cheal Kim*a
aDepartment of Fine Chemistry, Department of Interdisciplinary Bio IT Materials, Seoul National University of Science and Technology, Seoul 139-743, Republic of Korea. E-mail: iamjemin@naver.conm; chealkim@seoultech.ac.kr; Fax: +82-2-973-9149; Tel: +82-2-970-6693
bDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
First published on 31st May 2017
Abstract
A sulfonate-based chemosensor 1 was designed and synthesized for sensing various analytes: Cu2+, Fe2+/3+ and Al3+. Sensor 1 showed a high selectivity and sensitivity for the analytes in a near-perfect aqueous medium. Cu2+ and Fe2+/3+ could be monitored by fluorescence quenching of 1. It had sufficiently low detection limits (1.25 μM for Cu2+ and 3.96 μM for Fe3+), which were below the recommended levels of the World Health Organization for Cu2+ (31.5 μM) and the Environmental Protection Agency for Fe3+ (5.37 μM). 1 showed the high preferential selectivity for Cu2+ and Fe3+ in the presence of competitive metal ions without any interference. Importantly, pyrophosphate could be used to distinguish Fe3+ from Cu2+. In addition, this sensor could monitor Al3+ through fluorescence emission change. Moreover, 1 was successfully applied to quantify and image Al3+ in water samples and living cells. Based on photophysical studies and theoretical calculations, the sensing mechanisms of 1 for Cu2+ and Al3+ were explained, respectively.
1. Introduction
Development of chemosensors with high selectivity and sensitivity for specific metal ions has been important due to their significant roles in medical, industrial, environmental and biological processes.1 Among various metals, copper ions are the third abundant transition metal in the human body. They play diverse and significant roles in many biochemical processes in organisms from bacteria to mammals.2–7 However, a deficiency or overbalance of copper ions in human body can cause fatal diseases, such as Alzheimer's and Wilson's diseases.8–13As the first most abundant metal ions in human body, iron plays crucial roles in numerous biological processes, including electron transport function, synthesis of hemoglobin and immune system.14–18 On the contrary, disruption of iron-ion homeostasis can induce a number of severe neurological diseases with diverse clinical manifestations, ranging from anemia to iron overload.19 Aluminum is the third most abundant element on earth and extensively used in food additives, pharmaceutical synthesis, cosmetics and the manufacturing industry.20–22 Due to its extensive use, aluminum can be easily accumulated in human body. Such accumulation of the metal ion could lead neuronal disorders such as Parkinson's disease and Alzheimer's disease.23–30 Therefore, the development of the chemosensors that could successfully recognize and determine these metal ions should be urgently developed.
In recent years, multiple target sensors for metal ions are attracting considerable interest, because they could abbreviate the processes to synthesize multiple compounds and facilitate to detect multiple analytes with a single device.3–7,11–13,16–18,20–22,31–36 However, there are still challenges to develop the sensors that could simultaneously detect and distinguish different target metal ions.
Sulfonic acid groups have been used to improve the solubility of chemosensors and naphthol moiety is a well-known excellent fluorophore.36–46 Therefore, we expected that the presence of a sulfonic acid group and a naphthol moiety in a chemosensor might not only increase its water solubility, but also have good optical properties.
Herein, we report the synthesis, characterization and fluorescent sensing behaviors of sulfonate-based sensor 1, triethylammonium (E)-3-hydroxy-4-(((2-hydroxynaphthalen-1-yl)methylene)amino)naphthalene-1-sulfonate. Sensor 1 could recognize selectively Cu2+ and Fe2+/3+ by the fluorescence quenching and Al3+ by the obvious fluorescence emission change. Based on UV-vis and fluorescence titrations, Job plots, ESI-mass analyses, 1H NMR titration and theoretical calculations, the sensing properties and mechanisms of 1 toward the three analytes were explained.
2. Experimental section
2.1. General information
All chemicals (analytical grade and spectroscopic grade) were purchased from Sigma-Aldrich and used without further purification. Both 1H NMR and 13C NMR were recorded on a Varian spectrometer (400 MHz for 1H and 100 MHz for 13C). The chemical shifts (δ) were recorded in ppm. Absorption spectra were recorded at room temperature using a Perkin Elmer model Lambda 2S UV/vis spectrophotometer. Fluorescence measurements were performed on a Perkin Elmer model LS45 fluorescence spectrophotometer. Electrospray ionization mass spectra (ESI-mass) were collected on a Thermo Finnigan (San Jose, CA, USA) LCQ™ Advantage MAX quadrupole ion trap instrument. Elemental analysis for carbon, nitrogen and hydrogen was carried out by using a MICRO CUBE elemental analyzer (Germany) in Laboratory Center of Seoul National University of Science and Technology, Korea.2.2. Synthesis of 1
4-Amino-3-hydroxynaphthalene-1-sulfonic acid (0.24 g, 1 mmol) and triethylammonium (TEA) were dissolved in 10 mL of methanol (MeOH). 2-Hydroxy-1-naphthaldehyde (0.17 g, 1 mmol) was added into the resulting solution. Then, the reaction solution was stirred for 5 h at room temperature. After evaporation, the product was recrystallized by diethyl ether and methanol, filtered and dried under vacuum. The yield: 0.17 g (34.4%). 1H NMR (DMSO-d6, 400 MHz): δ 16.17 (s, 1H), 10.43 (s, 1H), 9.95 (d, J = 3.6 Hz, 1H), 8.83 (d, J = 8.4 Hz, 2H), 8.23 (d, J = 8.4 Hz, 1H), 7.99 (m, 3H), 7.86 (d, J = 8.4 Hz, 1H), 7.55 (q, J = 8.0 Hz, 2H), 7.39 (m, 2H), 7.13 (d, J = 9.2 Hz, 2H), 3.10 (m, 6H for TEA), 1.17 (t, J = 7.2, 9H for TEA). 13C NMR (DMSO-d6, 100 MHz): δ 155.46, 155.35, 154.17, 146.13, 129.14, 113.15, 106.55, 105.85, 49.77, 49.29, 26.92, 21.86, 21.04, 20.60 ppm. ESI-mass: m/z calcd for C21H15NO5S − H+ ([M − H+]), 392.06; found, 392.30.2.3. Fluorescent sensing for Cu2+, Fe2+/3+ and Al3+
For Cu2+, 20 μL of each metal-ion solution (9 mM) was diluted to Bis–Tris buffer solution, respectively. 20 μL of Cu2+ solution (9 mM) was added to the solutions prepared above. Then, 9 μL of sensor 1 solution (10 mM) was added to the mixed solutions. Each vial had a total volume of 3 mL. After stirring them for a few seconds, fluorescence spectra were recorded at room temperature.
For Fe3+, 40 μL of each metal-ion solution (9 mM) was diluted to Bis–Tris buffer solution, respectively. 40 μL of Fe3+ solution (9 mM) was added to the solutions prepared above. Then, 9 μL of sensor 1 solution (10 mM) was added to the mixed solutions. Each vial had a total volume of 3 mL. After stirring them for a few seconds, fluorescence spectra were recorded at room temperature.
For Al3+, 67.5 μL of each metal-ion solution (100 mM) was diluted to Bis–Tris buffer solution, respectively. 67.5 μL of Al3+ solution (100 mM) was added to the solutions prepared above. Then, 9 μL of sensor 1 solution (10 mM) was added to the mixed solutions. Each vial had a total volume of 3 mL. After stirring them for a few seconds, fluorescence spectra were recorded at room temperature.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per 1 mL and then incubated at 37 °C for 12 h. For fluorescence imaging experiments, cells were first treated with 1 (dissolved in DMSO; 1% v/v final DMSO concentration; 20 μM; at room temperature) for 10 min. After incubation with aluminum nitrite (dissolved in water; 1% v/v DMSO; 200 μM) for 10 min, cells were washed with 2 mL of 10 mM Bis–Tris buffer (pH 7.4, 150 mM NaCl) two times. Imaging was performed with an EVOS FL fluorescence microscope (Life technologies) using a GFP light cube [excitation 470 (±11) nm; emission 510 (±21) nm].
000 cells per 1 mL and then incubated at 37 °C for 12 h. For fluorescence imaging experiments, cells were first treated with 1 (dissolved in DMSO; 1% v/v final DMSO concentration; 20 μM; at room temperature) for 10 min. After incubation with aluminum nitrite (dissolved in water; 1% v/v DMSO; 200 μM) for 10 min, cells were washed with 2 mL of 10 mM Bis–Tris buffer (pH 7.4, 150 mM NaCl) two times. Imaging was performed with an EVOS FL fluorescence microscope (Life technologies) using a GFP light cube [excitation 470 (±11) nm; emission 510 (±21) nm].2.4. Theoretical calculations
All theoretical calculations based on the hybrid exchange correlation functional B3LYP47,48 applying the 6-31G** basis set49,50 was employed for the main group elements, whereas the Lanl2DZ effective core potential (ECP)51,52 was used for Cu. In vibrational frequency calculations, there was no imaginary frequency for the optimized geometries of 1, 1–Al3+ and 1–Cu2+ complex, suggesting that these geometries represented local minima. In order to investigate the transition energies for the optimized structures of 1, 1–Al3+ and 1–Cu2+ species, twenty lowest singlets were calculated with TD-DFT (B3LYP). The GaussSum 2.1 (ref. 53) was used to calculate the contributions of molecular orbitals in electronic transitions. All the calculations were carried out using Gaussian 03 program.543. Results and discussion
Chemosensor 1 was synthesized by the condensation reaction of 4-amino-3-hydroxynaphthalene-1-sulfonic acid with 2-hydroxy-1-naphthaldehyde in methanol (Scheme 1), and characterized by 1H (Fig. S1†) and 13C NMR, ESI-mass spectrometry and elemental analysis.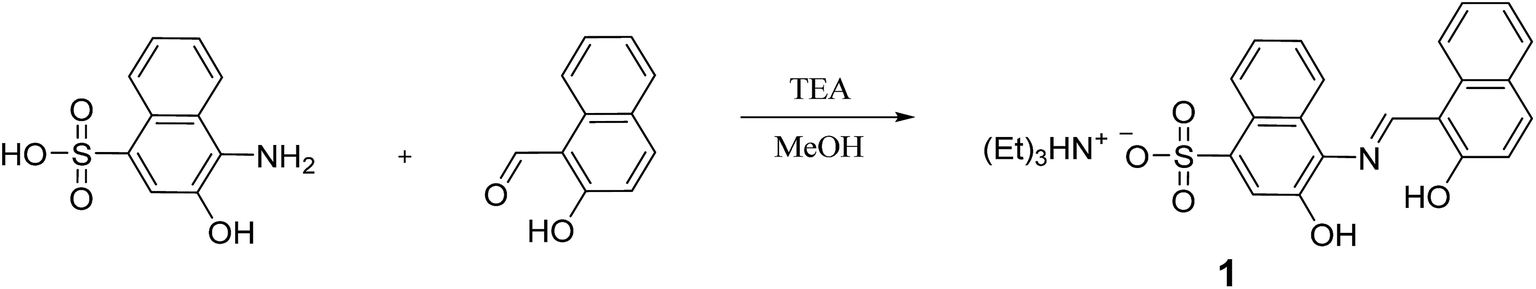 | ||
| Scheme 1 Synthetic procedure of 1. | ||
The fluorescence sensing ability of 1 was examined toward various metal ions such as Al3+, Ga3+, In3+, Zn2+, Cd2+, Cu2+, Fe2+, Fe3+, Mg2+, Cr3+, Co2+, Ni2+, Na+, K+, Ca2+, Mn2+ and Pb2+ in Bis–Tris buffer solution (10 mM, pH 7.0) (Fig. 1). Upon excitation at 380 nm, 1 exhibited a fluorescence emission at 442 nm. In the presence of most cations, there was no significant change in the fluorescence spectrum, whereas Cu2+, Fe2+, Fe3+ and Al3+ ions showed the notable spectral changes. In cases of Cu2+, Fe2+ and Fe3+, the emission intensity of 1 at 442 nm was completely quenched. Meanwhile, the fluorescence emission at 442 nm was red-shifted to 535 nm in the presence of Al3+. Therefore, 1 might be a potential fluorescence chemosensor that could detect copper and iron ions by fluorescence quenching and aluminum ion by fluorescence emission change.
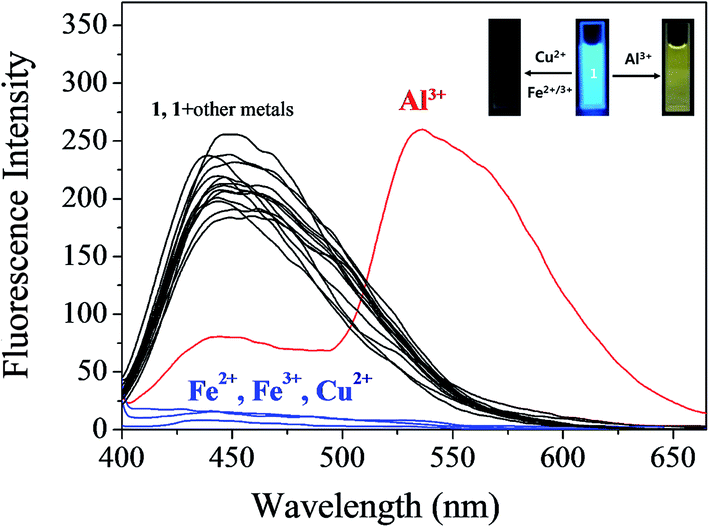 | ||
| Fig. 1 Fluorescence spectral change of 1 (30 μM) upon addition of 70 equiv. of different metal ions in Bis–Tris buffer (10 mM Bis–Tris, pH 7.0). | ||
3.1. Turn-off fluorescence detection of Cu2+ and Fe2+/3+
The fluorescence and UV-vis titration experiments were conducted to understand the binding property of 1 with Cu2+. Upon the addition of Cu2+, the fluorescence emission intensity of 1 at 442 nm steadily decreased and quenched until amount of Cu2+ reached 2 equiv. (Fig. 2). The UV-vis titration showed that the absorption bands of 1 at 320 nm and 450 nm decreased, while the bands at 270 nm and 467 nm increased gradually (Fig. S2†). Two isosbestic points at 302 nm and 458 nm indicated the formation of the only one species between 1 and Cu2+.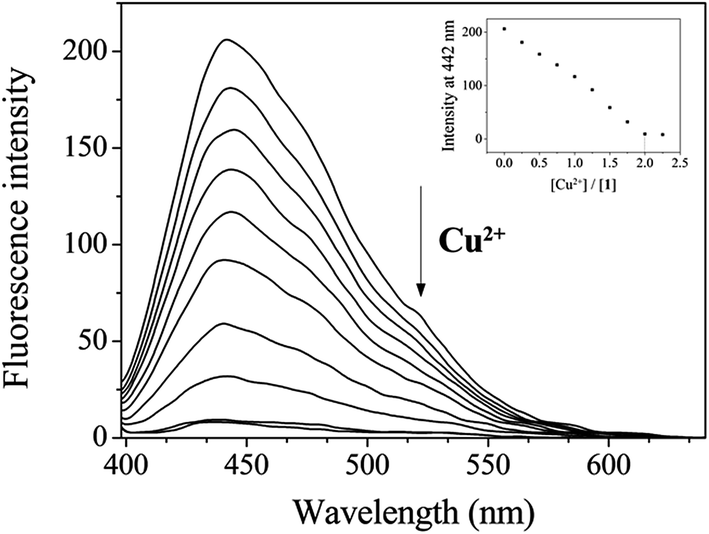 | ||
| Fig. 2 Fluorescence spectral change of 1 (30 μM) with Cu2+ ions (0–2.25 equiv.) in Bis–Tris buffer (10 mM, pH 7.0). | ||
In order to examine the binding stoichiometry of 1 with Cu2+, Job plot analysis was carried out (Fig. S3†). A maximum intensity appeared at the molar fraction of 0.5, which indicated a 1:1 binding mode between 1 and Cu2+. To further confirm the binding mode between 1 and Cu2+, ESI-mass spectrometry analysis was conducted (Fig. 3). The negative mass data of 1 for Cu2+ showed that the peak at m/z = 453.3 was assignable to 1–3H+ + Cu2+ [calcd, m/z: 453.0]. Based on the titration measurement, the association constant (K) of 1 with Cu2+ was calculated as 4.8 × 103 M−1 by Benesi–Hildebrand equation55 (Fig. S4†), which was within the range of those (103 to 1012) previously reported for chemosensors toward Cu2+.56–61 As shown in Fig. S5,† the detection limit (3σ/k)62 of 1 for Cu2+ was determined to be 1.25 μM which was much lower than that (31.5 μM) recommended by WHO in drinking water.63
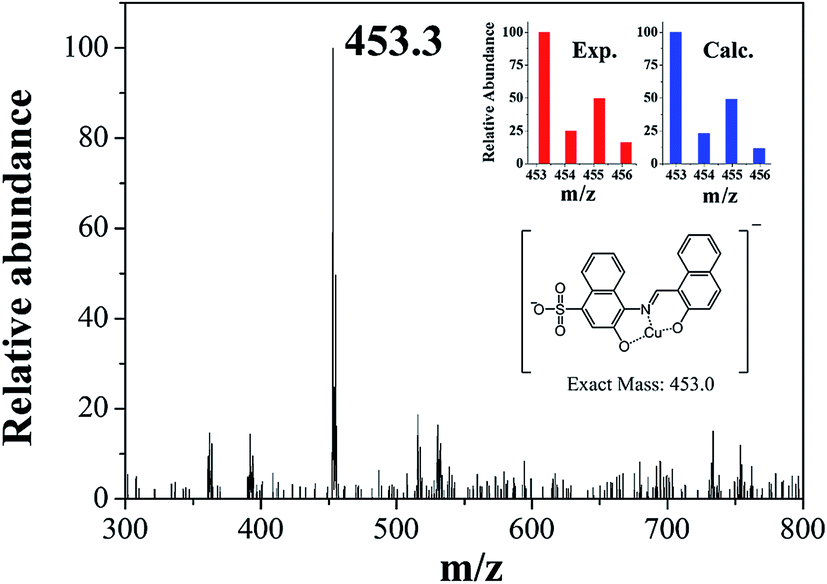 | ||
| Fig. 3 Negative-ion ESI-mass spectrum of 1 (100 μM) upon addition of 1 equiv. of Cu2+. | ||
A preferential selectivity of 1 toward Cu2+ was evaluated in the presence of other competitive species (Al3+, Ga3+, In3+, Zn2+, Cd2+, Fe2+, Fe3+, Mg2+, Cr3+, Co2+, Ni2+, Na+, K+, Ca2+, Mn2+ and Pb2+) (Fig. 4). There was no significant interference for sensing of Cu2+. For practical application, the pH effects of 1 in the absence and presence of Cu2+ were investigated at various pH range of 2 to 12. The fluorescence quenching of 1 by adding Cu2+ was observed between pH 6 and 10 (Fig. S6†), which warrants its application for detection of Cu2+ under physiological pH 7.0–8.4.64
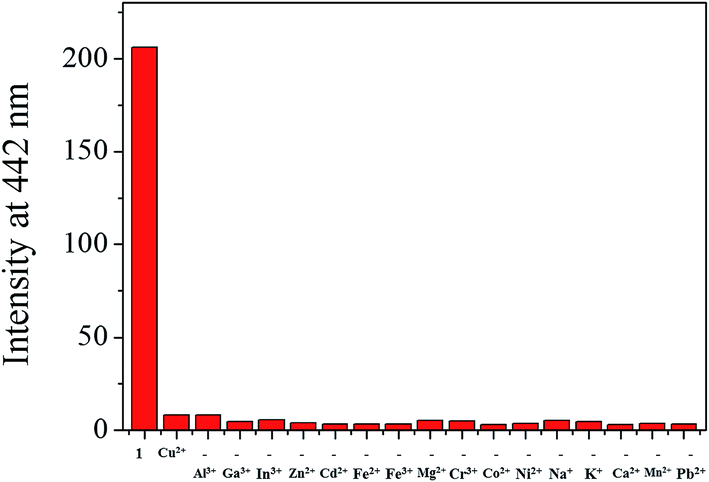 | ||
| Fig. 4 Fluorescence intensity (at 442 nm) of 1 upon addition of Cu2+ ions (2.0 equiv.) in the absence and presence of other metal ions (2.0 equiv.). | ||
Next, fluorescence titration of 1 toward Fe3+ was carried out to understand binding properties. Upon the addition of Fe3+, the fluorescence intensity at 442 nm was gradually reduced and completely quenched when 4 equiv. of Fe3+ were added (Fig. 5). UV-vis titration of 1 for Fe3+ showed that the absorption at 450 nm decreased and those at 300 and 580 nm increased with two clear isosbestic points at 405 and 527 nm (Fig. S7†). The peak at 580 nm, which has a high molar extinction coefficient (1.9 × 103 M−1 cm−1), are too large to be Fe-based d–d transitions. Thus, the new peak might be due to a metal-to-ligand charge-transfer (MLCT).65
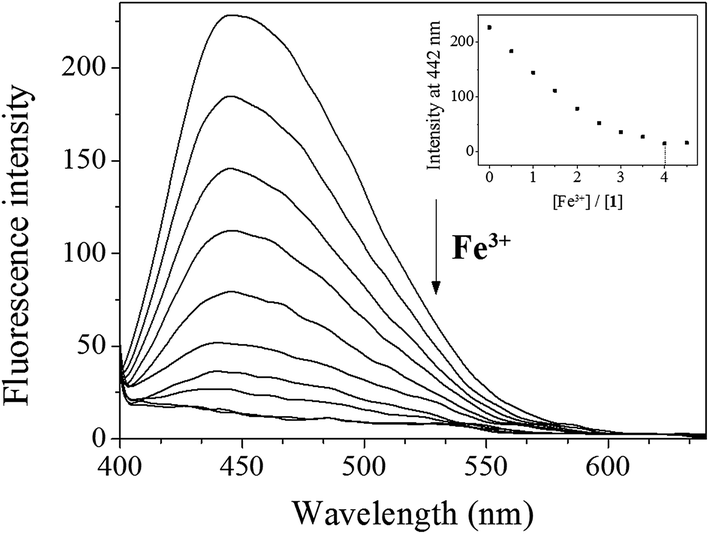 | ||
| Fig. 5 Fluorescence spectral change of 1 (30 μM) with Fe3+ ions (0–4.5 equiv.) in Bis–Tris buffer (10 mM, pH 7.0). | ||
Job plot analysis revealed a 1:1 stoichiometry for 1 and Fe3+ (Fig. S8†), which further was confirmed by ESI-mass spectrometry analysis. As shown in Fig. 6, the negative mass data of 1 for Fe3+ showed the major peak at m/z = 508.0, which was assigned to 1–3H+ + Fe3+ + NO3− [calcd, m/z: 508.0]. The association constant (K) for Fe3+ was determined to be 1.1 × 104 M−1 by using Benesi–Hildebrand equation55 (Fig. S9†), which was within the range of those (103 to 105) previously reported for chemosensors toward Fe3+.66 The detection limit (3σ/k)62 of 1 for Fe3+ was calculated to be 3.96 μM (Fig. S10†), which was lower than the guideline (5.37 μM) recommended by environmental protection agency guideline (EPA) for iron in drinking water.63 To examine the preferential selectivity for Fe3+, the interference experiments were evaluated in the presence of other competitive species (Fig. 7). Compared with the fluorescence intensity of 1–Fe3+, there was no distinct variation in the presence of other metal ions. These results suggested that sensing properties of 1 for Fe3+ was hardly affected from potentially competitive metal ions. The pH sensitivity of Fe3+ detection by 1 was examined at various pH range of 2 to 12 (Fig. S11†). The fluorescence quenching of the 1–Fe3+ complex was exhibited between pH 2 and 10, which warrants that Fe3+ could be clearly detected by fluorescence measurements using chemosensor 1 over a wide range of pH.
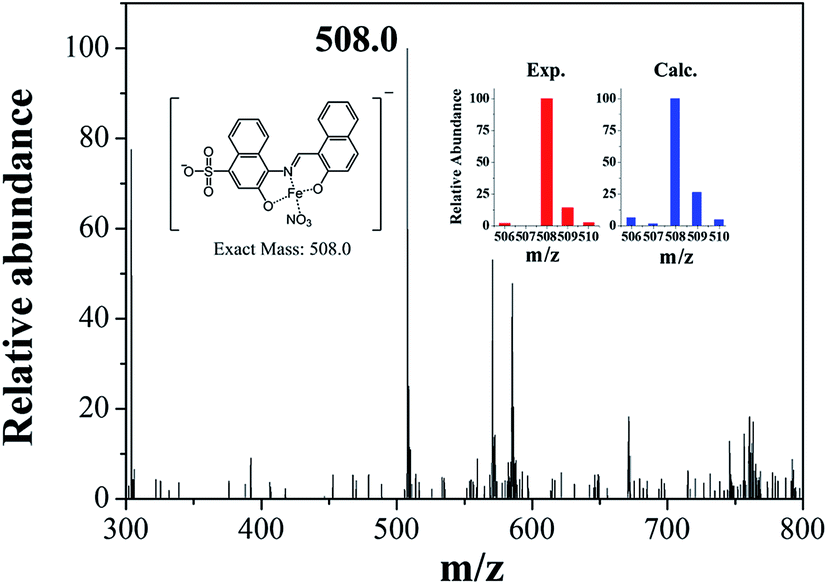 | ||
| Fig. 6 Negative-ion ESI-mass spectrum of 1 (100 μM) upon addition of 1 equiv. of Fe3+. | ||
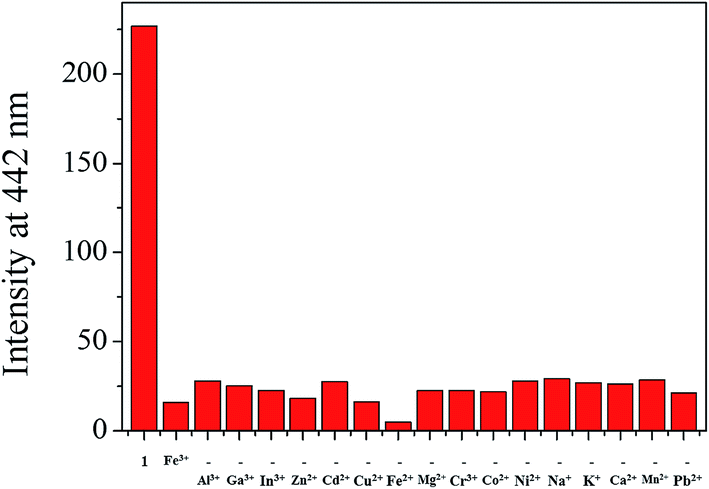 | ||
| Fig. 7 Fluorescence intensity (at 442 nm) of 1 upon addition of Fe3+ ions (4.0 equiv.) in the absence and presence of other metal ions (4.0 equiv.). | ||
On the other hand, we also conducted the UV-vis titration of 1 for Fe2+ (Fig. S12†). The titration for Fe2+ was nearly identical to that of Fe3+. These results could be explained by the rapid oxidation reaction of Fe2+ to Fe3+ in the 1–Fe2+ complex by oxygen molecule.28 To further verify our proposal, the UV-vis spectral changes of 1 for Fe2+ were observed under the degassed conditions (Fig. S13†). Upon the addition of Fe2+ into a solution of 1 under an anaerobic condition, there was no significant change in spectrum of 1. Upon the exposure of 1–Fe2+ complex to air, however, the spectrum of 1–Fe2+ complex was dramatically changed, which was nearly identical to that of 1–Fe3+ complex. These observations indicated that 1–Fe2+ complex formed under the degassed conditions might be rapidly oxidized to 1–Fe3+ complex in air.
Some metal complexes showed the selectivity toward specific anions in the systems such as Cu–S, Cu–CN, and Hg–I.67 Therefore, we also examined the selectivity of 1–Fe3+ and 1–Cu2+ complexes toward various anions, such as PPi (pyrophosphate), AMP, ADP, ATP, CN−, AcO−, F−, Cl−, Br−, I−, BzO−, N3−, SCN−, H2PO4−, S2−, NO3− and SO42− (Fig. S14†). Only PPi induced a recovery of fluorescence intensity toward 1–Fe3+ complex, while there was no change in fluorescence intensities of 1–Fe3+ and 1–Cu2+ complex solutions. It is worthwhile to mention that the fluorescence recovery of 1–Fe3+ complex by PPi is very useful, because it could distinguish 1–Fe3+ from 1–Cu2+ complex. As shown in Fig. 1, both 1–Fe3+ and 1–Cu2+ complexes exhibited the fluorescence quenching. If sensor 1 with the strong fluorescence intensity would show fluorescence quenching upon the addition of a certain metal ion, it can be Cu2+ or Fe ion. In such a case, the fluorescence recovery of 1 by PPi would indicate that the metal ion could be Fe ion, while in the absence of the fluorescence recovery of 1 it could be Cu2+.
3.2. Detection of Al3+ by fluorescence emission change
To investigate the sensing properties of 1 toward Al3+, UV-vis and fluorescence titrations were performed in a near-perfect aqueous solution. On the gradual addition of Al3+, the fluorescence intensity at 442 nm decreased and a new emission band at 535 nm appeared (Fig. 8). The UV-titration experiments showed that the absorbance band at 425 nm decreased gradually, while the absorbance at 280 nm and 467 nm increased upon the gradual addition of Al3+ (Fig. S15†). The isosbestic points at 301 nm and 448 nm were observed, indicating that the binding between 1 and Al3+ ions afforded only one species.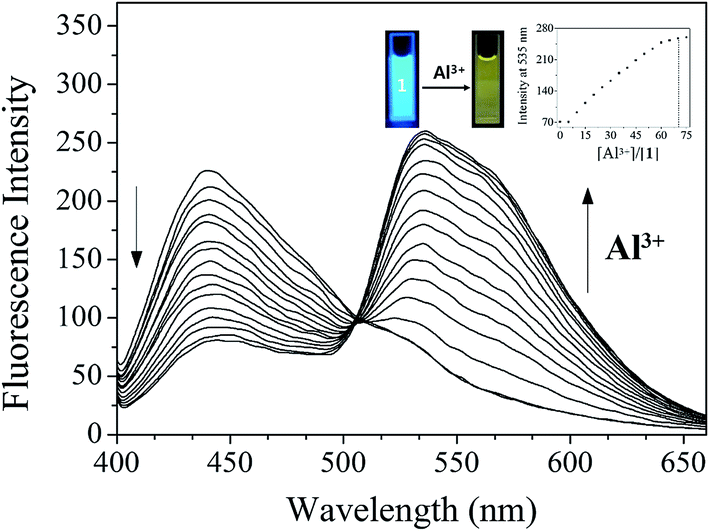 | ||
| Fig. 8 Fluorescence spectral change of 1 (30 μM) with Al3+ ions (0–80 equiv.) in Bis–Tris buffer (10 mM, pH 7.0). | ||
The Job plot analysis68 for 1 and Al3+ showed a 1:1 complexation (Fig. S16†). The binding mode of 1 and Al3+ was further confirmed by ESI-mass spectrometry analysis (Fig. S17†). The negative mass spectrum of 1 for Al3+ showed that the major peaks at m/z = 479.3 and 556.5 were assignable to 1–3H+ + Al3+ + NO3− [calcd, m/z: 479.0] and 1–3H+ + Al3+ + NO3− + DMSO [calcd, m/z: 557.0], respectively. Based on the fluorescence titration, the association constant (K) of 1 for Al3+ derived from Benesi–Hildebrand equation55 was calculated to be 8.9 × 10 M−1 (Fig. S18†), which was within the range of those (102 to 109) previously reported for Al3+-binding chemosensors.69 The detection limit (3σ/k)62 of 1 as a fluorescence sensor for Al3+ sensing was determined to be 18.07 μM (Fig. S19†).
In order to confirm the selectivity of 1 for Al3+ over competing metal ions, the interference experiments were carried out (Fig. S20†). In the presence of Cu2+, Fe2+, Fe3+ and Cr3+, the relative emission intensity was inhibited, whereas most of other metal ions did not interfere with the detection of Al3+. The practical application of 1 was evaluated through pH dependent study. As shown in Fig. S21,† 1 could clearly detect Al3+ over a wide range of pH 4.0–11.0. To further check the practical applicability of sensor 1, we conducted the real sample analysis for quantitative measurement of Al3+. A good calibration curve was constructed for the determination of Al3+ (Fig. S22†). Then, it was applied to determinate Al3+ ions in both artificial polluted and drinking water samples. As shown in Table 1, suitable recoveries and Relative Standard Deviation (R.S.D.) values of the water samples were obtained.
| Sample | Al(III) added (μmol L−1) | Al(III) found (μmol L−1) | Recovery (%) | R.S.D (n = 3) (%) |
|---|---|---|---|---|
| a Condition: [1] = 30 μmol L−1 in Bis–Tris buffer (10 mM, pH 7.0).b Prepared by deionized water, 300 μmol L−1: Zn2+, Cd2+, Pb2+, Hg2+, Na+, K+, Ca2+, Mg2+. | ||||
| Artificial polluted waterb | 300.0 | 303.8 | 101.3 | 1.40 |
| Drinking water | 300.0 | 296.9 | 99.0 | 0.96 |
To investigate the sensing mechanism of 1 toward Al3+, 1H NMR titration of 1 with Al3+ was carried out (Fig. 9). Upon the addition of Al3+, the protons H1 and H2 disappeared, and the protons H3, H5 and H8 shifted slightly up-field. When more than 1 equiv. of Al3+ were added, there was no shift in the position of proton signals, which implied the 1:1 complexation of 1 with Al3+.
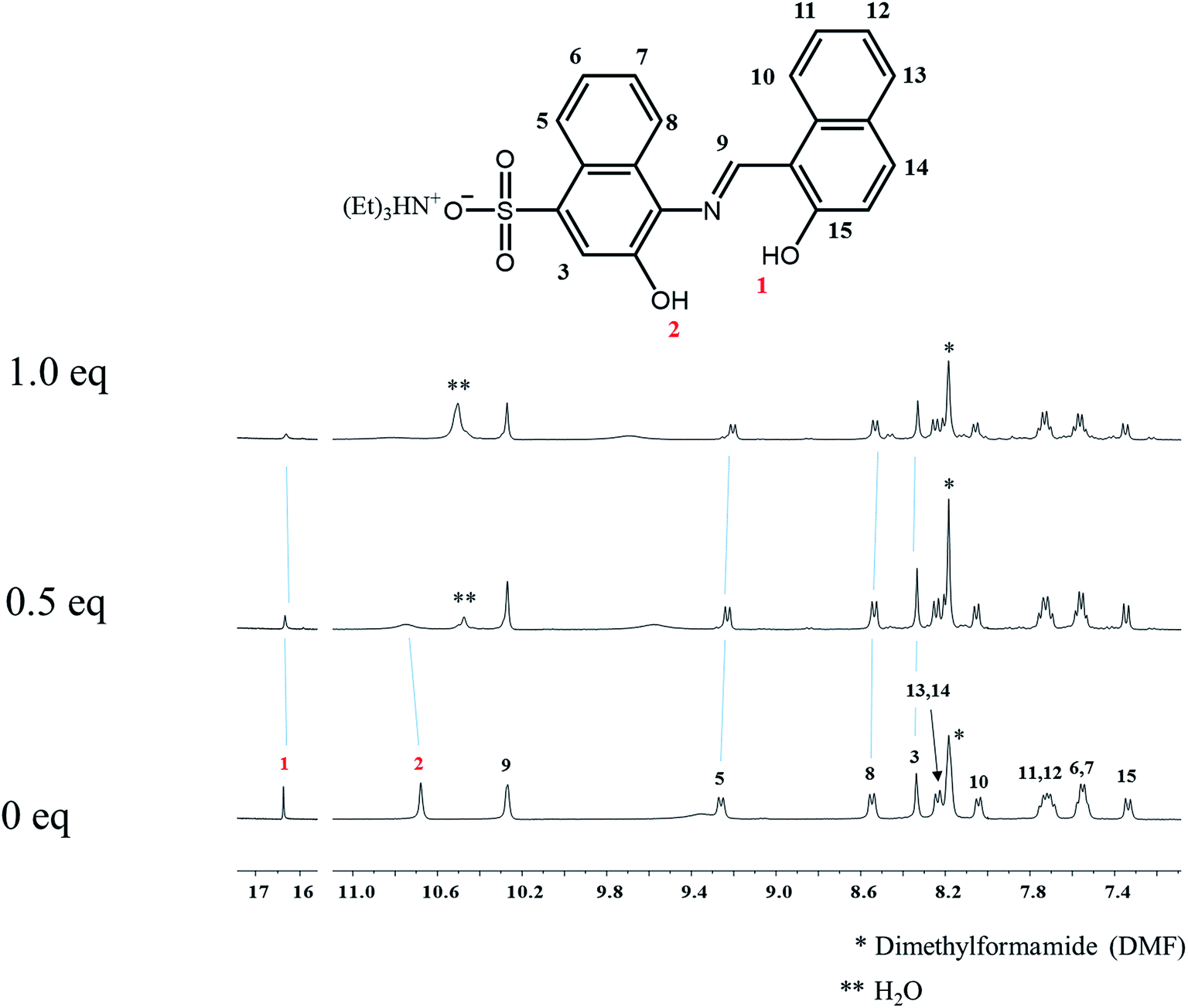 | ||
| Fig. 9 1H NMR titrations of 1 with Al3+ (0, 0.5 and 1.0 equiv.). | ||
To examine the potential of 1 to monitor Al3+ in living cells, fluorescence imaging experiments were conducted (Fig. 10). HeLa cells were first incubated with 1 for 10 min and then exposed to aqueous Al3+ solution for 10 min before imaging. The results showed that the HeLa cells without either Al3+ or 1 showed negligible intracellular fluorescence, while those cultured with both Al3+ and 1 exhibited fluorescence.
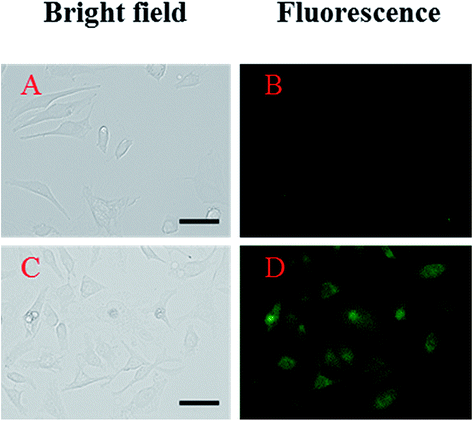 | ||
| Fig. 10 Fluorescent responses of 1 to Al3+ in HeLa cells. Cells (A and B) were preincubated with 1 for 10 min prior to addition of Al3+ (C and D). The left side images (A and C) were observed with the light microscope and the right side images (B and D) were taken with a fluorescence microscope. Conditions: [1] = 20 μM; [Al3+] = 200 μM; 37 °C; 5% CO2. | ||
3.3. Theoretical calculations
Theoretical calculations were carried out to get deep insight into fluorescence sensing mechanisms of 1 toward Cu2+ and Al3+ in parallel to the experimental studies. Based on Job plots and ESI-mass spectrometry analyses, all theoretical calculations were considered with the 1:1 stoichiometry for the 1–Cu2+ and 1–Al3+ complexes. Moreover, for the simplicity of the calculations, the triethylammonium salt of 1 was replaced by a hydrogen atom (Fig. 11). The energy-minimized structures for 1, 1–Cu2+ and 1–Al3+ complex were calculated by utilizing the density functional theory (DFT/B3LYP/main group atom and Al: 6-31G** and Cu: Lanl2DZ/ECP) (Fig. 11). The energy-minimized structure (1C, 2C, 3N, 4C = 128.9°) of 1 showed a twisted shape (Fig. 11a). After combined with Cu2+ or Al3+, the structures of the complexes 1–Cu2+ and 1–Al3+ were flatter than that of 1 (1C, 2C, 3N, 4C = 157.0° for Cu2+ and 1C, 2C, 3N, 4C = 162.6° for Al3+) (Fig. 11b and c). Both Cu2+ and Al3+ were coordinated to the N atom in the Schiff-base and the two O atoms in the hydroxyl groups of 1.
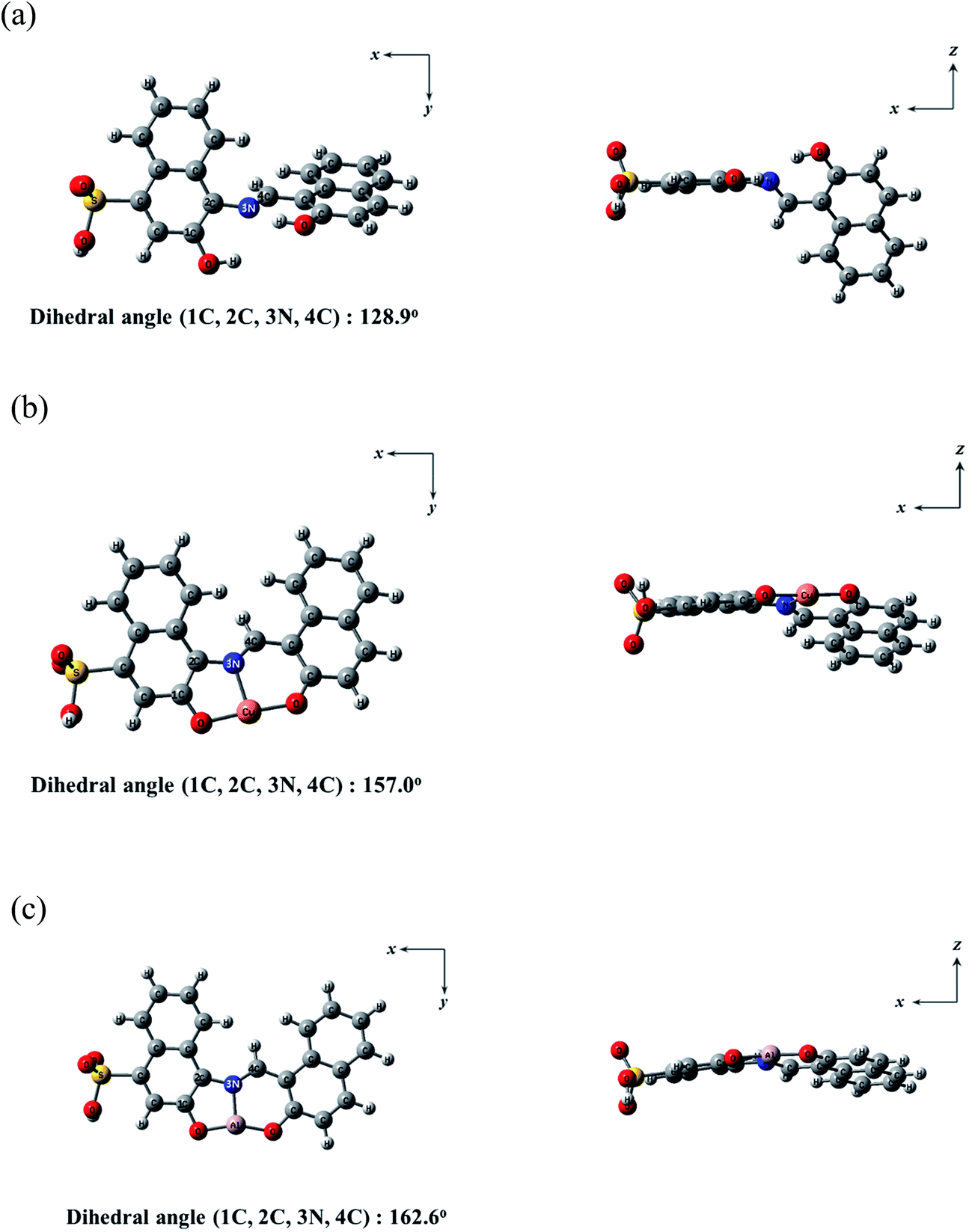 | ||
| Fig. 11 Energy-minimized structures of (a) 1, (b) 1–Cu2+ and (c) 1–Al3+. | ||
We further investigated the singlet excited states of 1, 1–Cu2+ and 1–Al3+ species by using the TD-DFT (time dependent-density functional theory) methods, which were compared with their UV-vis spectra. In case of 1, the main molecular orbital (MO) contribution of the first lowest excited state was determined for HOMO → LUMO transition (424.0 nm, Fig. S23†), which was characterized by intramolecular charge transfer (ICT) transition. For 1–Cu2+, the main MO contributions of the 10th excited state were determined for HOMO (α) → LUMO (α) and HOMO (β) → LUMO+1 (β) transitions with predominant ICT (474.0 nm, Fig. S24†). These results were consistent with the bathochromic shift in the UV-vis spectra of 1 and 1–Cu2+ complex. The remainder of the MO contributions was determined for ligand-to-metal charge-transfer (LMCT).70,71 The charge-transfer might provide a pathway for non-radiative decay of the excited state, which induced the quenching of fluorescence of 1. For 1–Al3+, the main MO contribution of the first lowest excited state was determined for HOMO → LUMO transitions (492.0 nm, Fig. S25†), which indicated ICT transition. There was no obvious change in the electronic transition between 1 and 1–Al3+ complex, while only the decrease of energy gap (424.0–492.0 nm) was observed upon chelating of 1 with Al3+. These results suggested that the sensing mechanism of 1 toward Al3+ was originated from enhancement of ICT, which caused the red-shift of the emission maximum of fluorescence from 442 nm to 535 nm.72 From the Job plots, ESI-mass spectrometry analyses and theoretical calculations, we proposed the binding structures of 1–Cu2+, 1–Fe3+ and 1–Al3+ complexes in Schemes 2 and 3.
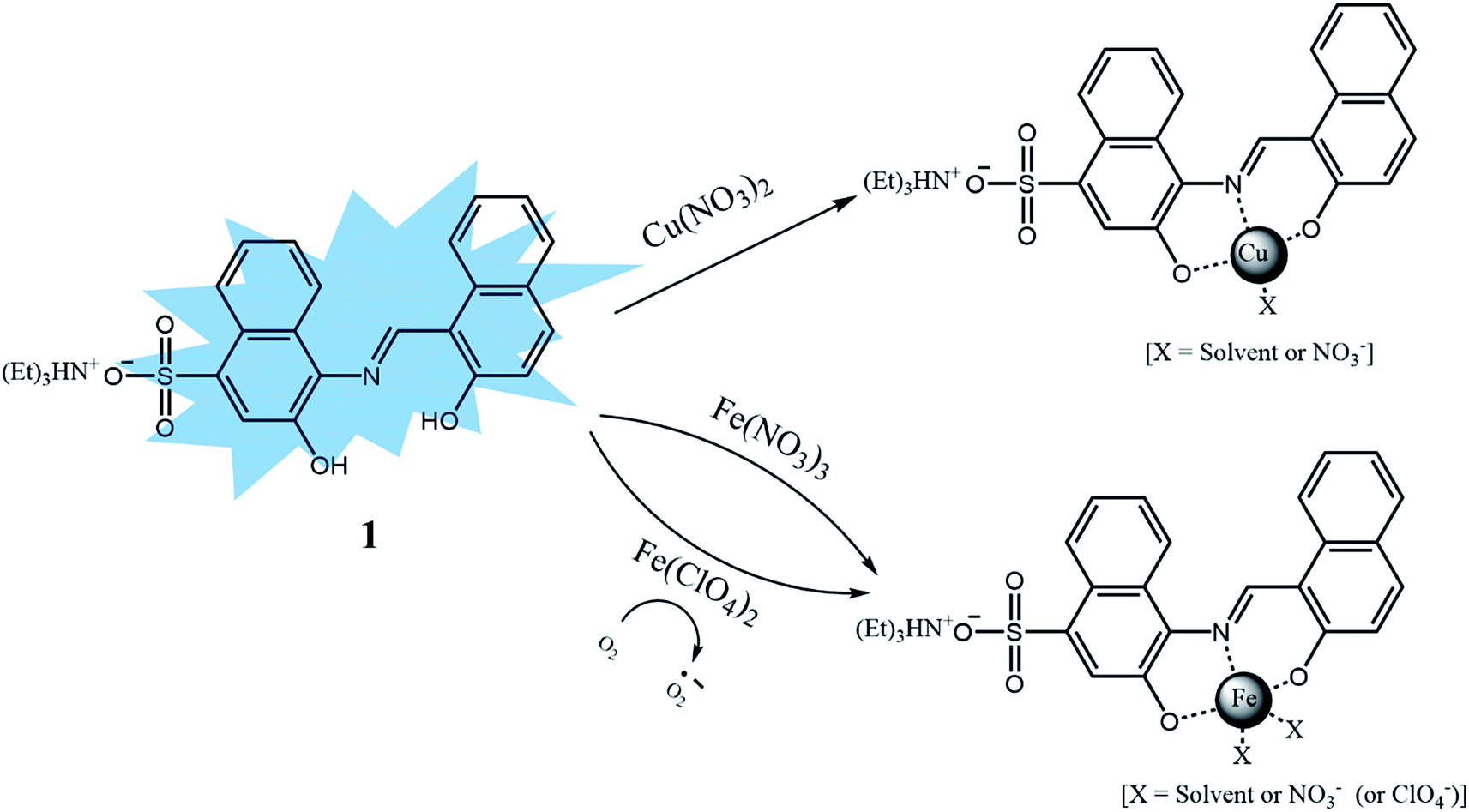 | ||
| Scheme 2 Proposed structures of 1–Cu2+ and 1–Fe2+/3+ complexes. | ||
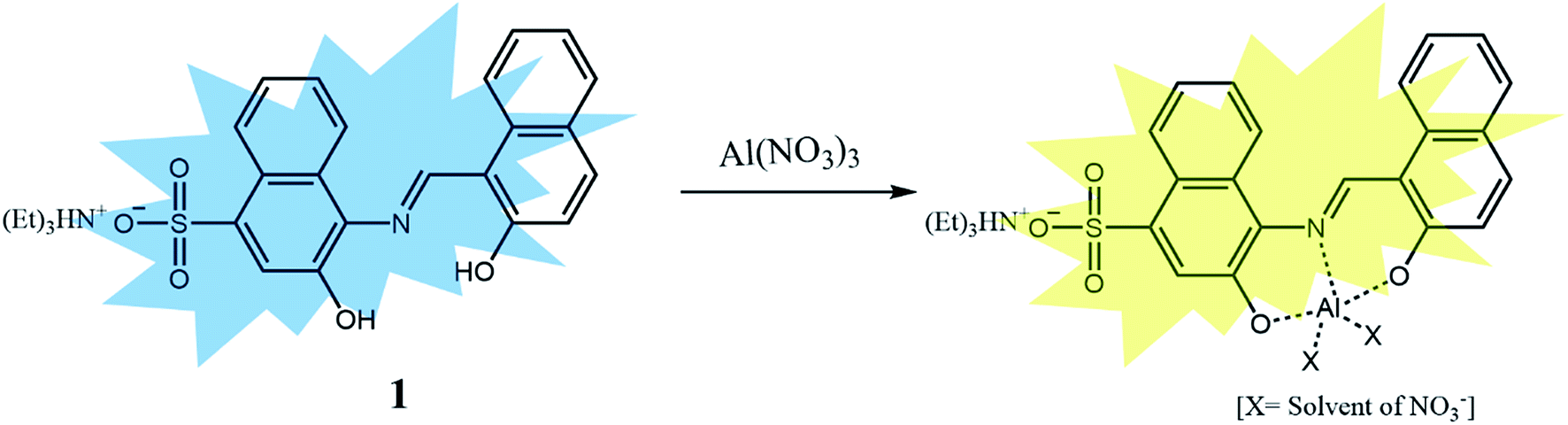 | ||
| Scheme 3 Proposed structure of 1–Al3+ complex. | ||
4. Conclusion
In this study, the sulfonate-based chemosensor 1 was developed for the detection of Cu2+ and Fe2+/3+ by fluorescence quenching and Al3+ by fluorescence change in a near-perfect aqueous solution. Sensor 1 enabled analysis of Cu2+ and Fe3+ with quite low detection limits (1.25 μM for Cu2+ and 3.96 μM for Fe3+), which were much lower than recommended values (31.5 μM for Cu2+ and 5.37 μM for Fe3+) of WHO and EPA. In addition, pyrophosphate could be used to distinguish Fe3+ ion from Cu2+ ion. Moreover, 1 showed a highly selective fluorescence emission change toward Al3+ over other metal ions including Ga3+ and In3+. Importantly, sensor 1 could be used to quantify Al3+ in living cells and real water samples. Furthermore, the sensing mechanisms of 1 for Cu2+ and Al3+ were explained by theoretical calculations, respectively. On the basis of the results, we believe that sensor 1 will offer an important guidance to the development of multiple target sensors by a fluorescent sensing method.Acknowledgements
Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2014R1A2A1A11051794 and NRF-2016M3D3A1A01913239 for the Korea C1 Gas Refinery R&D Center) are gratefully acknowledged. This work was also supported by Nine Bridges Program Research Fund (1.170051.01) of UNIST (Ulsan National Institute of Science & Technology) (to M. H. L.).References
- S. Bothra, Y. Upadhyay, R. Kumar and S. K. Sahoo, Spectrochim. Acta, Part A, 2017, 174, 1–6 CrossRef CAS PubMed.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS PubMed.
- Z. Xu, J. Pan, D. R. Spring, J. Cui and J. Yoon, Tetrahedron, 2010, 66, 1678–1683 CrossRef CAS.
- H. Zhang, L. Feng, Y. Jiang, Y.-T. Wong, Y. He, G. Zheng, J. He, Y. Tan, H. Sun and D. Ho, Biosens. Bioelectron., 2017, 94, 24–29 CrossRef CAS PubMed.
- D. Maity and T. Govindaraju, Chem.–Eur. J., 2011, 17, 1410–1414 CrossRef CAS PubMed.
- D. Maity, A. K. Manna, D. Karthigeyan, T. K. Kundu, S. K. Pati and T. Govindaraju, Chem.–Eur. J., 2011, 17, 11152–11161 CrossRef CAS PubMed.
- Q. Liu, R. Huo, D. Wei, X. Zhao, Z. Zhao and K. Cai, Dyes Pigm., 2017, 139, 1–6 CrossRef CAS.
- D. J. Waggoner, T. B. Bartnikas and J. D. Gitlin, Neurobiol. Dis., 1999, 6, 221–230 CrossRef CAS PubMed.
- C. Deraeve, C. Boldron, A. Maraval, H. Mazarguil, H. Gornitzka, L. Vendier, M. Pitiie and B. Meunier, Chem.–Eur. J., 2008, 14, 682–696 CrossRef CAS PubMed.
- S. Montes, S. Rivera-Mancia, A. Diaz-Ruiz, L. Tristan-Lopez and C. Rios, Oxid. Med. Cell. Longevity, 2014, 2014, 147251 Search PubMed.
- J. F. Zhang, Y. Zhou, J. Yoon, Y. Kim, S. J. Kim and J. S. Kim, Org. Lett., 2010, 12, 3852–3855 CrossRef CAS PubMed.
- J. Y. Noh, G. J. Park, Y. J. Na, H. Y. Jo, S. A. Lee and C. Kim, Dalton Trans., 2014, 43, 5652–5656 RSC.
- X. Xue, H. Fang, H. Chen, C. Zhang, C. Zhu, Y. Bai, W. He and Z. Guo, Dyes Pigm., 2016, 130, 116–121 CrossRef CAS.
- D. Duy, G. Wanner, A. R. Meda, N. von Wirén, J. Soll and K. Philippar, Plant Cell, 2007, 19, 986–1006 CrossRef CAS PubMed.
- M. W. Hentze, M. U. Muckenthaler and N. C. Andrews, Cell, 2004, 117, 285–297 CrossRef CAS PubMed.
- K. B. Kim, H. Kim, E. J. Song, S. Kim, I. S. Noh and C. Kim, Dalton Trans., 2013, 42, 16569–16577 RSC.
- F. Ge, H. Ye, H. Zhang and B.-X. Zhao, Dyes Pigm., 2013, 99, 661–665 CrossRef CAS.
- Z. Li, Y. Zhou, K. Yin, Z. Yu, Y. Li and J. Ren, Dyes Pigm., 2014, 105, 7–11 CrossRef CAS.
- P. T. Lieu, M. Heiskala, P. A. Peterson and Y. Yang, Mol. Aspects Med., 2001, 22, 1–87 CrossRef CAS PubMed.
- D. Maity and T. Govindaraju, Inorg. Chem., 2010, 49, 7229–7231 CrossRef CAS PubMed.
- D. Maity and T. Govindaraju, Eur. J. Inorg. Chem., 2011, 2011, 5479–5485 CrossRef CAS.
- S. Pu, C. Zhang, C. Fan and G. Liu, Dyes Pigm., 2016, 129, 24–33 CrossRef CAS.
- B. R. Stephens and J. S. Jolliff, Diet and Nutrition in Dementia and Cognitive Decline, 2014, pp. 553–562 Search PubMed.
- P. Nayak, Environ. Res., 2002, 89, 101–115 CrossRef CAS PubMed.
- D. Sarkar, P. Ghosh, S. Gharami, T. K. Mondal and N. Murmu, Sens. Actuators, B, 2017, 242, 338–346 CrossRef CAS.
- S. Y. Li, D. B. Zhang, J. Y. Wang, R. M. Lu, C. H. Zheng and S. Z. Pu, Sens. Actuators, B, 2017, 245, 263–267 CrossRef CAS.
- J. Jun Lee, G. Jin Park, Y. Sung Kim, S. Young Lee, H. Ji Lee, I. S. Noh and C. Kim, Biosens. Bioelectron., 2015, 69, 226–229 CrossRef CAS PubMed.
- Y. W. Choi, G. J. Park, Y. J. Na, H. Y. Jo, S. A. Lee, G. R. You and C. Kim, Sens. Actuators, B, 2014, 194, 343–352 CrossRef CAS.
- S. A. Lee, G. R. You, Y. W. Choi, H. Y. Jo, A. R. Kim, I. Noh, S.-J. Kim, Y. Kim, C. Kim, X. Bu and D. Das, Dalton Trans., 2014, 43, 6650–6659 RSC.
- Y. K. Jang, U. C. Nam, H. L. Kwon, I. H. Hwang and C. Kim, Dyes Pigm., 2013, 99, 6–13 CrossRef CAS.
- E. J. Song, J. Kang, G. R. You, G. J. Park, Y. Kim, S.-J. Kim, C. Kim and R. G. Harrison, Dalton Trans., 2013, 42, 15514–15520 RSC.
- H. J. Jung, N. Singh, D. Y. Lee and D. O. Jang, Tetrahedron Lett., 2010, 51, 3962–3965 CrossRef CAS.
- H. Yang, H. Song, Y. Zhu and S. Yang, Tetrahedron Lett., 2012, 53, 2026–2029 CrossRef CAS.
- V. K. Gupta, A. K. Singh and B. Gupta, Anal. Chim. Acta, 2006, 575, 198–204 CrossRef CAS PubMed.
- H. A. Tajmir-Riahi, Polyhedron, 1983, 2, 723–726 CrossRef CAS.
- B.-L. Wang, C. Jiang, K. Li, Y.-H. Liu, Y. Xie and X.-Q. Yu, Analyst, 2015, 140, 4608–4615 RSC.
- A. Atahan and S. Durmus, Spectrochim. Acta, Part A, 2015, 144, 61–67 CrossRef CAS PubMed.
- B. Shi, P. Zhang, T. Wei, H. Yao, Q. Lin and Y. Zhang, Chem. Commun., 2013, 49, 7812–7814 RSC.
- A. Syamal and M. M. Singh, React. Polym., 1993, 21, 149–158 CrossRef CAS.
- F. Y. Wu, H. Zhang, M. Xiao and B. X. Han, Spectrochim. Acta, Part A, 2013, 109, 221–225 CrossRef CAS PubMed.
- S. Goswami, A. Manna, S. Paul, C. Kheng Quah and H.-K. Fun, Chem. Commun., 2013, 49, 11656–11658 RSC.
- V. K. Gupta, A. K. Singh and L. K. Kumawat, Sens. Actuators, B, 2014, 195, 98–108 CrossRef CAS.
- K. Tiwari, M. Mishra, V. P. Singh, S. Lohar, D. Karak, B. Sarkar, S. K. Mukhopadhyay, A. K. Mukherjee, D. Das and L. Tian, RSC Adv., 2013, 3, 12124–12132 RSC.
- Z. Liu, Y. Li, Y. Ding, Z. Yang, B. Wang, Y. Li, T. Li, W. Luo, W. Zhu, J. Xie and C. Wang, Sens. Actuators, B, 2014, 197, 200–205 CrossRef CAS.
- B. Liu, P. Wang, J. Chai, X. Hu, T. Gao, J. Chao, T. Chen and B. Yang, Spectrochim. Acta, Part A, 2016, 168, 98–103 CrossRef CAS PubMed.
- H.-Y. Lin, T.-Y. Chen, C.-K. Liu and A.-T. Wu, Luminescence, 2016, 31, 236–240 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- N. M. O’boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- R. M. Yucel, Y. He and A. M. Zaslavsky, Stat. Med., 2011, 30, 3447–3460 CrossRef PubMed.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- Y. Hu, J. Zhang, Y.-Z. Lv, X.-H. Huang and S. Hu, Spectrochim. Acta, Part A, 2016, 157, 164–169 CrossRef CAS PubMed.
- S. Samanta, U. Manna, T. Ray and G. Das, Dalton Trans., 2015, 44, 18902–18910 RSC.
- G. R. You, G. J. Park, J. J. Lee and C. Kim, Dalton Trans., 2015, 44, 9120–9129 RSC.
- T. G. Jo, Y. J. Na, J. J. Lee, M. M. Lee, S. Y. Lee and C. Kim, Sens. Actuators, B, 2015, 211, 498–506 CrossRef CAS.
- L. Tang, J. Zhao, M. Cai, P. Zhou, K. Zhong, S. Hou and Y. Bian, Tetrahedron Lett., 2013, 54, 6105–6109 CrossRef CAS.
- S. Goswami, S. Maity, A. K. Das and A. C. Maity, Tetrahedron Lett., 2013, 54, 6631–6634 CrossRef CAS.
- Y. K. Tsui, S. Devaraj and Y. P. Yen, Sens. Actuators, B, 2012, 161, 510–519 CrossRef CAS.
- N. Graham, Urban Water, 1999, 1, 183 Search PubMed.
- R. M. Harrison, D. P. H. Laxen and S. J. Wilson, Environ. Sci. Technol., 1981, 15, 1378–1383 CrossRef CAS.
- M. A. R. Meier and U. S. Schubert, Chem. Commun., 2005, 4610–4612 RSC.
- G. R. You, G. J. Park, S. A. Lee, K. Y. Ryu and C. Kim, Sens. Actuators, B, 2015, 215, 188–195 CrossRef CAS.
- G. J. Park, Y. J. Na, H. Y. Jo, S. A. Lee and C. Kim, Dalton Trans., 2014, 43, 6618–6622 RSC.
- P. Job, Ann. Chim, 1928, 9, 113–203 CAS.
- L. Wang, H. Li and D. Cao, Sens. Actuators, B, 2013, 181, 749–755 CrossRef CAS.
- H. S. Jung, P. S. Kwon, J. W. Lee, J. Il Kim, C. S. Hong, J. W. Kim, S. Yan, J. Y. Lee, J. H. Lee, T. Joo and J. S. Kim, J. Am. Chem. Soc., 2009, 131, 2008–2012 CrossRef CAS PubMed.
- O. García-Beltrán, B. K. Cassels, C. Pérez, N. Mena, M. T. Núñez, N. P. Martínez, P. Pavez and M. E. Aliaga, Sensors, 2014, 14, 1358–1371 CrossRef PubMed.
- R. Patil, A. Moirangthem, R. Butcher, N. Singh, A. Basu, K. Tayade, U. Fegade, D. Hundiwale and A. Kuwar, Dalton Trans., 2014, 43, 2895–2899 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional experimental data. See DOI: 10.1039/c7ra05565j |
| This journal is © The Royal Society of Chemistry 2017 |