
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly oxygenated caryophyllene-type and drimane-type sesquiterpenes from Pestalotiopsis adusta, an endophytic fungus of Sinopodophyllum hexandrum†
Jian Xiao‡
 *a,
Libin Lin‡a,
Jiayao Hua,
Furong Jiaoa,
Dongzhu Duana,
Qiang Zhangb,
Haoyu Tangc,
Jinming Gaob,
Le Wanga and
Xiaoling Wang*a
*a,
Libin Lin‡a,
Jiayao Hua,
Furong Jiaoa,
Dongzhu Duana,
Qiang Zhangb,
Haoyu Tangc,
Jinming Gaob,
Le Wanga and
Xiaoling Wang*a
aShaanxi Key Laboratory of Phytochemistry, College of Chemistry and Chemical Engineering, Baoji University of Arts and Sciences, Baoji 721013, Shaanxi, P. R. China. E-mail: xjxs163@126.com; xlwangwang@163.com; Tel: +86-917-3566233
bShaanxi Key Laboratory of Natural Products & Chemical Biology, College of Chemistry & Pharmacy, Northwest A&F University, Yangling 712100, Shaanxi, P. R. China
cState Key Laboratory of Bioorganic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, P. R. China
First published on 5th June 2017
Abstract
Four new highly oxidized caryophyllene-type sesquiterpenes (1–4), one new caryophyllene-type natural product (5), and two new drimane-type sesquiterpenes (6, 7), along with eight known compounds have been isolated from an endophytic fungus Pestalotiopsis adusta. Their structures were elucidated on the basis of HRESIMS, 1D and 2D NMR spectroscopic data analyses. The absolute configurations for 1 and 5 were determined by single-crystal X-ray crystallographic analysis using Cu Kα radiation. The absolute configuration of 6 was determined by CD spectrum associated with TD-DFT calculation of its benzoylated derivative 6a. The in vitro bioassay revealed that 8 and 9 presented cytotoxicity against cancer cell lines A549, HeLa, and SMMC-7721, in which 8 showed comparable activity (IC50 = 28.3 μM) to the positive control etoposide (IC50 = 23.2 μM) against SMMC-7721.
Introduction
Plant endophytic fungi are well known for their potential to produce abundant secondary metabolites and contribute greatly to the discovery of new medicines.1,2 The fungal genus of Pestalotiopsis is one of the most commonly found endophytes on the planet, 234 species of which are listed in Index Fungorum.3 Since a broad-spectrum antitumor drug taxol identified from P. microspora, a plant endophyte of Taxus wallachiana, exploring for the bioactive secondary metabolites from endophytes of the Pestalotiopsis genus of different plants has attracted much attention.4 Species of Pestalotiopsis are proved to be a rich source for natural products, and the chemical investigations of Pestalotiopsis spp. have afforded over 300 bioactive secondary metabolites5,6 including terpenoids, alkaloids, quinonoids, phenylpropanoids and lactones with diverse bioactivities such as antitumor, antibacterial, acetylcholinesterase inhibitory, antioxidant and anti-HIV.7–10β-Caryophyllene, a sesquiterpene characteristic of a strained four-membered ring and a flexible nine-membered macrocyclic moiety, exists popularly in the essential oils from medicinal plants. It acts as a key precursor in nature to form tricyclic and tetracyclic highly oxygenated caryophyllene-type sesquiterpenes by transannular rearrangements.11 Its highly oxygenated derivatives were proved to possess significant bioactivities, including immunosuppressive activity,7 affecting growth and proliferation of numerous cancer cells,12 antibacterial activity,13 and anti-inflammatory activity.14 Although β-caryophyllene is widely distributed, its highly oxygenated derivatives are not common.7 Until now, only a few highly oxygenated caryophyllene-type sesquiterpenes have been reported from terrestrial plant endophyte, including three unprecedented novel compounds cytosporinols A–C.15–24 The highly oxygenated caryophyllene-type sesquiterpenes also attracted the interests of synthetic chemists due to their structural characteristics and special bioactivities.25
In our continuous screening for bioactive natural products from plant endophytes,26–28 we investigated an endophytic fungus Pestalotiopsis adusta collected from the stem bark of wild rare medicinal plant Sinopodophyllum hexandrum (Royle) Ying in Qinling Mountains. The chemical investigation on the solid culture of P. adusta led to the isolation of four new highly oxidized caryophyllene-type sesquiterpenes pestalotiopsins D–G (1–4), two new drimane-type sesquiterpenes pestalotiophol A and B (6, 7), along with nine known compounds pestalotiopsin H (5),25 pestalotiopsin C (8),29 pestalotiopsin A (9),29 pestalotiopsin B (10),29 punctaporonin H (11),30 punctatin A (12),31 punctaporonin B (13),30 2α-hydroxydimeninol (14),32 and 7-hydroxy-5-methoxy-4,6-dimethylphthalide (15),33 of which 5 was a new natural product (Fig. 1). Details of the isolation, structure elucidation, and cytotoxicity of these secondary metabolites are reported herein.
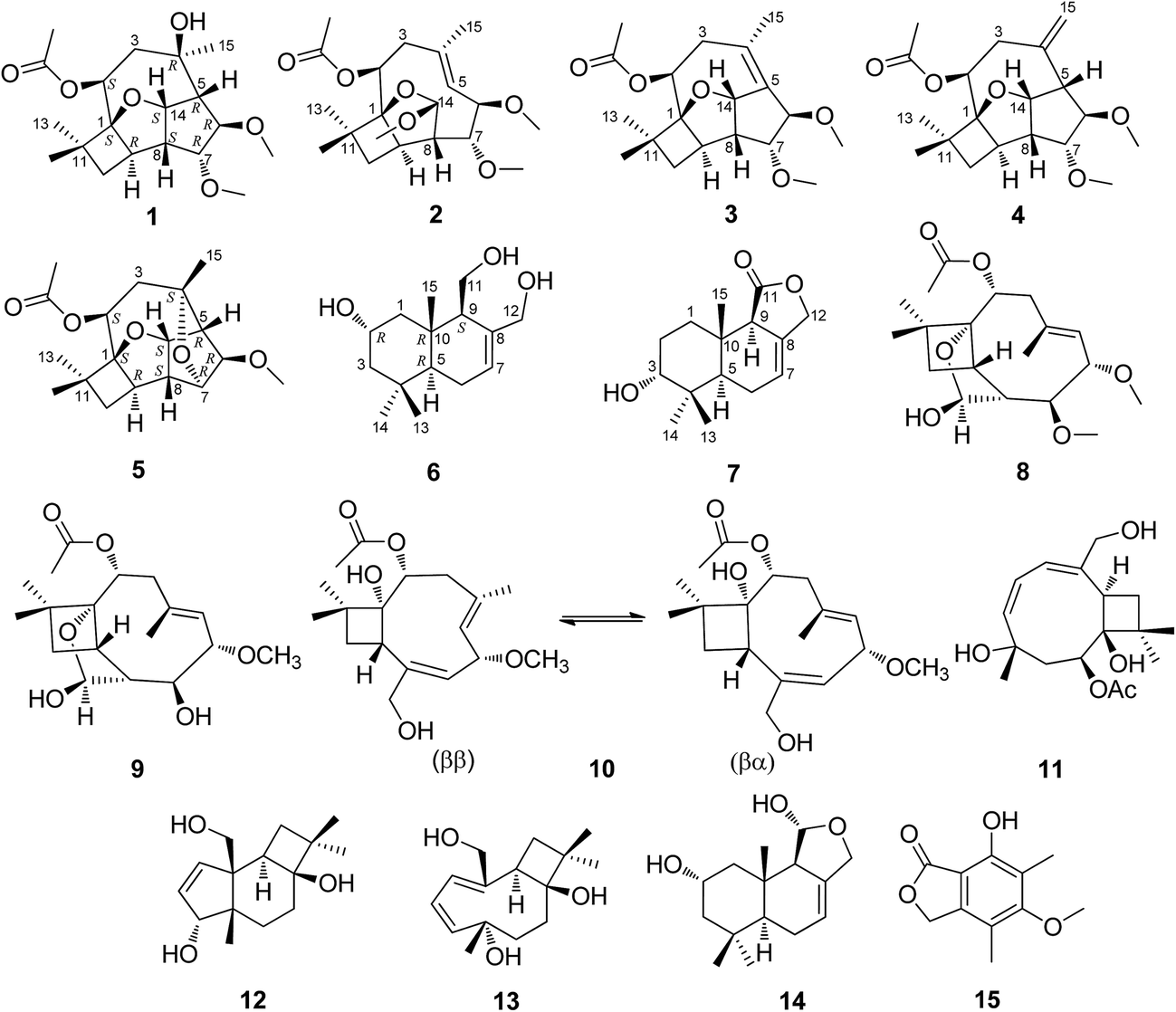 | ||
| Fig. 1 Structures of compounds 1–15. | ||
Results and discussions
Pestalotiopsin D (1) was obtained as a white amorphous powder, its molecular formula was deduced to be C19H30O6 with five degrees of unsaturation based on HRESIMS at m/z 377.1931 ([M + Na]+, calcd for 377.1940). The 1H NMR (Table 1) spectrum revealed that 1 had two methoxyls [δH 3.36 (3H, s, OCH3-6), 3.27 (3H, s, OCH3-7)], four tertiary methyls [δH 1.06 (3H, s, H3-12), 1.01 (3H, s, H3-13), 1.13 (3H, s, H3-15), 2.05 (3H, s, OAc)], and four oxygenated methines [δH 5.25, (1H, dd, J = 4.1/2.6 Hz, H-2), 3.24 (1H, t, J = 6.6 Hz, H-6), 3.66 (1H, t, J = 6.7 Hz, H-7), 5.02 (1H, dd, J = 9.9/6.9 Hz, H-14)]. The 13C NMR spectrum (Table 2), together with DEPT135 and HSQC, revealed the existence of nineteen carbon atoms including one acetoxyl (for its methyl and carbonyl), two methoxyls, three tertiary methyls, two methylenes, seven methines (four oxygenated) and three quaternary carbons. The proton resonances were well assigned to relevant carbon atoms by HSQC. The NMR signals of protons and carbons were characteristic for caryophyllene-type sesquiterpene.29 One acetoxy group that accounted for one of the five indices of unsaturation implied that 1 had a tetracyclic ring system. The 1H–1H COSY correlations established two isolated systems (Fig. 2). 1H–1H COSY correlations of H-14 with H-5, H-5 with H-6, H-6 with H-7, H-7 with H-8 and H-8 with H-14 constructed a five-membered carbon ring of C-5/C-6/C-7/C-8/C-14. 1H–1H COSY correlations of H-14 with H-8 and H-8 with H-9, HMBC correlations of H-8 with C-9/C-1 and H-14 with C-1, and consideration of the downfield chemical shifts of C-1 and C-14, constructed a five membered tetrahydrofuran ring of C-8/C-9/C-1/O/C-14. 1H–1H COSY correlation of H-9 with H-10, HMBC correlations of H-9 with C-1/C-10/C-11, and both H3-12 and H3-13 with C-1/C-10/C-11, produced a four membered carbon ring of C-1/C-9/C-10/C-11 with two tertiary methyls attached. The last ring, the nine-membered macrocyclic moiety for caryophyllene-type sesquiterpene, was deduced by 1H–1H COSY correlation of H-2 with H-3, HMBC correlations of H-9 with C-2 and H-3 with C-4/C-5, and some of the above 1H–1H COSY and HMBC signals. Subsequently, an acetoxyl was attached to C-2 by H-2 correlating with carbonyl at δC 170.4 in HMBC. A methyl was attached to C-4 by H3-15 correlating with C-3/C-4/C-5. The HMBC correlations of methoxyl at δH 3.36 with C-6 and methoxyl at δH 3.27 with C-7 located two methoxyls at C-6 and C-7, respectively. Taking into account the molecular formula and the chemical shift of C-4, there should be a hydroxyl at C-4. The relative configuration of 1 was determined by ROESY experiment (Fig. 2). The ROESY correlations of H-2 with H-9/H3-13, H-9 with H-6, and H-6 with H3-15 suggested that H-2, H-6, H-9, H3-13 and H3-15 had the relative configuration of α. Whereas, the ROESY correlations of H-5 with H-14/H-7, H-14 with H-8, H-7 with CH3O-6 revealed that H-5, H-7, H-8 and H-14 had the relative configuration of β (Fig. 2). The absolute configuration of 1 was determined by single crystal X-ray diffraction, a suitable low Flack parameter 1.1(10) effectively suggested 1 as 1S, 2S, 4R, 5R, 6R, 7R, 8S, 9R, 14S (Fig. 3). Therefore, 1 was elucidated and named as pestalotiopsin D.| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| 2 | 5.25, dd (4.1, 2.6) | 5.22, dd (10.7, 5.6) | 5.21, dd (10.6, 5.2) | 5.17, s | 5.18, dd (7.1, 6.1) |
| 3a | 1.99, m | 2.44, t (10.7) | 1.95, t (6.7) | 2.49, m | 1.82, m |
| 3b | 2.52, m | 3.15, d (11.7) | 2.27, m | ||
| 5 | 2.28, dd (9.9, 6.9) | 5.14, d (11.8) | 3.04, d (9.5) | 2.26, m | |
| 6 | 3.24, t (6.6) | 3.94, dd (11.8, 5.1) | 3.91, s | 3.63, s | 4.25, t (1.8) |
| 7 | 3.66, t (6.7) | 3.52, dd (5.2, 2.8) | 3.63, d (7.6) | 3.65, d (6.6) | 3.97, t (2.5) |
| 8 | 2.92, td (6.9, 3.7) | 2.50, m | 2.57, td (7.0, 3.6) | 2.92, s | 2.57, dd (8.3, 2.7) |
| 9 | 2.58, ddd (9.5, 5.8, 3.7) | 2.53, m | 2.84, m | 2.65, s | 2.72, t (8.6) |
| 10a | 1.40, dd (11.9, 5.8) | 1.53, dd (12.1, 5.8) | 1.49, dd (11.5, 6.5) | 1.42, dd (11.4, 6.3) | 1.59, m |
| 10b | 2.01, m | 1.94, dd (12.1, 9.7) | 1.98, m | 1.97, t (10.6) | 1.72, dd (10.7, 5.8) |
| 12 | 1.06, s | 1.09, s | 1.11, s | 1.04, s | 1.07, s |
| 13 | 1.01, s | 1.01, s | 1.09, s | 1.03, s | 1.11, s |
| 14 | 5.02, dd (9.9, 6.9) | 5.28, d (2.4) | 5.25, d (6.3) | 5.03, t (8.4) | 5.22, m |
| 15a | 1.13, s | 1.87, s | 1.79, s | 4.80, s | 1.19, s |
| 15b | 4.95, s | ||||
| 2-OAc | 2.05, s | 2.05, s | 2.01, s | 2.08, s | 2.01, s |
| 6-OCH3 | 3.36, s | 3.27, s | 3.20, s | 3.32, s | 3.28, s |
| 7-OCH3 | 3.27, s | 3.38, s | 3.25, s | 3.31, s | |
| 14-OCH3 | 3.50, s |
| Position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 94.0, C | 98.0, C | 92.2, C | 94.0, C | 94.6, C |
| 2 | 74.0, CH | 73.7, CH | 78.5, CH | 72.1, CH | 75.9, CH |
| 3 | 37.9, CH2 | 41.2, CH2 | 34.4, CH2 | 34.5, CH2 | 41.4, CH2 |
| 4 | 74.6, C | 136.3, C | 129.7, C | 142.5, C | 79.8, C |
| 5 | 56.8, CH | 125.4, CH | 133.8, C | 56.9, CH | 51.7, CH |
| 6 | 87.4, CH | 82.0, CH | 81.9, CH | 89.2, CH | 87.9, CH |
| 7 | 85.9, CH | 87.5, CH | 83.5, CH | 85.4, CH | 77.0, CH |
| 8 | 56.2, CH | 61.1, CH | 54.2, CH | 56.1, CH | 49.9, CH |
| 9 | 34.3, CH | 38.3, CH | 39.5, CH | 34.4, CH | 38.6, CH |
| 10 | 40.2, CH2 | 42.5, CH2 | 42.1, CH2 | 40.3, CH2 | 37.5, CH2 |
| 11 | 37.5, C | 39.8, C | 38.1, C | 37.1, C | 38.6, C |
| 12 | 24.5, CH3 | 24.1, CH3 | 23.9, CH3 | 24.1, CH3 | 23.1, CH3 |
| 13 | 26.1, CH3 | 27.4, CH3 | 26.8, CH3 | 26.2, CH3 | 25.1, CH3 |
| 14 | 84.5, CH | 115.1, CH | 89.0, CH | 85.5, CH | 88.8, CH |
| 15 | 29.4, CH3 | 17.5, CH3 | 22.1, CH3 | 118.3, CH2 | 31.6, CH3 |
| 2-OAc | 170.4, C | 170.6, C | 170.3, C | 170.7, C | 170.4, C |
| 21.6, CH3 | 21.7, CH3 | 21.7, CH3 | 21.5, CH3 | 21.5, CH3 | |
| 6-OCH3 | 57.7, CH3 | 56.1, CH3 | 55.3, CH3 | 57.5, CH3 | 57.5, CH3 |
| 7-OCH3 | 57.4, CH3 | 57.7, CH3 | 57.2, CH3 | 57.8, CH3 | |
| 14-OCH3 | 56.3, CH3 |
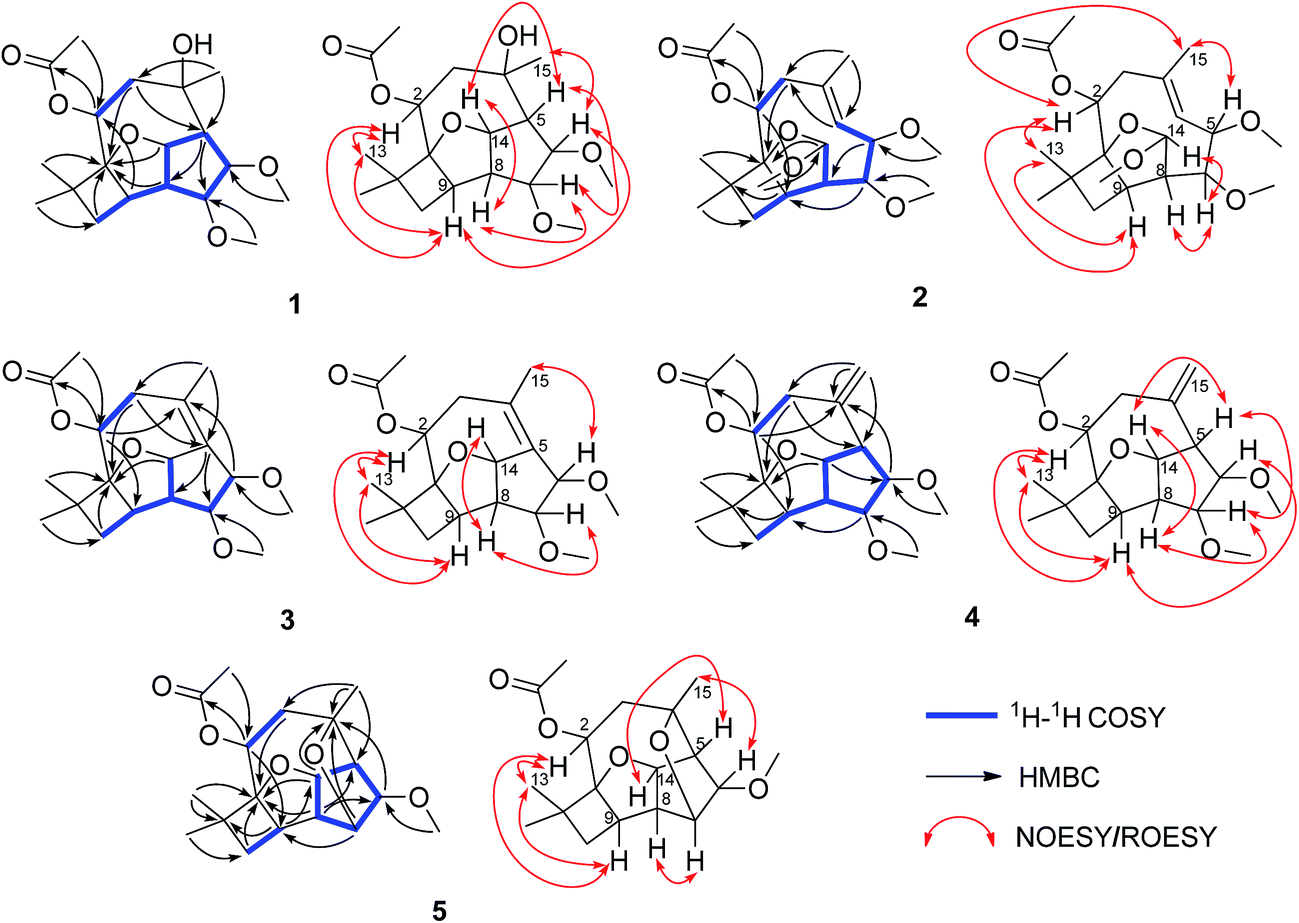 | ||
| Fig. 2 The 1H–1H COSY, key HMBC and NOESY/ROESY correlations of compounds 1–5. | ||
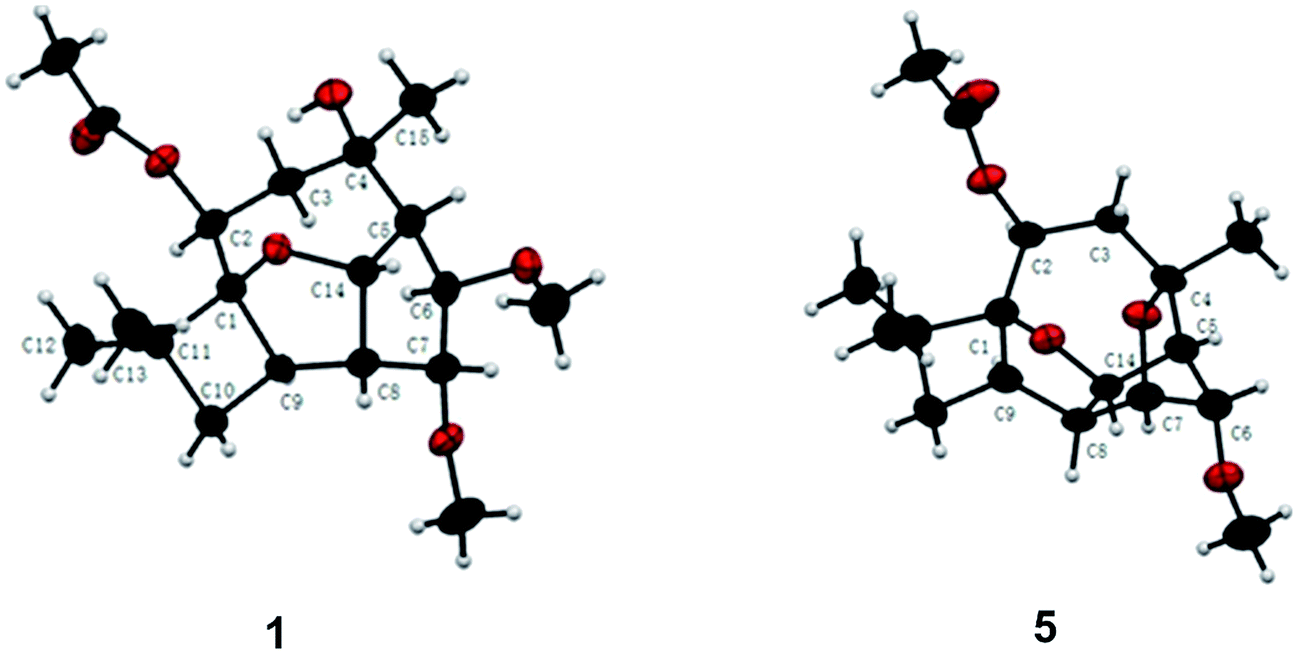 | ||
| Fig. 3 The ORTEP diagrams of compounds 1 and 5. | ||
Pestalotiopsin E (2), obtained as a colorless oil, had a molecular formula of C20H32O6 with five degrees of unsaturation based on its HRESIMS analysis (m/z 391.2090 [M + Na]+, calcd for 391.2096). The 1H NMR (Table 1) spectrum revealed that 2 had three methoxyls [δH 3.27 (3H, s, OCH3-6), 3.38 (3H, s, OCH3-7), 3.50 (3H, s, OCH3-14)], four tertiary methyls [δH 1.09 (3H, s, H3-12), 1.01 (3H, s, H3-13), 1.87 (3H, s, H3-15), 2.05 (3H, s, OAc)], one olefinic methine [δH 5.14 (1H, d, J = 11.8 Hz, H-5)] and four oxygenated methines [δH 5.22 (1H, dd, J = 10.7/5.6 Hz, H-2), 3.94 (1H, dd, J = 11.8/5.1 Hz, H-6), 3.52 (1H, dd, J = 5.2/2.8 Hz, H-7), 5.28 (1H, d, J = 2.4 Hz, H-14)]. The 13C NMR (Table 2), along with DEPT 135 and HSQC, revealed the existence of twenty carbon atoms including one acetoxyl (for its methyl and carbonyl), three methoxyls, three tertiary methyls, two methylenes, seven methines (one olefinic and four oxygenated), and three quaternary carbons (one olefinic). One acetoxyl and one double bond accounting for two of the five indices of unsaturation illustrated the presence of a tricyclic ring system. All the proton signals were well assigned to their relevant carbons by HSQC experiment. The 1H–1H COSY spectrum established two isolated systems (Fig. 2). Comparing the NMR data of 2 with those of 1, revealed that they possessed identical carbon skeleton, with the only difference for C-4 (δC 136.3 in 2, 74.6 in 1), C-5 and H-5 (δC 125.4 and δH 5.14 in 2, δC 56.8 and δH 2.28 in 1), and C-14 and H-14 (δC 115.1 and δH 5.28 in 2, δC 84.5 and δH 5.02 in 1). Clearly, there should be a double bond produced between C-4 and C-5 in 2. The much downfield chemical shift of C-14 indicated that it was di-oxygenated. The HMBC correlation of H-2 with carbonyl located the acetoxyl at C-2. The HMBC correlations of methoxyl at δH 3.27 with C-6, methoxyl at δH 3.38 with C-7, and methoxyl at δH 3.50 with C-14 located three methyls at C-6, C-7 and C-14, respectively. H3-12, H3-13, and H3-15 were also located by HMBC correlations (Fig. 2). ROESY correlations of H-2 with H3-13/H-9/H3-15, and H3-15 with H-6 suggested that H-2, H-6, H-9, H3-13 and H3-15 had the relative configuration of α. ROESY correlations of H-14 with H-7/H-8 revealed that H-7, H-8 and H-14 had the relative configuration of β. No observation of ROESY correlation between H-5 with H3-15 indicated a E-geometry for the double bond7 (Fig. 2). At last, 2 was elucidated and named as pestalotiopsin E.
Pestalotiopsin F (3), obtained as a colorless oil, had a molecular formula of C19H28O5 with six degrees of unsaturation based on its HRESIMS analysis (m/z 359.1834 [M + Na]+, calcd for 359.1834), which was 18 atomic mass units less than that of pestalotiopsin D (1). Comparing the NMR data of 3 (Tables 1 and 2) with those of 1 and on the basis of its 1H–1H COSY and HMBC (Fig. 2) revealed that they possessed identical carbocyclic cores. The difference was the one side oxygenated single bond between C-4 (δC 74.6) and C-5 (δC 56.8 and δH 2.88) in 1 transferred to a non-oxygenated double bond (δC 129.7 for C-4 and 133.8 for C-5) in 3, it was clear that 1 experienced the loss of a H2O, this was also supported by the molecular weight difference between 1 and 3. ROESY correlations of H-2 with H3-13/H-9 and H3-15 with H-6 but no correlation of H-6 with H-7 suggested that H-2, H-6, H-9, H3-13 and H-15 had the relative configuration of α. ROESY correlations of H-14 with H-7/H-8 revealed that H-7, H-8 and H-14 had the relative configuration of β (Fig. 2). Finally, 3 was elucidated and named as pestalotiopsin F.
Pestalotiopsin G (4), obtained as white power. Based on the HRESIMS analysis (m/z 359.1833 [M + Na]+, calcd for 359.1834) and the 13C NMR data, the molecular formula of was established as C19H28O5 with six degrees of unsaturation. The NMR data of 4 (Tables 1 and 2) revealed that it had the similar structure with 3, the largest variation being at C-5 (from δC 133.8 in 3 to δC 56.9 in 4) and C-15 (from δC 22.1 for a methyl in 3 to δC 118.3 for a terminal vinyl in 4), implying that the double bond between C-4 and C-5 in 3 fused to a terminal double bond between C-4 and C-15 in 4 (Fig. 2), this was also supported by HMBC correlations (Fig. 2). The NOESY experiment analysis (Fig. 2) indicated that H-2, H-9, H-6 and H3-13 had the relative configuration of α, whereas H-5, H-7, H-8 and H-14 had the relative configuration of β. Finally, 4 was elucidated and named as pestalotiopsin G.
Pestalotiopsin H (5) was isolated as colorless oil, its molecular formula was established as C18H26O5 with six degrees of unsaturation by the quasi-molecular ion at m/z 345.1674 [M + Na]+ (calcd for 345.1678) in the HRESIMS, which was 32 atomic mass units fewer than 1. The 1H and 13C NMR spectroscopic data (Tables 1 and 2) revealed 5 had a very similar structural feature to 1. The difference of the downfield shift for C-4 (from δC 74.6 in 1 to δC 79.8 in 5), the upfield shift for C-7 (from δC 85.9 in 1 to δC 77.0 in 5), and the downfield shift for H-7 (from δH 3.66 in 1 to δH 3.97 in 5) were observed. For 5, the HMBC (Fig. 2) correlation between H-7 and C-4, and 32 atomic mass units less than 1, indicated that 1 experienced the loss of a CH3OH molecule to produce 5. The relative configuration of 5 was assigned by NOESY experiment (Fig. 2). The structure and spectral data of 5 were consistent with a synthetic intermediate in the literature,25 and therefore it was deduced as a new natural product and named as pestalotiopsin H. Its absolute configuration (1S, 2S, 4S, 5R, 6R, 7R, 8S, 9R, 14S) was identified by single-crystal X-ray crystallographic analysis with a suitable low Flack parameter 0.02(11) (Fig. 3).
Pestalotiophol A (6), obtained as a colorless oil, had a molecular formula of C15H26O3 with three degrees of unsaturation based on its HRESIMS analysis (m/z 277.1779 [M + Na]+, calcd for 277.1779) and 13C NMR data. The 1H NMR (Table 3) spectrum revealed that 6 had three tertiary methyls [δH 0.95 (3H, s, H3-14), 0.96 (3H, s, H3-13), 0.86 (3H, s, H3-15)], two mono-oxygenated methylenes [δH 3.66 (1H, dd, J = 11.1/7.3 Hz, H-11α), 3.88 (1H, dd, J = 11.1/2.8 Hz, H-11β); 4.26 (1H, d, J = 12.6 Hz, H-12α), 3.98 (1H, d, J = 12.7 Hz, H-12β)], one olefinic proton [δH 5.81 (1H, d, J = 4.6 Hz, H-7)], one mono-oxygenated methine [δH 3.82 (1H, ddd, J = 11.6/7.7/3.8 Hz, H-2α)], and two non-oxygenated methines [δH 2.12 (1H, dd, J = 11.2/4.4 Hz, H-9α), 1.22 (1H, dd, J = 12.3/4.5 Hz, H-5α)]. The 13C NMR (Table 3) spectrum showed fifteen carbon signals. Combined with the DEPT 135 and HSQC experiments, these carbons were classified into three methyls, five methylenes (two mono-oxygenated), four methines (one mono-oxygenated and one olefinic), and three quaternary carbons (one olefinic). The proton resonances were well assigned to relevant carbon atoms by HSQC. The 1H and 13C NMR signals were characteristic for the structures of drimane-type sesquiterpenes with 6/6-fused ring system.32 The 1H–1H COSY correlations established two isolated systems (Fig. 4). The downfield chemical shifts for C-2 at δC 65.3, C-11 at δC 61.2, and C-12 at δC 66.8 implied that there should be three hydroxyls attached to C-2, C-13 and C-14, respectively, this result was also supported by the HRESIMS data. NOESY signals (Fig. 4) of H3-15 with H2-11/H3-13 and H3-13 with H-2 indicated that H3-15, H2-11, H3-13 and H-2 had the relative configuration of β. NOESY signals (Fig. 4) of H-9 with H-5/H3-14 indicated that H-9, H-5 and H3-14 had the relative configuration of α. To determine the absolute configuration of 6, its fully benzoylated derivative 6a (Fig. 4) was prepared. The absolute configuration of 6a was determined to be 2R, 5R, 9S and 10R by its CD spectrum (Fig. 5) associated with TD-DFT calculation. So, the absolute configuration of 6 was 2R, 5R, 9S and 10R, same to 6a. Thus, 6 was elucidated and named as pestalotiophol A.
| Position | 6 | 7 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1a | 49.5, CH2 | 1.07, dd (16.1, 7.5) | 30.5, CH2 | 1.77, m |
| 1b | 2.28, m | 2.18, dt (13.5, 3.3) | ||
| 2a | 65.3, CH | 3.82, ddd (11.6, 7.7, 3.8) | 24.9, CH2 | 1.61, m |
| 2b | 1.90, m | |||
| 3a | 51.7, CH2 | 1.16, t (12.0) | 75.8, CH | 3.48, m |
| 3b | 1.74, m | |||
| 4 | 35.4, C | 37.3, C | ||
| 5 | 50.5, CH | 1.22, dd (12.3, 4.5) | 42.9, CH | 1.82, m |
| 6a | 24.4, CH2 | 1.92, m | 23.0, CH2 | 2.03, m |
| 6b | 2.12, dd (11.2, 4.4) | |||
| 7 | 126.4, CH | 5.81, d (4.6) | 121.3, CH | 5.72, s |
| 8 | 138.4, C | 129.9, C | ||
| 9 | 55.9, CH | 2.12, dd (11.2, 4.4) | 53.5, CH | 2.85, s |
| 10 | 38.5, C | 34.0, C | ||
| 11a | 61.2, CH2 | 3.66, dd (11.1, 7.3) | 175.6, C | |
| 11b | 3.88, dd (11.1, 2.8); | |||
| 12a | 66.8, CH2 | 3.98, d (12.7) | 70.0, CH2 | 4.63, m |
| 12b | 4.26, d (12.6) | |||
| 13 | 23.2, CH3 | 0.96, s | 28.0, CH3 | 0.94, s |
| 14 | 33.8, CH3 | 0.95, s | 21.8, CH3 | 0.90, s |
| 15 | 15.9, CH3 | 0.86, s | 14.0, CH3 | 0.85, s |
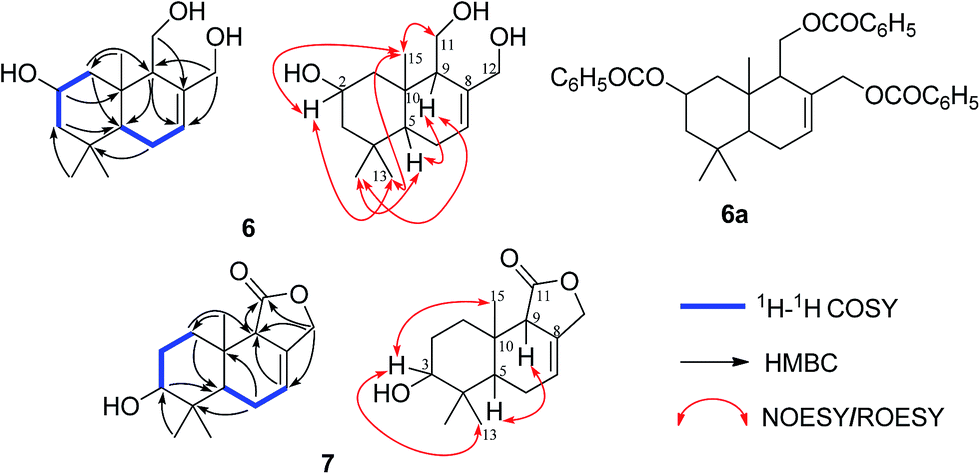 | ||
| Fig. 4 The 1H–1H COSY, key HMBC and NOESY/ROESY correlations of compounds 6 and 7 and the structure of compound 6a. | ||
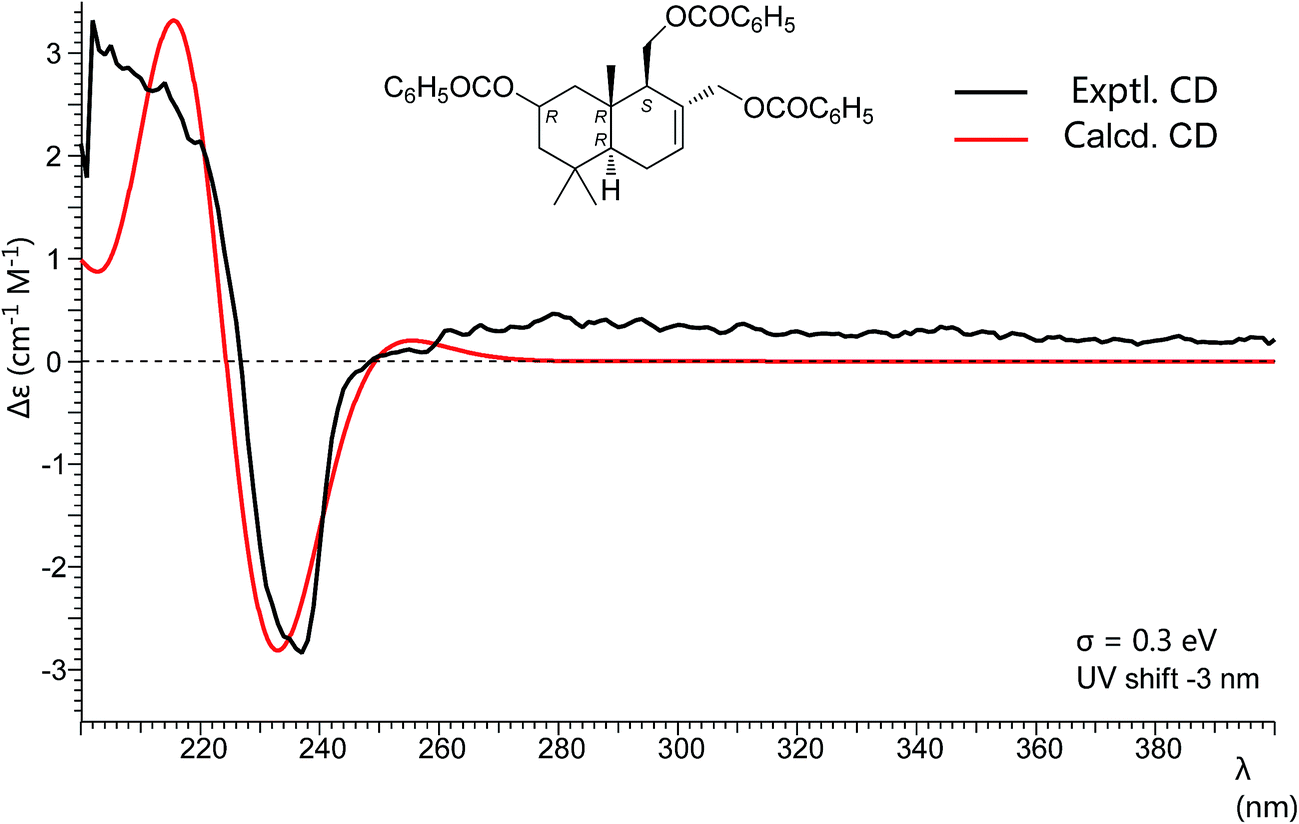 | ||
| Fig. 5 Experimental and calculated CD spectra of compound 6a. | ||
Pestalotiophol B (7) was isolated as colorless oil. The quasi-molecular ion at m/z 273.1464 [M + Na]+ (calcd for 273.1466) by HRESIMS indicated the molecular formula of 7 was C15H22O3 with five degrees of unsaturation. The 13C NMR (Table 3), with DEPT 135 together, showed fifteen carbons including one carbonyl, two olefinic carbons, four aliphatic methylenes, three aliphatic methines, and three methyl carbons. The 1H NMR spectrum (Table 3) revealed that 7 had one olefinic proton [δH 5.72 (1H, s, H-7)], one oxygenated methine [δH 3.48 (1H, m, H-3)], one oxygenated methylene [δH 4.63 (2H, m, H-12)] and three tertiary methyls [δH 0.94 (3H, s, H3-13), 0.90 (3H, s, H3-14), 0.85 (3H, s, H3-15)]. The proton resonances were distinctly assigned to relevant carbon atoms by HSQC. The 1H NMR and 13C NMR data indicated that 7 was characteristic of the drimane-type sesquiterpene with 6/6/5-fused ring system.34 The 1H–1H COSY (Fig. 4) showed two isolated spin systems (Fig. 4). The HMBC correlations of H-9 with C-7/C-8, and H-7 with C-8/C-9, located a double bond of C-7/C-8, a hydroxyl was located at C-3 by considering the molecular formula and the downfield chemical shift of C-3. OH-3 and double bond of C-7 and C-8 in 7 were two differences between 7 and one reported drimane-type sesquiterpene in the literature.34 ROESY correlations (Fig. 4) of H3-15 with H-3/H3-13 indicated that H3-15, H3-13 and H-3 had the relative configuration of β. ROESY correlation (Fig. 4) of H-9 with H-5 indicated that H-9 and H-5 had the relative configuration of α. Finally, 7 was elucidated and named as pestalotiophol B.
Compounds 1–15 were evaluated in vitro against cancer cell lines A549, HeLa, and SMMC-7721 by MTT assay. Compound 8 exhibited the strongest cytotoxicity against SMMC-7721 with an IC50 value of 28.3 μM, which is comparable to that of the positive control etoposide (IC50 of 23.2 μM), while it displayed moderate activity against A459 and Hela with IC50 values of 49.3 and 33.3 μM, respectively. Compound 9 showed the moderate activity against all three cell lines of A549, HeLa, and SMMC-7721 with IC50 values of 61.9, 42.9, and 44.7 μM, respectively. Unfortunately, the remaining compounds were inactive.
Experimental
General experimental procedures
1H, 13C, and 2D NMR data were measured on an Agilent DD2 400-MR NMR spectrometer (Agilent Technologies Inc, Santa Clara, United States), and the chemical shifts were referenced to the residual solvent peaks. IR spectra were measured with a Bruker Tensor 27 spectrophotometer in KBr pellets. HR-ESI-MS data were obtained by an AB Sciex Triple TOF® 4600 system. UV spectra were recorded on an UV-2550 UV/Vis spectrophotometer (Shimadzu Corporation, Tokyo, Japan). CD spectra were measured on a JASCO J-810 spectrometer in n-hexane and CD calculation was carried out by TD-DFT method with cam-B3LYP/TZVP level. The analytical and semi-preparative HPLC were performed on a Waters HPLC system, which was equipped with a W1525 Binary pump, a W2998 photodiode array detector, a W1500-CH column compartment, and a W2707 auto sampler using a Waters XBridge C18 column (4.6 × 250 mm, 5 μm) and Waters XBridge C18 column (10 × 250 mm, 5 μm) (Waters Corporation, Milford, MA, USA). Preparative HPLC was performed on an Agilent 1260 series HPLC (Agilent Technologies Inc, Santa Clara, United States), which was equipped with a 1260 pump and a 1260 diode array detector using a Waters XBridge C18 column (19 mm × 250 mm, 5 μm) eluted by MeCN/H2O system, flow rate was 15 mL min−1. The medium-pressure liquid chromatography (MPLC) apparatus used was equipped with a dual-pump gradient system, and a UV preparative detector (Buchi Inc, Swiss Confederation). Silica gels (200–300, 300–400 and 230–400 mesh) used for column chromatographies were from Qingdao Haiyang Chemical Company Ltd., Qingdao, China, and from Merck, Darmstadt, Germany. Sephadex LH-20 used for column chromatographies was from GE Healthcare Life Sciences (GE).Fungal material
The strain numbered HM-5 was isolated from stem bark of Sinopodophyllum hexandrum (Royle) Ying. collected from Qinling mountains, and was deposited at Shaanxi Key Laboratory of Phytochemistry, Baoji University of Arts and Sciences. The strain was identified as Pestalotiopsis adusta based on the morphological characteristics.Fermentation and cultivation
The strain was grown on a plate of potato dextrose medium consisting of 20 g glucose, 200 g potato, and 20 g agar in 1000 mL distilled water. After 7 days inoculation at 28 °C, a mycelium was inoculated into each of the 100 mL erlenmeyer flasks containing 30 mL of liquid PD medium consisting of 20 g glucose and 200 g potato in 1000 mL distilled water to obtain the seed strain. The flasks were incubated at 28 °C on a rotary shaker (120 rpm) for 3 days. A suspension (500 μL) of the seed liquid was inoculated aseptically to 500 mL Erlenmeyer flasks each containing 40 g rice and 60 mL distilled water, a total of 200 flasks. The flasks were incubated at 28 °C for 30 days.Extraction and isolation
After fermentation, the culture was extracted by ultrasonic with methanol. The extracting solution was evaporated to remaining 10 L aqueous extract. Then the aqueous extract was degreased with petroleum ether, concentrated and extracted thoroughly with EtOAc. Combined the EtOAc solvent and then evaporated under vacuum to afford a crude extract (111.8 g). Then the crude extract (111.8 g) was subjected to silica gel CC using a successive elution of petroleum ether–ethyl acetate (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 50:1, 20:1; 10:1, 8:1, 4:1, 2:1, 1:1, 1:2 and 0:100, v/v) to afford eight fractions (Fr1 to Fr8). Fr3 (10.6 g) was separated by repeated CC on silica gel, Sephadex LH-20, to yield 10 (3.2 g). Fr4 (6.3 g) was subjected to Sephadex LH-20 CC using MeOH to give three fractions (Fr4-1 to Fr4-3). Fr4-2 (3.6 g) was further subjected to MPLC on ODS RP-C18 CC, then purified with silica gel CC and Sephadex LH-20 gel, to afford 1 (9.8 mg), 2 (8.0 mg), 3 (20.1 mg), 8 (100.6 mg), and 9 (305.7 mg). Fr5 (24.9 g) was subjected to repeated CC on silica gel and Sephadex LH-20, then followed by recrystallization, to yield 4 (100.8 mg), 5 (20.4 mg), 12 (30.2 mg), 13 (150.2 mg), and 15 (26.0 mg). Fr6 (7.7 g) was subjected to MPLC on ODS RP-C18 CC, and then subjected to HPLC using MeCN–H2O (30:70, v/v) at a flow rate of 15 mL min−1 to yield 6 (tR: 10.1 min, 12.6 mg), 14 (tR: 12.7 min 50.3 mg), and 7 (tR: 16.0 min, 3.0 mg). Fr7 (5.3 g) was separated by repeated CC on silica gel and Sephadex LH-20, then purified on semi-preparative RP-HPLC using CH3CN–H2O (40:60, v/v) at a flow rate of 3 mL min−1 to yield 11 (tR:17.2 min, 26.2 mg).
ε) 244 (2.25), 290 (2.85), 652 (3.29) nm; IR (KBr) νmax 3012, 2970, 2924, 1719, 1439, 1244, 1122, 1028, 908 cm−1; 1H and 13C NMR data (CDCl3), see Tables 1 and 2; HRESIMS m/z 377.1931 [M + Na]+ (calcd for C19H30O6Na+, 377.1940).ε) 290 (2.80), 652 (3.35) nm; IR (KBr) νmax 2928, 1736, 1454, 1375, 1242, 1091, 1018, 969 cm−1; 1H and 13C NMR data (CDCl3), see Tables 1 and 2; HRESIMS m/z 391.2090 [M + Na]+ (calcd for C20H32O6Na+, 391.2096).ε) 290 (2.67), 652 (3.30) nm; IR (KBr) νmax 2930, 2867, 1734, 1455, 1370, 1241, 1098, 1021, 955 cm−1; 1H and 13C NMR data (CDCl3), see Tables 1 and 2; HRESIMS m/z 359.1834 [M + Na]+ (calcd for C19H28O5Na+, 359.1834).ε) 290 (2.76), 652 (3.31) nm; IR (KBr) νmax 2955, 2927, 2867, 1725, 1442, 1372, 1246, 1102, 1014, 952 cm−1; 1H and 13C NMR data (CDCl3), see Tables 1 and 2; HRESIMS m/z 359.1833 [M + Na]+ (calcd for C19H28O5Na+, 359.1834).ε) 290 (2.68), 652 (3.29) nm; IR (KBr) νmax 2959, 2925, 2858, 1776, 1736, 1456, 1367, 1247, 1085, 1022, 801 cm−1; 1H and 13C NMR data (CDCl3), see Tables 1 and 2; HRESIMS m/z 345.1674 [M + Na]+ (calcd for C18H26O5Na+, 345.1678).ε) 290 (2.79), 652 (3.29) nm; IR (KBr) νmax 3333, 2928, 2856, 1460, 1376, 1212, 1147, 1033, 997 cm−1; 1H and 13C NMR data (CD3OD), see Table 3; HRESIMS m/z 277.1779 [M + Na]+ (calcd for C15H26O3Na+, 277.1779).ε) 290 (2.65), 652 (3.17) nm; IR (KBr) νmax 2954, 2923, 2862, 1756, 1687, 1456, 1375, 1018 cm−1; 1H and 13C NMR data (CDCl3), see Table 3; HRESIMS m/z 273.1464 [M + Na]+ (calcd for C15H22O3Na+, 273.1466).
:1, 50:1, 20:1; 10:1, 8:1, 4:1, 2:1, 1:1, 1:2 and 0:100, v/v) to afford eight fractions (Fr1 to Fr8). Fr3 (10.6 g) was separated by repeated CC on silica gel, Sephadex LH-20, to yield 10 (3.2 g). Fr4 (6.3 g) was subjected to Sephadex LH-20 CC using MeOH to give three fractions (Fr4-1 to Fr4-3). Fr4-2 (3.6 g) was further subjected to MPLC on ODS RP-C18 CC, then purified with silica gel CC and Sephadex LH-20 gel, to afford 1 (9.8 mg), 2 (8.0 mg), 3 (20.1 mg), 8 (100.6 mg), and 9 (305.7 mg). Fr5 (24.9 g) was subjected to repeated CC on silica gel and Sephadex LH-20, then followed by recrystallization, to yield 4 (100.8 mg), 5 (20.4 mg), 12 (30.2 mg), 13 (150.2 mg), and 15 (26.0 mg). Fr6 (7.7 g) was subjected to MPLC on ODS RP-C18 CC, and then subjected to HPLC using MeCN–H2O (30:70, v/v) at a flow rate of 15 mL min−1 to yield 6 (tR: 10.1 min, 12.6 mg), 14 (tR: 12.7 min 50.3 mg), and 7 (tR: 16.0 min, 3.0 mg). Fr7 (5.3 g) was separated by repeated CC on silica gel and Sephadex LH-20, then purified on semi-preparative RP-HPLC using CH3CN–H2O (40:60, v/v) at a flow rate of 3 mL min−1 to yield 11 (tR:17.2 min, 26.2 mg).
ε) 244 (2.25), 290 (2.85), 652 (3.29) nm; IR (KBr) νmax 3012, 2970, 2924, 1719, 1439, 1244, 1122, 1028, 908 cm−1; 1H and 13C NMR data (CDCl3), see Tables 1 and 2; HRESIMS m/z 377.1931 [M + Na]+ (calcd for C19H30O6Na+, 377.1940).ε) 290 (2.80), 652 (3.35) nm; IR (KBr) νmax 2928, 1736, 1454, 1375, 1242, 1091, 1018, 969 cm−1; 1H and 13C NMR data (CDCl3), see Tables 1 and 2; HRESIMS m/z 391.2090 [M + Na]+ (calcd for C20H32O6Na+, 391.2096).ε) 290 (2.67), 652 (3.30) nm; IR (KBr) νmax 2930, 2867, 1734, 1455, 1370, 1241, 1098, 1021, 955 cm−1; 1H and 13C NMR data (CDCl3), see Tables 1 and 2; HRESIMS m/z 359.1834 [M + Na]+ (calcd for C19H28O5Na+, 359.1834).ε) 290 (2.76), 652 (3.31) nm; IR (KBr) νmax 2955, 2927, 2867, 1725, 1442, 1372, 1246, 1102, 1014, 952 cm−1; 1H and 13C NMR data (CDCl3), see Tables 1 and 2; HRESIMS m/z 359.1833 [M + Na]+ (calcd for C19H28O5Na+, 359.1834).ε) 290 (2.68), 652 (3.29) nm; IR (KBr) νmax 2959, 2925, 2858, 1776, 1736, 1456, 1367, 1247, 1085, 1022, 801 cm−1; 1H and 13C NMR data (CDCl3), see Tables 1 and 2; HRESIMS m/z 345.1674 [M + Na]+ (calcd for C18H26O5Na+, 345.1678).ε) 290 (2.79), 652 (3.29) nm; IR (KBr) νmax 3333, 2928, 2856, 1460, 1376, 1212, 1147, 1033, 997 cm−1; 1H and 13C NMR data (CD3OD), see Table 3; HRESIMS m/z 277.1779 [M + Na]+ (calcd for C15H26O3Na+, 277.1779).ε) 290 (2.65), 652 (3.17) nm; IR (KBr) νmax 2954, 2923, 2862, 1756, 1687, 1456, 1375, 1018 cm−1; 1H and 13C NMR data (CDCl3), see Table 3; HRESIMS m/z 273.1464 [M + Na]+ (calcd for C15H22O3Na+, 273.1466).X-ray single-crystal diffraction
Crystals of compounds 1 and 5 were grown in methanol at room temperature. Crystallographic data for 1 were collected on a SuperNova Eos diffractometer with graphite monochromatic Cu Kα radiation at 297 (2) K. All calculations were performed using SHELXL-97 program. Crystallographic data for 5 were collected on a SuperNova Eos diffractometer with graphite monochromatic Cu Kα radiation at 293 K. All calculations were performed using SHELXL-97 program.35 Parameters in CIF format are available as ESI† from Cambridge Crystallographic Data Centre (pestalotiopsin D (1), CCDC1534582; pestalotiopsin H (5), CCDC1534584).CD spectrum associated with TD-DFT calculation analysis
A preliminary conformational search was performed in Conflex6.7 using MMFF94s forcefield.36 Conformers were saved and further optimized using the density functional theory (DFT) method and CPCM solvent model at B3LYP/6-31+G (d,p) level in Gaussian 09 software package.37 Frequency was calculated at the same level of theory to check optimized results. The stable conformers with populations greater than 1% and without imaginary frequencies were submitted to ECD calculation by the TDDFT method associated at cam-B3LYP/TZVP level. The excitation energies (E), oscillator strength (f), rotatory strength in velocity form (Rvel), and rotatory strength in length form (Rlen) of the lowest 32 excited states were calculated. ECD spectra of different conformers were summated in SpecDis according to their Boltzmann-calculated distributions.38Cytotoxicity assay
The cell viability was determined by the MTT assay. 8 × 103 cells were incubated with the tested compounds in triplicate in 96-well plates for 68 h at 37 °C in a final volume of 100 μL. Cells treated with DMSO alone were set as controls. At the end of the treatment, 10 μL of MTT (5 mg mL−1) was added to each well and incubated for an additional 4 h. An extraction buffer (100 μL, 10% SDS, 5% iso-butanol, 0.1% HCl) was added, and the cells were incubated overnight at 37 °C. The viability was calculated by measuring the absorbance at 570 nm using a microplate reader (BioTek Powerwave XS2, USA).Acknowledgements
We gratefully acknowledge financial support from National Natural Science Foundation of China (No. 31600268), Shaanxi Science and Technology Commission (No. 2014JM2058), Shaanxi Education Commission (No. 13JS005), Baoji University of Arts and Sciences (No. ZK15043) and Shaanxi Association for Science and Technology (No. 20150202).Notes and references
- H. B. Bode, B. Bethe, R. Höfs and A. Zeeck, ChemBioChem, 2002, 3, 619–627 CrossRef CAS PubMed.
- J. M. Gao, S. X. Yan and J. C. Qin, Chem. Rev., 2013, 113, 4755–4811 CrossRef CAS PubMed.
- X. L. Yang, J. Z. Zhang and D. Q. Luo, Nat. Prod. Rep., 2012, 29, 622–641 RSC.
- G. Strobel, X. Yang, J. Sears, R. Kramer, R. S. Sidhu and W. M. Hess, Microbiology, 1996, 142, 435–440 CrossRef CAS PubMed.
- S. S. Maharachchikumbura, K. D. Hyde, J. Z. Groenewald, J. Xu and P. W. Crous, Stud. Mycol., 2014, 79, 121–186 CrossRef CAS PubMed.
- K. Wang, J. Lei, J. Wei and N. Yao, Mini-Rev. Med. Chem., 2012, 12, 1382–1393 CAS.
- M. Pulici, F. Sugawara, H. Koshino, J. Uzawa, S. Yoshida, E. Lobkovsky and J. Clardy, J. Org. Chem., 1996, 61, 2122–2124 CrossRef CAS.
- G. Ding, S. Liu, L. Guo, Y. Zhou and Y. Che, J. Nat. Prod., 2008, 71, 615–618 CrossRef CAS PubMed.
- L. Liu, S. Liu, X. Chen, L. Guo and Y. Che, Bioorg. Med. Chem., 2009, 17, 606–613 CrossRef CAS PubMed.
- L. Liu, T. Bruhn, L. Guo, D. C. Goetz, R. Brun, A. Stich and G. Bringmann, Chem.–Eur. J., 2011, 17, 2604–2613 CrossRef CAS PubMed.
- H. Y. Tang, J. M. Gao and Q. Zhang, RSC Adv., 2015, 5, 72433–72436 RSC.
- I. G. Collado, J. R. Hanson and A. J. Macías-Sánchez, Nat. Prod. Rep., 1998, 15, 187–204 RSC.
- J. P. Poyser, R. L. Edwards, J. R. Anderson, M. B. Hursthouse, N. P. C. Walker, G. M. Sheldrick and A. J. S. Whalley, J. Antibiot., 1986, 39, 167–169 CrossRef CAS PubMed.
- J. Gertsch, M. Leonti, S. Raduner, I. Racz, J. Z. Chen, X. Q. Xie and A. P. Zimmer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9099–9104 CrossRef CAS PubMed.
- H. Yu, W. X. Li, J. C. Wang, Q. Yang, H. J. Wang, C. C. Zhang and H. J. Zhu, Tetrahedron, 2015, 71, 3491–3494 CrossRef CAS.
- Y. Li, S. Niu, B. Sun, S. Liu, X. Liu and Y. Che, Org. Lett., 2010, 12, 3144–3147 CrossRef CAS PubMed.
- R. F. Magnan, E. Rodrigues-Fo, C. Daolio, A. G. Ferreira and A. Q. Z. de Souza, Z. Naturforsch., C: J. Biosci., 2003, 58, 319–324 Search PubMed.
- Y. Li, C. W. Li, C. B. Cui, X. Z. Liu and Y. S. Che, Nat. Prod. Bioprospect., 2012, 2, 70–75 CrossRef CAS.
- S. T. Deyrup, D. C. Swenson, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 2006, 69, 608–611 CrossRef CAS PubMed.
- I. H. Hwang, J. B. Gloer and D. T. Wicklow, Planta Med., 2013, 79, PL11 Search PubMed.
- S. W. Yang, T. M. Chan, J. Terracciano, E. Boehm, R. Patel, G. Chen and M. Chu, J. Nat. Prod., 2009, 72, 484–487 CrossRef CAS PubMed.
- M. Wichlacz, W. A. Ayer, L. S. Trifonov, P. Chakravarty and D. Khasa, J. Nat. Prod., 1999, 62, 484–486 CrossRef CAS PubMed.
- J. R. Anderson, R. L. Edwards, J. P. Poyser and A. J. Whalley, J. Chem. Soc., Perkin Trans. 1, 1988, 4, 823–831 RSC.
- J. R. Anderson, R. L. Edwards, A. A. Freer, R. P. Mabelis, J. P. Poyser, H. Spencer and A. J. Whalley, J. Chem. Soc., Chem. Commun., 1984, 14, 917–919 RSC.
- T. M. Baker, D. J. Edmonds, D. Hamilton, C. J. O'Brien and D. J. Procter, Angew. Chem., Int. Ed., 2008, 47, 5631–5633 CrossRef CAS PubMed.
- J. Xiao, Q. Zhang, Y. Q. Gao, J. J. Tang, A. L. Zhang and J. M. Gao, J. Agric. Food Chem., 2014, 62, 3584–3590 CrossRef CAS PubMed.
- H. Li, J. Xiao, Y. Q. Gao, J. J. Tang, A. L. Zhang and J. M. Gao, J. Agric. Food Chem., 2014, 62, 3734–3741 CrossRef CAS PubMed.
- J. Xiao, Q. Zhang, Y. Q. Gao, X. W. Shi and J. M. Gao, Nat. Prod. Res., 2014, 28, 1388–1392 CrossRef CAS PubMed.
- M. Pulici, F. Sugawara, H. Koshino, G. Okada, Y. Esumi, J. Uzawa and S. Yoshida, Phytochemistry, 1997, 46, 313–319 CrossRef CAS.
- Z. Wu, D. Liu, P. Proksch, P. Guo and W. Lin, Mar. Drugs, 2014, 12, 3904–3916 CrossRef PubMed.
- J. R. Anderson, C. E. Briant, R. L. Edwards, R. P. Mabelis, J. P. Poyser, H. Spencer and A. J. Whalley, Chem. Commun., 1984, 7, 405–406 RSC.
- M. Pulici, F. Sugawara, H. Koshino, J. Uzawa, S. Yoshida, E. Lobkovsky and J. Clardy, J. Nat. Prod., 1996, 59, 47–48 CrossRef CAS.
- J. G. Xing, H. Y. Deng and D. Q. Luo, J. Asian Nat. Prod. Res., 2011, 13, 1069–1073 CrossRef CAS PubMed.
- V. J. Paul, Y. Seo, K. W. Cho, J. R. Rho, J. Shin and P. R. Bergquist, J. Nat. Prod., 1997, 60, 1115–1120 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXTL-97 – A Program for Refining Crystal Structure, University of Goettingen, Goettingen, Germany, 1997 Search PubMed.
- Conflex 6.7, Conflex Corp., Tokyo Yokohama, Japan, 2010 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, O. J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 revision D.01, Gaussian Inc., Wallingford, CT, 2013 Search PubMed.
- T. Bruhn, A. Schaumloeffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1534582 and 1534584. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra04267a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |