
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioactive composition of Reevesia formosana root and stem with cytotoxic activity potential†
Hsun-Shuo Changabc,
Chu-Hung Lina,
Pei-Yu Hsiaod,
Hung-Ti Pengb,
Shiow-Ju Leee,
Ming-Jen Chengf and
Ih-Sheng Chen *ab
*ab
aSchool of Pharmacy, College of Pharmacy, Kaohsiung Medical University, Kaohsiung 807, Taiwan. E-mail: m635013@kmu.edu.tw; Fax: +886-7321068; Tel: +886-73121101ext. 2191
bGraduate Institute of Natural Products, College of Pharmacy, Kaohsiung Medical University, Kaohsiung 807, Taiwan
cCenter for Infectious Disease and Cancer Research (CICAR), Kaohsiung Medical University, Kaohsiung 807, Taiwan
dDepartment of Pharmacy, Kaohsiung Veterans General Hospital, Kaohsiung 813, Taiwan
eDivision of Biotechnology and Pharmaceutical Research, National Health Research Institutes, Miaoli 350, Taiwan
fBioresource Collection and Research Center, Food Industry Research and Development Institute, Hsinchu 300, Taiwan
First published on 22nd May 2017
Abstract
Six new compounds including three lignanoids: reevesiacoumarin (1), reevesic acid (2), and reevesilignan (3), and three terpenoids: reevesiterpenol A (4), reevesiterpenol B (5), and 3α,27-di-O-trans-caffeoylbetulinic acid (6), along with 40 known compounds were isolated from the root and stem of Reevesia formosana (Sterculiaceae). The structures of 1–6 were determined by spectroscopic techniques. Bioassays for the cytotoxicities of MCF-7, NCI–H460, and HepG2 cancer cell lines led to finding three cardenolides: strophanthojavoside (31) and ascleposide (32) with IC50 < 1 μM and strophalloside (33) displayed selective cytotoxicity to NCI–H460 with IC50 0.62 ± 0.06 μM as well. 3α,27-Di-O-trans-caffeoylbetulinic acid (6) and secoisolariciresinol (13) also showed weak but selective cytotoxicity to NCI–H460 and HepG2 cancer cell lines, respectively.
Introduction
For decades, the role of cardenolides had transformed from the traditional use, treatment of anti-arrhythmia and heart failure, into the new prospect of anticancer. Reevesia formosana Sprague (Sterculiaceae) was found to be cytotoxic in the root, stem, and fruits, and also as the most bioactive one among 1400 species of Formosan plants through the cytotoxic assay for MCF-7, NCI–H460, and HepG2 in vitro. With our previous investigation of the root of R. formosana, individual cardenolides have been isolated,1 including reevesiosides A–I and epi-reevesiosides F–I. Continuing these rigorous studies, three cardenolides: reevesioside J, reevesioside K, and epi-reevesioside K, three sesquiterpenoids: reevesiterpenols C–E, and two glycosides: reevesianins A and B, along with 46 known compounds were also isolated from the fruits of R. formosana.2 Among these isolates, all cardenolides showed significant cytotoxicity against MCF-7, NCI–H460, and HepG2 cancer cell lines and reevesiterpenol E also exhibited the best selective cytotoxicity to the NCI–H460 cell line. Furthermore, reevesioside A,3 reevesioside F,4 and epi-reevesioside F5 had already been discussed for the mechanism of several cancer cells. In this study, we investigated the stem of R. formosana and the remaining fractions of the root of R. formosana. From these two parts led to the isolation of six new compounds including three lignanoids: reevesiacoumarin (1), reevesic acid (2), and reevesilignan (3), and three terpenoids: reevesiterpenol A (4), reevesiterpenol B (5), and 3α,27-di-O-trans-caffeoylbetulinic acid (6) (Fig. 1), along with 40 known compounds.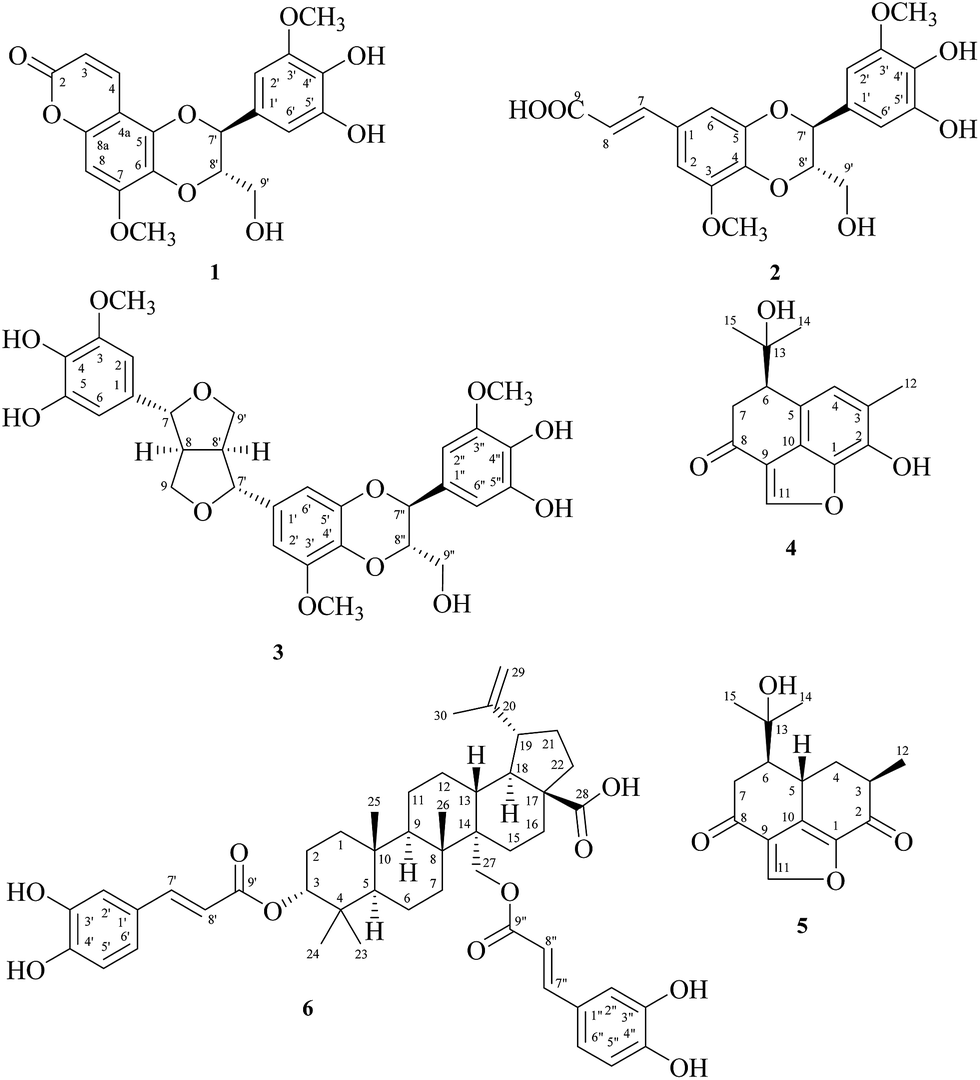 | ||
| Fig. 1 Chemical structures of new compounds 1–6. | ||
The bioassay indicated three cardenolides: strophanthojavoside (31) and ascleposide (32) with IC50 < 1 μM and strophalloside (33) displayed selective cytotoxicity to NCI–H460 with IC50 0.62 ± 0.06 μM as well. 3α,27-Di-O-trans-caffeoylbetulinic acid (6) and secoisolariciresinol (13) also showed weak but selective cytotoxicity to NCI–H460 and HepG2 cancer cell lines, respectively. All the structures were elucidated and confirmed through the 1D and 2D spectroscopic techniques.
Results and discussion
The root and stem of R. formosana were extracted with methanol, and the produced extracts were partitioned into the EtOAc and H2O soluble layers. Both of the EtOAc layers were purified by conventional chromatographic techniques to obtain forty-six compounds (1–46), and the structures were elucidated by 1D and 2D NMR spectra and comparison with literature data.Compound 1 was isolated as a yellowish powder with a molecular formula of C20H18O9 as determined by positive-ion HRESIMS, showing a [M + Na]+ ion at m/z 425.0845 (calcd for C20H18O9Na, m/z 425.0848). The presence of hydroxy and carbonyl groups in 1 was shown by the bands at 3420 and 1708 cm−1, respectively, in the IR spectrum. The 1H NMR spectrum showed two meta-coupled protons of an aromatic ring at δH 6.70 (1H, dd, J = 2.0, 0.6 Hz, H-6′) and 6.73 (1H, d, J = 2.0 Hz, H-2′), one singlet proton of another aromatic ring at δH 6.61 (1H, s, H-8), two oxymethine protons at δH 4.08 (1H, ddd, J = 8.0, 6.4, 3.6 Hz, H-8′) and 5.08 (1H, d, J = 8.0 Hz, H-7′), two non-equivalent oxymethylene protons at δH 3.57 (1H, dd, J = 12.0, 3.6 Hz, H-9′b) and 3.85 (1H, dd, J = 12.0, 6.4 Hz, H-9′a), two methoxy groups at δH 3.84 (3H, s, OCH3-3′) and 3.96 (3H, s, OCH3-7). Also, a pair of mutually coupled protons at δH 6.15 (1H, d, J = 9.6 Hz, H-3) and 7.96 (1H, dd, J = 9.6, 0.6 Hz, H-4), assigned to the vinylic protons. The HMBC correlations from H-3 to C-2 (δC 161.8) and C-4a (δC 104.5), from H-4 to C-2, C-5 (δC 141.4), and C-8a (δC 151.1), from H-8 to C-4a, C-6 (δC 131.3), C-7 (δC 154.2), and C-8a and from OCH3-7 to C-7 were further confirmed the 5,6-dioxo-7-methoxycoumarin moiety.6 Furthermore, the location of the another methoxy group of a tetrasubstituted aromatic ring at C-3′ (δC 149.8) was further confirmed by the HMBC cross-peaks of H-2′ to C-3′ and C-4′ (δC 136.1), H-6′ to C-4′ and C-5′ (δC 147.1), and OCH3-3′ to C-3′. The fragments of C-7′ (δC 78.8)-C-8′ (δC 79.9)-C-9′ (δC 62.3) were observed by COSY analysis (Fig. 2) as well as the phenylpropanoid moiety (C-1′–C-9′) was confirmed by correlations in the HMBC spectrum from H-7′ to C-1′, C-2′, and C-6′. According to the molecular formula of 1 with 12 indices of hydrogen deficiency (IHD) indicated the presence of a 1,4-dioxane ring between the 5,6-dioxo-7-methoxycoumarin moiety and the phenylpropanoid moiety (C-1′–C-9′). The O-linkages between C-5–O–C-7′ and C-6–O–C-8′ were confirmed by the NOESY spectrum (Fig. 3) showed correlations between H-9′ and OCH3-7. The coupling constant (J = 8.0 Hz) between H-7′ and H-8′ approved the trans-form.7 The absolute configurations at C-7′ and C-8′ were determined as 7′S,8′S by CD spectral comparison with the analogous neolignan 7S,8S-nitidanin.8 By the above data, the structure of 1 was further confirmed by DEPT, HSQC, COSY, NOESY, and HMBC experiments and named reevesiacoumarin.
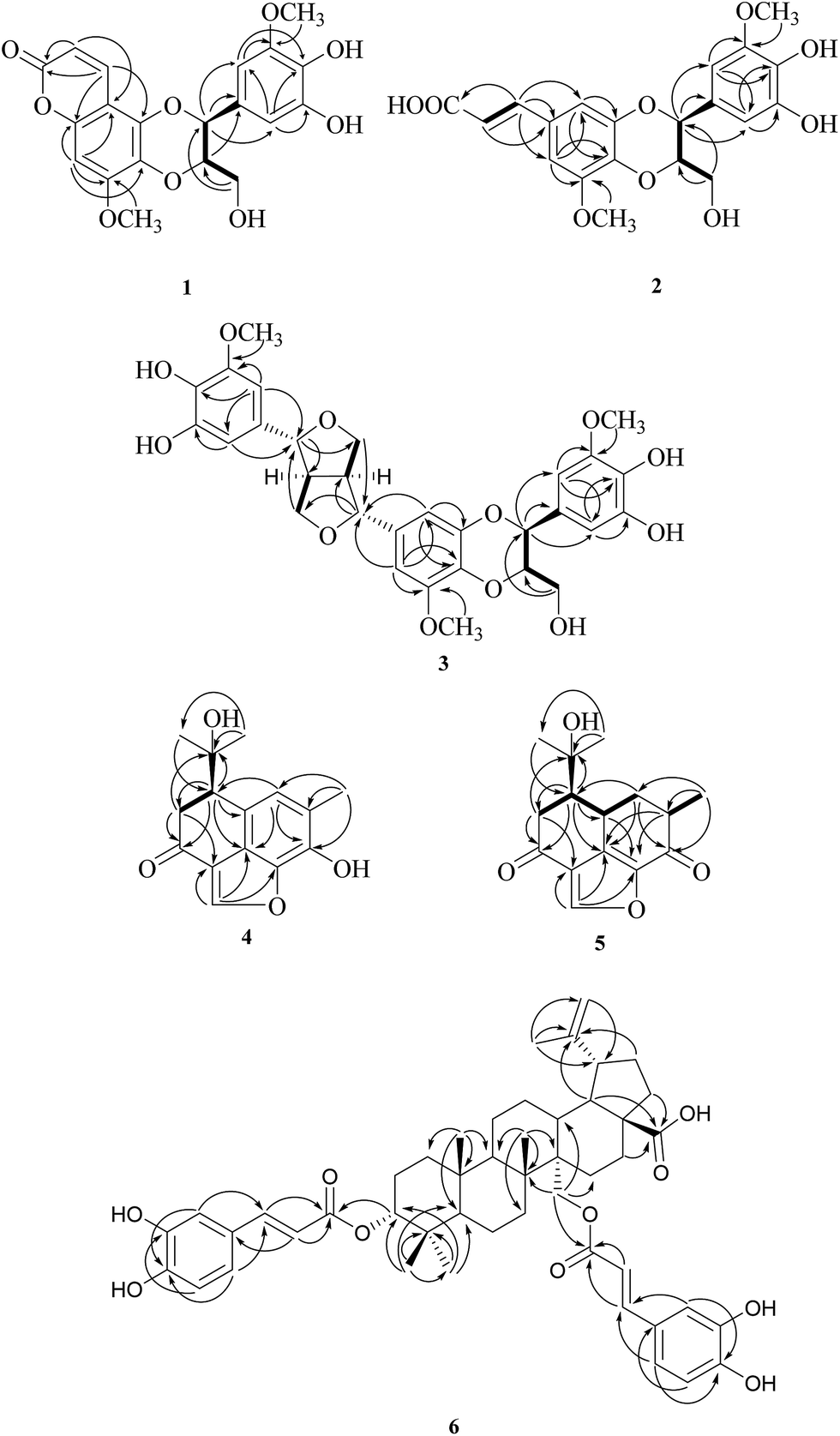 | ||
| Fig. 2 Key HMBC (H → C), COSY (bold line) of compounds 1–6. | ||
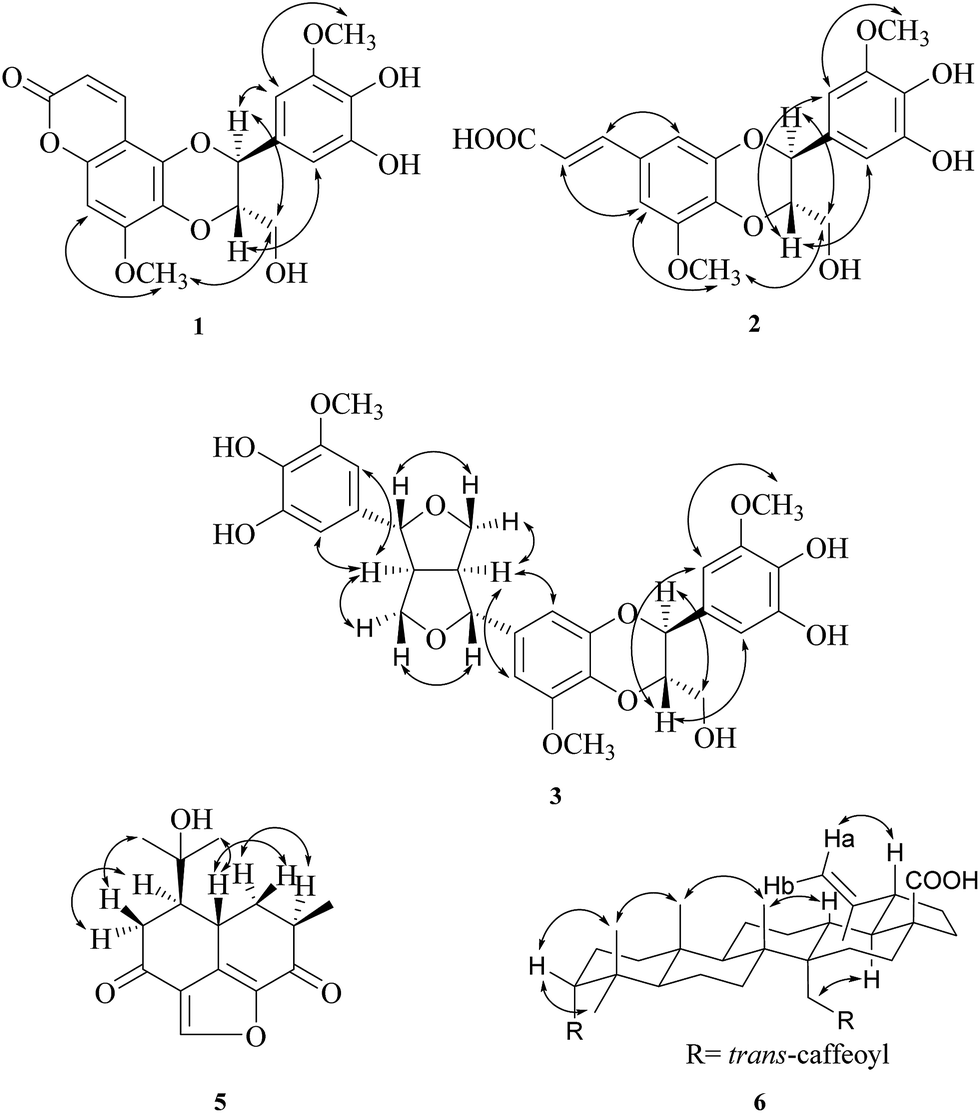 | ||
| Fig. 3 Key NOESY (H ↔ H) correlations of compounds 1–6. | ||
Compound 2 was obtained as an optically colorless oil with [α]25D −8.1 (c 0.14, MeOH), and the molecular formula was calculated as C20H20O9 by ESIMS and HRSIMS analyses with 11 degrees of unsaturation. UV and IR spectra were similar to those of simplidin (7)8 also isolated in this study, except one additional carbonyl (1731 cm−1) was appeared in IR spectrum. Analyses of 1D and 2D NMR [COSY (Fig. 2), HSQC, and HMBC (Fig. 2)] data established a neolignan-based gross structure, which was also closely related to simplidin (7).9 The difference was attributed to a carboxylic acid (δC 173.8) at C-8 of 2 to replace a hydroxy group of simplidin (7), as evident from the 3J-correlation of HMBC between H-7 to a carbonyl carbon (δC 173.8, C-9) and IR plot. Thus, the structure of 2 was determined and named reevesic acid.
Compound 3 was yielded as a colorless oil, with [α]25D −10.5 (c 0.06, MeOH), and the ESIMS and HRESIMS established the molecular formula as C30H32O12, and the phenolic moiety was present by the bathochromic shift of UV spectrum. From the 1H NMR spectrum, four methines [δH 3.11 (2H, m, H-8, H-8′)] including two oxygen-bearing [δH 4.64 (1H, br d, J = 4.2 Hz, H-7′) and 4.71 (1H, dd, J = 4.8, 1.8 Hz, H-7)], two oxymethylene groups [δH 3.86 (2H, m, H-9b, H-9′b) and 4.25 (2H, m, H-9a, H-9′a)], two pairs of meta-coupled aromatic protons [δH 6.49 (1H, br d, J = 1.8 Hz, H-6), 6.51 (1H, br d, J = 1.8 Hz, H-2)/δH 6.60 (1H, br t, J = 1.8 Hz, H-6′), 6.64 (1H, br t, J = 1.8 Hz, H-2′)], and the connection of two methoxy groups (δH 3.85, 3.88) to C-3 and C-3′, respectively, by HMBC (Fig. 2) correlations, pointed out the existence of 4′,5′-dioxo-5-hydroxypinoresinol moiety. While the rest of the 1H NMR signals of 3 were identical to a phenylpropanoid moiety [δH 3.51 (1H, dd, J = 12.6, 4.2 Hz, H-9′′b), 3.71 (1H, dd, J = 12.6, 2.4 Hz, H-9′′a), 3.98 (1H, ddd, J = 7.8, 4.2, 2.4 Hz, H-8′′), 4.80 (1H, d, J = 7.8 Hz, H-7′′), 6.55 (1H, br d, J = 2.4 Hz, H-6′′), and 6.58 (1H, br d, J = 1.8 Hz, H-2′′)] alike C-1′–C-9′ of 1. The coupling constant (J = 7.8 Hz) between H-7′′ and H-8′′ of 3 approved the trans-form.7 The H-7′′ showed correlation with H-9′′ and showed no correlation to H-8′′ also confirmed the trans-form of H-7′′ and H-8′′. Furthermore, 1,4-dioxane ring between the 4′,5′-dioxo-5-hydroxypinoresinol moiety and the phenylpropanoid moiety (C-1′′–C-9′′) was also confirmed the same as 1. Thus, the planar structure of 3 was decided and the relative configuration was determined by NOESY (Fig. 3) correlations. According to the above evidence, compound 3 as a new substance named reevesilignan.
Compound 4 was obtained as an optically active colorless oil, with [α]25D +20.0 (c 0.10, CHCl3). The molecular formula was obtained as C15H16O4 with ESIMS and HRESIMS analyses, with the observation of HSQC and DEPT spectra, the substance was suggested to be sesquiterpenoid. The UV spectrum displayed the maxima absorptions at 211, 223 sh, and 249 sh nm then with the bathochromic shift by the addition of KOH aqueous solution further provided the presence of phenolic moiety. The 1H NMR spectrum showed three singlet methyl groups at δH 1.16, 1.21, and 2.42, one methylene group [δH 2.84 (1H, dd, J = 16.6, 6.9 Hz, H-7b), 3.06 (1H, dd, J = 16.6, 1.7 Hz, H-7a)], one methine [δH 3.31 (1H, dd, J = 6.9, 1.7 Hz, H-6)], one aromatic proton [δH 7.08 (1H, s, H-4)], one oxoolefinic proton [δH 7.99 (1H, s, H-11)], and two broad singlets of hydroxy group at δH 3.60 and 5.60 as well. As eight degrees of unsaturation, the indication of conjugated carbonyl group (1682 cm−1) and phenolic moiety, and the oxoolefinic proton (H-11) presented the 2,3J-correlations to δC 118.8 (C-9), 128.2 (C-10), 141.6 (C-1), suggested the presence of a furan ring, thus the structure of 4 was further confirmed as a furanosesquiterpenoid. The above 1H NMR and physical data of 4 resembled hibiscone D10 while the downfield shift of the quaternary carbon [δC 73.5 (C-13)] proposed a hydroxyisopropyl group [δH 1.16 (3H, s, H-14), 1.21 (3H, s, H-15); δC 73.5 (C-13), 27.2 (C-14), and 27.7 (C-15)] in 4 replaced an isopropyl group in hibiscone D. This was also proved by the HRESIMS m/z 283.0947 [M + Na]+ (calcd for C15H16O4Na, 283.0946). Therefore, the planar structure of 4 was determined and its relative configuration of 4 is the same as hibiscone D10 according to the positive optical rotation ([α]25D +20.0), similar to hibiscone D ([α]26D +37). Compound 5, as an optically active colorless oil with [α]25D −6.9 (c 0.05, CHCl3). The molecular formula calculated for C15H18O4 by HRESIMS, then further combined to the observation of 13C and DEPT spectra, 5 was suggested to share the similar skeletone with 4 as furanosesquiterpenoid. Comparison of 5 to hibiscone C,10 isolated from Hibiscus elatus, showed similarities in both the physical data and the 1H NMR spectra while the difference appeared at the HRESIMS analysis for one more oxygen atom. The disappearance of one methine and presence of a quaternary carbon at δC 73.1 (C-13) were implied that the hydroxyisopropyl group [δH 1.34 (3H, s, H-14), 1.35 (3H, s, H-15); δC 73.1 (C-13), 24.9 (C-14), and 30.7 (C-15)] at C-6 in 5 was in place of isopropyl group at C-6 in hibiscone C. The relative configuration of 5 was confirmed with the NOESY correlations and the optical rotation ([α]25D −6.9), similar to hibiscone C ([α]27D −23). As determined by the above observations, 4 and 5 were recommended as the structures in Fig. 1 and named reevesiterpenol A and reevesiterpenol B, respectively, which were further confirmed by DEPT, HSQC, COSY (Fig. 2), and HMBC (Fig. 2) experiments.
Compound 6 was obtained as a yellowish oil. ESIMS and HRESIMS (m/z 819.4089 [M + Na]+) analyses established the molecular formula of 6 as C48H60O10. The IR absorption bands suggested the presence of hydroxy (3335 cm−1), conjugated carbonyl ester (1697, 1683 cm−1), and 13C NMR data supported the presences of carboxylic (δC 179.9) and ester carbonyl (δC 169.5 and 168.9) groups. The 1H NMR spectrum of 6 indicated five methyl singlets at δH 0.86, 0.93, 0.96, 1.06, and 1.73; the presence of two typical trans-caffeoyl groups were deduced by four olefinic protons at δH 6.287 (1H, d, J = 16.0 Hz, H-8′′), 6.291 (1H, d, J = 16.0 Hz, H-8′), 7.56 (1H, d, J = 16.0 Hz, H-7′′), 7.58 (1H, d, J = 16.0 Hz, H-7′) and by two 1,3,4-trisubstituted benzene rings at δH 6.75 (1H, d, J = 8.4 Hz, H-5′), 6.80 (1H, d, J = 8.4 Hz, H-5′′), 6.90 (1H, dd, J = 8.4, 2.0 Hz, H-6′), 7.015 (1H, dd, J = 8.4, 2.0 Hz, H-6′′), 7.018 (1H, d, J = 2.0 Hz, H-2′), and 7.11 (1H, d, J = 2.0 Hz, H-2′′). The 13C NMR data of 6 resembles 27-O-trans-caffeoylcylicodiscic acid with lupane type skeleton.11 The major differences between 6 and 27-O-trans-caffeoylcylicodiscic acid were one additional trans-caffeoyl group at C-3 in 6 instead of the hydroxy group at C-3 in 27-O-trans-caffeoylcylicodiscic acid. The HMBC correlations from H-3 (δH 4.69) to C-9′ (δC 168.9); from H-27a (δH 4.88) and H-27b (δH 4.52) to C-9′′ (δC 169.5) suggested two trans-caffeoyl groups linkage at C-3 and C-27, respectively. Moreover, the HMBC (Fig. 2) correlations from H-18 (δH 1.80) to C-28 (δC 179.9) indicated that a carboxylic group is attached to C-17. The 3α-configuration of the trans-caffeoyl group was deduced from the H-3 signal pattern at the downfield shifts at δH 4.69 (br s) and its 13C NMR signal at δC 79.5.12,13 The relative configurations of 6 were determined through inspection of the NOESY spectrum (Fig. 3). The several key NOESY correlations (H-3/H-23; H-3/H-24; H-24/H-25; H-25/H-26; H-13/H-26; H-18/H-27) suggested that the α-equatorial orientation of H-3 in trans A/B ring junction (Fig. 3). As a result, 6 was established as 3α,27-di-O-trans-caffeoylbetulinic acid and was further confirmed by DEPT, HSQC, COSY, and HMBC (Fig. 2) experiments.
The known compounds, simplidin (7),9 5-O-demethylbilagrewin (8),14 malloapelin C (9),15 syringaresinol (10),16 pinoresinol (11),16 3-(α,4-dihydroxy-3-methoxy-benzyl)-4-(4-hydroxy-3-methoxybenzyl)tetrahydrofuran (12),17 secoisolariciresinol (13),18 rosmarinic acid (14),19 clinopodic acid A (15),19 cis-7-hydroxycalamenene (16),20 trans-7-hydroxycalamenene (17),20 7-hydroxycadalene (18),21 4,5-dihydroblumenol A (19),22 scopoletin (20),23 fraxetin (21),23 isofraxidin (22),24 trans-ferulic acid (23),18 vanillic acid (24),25 a mixture of β-sitosterol (25) & stigmasterol (26),26 a mixture of (24R)-stigmast-4-en-3-one (27) & (22E,24S)-stigmast-4,22-dien-3-one (28),27 Q10 (29),28 proanthocyanidin A2 (30),29 strophanthojavoside (31),30 ascleposide (32),31 and strophalloside (33)30 from the root of R. formosana, and 7, 8, 10, 20, a mixture of 25 & 26, 3β-trans-caffeoylbetulinic acid (34),32 3β-trans-caffeoylbetulin (35),33 27-O-trans-caffeoylcylicodiscic acid (36),11 3-epi-betulinic acid (37),34 3-epi-betulinic acid acetate (38),35 betulonic acid (39),36 lupeol (40),37 oleanolic acid (41),38 3β-hydroxysitost-5-en-7-one (42),39 ergosterol peroxide (43),40 reevesioside A (44),1 and a mixture of reevesioside G (45), and epi-reevesioside G (46)1 from the stem of R. formosana were identified by comparison of their physical and spectroscopic data with values reported in the literatures.
Among the 46 compounds isolated, 25 compounds (1–10, 12–15, 19–24, and 29–33) had been tested for their cytotoxicity against the MCF-7, NCI–H460, and HepG2 cancer cell lines. The results for the active compounds are listed in Table 1. The results indicated that 3α,27-di-O-trans-caffeoylbetulinic acid (6) and secoisolariciresinol (13) displayed weak but selective cytotoxicity toward NCI–H460 and HepG2 cancer cell lines, respectively. While three cardenolides: strophanthojavoside (31) and ascleposide (32) with IC50 < 1 μM and strophalloside (33) displayed selective cytotoxicity to NCI–H460 with IC50 0.62 ± 0.06 μM as well. The bioactive compounds were provided as cardenolides, with the results corresponded to the previous reports of the root1 and fruits,2 suggested that cardenolides played an important role and contributed mainly to the cytotoxicity of this species as the major component.
| Compounds | IC50 (μM) | ||
|---|---|---|---|
| MCF-7 | NCI–H460 | HepG2 | |
| a Positive control. | |||
| 3α,27-Di-O-trans-caffeoylbetulinic acid (6) | 13.20 ± 0.90 | 7.60 ± 1.70 | 29.00 ± 0.80 |
| 5-O-Demethylbilagrewin (8) | 21.20 ± 1.10 | 39.80 ± 1.00 | 41.8 ± 2.20 |
| Secoisolariciresinol (13) | >50 | >50 | 31.94 ± 0.93 |
| Strophanthojavoside (31) | 0.77 ± 0.03 | 0.17 ± 0.01 | 0.65 ± 0.06 |
| Ascleposide (32) | 0.16 ± 0.02 | 0.03 ± 0.01 | 0.37 ± 0.02 |
| Strophalloside (33) | 3.46 ± 0.13 | 0.62 ± 0.06 | 2.59 ± 0.13 |
| Actinomycin Da | 0.01 ± 0.001 | 0.02 ± 0.005 | 0.10 ± 0.015 |
Through the bioassay screening among 1400 species of Formosan plants, R. formosana was found to be the most active one with the discovery to the new cytotoxic cardenolides. The phytochemistry of genus “Reevesia” hasn't been studied before our studies from the root1 and fruits2 of R. formosana, except for a report with five known compounds isolated from R. longipetiolata.41 The results of the investigation this time were coherent with the previous reports, both led to the isolation and identification of cytotoxic cardenolides. So far, 27 new compounds including 16 cardenolides (reevesiosides A–K and epi-reevesiosides F–I, K), five sesquiterpenoids (reevesiterpenols A–E), two glycosides (reevesianins A and B), three lignanoids, (reevesiacoumarin, reevesic acid, and reevesilignan), and one triterpenoid (3α,27-di-O-trans-caffeoylbetulinic acid), along with 65 known compounds were identified from the root, stem, and fruits of R. formosana. Three new sugar moieties 4,6-dideoxy-2,3-methylenedioxy-β-D-allopyranosyl, 4,6-dideoxy-2-O-methyl-β-D-allopyranosyl, and 6-deoxy-2,3-methylenedioxy-β-D-glucopyranosyl together with some rare sugar moieties are also found as the glycones of cardenolides in this species. Among these isolates, all cardenolides presented prominent cytotoxicities against the MCF-7, NCI–H460, and HepG2 cancer cell lines, and particularly, reevesiosides A, F, and epi-reevesioside F were in the nanomolar level.1 Reevesiterpenol E also displayed the best selective cytotoxicity to the NCI–H460 cell line.2
Therefore, the cardenolides and furanosesquiterpenoids from R. formosana are hopeful to be candidates for the discovery of anticancer compounds, primarily, the anti-cancer mechanisms had been studied by our research group. Such as reevesioside A induced G1 arrest and suppressed the expression of c-myc of human hormone-refractory prostate cancer,3 and the anti-proliferative activity of reevesioside F was confirmed to be Na+/K+-ATPase α3 subunit-dependent4 whereas the function of epi-reevesioside F was further identified to be PI3-kinase/Akt pathway related.5 The new compounds, reevesiterpenols A–D were isolated from R. formosana in our previous study2 and this study, were identified as furanosesquiterpenoids, which type was commonly found in genus Hibiscus (Malvaceae) before, and occurred in Sterculiaceae for the first time. Though the family of Sterculiaceae shared an intimate relationship with Malvaceae in plant taxonomy, there were no cardenolides revealed in Malvaceae. With entirely studied on the constituents of R. formosana, this species was standing as a unique one in the family of Sterculiaceae for the presence of cardenolides.
Experimental
General experimental procedures
The UV spectra were measured on a Jasco V-530 UV/VIS spectrophotometer; the IR spectra were recorded on a Jasco FTIR-4200 spectrophotometer (KBr or neat or ATR); optical rotations data were obtained with a JASCO P-2000 polarimeter; CD experiments were performed by a Jasco J-810 circular dichroism spectrophotometer. Silica gel (70–230 or 230–400 mesh, Merck) were used for column chromatography; TLC was carried out on precoated silica gel 60 F254 (Merck) for analytics and preparation; a spherical C18 100 Å (particle size: 20–40 μm) (Silicycle) was used for medium-pressure liquid chromatography. The NMR spectra were used methanol-d4 (1H, δ 3.31; 13C, δ 49.0), acetone-d6 (1H, δ 2.05; 13C, δ 30.5) or CDCl3 (1H, δ 7.26; 13C, δ 77.0) as solvent were recorded on Varian Gemini 2000–200 (200 MHz for 1H and 50 MHz for 13C NMR), Varian Unity Plus 400 (400 MHz for 1H and 100 MHz for 13C NMR) and Varian VNMRS-600 (600 MHz for 1H NMR and 150 MHz for 13C NMR) spectrometers. The EIMS data obtained on a VG-Biotech Quatro-5022 mass spectrometer: in m/z (rel.%). The HREIMS data were recorded on a Finnigan/Thermo Quest NAT mass spectrometer. The ESI/HRESIMS data obtained from a Bruker APEX-II mass spectrometer; in m/z.Plant material
The root and stem of R. formosana were collected from the Mudan Village, Pingtung County, Taiwan, in September 2009 and August 2010, respectively. They were identified by Prof. Ih-Sheng Chen, one of the authors, Kaohsiung Medical University, Kaohsiung, Taiwan. A voucher specimen (Chen 6117) was deposited in the Herbarium of the School of Pharmacy, College of Pharmacy, Kaohsiung Medical University.Extraction and isolation
The dried root of R. formosana (6.5 kg) was sliced and extracted with MeOH (30 L) at room temperature repeated three times, three days for each time. Evaporated in vacuo to obtain the methanolic extract (150 g), then partitioned into the EtOAc-soluble (45 g) and H2O-soluble parts (100 g). As the bioassay shown cytotoxicity toward both parts, the EtOAc-soluble part (45 g) eluted with n-hexane–EtOAc by silica gel CC (70–230 mesh) in the gradient to 12 fractions (A-1–A-12). The bioactive fractions are A-9–A-12 mentioned previously.1 Fraction A-2 (217 mg) was subjected to MPLC with n-hexane–EtOAc (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford seven fractions (A-2-1–A-2-7). Fraction A-2-4 (9.2 mg) purified with PTLC (RP-18, MeOH–CH2Cl2, 2:1) to obtain 29 (2.2 mg, Rf 0.29). Fraction A-2-6 (10.0 mg) treated through PTLC (n-hexane–acetone, 10:1) for three times then afforded a mixture of 16&17 (1.0 mg, Rf 0.33) and 18 (2.2 mg, Rf 0.57). Fraction A-3 (410 mg) subjected to MPLC (n-hexane–CH2Cl2–EtOAc, 20:1:1) to yield 12 fractions (A-3-1–A-3-12), and fraction A-3-7 (164 mg) was purified by MPLC (RP-18, acetone–MeOH, 1:3) to obtain a mixture of 27 & 28 (56 mg). Fraction A-5 (1.8 g) was crystallized from MeOH and afforded a mixture of 25 & 26 (722 mg). Fraction A-9 (3.6 g) went through MPLC (RP-18, MeOH–H2O, 1:1) and provided 10 fractions (A-9-1–A-9-10). Fraction A-9-2 (214 mg) eluted with CH2Cl2–MeOH (25:1) by MPLC to gain 10 fractions (A-9-2-1–A-9-2-10), and fraction A-9-2-4 (48.4 mg) further purified by PTLC (CH2Cl2–MeOH, 10:1) to give 23 (3.0 mg, Rf 0.32) and 24 (4.9 mg, Rf 0.45). Fraction A-9-2-5 (14.2 mg) further purified by PTLC (acetone–H2O, 1:2) to give 21 (3.0 mg, Rf 0.32). The application of PTLC (CH2Cl2–EtOAc, 40:1) to fraction A-9-4 (119 mg), then repeated four times to yield 20 (4.9 mg, Rf 0.59) and 22 (8.5 mg, Rf 0.43), respectively. Eluting with n-hexane–CH2Cl2–acetone (4:1:1) by MPLC, fraction A-9-5 (67.7 mg) afforded six fractions (A-9-5-1–A-9-5-6). Fraction A-9-5-4 (13.7 mg) purified with PTLC (CH2Cl2–EtOAc, 6:1) to give 5 (4.3 mg, Rf 0.26) and 19 (4.2 mg, Rf 0.15). Fraction A-9-5-6 (39.3 mg) eluted with MeOH–H2O (1:2) through MPLC to afford 10 (1.4 mg) and 12 (1.4 mg). Fraction A-9-6 (344 mg), eluted with n-hexane–CH2Cl2–acetone (2:1:1) by MPLC to gain nine fractions (A-9-6-1–A-9-6-9), and fraction A-9-6-7 (27.4 mg) further purified by PTLC (CH2Cl2–EtOAc, 5:1) to give 4 (2.0 mg, Rf 0.21). Fraction A-10 (3.6 g) went through Sephadex LH-20 column eluted with MeOH and separated into 13 fractions (A-10-1–A-10-13). Fraction A-10-4 (680 mg) through the elution of MeOH–H2O (3:2) with MPLC (RP-18) was parted into 10 fractions (A-10-4-1–A-10-4-10), and with the further purification of MPLC under the same solvent system to give 9 (2.4 mg), 11 (2.0 mg) and 13 (2.1 mg). Fraction A-10-7 (521 mg) separated to nine fractions via the application of MPLC (RP-18, MeOH–H2O, 1:1). Fraction A-10-7-1 (38.5 mg) was applied to PTLC (RP-18, acetone–H2O, 1:2) for three times repeatedly to yield 14 (19.0 mg, Rf 0.46) and fraction A-10-7-4 (143 mg) further followed the same steps of purification to obtain 2 (2.9 mg, Rf 0.25). As for fraction A-10-7-5 (49 mg) was purified by PTLC (RP-18) with solvent system MeOH–H2O (1:1) to provide 8 (14.6 mg, Rf 0.14) and 15 (3.6 mg, Rf 0.25). Fraction A-10-9 (26.9 mg) treated with PTLC (RP-18, acetone–H2O, 1:2) then 30 (11.6 mg, Rf 0.38) was yielded. Fraction A-11 (9.0 g) submitted to Sephadex LH-20 and eluted with MeOH to gain nine fractions (A-11-1–A-11-9). Fraction A-11-2 (741.1 mg) through the elution of MeOH–H2O (1:1) with MPLC (RP-18) was parted into 14 fractions (A-11-2-1–A-11-2-14), and fraction A-11-2-6 was further purified by MPLC under the same solvent system to give 31 (3.9 mg), 32 (34 mg), and 33 (2.0 mg). Fraction A-11-4 (1.5 g) was applied to MPLC (RP-18, MeOH–H2O, 1:2) and further purified by PTLC (RP-18, acetone–MeOH–H2O, 1:1:2) to provide 7 (13.1 mg, Rf 0.26), and continuing via PTLC (CH2Cl2–EtOAc, 1:3) to afford 3 (4.4 mg, Rf 0.37) eventually.
:1) to afford seven fractions (A-2-1–A-2-7). Fraction A-2-4 (9.2 mg) purified with PTLC (RP-18, MeOH–CH2Cl2, 2:1) to obtain 29 (2.2 mg, Rf 0.29). Fraction A-2-6 (10.0 mg) treated through PTLC (n-hexane–acetone, 10:1) for three times then afforded a mixture of 16&17 (1.0 mg, Rf 0.33) and 18 (2.2 mg, Rf 0.57). Fraction A-3 (410 mg) subjected to MPLC (n-hexane–CH2Cl2–EtOAc, 20:1:1) to yield 12 fractions (A-3-1–A-3-12), and fraction A-3-7 (164 mg) was purified by MPLC (RP-18, acetone–MeOH, 1:3) to obtain a mixture of 27 & 28 (56 mg). Fraction A-5 (1.8 g) was crystallized from MeOH and afforded a mixture of 25 & 26 (722 mg). Fraction A-9 (3.6 g) went through MPLC (RP-18, MeOH–H2O, 1:1) and provided 10 fractions (A-9-1–A-9-10). Fraction A-9-2 (214 mg) eluted with CH2Cl2–MeOH (25:1) by MPLC to gain 10 fractions (A-9-2-1–A-9-2-10), and fraction A-9-2-4 (48.4 mg) further purified by PTLC (CH2Cl2–MeOH, 10:1) to give 23 (3.0 mg, Rf 0.32) and 24 (4.9 mg, Rf 0.45). Fraction A-9-2-5 (14.2 mg) further purified by PTLC (acetone–H2O, 1:2) to give 21 (3.0 mg, Rf 0.32). The application of PTLC (CH2Cl2–EtOAc, 40:1) to fraction A-9-4 (119 mg), then repeated four times to yield 20 (4.9 mg, Rf 0.59) and 22 (8.5 mg, Rf 0.43), respectively. Eluting with n-hexane–CH2Cl2–acetone (4:1:1) by MPLC, fraction A-9-5 (67.7 mg) afforded six fractions (A-9-5-1–A-9-5-6). Fraction A-9-5-4 (13.7 mg) purified with PTLC (CH2Cl2–EtOAc, 6:1) to give 5 (4.3 mg, Rf 0.26) and 19 (4.2 mg, Rf 0.15). Fraction A-9-5-6 (39.3 mg) eluted with MeOH–H2O (1:2) through MPLC to afford 10 (1.4 mg) and 12 (1.4 mg). Fraction A-9-6 (344 mg), eluted with n-hexane–CH2Cl2–acetone (2:1:1) by MPLC to gain nine fractions (A-9-6-1–A-9-6-9), and fraction A-9-6-7 (27.4 mg) further purified by PTLC (CH2Cl2–EtOAc, 5:1) to give 4 (2.0 mg, Rf 0.21). Fraction A-10 (3.6 g) went through Sephadex LH-20 column eluted with MeOH and separated into 13 fractions (A-10-1–A-10-13). Fraction A-10-4 (680 mg) through the elution of MeOH–H2O (3:2) with MPLC (RP-18) was parted into 10 fractions (A-10-4-1–A-10-4-10), and with the further purification of MPLC under the same solvent system to give 9 (2.4 mg), 11 (2.0 mg) and 13 (2.1 mg). Fraction A-10-7 (521 mg) separated to nine fractions via the application of MPLC (RP-18, MeOH–H2O, 1:1). Fraction A-10-7-1 (38.5 mg) was applied to PTLC (RP-18, acetone–H2O, 1:2) for three times repeatedly to yield 14 (19.0 mg, Rf 0.46) and fraction A-10-7-4 (143 mg) further followed the same steps of purification to obtain 2 (2.9 mg, Rf 0.25). As for fraction A-10-7-5 (49 mg) was purified by PTLC (RP-18) with solvent system MeOH–H2O (1:1) to provide 8 (14.6 mg, Rf 0.14) and 15 (3.6 mg, Rf 0.25). Fraction A-10-9 (26.9 mg) treated with PTLC (RP-18, acetone–H2O, 1:2) then 30 (11.6 mg, Rf 0.38) was yielded. Fraction A-11 (9.0 g) submitted to Sephadex LH-20 and eluted with MeOH to gain nine fractions (A-11-1–A-11-9). Fraction A-11-2 (741.1 mg) through the elution of MeOH–H2O (1:1) with MPLC (RP-18) was parted into 14 fractions (A-11-2-1–A-11-2-14), and fraction A-11-2-6 was further purified by MPLC under the same solvent system to give 31 (3.9 mg), 32 (34 mg), and 33 (2.0 mg). Fraction A-11-4 (1.5 g) was applied to MPLC (RP-18, MeOH–H2O, 1:2) and further purified by PTLC (RP-18, acetone–MeOH–H2O, 1:1:2) to provide 7 (13.1 mg, Rf 0.26), and continuing via PTLC (CH2Cl2–EtOAc, 1:3) to afford 3 (4.4 mg, Rf 0.37) eventually.
The dried stem of R. formosana (7.0 kg) was sliced and extracted with MeOH (20 L) at room temperature repeated three times, three days for each time. Evaporated in vacuo to obtain the methanolic extract (160 g), then partitioned into the EtOAc-soluble (30 g) and H2O-soluble parts (100 g). As the bioassay shown cytotoxicity toward both parts, the EtOAc-soluble part (45 g) eluted with n-hexane–EtOAc by silica gel CC (70–230 mesh) in gradient to 19 fractions (B-1–B-19). The bioactive fractions B-7, B-12–B-19 tended to be polar and against the cancer cell lines mentioned previously. Fraction B-6 (3.0 g) was subjected to MPLC with n-hexane–acetone (12:1) to yield 11 fractions (B-6-1–B-6-11). Fraction B-6-5 (1.2 g) was crystallized from MeOH to afford a mixture of 25 & 26 (1.0 g). Fraction B-7 (531 mg) subjected to MPLC (n-hexane–EtOAc, 5:1) to yield nine fractions (B-7-1–B-7-9). Fraction B-7-4 (47.3 mg) purified with PTLC (CH2Cl2–EtOAc, 30:1) to obtain 38 (5.8 mg, Rf 0.69) and 40 (2.0 mg, Rf 0.26). Fraction B-7-5 (250 mg) eluted with n-hexane–acetone, 10:1 by MPLC to gain six fractions (B-7-5-1–B-7-5-6), and fraction B-7-5-3 (44.7 mg) purified with PTLC (CH2Cl2–EtOAc, 80:1) to obtain 39 (14.9 mg, Rf 0.50), and fraction B-7-5-4 (49 mg) purified with PTLC (CH2Cl2–EtOAc, 60:1) to give 37 (10.5 mg, Rf 0.66). Eluting with n-hexane–EtOAc (3:1) by MPLC to fraction B-9 (409 mg) afforded 10 fractions (B-9-1–B-9-10). Fraction B-9-3 (155 mg) went through MPLC (CH2Cl2–EtOAc, 30:1) and provided nine fractions (B-9-3-1–B-9-3-9). Fraction A-9-3-9 was to obtain 36 (13.5 mg). Fraction B-9-4 (42.7 mg) treated with PTLC (CH2Cl2–acetone, 15:1) then 41 (8.8 mg, Rf 0.24) was yielded. Fraction B-9-6 (48.4 mg) purified with PTLC (CH2Cl2–acetone, 15:1) to give 42 (5.4 mg, Rf 0.24) and 43 (7.5 mg, Rf 0.32). Fraction B-12 (1.64 g) went through MPLC (n-hexane–EtOAc, 3:1) and provided eight fractions (B-12-1–B-12-8). Fraction B-12-4 (233 mg) eluted with CH2Cl2–EtOAc (20:1) through MPLC to afford 10 fractions (B-12-4-1–B-12-4-10). Fraction B-12-4-5 (37.5 mg) further purified by PTLC (n-hexane–EtOAc, 2:1) to give 34 (15.2 mg, Rf 0.26). Fraction B-12-4-6 (93 mg) separated to seven fractions with the application of MPLC (n-hexane–EtOAc, 2:1), then fraction B-12-4-6-4 (38.8 mg) was applied to PTLC (n-hexane–acetone, 1:1) to yield 35 (7.7 mg, Rf 0.53). Fraction B-12-5 (441 mg) was subjected to MPLC with CH2Cl2–acetone (5:1) to afford 11 fractions (B-12-5-1–B-12-5-11). Fraction B-12-5-3 (78.9 mg) eluted with CH2Cl2–acetone (3:1) by MPLC to gain nine fractions (B-15-5-3-1–B-12-5-3-9), and fraction B-12-5-3-4 (9.0 mg) further purified by PTLC (CH2Cl2–acetone, 6:1) to give 20 (3.2 mg, Rf 0.48). Fraction B-13 (1.7 g) went through MPLC (CH2Cl2–acetone, 8:1) and provided 10 fractions (B-13-1–B-13-10). Fraction B-13-8 (36.4 mg) subjected to MPLC (CH2Cl2–MeOH, 20:1) to yield eight fractions (B-13-8-1–B-13-8-8). Fraction B-13-8-1 was to obtain 10 (15.7 mg). Fraction B-13-10 (1.4 g) eluted with CH2Cl2–MeOH (20:1) by MPLC to gain eight fractions (B-13-10-1–B-13-10-8), then fraction B-13-10-2 (271 mg) further purified by MPLC (RP-18, H2O–acetone, 1:1) to yield 10 fractions (B-13-10-2-1–B-13-2-10-10), then fraction B-13-10-2-8 was to afford a mixture of 45 and 46 (92.7 mg) and fraction B-13-10-2-9 was to give 44 (83 mg). Fraction B-14 (813 mg) submitted to Sephadex LH-20 eluted with MeOH and six fractions (B-14-1–B-14-6) were separated. Fraction B-14-2 were further applied to MPLC (RP-18, H2O–acetone, 2:1) to provide 6 (7.0 mg, Rf 0.24). Fraction B-15 (712 mg) submitted to Sephadex LH-20 with seven fractions (B-15-1–B-15-7). Fraction B-15-4 (145 mg) separated to nine fractions with the application of MPLC (RP-18, H2O–MeOH, 1.5:1) to afford 1 (9.5 mg, Rf 0.20). Fraction B-16 (1.63 g) went through Sephadex LH-20 column eluted with MeOH and separated into seven fractions (B-16-1–B-16-7). Fraction B-16-6 (600 mg) eluted with H2O–MeOH–acetone (1:1) by MPLC (RP-18) to gain eight fractions (B-16-6-1–B-16-6-10), and fraction B-16-6-1 was to afford 8 (32 mg, Rf 0.38). Fraction B-16-9 (210 mg) further purified by MPLC (RP-18, H2O–acetone, 2:1) to give 7 (3.0 mg, Rf 0.51).
ε) 237 sh (4.32), 320 (4.10) nm; UV (MeOH + KOH) λmax (logε) 322 (4.11) nm; CD (MeOH, Δε) 224 (−0.59), 236 (+0.39), 286 (+0.60) nm; IR (KBr) νmax 3420 (OH), 1708 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) cm−1; 1H NMR (acetone-d6, 400 MHz) δ 3.57 (1H, dd, J = 12.0, 3.6 Hz, H-9′b), 3.84 (3H, s, OCH3-3′), 3.85 (1H, dd, J = 12.0, 6.4 Hz, H-9′a), 3.96 (3H, s, OCH3-7), 4.08 (1H, ddd, J = 8.0, 6.4, 3.6 Hz, H-8′), 5.08 (1H, d, J = 8.0 Hz, H-7′), 6.15 (1H, d, J = 9.6 Hz, H-3), 6.61 (1H, s, H-8), 6.70 (1H, dd, J = 2.0, 0.6 Hz, H-6′), 6.73 (1H, d, J = 2.0 Hz, H-2′), 7.79 (1H, br s, OH, D2O exchangeable), 7.96 (1H, dd, J = 9.6, 0.6 Hz, H-4); 13C NMR (acetone-d6, 100 MHz) δ 57.2 (OCH3-3′), 57.4 (OCH3-7), 62.3 (C-9′), 78.8 (C-7′), 79.9 (C-8′), 94.1 (C-8), 104.5 (C-4a), 104.6 (C-2′), 110.1 (C-6′), 113.1 (C-3), 128.6 (C-1′), 131.3 (C-6), 136.1 (C-4′), 139.4 (C-4), 141.4 (C-5), 147.1 (C-5′), 149.8 (C-3′), 151.1 (C-8a), 154.2 (C-7), 161.8 (C-2); ESIMS m/z 403 [M + H]+; HRESIMS m/z 425.0845 [M + Na]+ (calcd for C20H18O9Na, 425.0848).ε) 210 (4.09), 229 sh (3.93), 299 (3.63) nm; UV (MeOH + KOH) λmax (logε) 220 (4.73), 305 (3.66) nm; IR (neat) νmax 3483 (OH), 1731 (CO) cm−1; 1H NMR (CD3OD, 600 MHz) δ 3.52 (1H, dd, J = 12.2, 5.7 Hz, H-9′b), 3.73 (1H, dd, J = 12.2, 2.7 Hz, H-9′a), 3.86 (3H, s, OCH3-3′), 3.91 (3H, s, OCH3-3), 4.03 (1H, ddd, J = 7.8, 5.7, 2.7 Hz, H-8′), 4.83 (1H, d, J = 7.8 Hz, H-7′), 6.37 (1H, d, J = 15.6 Hz, H-8), 6.57 (1H, dd, J = 1.8, 0.6 Hz, H-6′), 6.59 (1H, d, J = 1.8 Hz, H-2′), 6.79 (1H, dd, J = 1.8, 0.6 Hz, H-6), 6.84 (1H, d, J = 1.8 Hz, H-2), 7.37 (1H, d, J = 15.6 Hz, H-7); 13C NMR (CD3OD, 150 MHz) δ 56.68 (OCH3-3′), 56.73 (OCH3-3), 62.0 (C-9′), 77.7 (C-7′), 80.3 (C-8′), 104.0 (C-2′), 104.9 (C-2), 109.3 (C-6′), 110.9 (C-6), 122.8 (C-8), 128.5 (C-1′), 129.4 (C-1), 135.9 (C-4 and C-4′), 142.7 (C-7), 145.9 (C-5), 146.8 (C-5′), 149.8 (C-3′), 150.4 (C-3), 173.8 (CO); ESIMS m/z 427 [M + Na]+; HRESIMS m/z 427.09977 [M + Na]+ (calcd for C20H20O9Na, 427.09995).ε) 214 (4.75), 239 sh (4.26), 277 (3.61) nm; UV (MeOH + KOH) λmax (logε) 223 (4.90), 262 (4.03) nm; IR (neat) νmax 3407 (OH) cm−1; 1H NMR (CD3OD, 600 MHz) δ 3.11 (2H, m, H-8, H-8′), 3.51 (1H, dd, J = 12.6, 4.2 Hz, H-9′′b), 3.71 (1H, dd, J = 12.6, 2.4 Hz, H-9′′a), 3.84 (3H, s, OCH3-3′′), 3.85 (3H, s, OCH3-3), 3.86 (2H, m, H-9b, H-9′b), 3.88 (3H, s, OCH3-3′), 3.98 (1H, ddd, J = 7.8, 4.2, 2.4 Hz, H-8′′), 4.25 (2H, m, H-9a, H-9′a), 4.64 (1H, br d, J = 4.2 Hz, H-7′), 4.71 (1H, br dd, J = 4.8, 1.8 Hz, H-7), 4.80 (1H, d, J = 7.8 Hz, H-7′′), 6.49 (1H, br d, J = 1.8 Hz, H-6), 6.51 (1H, br d, J = 1.8 Hz, H-2), 6.55 (1H, br d, J = 2.4 Hz, H-6′′), 6.58 (1H, br d, J = 1.8 Hz, H-2′′), 6.60 (1H, br t, J = 1.8 Hz, H-6′), 6.64 (1H, br t, J = 1.8 Hz, H-2′); 13C NMR (CD3OD, 150 MHz) δ 55.4 (C-8), 55.5 (C-8′), 56.63 (OCH3-3′′), 56.67 (OCH3-3), 56.7 (OCH3-3′), 62.1 (C-9′′), 72.7 (C-9), 72.8 (C-9′), 77.8 (C-7′′), 80.0 (C-8′′), 87.3 (C-7′), 87.6 (C-7), 102.6 (C-2), 103.6 (C-2′), 104.0 (C-2′′), 107.8 (C-6), 108.5 (C-6′), 109.3 (C-6′′), 128.6 (C-1′ and C-1′′), 133.1 (C-1), 133.9 (C-4′), 134.8 (C-4), 135.9 (C-4′′), 145.7 (C-5′), 146.6 (C-5), 149.7 (C-3), 146.8 (C-5′′), 149.8 (C-3′′), 150.3 (C-3′); ESIMS m/z 607 [M + Na]+; HRESIMS m/z 607.1787 [M + Na]+ (calcd for C30H32O12Na, 607.1791).ε) 211 (4.30), 223 sh (4.01), 249 sh (3.88) nm; UV (MeOH + KOH) λmax (logε) 219 (4.57), 236 sh (4.07), 274 sh (3.85) nm; IR (neat) νmax 3417 (OH), 1682 (CO), 1557, 1538, 1516 (aromatic ring) cm−1; 1H NMR (CDCl3, 400 MHz) δ 1.16 (3H, s, H-14), 1.21 (3H, s, H-15), 2.42 (3H, s, H-12), 2.84 (1H, dd, J = 16.6, 6.9 Hz, H-7b), 3.06 (1H, dd, J = 16.6, 1.7 Hz, H-7a), 3.31 (1H, dd, J = 6.9, 1.7 Hz, H-6), 3.60 (1H, br s, OH-13, D2O exchangeable), 5.60 (1H, br s, OH-2, D2O exchangeable), 7.08 (1H, s, H-4), 7.99 (1H, s, H-11); 13C NMR (CDCl3, 100 MHz) δ 15.7 (C-12), 27.2 (C-14), 27.7 (C-15), 43.9 (C-7), 50.9 (C-6), 73.5 (C-13), 118.8 (C-9), 120.9 (C-5), 121.7 (C-3), 126.9 (C-4), 128.2 (C-10), 137.8 (C-2), 141.6 (C-1), 142.4 (C-11), 193.8 (C-8); ESIMS m/z 283 [M + Na]+; HRESIMS m/z 283.0947 [M + Na]+ (calcd for C15H16O4Na, 283.0946).ε) 230 (4.49), 264 (4.42) nm; IR (neat) νmax 3440 (OH), 1679 (CO) cm−1; 1H NMR (CDCl3, 400 MHz) δ 1.34 (3H, s, H-14), 1.35 (3H, s, H-15), 1.36 (3H, d, J = 7.6 Hz, H-12), 2.06 (1H, ddd, J = 13.6, 11.2, 3.2 Hz, H-6), 2.16 (1H, ddd, J = 14.9, 11.4, 4.4 Hz, H-4b), 2.36 (1H, dd, J = 16.8, 13.6 Hz, H-7b), 2.64 (1H, ddd, J = 14.9, 4.6, 2.4 Hz, H-4a), 2.76 (1H, dd, J = 16.8, 3.2 Hz, H-7a), 2.77 (1H, qdd, J = 7.6, 4.4, 2.4 Hz, H-3), 3.22 (1H, ddd, J = 11.4, 11.2, 4.6 Hz, H-5), 8.12 (1H, s, H-11); 13C NMR (CDCl3, 100 MHz) δ 16.1 (C-12), 24.9 (C-14), 30.6 (C-5), 30.7 (C-15), 38.8 (C-4), 42.9 (C-3), 43.7 (C-7), 52.4 (C-6), 73.1 (C-13), 123.0 (C-9), 143.9 (C-10), 145.1 (C-1), 147.5 (C-11), 189.0 (C-2), 192.3 (C-8); ESIMS m/z 285 [M + Na]+; HRESIMS m/z 285.1102 [M + Na]+ (calcd for C15H18O4Na, 285.1103).ε) 215 (4.49), 243 (4.32), 300 (4.43), 327 (4.55) nm; UV (MeOH + KOH) λmax (logε) 258 (4.25), 308 (4.17), 369 (4.64) nm; IR (neat) νmax 3335 (OH), 1697 (OCOCH), 1683 (COOH) cm−1; 1H NMR (CD3OD, 400 MHz) δ 0.86 (3H, s, H-23), 0.93 (3H, s, H-24), 0.96 (3H, s, H-25), 1.02 (1H, m, H-12b), 1.06 (3H, s, H-26), 1.29 (1H, m, H-11b), 1.30 (1H, m, H-1b), 1.34 (1H, m, H-16b), 1.41 (1H, m, H-5), 1.42 (1H, m, H-21b), 1.43 (1H, m, H-6b), 1.45 (1H, m, H-22b), 1.46 (1H, m, H-7b), 1.47 (1H, m, H-15b), 1.48 (1H, m, H-6a), 1.54 (1H, m, H-1a), 1.56 (1H, m, H-11a), 1.58 (1H, m, H-7a), 1.61 (1H, m, H-9), 1.64 (1H, m, H-2b), 1.73 (3H, s, H-30), 1.79 (1H, m, H-18), 1.80 (1H, m, H-12a), 1.88 (1H, m, H-15a), 1.95 (1H, m, H-22a), 1.96 (1H, m, H-21a), 2.00 (1H, m, H-2a), 2.32 (1H, br d, J = 12.4 Hz, H-16a), 2.54 (1H, td, J = 12.6, 3.6 Hz, H-13), 3.07 (1H, td, J = 10.4, 4.8 Hz, H-19), 4.52 (1H, d, J = 12.8 Hz, H-27b), 4.62 (1H, br s, H-29b), 4.69 (1H, br s, H-3), 4.75 (1H, br s, H-29a), 4.88 (1H, d, J = 12.8 Hz, H-27a), 6.287 (1H, d, J = 16.0 Hz, H-8′′), 6.291 (1H, d, J = 16.0 Hz, H-8′), 6.75 (1H, d, J = 8.4 Hz, H-5′), 6.80 (1H, d, J = 8.4 Hz, H-5′′), 6.90 (1H, dd, J = 8.4, 2.0 Hz, H-6′), 7.015 (1H, dd, J = 8.4, 2.0 Hz, H-6′′), 7.018 (1H, d, J = 2.0 Hz, H-2′), 7.11 (1H, d, J = 2.0 Hz, H-2′′), 7.56 (1H, d, J = 16.0 Hz, H-7′′), 7.58 (1H, d, J = 16.0 Hz, H-7′); 13C NMR (CD3OD, 100 MHz) δ 16.9 (C-25), 17.2 (C-26), 19.2 (C-6), 19.6 (C-30), 22.0 (C-11), 22.2 (C-24), 24.0 (C-2), 25.3 (C-15), 26.6 (C-12), 28.5 (C-23), 31.6 (C-21), 33.8 (C-16), 35.5 (C-1), 36.5 (C-7), 37.9 (C-22), 38.1 (C-4), 38.7 (C-10), 40.3 (C-13), 42.9 (C-8), 46.9 (C-14), 48.4 (C-19), 50.6 (C-18), 52.0 (C-5), 53.2 (C-9), 57.3 (C-17), 64.4 (C-27), 79.5 (C-3), 110.5 (C-29), 115.1 (C-2′′), 115.2 (C-2′), 115.2 (C-8′), 115.7 (C-8′′), 116.5 (C-5′, C-5′′), 123.1 (C-6′), 123.4 (C-6′′), 127.6 (C-1′), 127.8 (C-1′′), 146.7 (C-7′′), 146.8 (C-3′′), 147.2 (C-3′), 147.2 (C-7′), 149.5 (C-4′′), 149.7 (C-4′), 151.7 (C-20), 168.9 (C-9′), 169.5 (C-9′′), 179.9 (C-28); ESIMS m/z 797 [M + H]+; HRESIMS m/z 819.4089 [M + Na]+ (calcd for C48H60O10Na, 819.4089).
O) cm−1; 1H NMR (acetone-d6, 400 MHz) δ 3.57 (1H, dd, J = 12.0, 3.6 Hz, H-9′b), 3.84 (3H, s, OCH3-3′), 3.85 (1H, dd, J = 12.0, 6.4 Hz, H-9′a), 3.96 (3H, s, OCH3-7), 4.08 (1H, ddd, J = 8.0, 6.4, 3.6 Hz, H-8′), 5.08 (1H, d, J = 8.0 Hz, H-7′), 6.15 (1H, d, J = 9.6 Hz, H-3), 6.61 (1H, s, H-8), 6.70 (1H, dd, J = 2.0, 0.6 Hz, H-6′), 6.73 (1H, d, J = 2.0 Hz, H-2′), 7.79 (1H, br s, OH, D2O exchangeable), 7.96 (1H, dd, J = 9.6, 0.6 Hz, H-4); 13C NMR (acetone-d6, 100 MHz) δ 57.2 (OCH3-3′), 57.4 (OCH3-7), 62.3 (C-9′), 78.8 (C-7′), 79.9 (C-8′), 94.1 (C-8), 104.5 (C-4a), 104.6 (C-2′), 110.1 (C-6′), 113.1 (C-3), 128.6 (C-1′), 131.3 (C-6), 136.1 (C-4′), 139.4 (C-4), 141.4 (C-5), 147.1 (C-5′), 149.8 (C-3′), 151.1 (C-8a), 154.2 (C-7), 161.8 (C-2); ESIMS m/z 403 [M + H]+; HRESIMS m/z 425.0845 [M + Na]+ (calcd for C20H18O9Na, 425.0848).ε) 210 (4.09), 229 sh (3.93), 299 (3.63) nm; UV (MeOH + KOH) λmax (logε) 220 (4.73), 305 (3.66) nm; IR (neat) νmax 3483 (OH), 1731 (CO) cm−1; 1H NMR (CD3OD, 600 MHz) δ 3.52 (1H, dd, J = 12.2, 5.7 Hz, H-9′b), 3.73 (1H, dd, J = 12.2, 2.7 Hz, H-9′a), 3.86 (3H, s, OCH3-3′), 3.91 (3H, s, OCH3-3), 4.03 (1H, ddd, J = 7.8, 5.7, 2.7 Hz, H-8′), 4.83 (1H, d, J = 7.8 Hz, H-7′), 6.37 (1H, d, J = 15.6 Hz, H-8), 6.57 (1H, dd, J = 1.8, 0.6 Hz, H-6′), 6.59 (1H, d, J = 1.8 Hz, H-2′), 6.79 (1H, dd, J = 1.8, 0.6 Hz, H-6), 6.84 (1H, d, J = 1.8 Hz, H-2), 7.37 (1H, d, J = 15.6 Hz, H-7); 13C NMR (CD3OD, 150 MHz) δ 56.68 (OCH3-3′), 56.73 (OCH3-3), 62.0 (C-9′), 77.7 (C-7′), 80.3 (C-8′), 104.0 (C-2′), 104.9 (C-2), 109.3 (C-6′), 110.9 (C-6), 122.8 (C-8), 128.5 (C-1′), 129.4 (C-1), 135.9 (C-4 and C-4′), 142.7 (C-7), 145.9 (C-5), 146.8 (C-5′), 149.8 (C-3′), 150.4 (C-3), 173.8 (CO); ESIMS m/z 427 [M + Na]+; HRESIMS m/z 427.09977 [M + Na]+ (calcd for C20H20O9Na, 427.09995).ε) 214 (4.75), 239 sh (4.26), 277 (3.61) nm; UV (MeOH + KOH) λmax (logε) 223 (4.90), 262 (4.03) nm; IR (neat) νmax 3407 (OH) cm−1; 1H NMR (CD3OD, 600 MHz) δ 3.11 (2H, m, H-8, H-8′), 3.51 (1H, dd, J = 12.6, 4.2 Hz, H-9′′b), 3.71 (1H, dd, J = 12.6, 2.4 Hz, H-9′′a), 3.84 (3H, s, OCH3-3′′), 3.85 (3H, s, OCH3-3), 3.86 (2H, m, H-9b, H-9′b), 3.88 (3H, s, OCH3-3′), 3.98 (1H, ddd, J = 7.8, 4.2, 2.4 Hz, H-8′′), 4.25 (2H, m, H-9a, H-9′a), 4.64 (1H, br d, J = 4.2 Hz, H-7′), 4.71 (1H, br dd, J = 4.8, 1.8 Hz, H-7), 4.80 (1H, d, J = 7.8 Hz, H-7′′), 6.49 (1H, br d, J = 1.8 Hz, H-6), 6.51 (1H, br d, J = 1.8 Hz, H-2), 6.55 (1H, br d, J = 2.4 Hz, H-6′′), 6.58 (1H, br d, J = 1.8 Hz, H-2′′), 6.60 (1H, br t, J = 1.8 Hz, H-6′), 6.64 (1H, br t, J = 1.8 Hz, H-2′); 13C NMR (CD3OD, 150 MHz) δ 55.4 (C-8), 55.5 (C-8′), 56.63 (OCH3-3′′), 56.67 (OCH3-3), 56.7 (OCH3-3′), 62.1 (C-9′′), 72.7 (C-9), 72.8 (C-9′), 77.8 (C-7′′), 80.0 (C-8′′), 87.3 (C-7′), 87.6 (C-7), 102.6 (C-2), 103.6 (C-2′), 104.0 (C-2′′), 107.8 (C-6), 108.5 (C-6′), 109.3 (C-6′′), 128.6 (C-1′ and C-1′′), 133.1 (C-1), 133.9 (C-4′), 134.8 (C-4), 135.9 (C-4′′), 145.7 (C-5′), 146.6 (C-5), 149.7 (C-3), 146.8 (C-5′′), 149.8 (C-3′′), 150.3 (C-3′); ESIMS m/z 607 [M + Na]+; HRESIMS m/z 607.1787 [M + Na]+ (calcd for C30H32O12Na, 607.1791).ε) 211 (4.30), 223 sh (4.01), 249 sh (3.88) nm; UV (MeOH + KOH) λmax (logε) 219 (4.57), 236 sh (4.07), 274 sh (3.85) nm; IR (neat) νmax 3417 (OH), 1682 (CO), 1557, 1538, 1516 (aromatic ring) cm−1; 1H NMR (CDCl3, 400 MHz) δ 1.16 (3H, s, H-14), 1.21 (3H, s, H-15), 2.42 (3H, s, H-12), 2.84 (1H, dd, J = 16.6, 6.9 Hz, H-7b), 3.06 (1H, dd, J = 16.6, 1.7 Hz, H-7a), 3.31 (1H, dd, J = 6.9, 1.7 Hz, H-6), 3.60 (1H, br s, OH-13, D2O exchangeable), 5.60 (1H, br s, OH-2, D2O exchangeable), 7.08 (1H, s, H-4), 7.99 (1H, s, H-11); 13C NMR (CDCl3, 100 MHz) δ 15.7 (C-12), 27.2 (C-14), 27.7 (C-15), 43.9 (C-7), 50.9 (C-6), 73.5 (C-13), 118.8 (C-9), 120.9 (C-5), 121.7 (C-3), 126.9 (C-4), 128.2 (C-10), 137.8 (C-2), 141.6 (C-1), 142.4 (C-11), 193.8 (C-8); ESIMS m/z 283 [M + Na]+; HRESIMS m/z 283.0947 [M + Na]+ (calcd for C15H16O4Na, 283.0946).ε) 230 (4.49), 264 (4.42) nm; IR (neat) νmax 3440 (OH), 1679 (CO) cm−1; 1H NMR (CDCl3, 400 MHz) δ 1.34 (3H, s, H-14), 1.35 (3H, s, H-15), 1.36 (3H, d, J = 7.6 Hz, H-12), 2.06 (1H, ddd, J = 13.6, 11.2, 3.2 Hz, H-6), 2.16 (1H, ddd, J = 14.9, 11.4, 4.4 Hz, H-4b), 2.36 (1H, dd, J = 16.8, 13.6 Hz, H-7b), 2.64 (1H, ddd, J = 14.9, 4.6, 2.4 Hz, H-4a), 2.76 (1H, dd, J = 16.8, 3.2 Hz, H-7a), 2.77 (1H, qdd, J = 7.6, 4.4, 2.4 Hz, H-3), 3.22 (1H, ddd, J = 11.4, 11.2, 4.6 Hz, H-5), 8.12 (1H, s, H-11); 13C NMR (CDCl3, 100 MHz) δ 16.1 (C-12), 24.9 (C-14), 30.6 (C-5), 30.7 (C-15), 38.8 (C-4), 42.9 (C-3), 43.7 (C-7), 52.4 (C-6), 73.1 (C-13), 123.0 (C-9), 143.9 (C-10), 145.1 (C-1), 147.5 (C-11), 189.0 (C-2), 192.3 (C-8); ESIMS m/z 285 [M + Na]+; HRESIMS m/z 285.1102 [M + Na]+ (calcd for C15H18O4Na, 285.1103).ε) 215 (4.49), 243 (4.32), 300 (4.43), 327 (4.55) nm; UV (MeOH + KOH) λmax (logε) 258 (4.25), 308 (4.17), 369 (4.64) nm; IR (neat) νmax 3335 (OH), 1697 (OCOCH), 1683 (COOH) cm−1; 1H NMR (CD3OD, 400 MHz) δ 0.86 (3H, s, H-23), 0.93 (3H, s, H-24), 0.96 (3H, s, H-25), 1.02 (1H, m, H-12b), 1.06 (3H, s, H-26), 1.29 (1H, m, H-11b), 1.30 (1H, m, H-1b), 1.34 (1H, m, H-16b), 1.41 (1H, m, H-5), 1.42 (1H, m, H-21b), 1.43 (1H, m, H-6b), 1.45 (1H, m, H-22b), 1.46 (1H, m, H-7b), 1.47 (1H, m, H-15b), 1.48 (1H, m, H-6a), 1.54 (1H, m, H-1a), 1.56 (1H, m, H-11a), 1.58 (1H, m, H-7a), 1.61 (1H, m, H-9), 1.64 (1H, m, H-2b), 1.73 (3H, s, H-30), 1.79 (1H, m, H-18), 1.80 (1H, m, H-12a), 1.88 (1H, m, H-15a), 1.95 (1H, m, H-22a), 1.96 (1H, m, H-21a), 2.00 (1H, m, H-2a), 2.32 (1H, br d, J = 12.4 Hz, H-16a), 2.54 (1H, td, J = 12.6, 3.6 Hz, H-13), 3.07 (1H, td, J = 10.4, 4.8 Hz, H-19), 4.52 (1H, d, J = 12.8 Hz, H-27b), 4.62 (1H, br s, H-29b), 4.69 (1H, br s, H-3), 4.75 (1H, br s, H-29a), 4.88 (1H, d, J = 12.8 Hz, H-27a), 6.287 (1H, d, J = 16.0 Hz, H-8′′), 6.291 (1H, d, J = 16.0 Hz, H-8′), 6.75 (1H, d, J = 8.4 Hz, H-5′), 6.80 (1H, d, J = 8.4 Hz, H-5′′), 6.90 (1H, dd, J = 8.4, 2.0 Hz, H-6′), 7.015 (1H, dd, J = 8.4, 2.0 Hz, H-6′′), 7.018 (1H, d, J = 2.0 Hz, H-2′), 7.11 (1H, d, J = 2.0 Hz, H-2′′), 7.56 (1H, d, J = 16.0 Hz, H-7′′), 7.58 (1H, d, J = 16.0 Hz, H-7′); 13C NMR (CD3OD, 100 MHz) δ 16.9 (C-25), 17.2 (C-26), 19.2 (C-6), 19.6 (C-30), 22.0 (C-11), 22.2 (C-24), 24.0 (C-2), 25.3 (C-15), 26.6 (C-12), 28.5 (C-23), 31.6 (C-21), 33.8 (C-16), 35.5 (C-1), 36.5 (C-7), 37.9 (C-22), 38.1 (C-4), 38.7 (C-10), 40.3 (C-13), 42.9 (C-8), 46.9 (C-14), 48.4 (C-19), 50.6 (C-18), 52.0 (C-5), 53.2 (C-9), 57.3 (C-17), 64.4 (C-27), 79.5 (C-3), 110.5 (C-29), 115.1 (C-2′′), 115.2 (C-2′), 115.2 (C-8′), 115.7 (C-8′′), 116.5 (C-5′, C-5′′), 123.1 (C-6′), 123.4 (C-6′′), 127.6 (C-1′), 127.8 (C-1′′), 146.7 (C-7′′), 146.8 (C-3′′), 147.2 (C-3′), 147.2 (C-7′), 149.5 (C-4′′), 149.7 (C-4′), 151.7 (C-20), 168.9 (C-9′), 169.5 (C-9′′), 179.9 (C-28); ESIMS m/z 797 [M + H]+; HRESIMS m/z 819.4089 [M + Na]+ (calcd for C48H60O10Na, 819.4089).Cytotoxicity assay
HepG2 (liver hepatocellular cells, [ATCC HB-8065]), NCI–H460 (nonsmall-cell lung cancer, [ATCCHTB-177]), and MCF-7 (human breast adenocarcinoma, [ATCC HTB-22]) cancer cells were seeded in 96-well microtiter plates in 100 μL culture medium per well at cell numbers of 10000, 2500, and 6500, respectively. HepG2 and MCF-7 were cultured in Dulbeccos modified Eagles medium (Hyclone Laboratory Inc.), NCI–H640 was cultured in RPMI-1640 medium (GIBCO-Life Technologies, Inc.), supplemented with 10% fetal calf serum (Biological Industries Inc.) and nonessential amino acid (Biological Industries, Inc.) and maintained at 37 °C in a humidified incubator with an atmosphere of 5% CO2. The cytotoxicity assay was performed as described.
Conclusions
In summary, we investigated the stem of R. formosana and the remaining fractions of the root of R. formosana led to the isolation of six new compounds including three lignanoids: reevesiacoumarin (1), reevesic acid (2), and reevesilignan (3), and three terpenoids: reevesiterpenol A (4), reevesiterpenol B (5), and 3α,27-di-O-trans-caffeoylbetulinic acid (6), along with 40 known compounds. In our serious studies found that all cardenolides presented prominent cytotoxicities against the MCF-7, NCI–H460, and HepG2 cancer cell lines and some terpenoids and lignans showed selective cytotoxic activities. Therefore, compounds isolated from R. formosana could potentially support the development of anticancer therapies.Acknowledgements
This work was supported by the Ministry of Science and Technology (MOST 105-2320-B-037-002) and was supported partially by the Kaohsiung Medical University “Aim for the Top Universities Grant,” Grant No. KMU-TP105E30. We thank Miss Chyi-Jia Wang and Mr Min-Yuan Hung of the Center for Research Resources and Development of Kaohsiung Medical University, Kaohsiung, Taiwan, for NMR and ESIMS measurements, respectively.Notes and references
- H. S. Chang, M. Y. Chiang, H. Y. Hsu, C. W. Yang, C. H. Lin, S. J. Lee and I. S. Chen, Phytochemistry, 2013, 87, 86–95 CrossRef CAS PubMed.
- P. Y. Hsiao, S. J. Lee, I. S. Chen, H. Y. Hsu and H. S. Chang, Phytochemistry, 2016, 130, 282–290 CrossRef CAS PubMed.
- W. J. Leu, H. S. Chang, S. H. Chan, J. L. Hsu, C. C. Yu, L. C. Hsu, I. S. Chen and J. H. Guh, PLoS One, 2014, 9, e87323 Search PubMed.
- S. H. Chan, W. J. Leu, L. C. Hsu, H. S. Chang, T. L. Hwang, I. S. Chen, C. S. Chen and J. H. Guh, Biochem. Pharmacol., 2013, 86, 1564–1575 CrossRef CAS PubMed.
- J. L. Hsu, F. L. Liu, L. C. Hsu, H. S. Chang, W. J. Leu, C. C. Yu, W. L. Chang, I. S. Chen, F. L. Kung and J. H. Guh, OncoTargets Ther., 2015, 6, 24032–24046 CrossRef PubMed.
- D. Zhou, Y. Zhang, Z. Jiang, Y. Hou, C. Yan and N. Li, Bioorg. Med. Chem. Lett., 2017, 27, 248–253 CrossRef CAS PubMed.
- S. Kumar, A. B. Ray, C. Konno, Y. Oshima and H. Hikino, Phytochemistry, 1988, 27, 636–638 CrossRef CAS.
- T. H. Kim, H. Ito, K. Hayashi, T. Hasegawa, T. Machiguchi and T. Yoshida, Chem. Pharm. Bull., 2005, 53, 641–644 CrossRef CAS PubMed.
- Y. K. Son, M. H. Lee and Y. N. Han, Arch. Pharmacal Res., 2005, 28, 34–38 CrossRef CAS.
- M. A. Ferreira, T. J. King, S. Ali and R. H. Thomson, J. Chem. Soc., Perkin Trans. 1, 1980, 249–256 RSC.
- P. Pailee, V. Prachyawarakorn, C. Mahidol, S. Ruchirawat and P. Kittakoop, Eur. J. Org. Chem., 2011, 2011, 3809–3814 CrossRef CAS.
- B. M. Shashi and P. K. Asish, Phytochemistry, 1994, 37, 1517–1575 CrossRef.
- P. Puapairoj, W. Naengchomnong, A. Kijjoa, M. M. Pinto, M. Pedro, M. S. Nascimento, A. M. Silva and W. Herz, Planta Med., 2005, 71, 208–213 CrossRef CAS PubMed.
- Y. Matsuo and Y. Mimaki, Chem. Pharm. Bull., 2010, 58, 587–590 CrossRef CAS PubMed.
- J. F. Xu, Z. M. Feng, J. Liu and P. C. Zhang, Chem. Biodiversity, 2008, 5, 591–597 CAS.
- F. Abe and T. Yamauchi, Phytochemistry, 1988, 27, 575–577 CrossRef CAS.
- A. Bardon, S. Montanaro, C. A. N. Catalan, J. G. Diaz and W. Herz, Phytochemistry, 1993, 34, 253–259 CrossRef CAS.
- L. B. Davin, D. L. Bedgar, T. Katayama and N. G. Lewis, Phytochemistry, 1992, 31, 3869–3874 CrossRef CAS PubMed.
- T. Murata, K. Sasaki, K. Sato, F. Yoshizaki, H. Yamada, H. Mutoh, K. Umehara, T. Miyase, T. Warashina, H. Aoshima, H. Tabata and K. Matsubara, J. Nat. Prod., 2009, 72, 1379–1384 CrossRef CAS PubMed.
- G. Davila-Huerta, H. Hamada, G. D. Davis, R. D. Stipanovic, C. M. Adams and M. Essenberg, Phytochemistry, 1995, 39, 531–536 CrossRef CAS.
- A. V. B. Sankaram, N. S. Reddy and J. N. Shoolery, Phytochemistry, 1981, 20, 1877–1881 CrossRef CAS.
- S. De Marino, N. Borbone, F. Zollo, A. Ianaro, P. Di Meglio and M. Iorizzi, J. Agric. Food Chem., 2004, 52, 7525–7531 CrossRef CAS PubMed.
- L. Li and N. P. Seeram, J. Agric. Food Chem., 2010, 58, 11673–11679 CrossRef CAS PubMed.
- E. Okuyama, T. Hasegawa, T. Matsushita, H. Fujimoto, M. Ishibashi and M. Yamazaki, Chem. Pharm. Bull., 2001, 49, 154–160 CrossRef CAS PubMed.
- Y. F. Kang, C. M. Liu, C. L. Kao and C. Y. Chen, Molecules, 2014, 19, 4234–4245 CrossRef PubMed.
- S. Xu, M. Y. Shang, G. X. Liu, F. Xu, X. Wang, C. C. Shou and S. Q. Cai, Molecules, 2013, 18, 5265–5287 CrossRef CAS PubMed.
- H. Choi and M. P. Doyle, Org. Lett., 2007, 9, 5349–5352 CrossRef CAS PubMed.
- B. H. Lipshutz, P. Mollard, S. S. Pfeiffer and W. Chrisman, J. Am. Chem. Soc., 2002, 124, 14282–14283 CrossRef CAS PubMed.
- H. Lou, Y. Yamazaki, T. Sasaki, M. Uchida, H. Tanaka and S. Oka, Phytochemistry, 1999, 51, 297–308 CrossRef CAS.
- P. Müehlradt, E. Weiss and T. Reichstein, Helv. Chim. Acta, 1964, 47, 2164–2186 CrossRef.
- A. M. Kuritzkes, C. Tamm, H. Jäger and T. Reichstein, Helv. Chim. Acta, 1963, 46, 8–23 CrossRef.
- Rudiyansyah, L. K. Lambert and M. J. Garson, J. Nat. Prod., 2010, 73, 1649–1654 CrossRef CAS PubMed.
- H. Pan, L. N. Lundgren and R. Andersson, Phytochemistry, 1994, 37, 795–799 CrossRef CAS.
- W. Herz, P. S. Santhanam and I. Wahlberg, Phytochemistry, 1972, 11, 3061–3063 CrossRef CAS.
- H. C. Wu, M. J. Cheng, C. F. Peng, S. C. Yang, H. S. Chang, C. H. Lin, C. J. Wang and I. S. Chen, Phytochemistry, 2012, 82, 118–127 CrossRef CAS PubMed.
- A. Barthel, S. Stark and R. Csuk, Tetrahedron, 2008, 64, 9225–9229 CrossRef CAS.
- T. K. Razdan, S. Harkar, B. Qadri, M. A. Qurishi and M. A. Khuroo, Phytochemistry, 1988, 27, 1890–1892 CrossRef CAS.
- H. Saimaru, Y. Orihara, P. Tansakul, Y. H. Kang, M. Shibuya and Y. Ebizuka, Chem. Pharm. Bull., 2007, 55, 784–788 CrossRef CAS PubMed.
- X. Zhang, P. Geoffroy, M. Miesch, J. D. Diane, F. Raul, A. W. Dalal and E. Marchioni, Steroids, 2005, 70, 886–895 CrossRef CAS PubMed.
- H. S. Chang, M. J. Cheng and I. S. Chen, Helv. Chim. Acta, 2011, 94, 703–710 CrossRef CAS.
- H. Zhu, P. F. Tu, Q. Chen and A. L. Xu, Zhongcaoyao, 2003, 34, 976–978 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04255h |
| This journal is © The Royal Society of Chemistry 2017 |