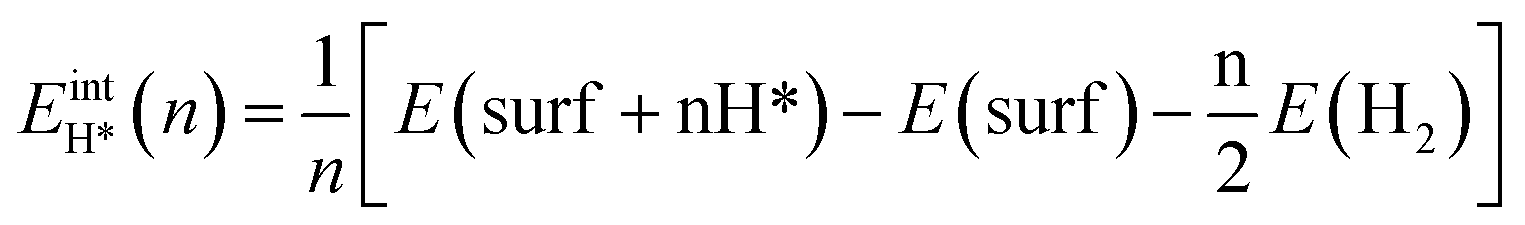DOI:
10.1039/C7RA04115B
(Paper)
RSC Adv., 2017,
7, 27033-27039
Theoretical evaluation of the structure–activity relationship in graphene-based electrocatalysts for hydrogen evolution reactions†
Received
11th April 2017
, Accepted 15th May 2017
First published on 22nd May 2017
Abstract
We systematically analyzed the relationship between structure and electrocatalytic activity of heteroatom-doped graphenes (GXs, where G and X represent graphene and the heteroatom dopant) for the hydrogen evolution reaction (HER). We compared the doping effects on the electronic structure and HER activity with the second row elements (B and N) and third row elements (Si, P and S) in the periodic table. In this work, we present evidence that structural deformation and periodic lattice defects play a fundamental role in the HER activity of GXs by adjusting the electronic properties of graphene. We found that graphene doped with third row elements has higher HER activity with out-of-plane structural deformation compared to graphene doped with second row elements, in which graphene tends to maintain its planar structure. In addition, the third row element-doped graphenes (GSi, GP and GS) show an interesting physical regularity described by a simple 3N rule: GXs provide outstanding HER activity with a sustained metallic property when its primitive cell size is a 3N × 3N (N is integral) multiple of that of pure graphene. Therefore, we discuss how a comprehensive understanding of the structure–activity relationship can explain the behavior of new electrocatalytic materials.
Introduction
Molecular hydrogen is an alternative to fossil fuels.1 However, efficient and sustainable H2 production technologies are challenging because hydrogen does not exist as a free molecule, although hydrogen is the most abundant element on earth. The steam reforming process of natural gases now commercially produces the most H2.2 However, this H2 production method still emits CO2 by consuming fossil fuels. One of the more environmentally kind methods is electrolysis of water using renewable energy, in particular solar energy.3,4 Water splitting can provide a sustainable energy supply via electrocatalytic reduction of water to molecular hydrogen; however it requires a favorable catalyst to support a sufficient reaction rate in order to have practical applications. Developing catalysts to facilitate the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) is an area of ongoing research. Among the available catalysts, the most popular HER electrocatalyst is platinum (Pt), which has a particularly high exchange current density and low Tafel slope.5 However, the main drawbacks of Pt catalysts are their high cost and limited availability. To assure sustainable hydrogen production, cost-effective alternatives to precious, rare Pt catalysts should be developed with similar electrocatalytic activities and robust stabilities.6–12
A large number of candidates have been developed based on various earth–abundant transition metals13–20 and their compounds, such as transition metal chalcogenide (TMD).21–28 MoS2 has also been considered as a potential HER electrocatalyst. Although only the edges of the MoS2 domain sheet are active for HER, comparatively high HER activity compared to Pt catalyst has been achieved through edge engineering,22,23 nanostructuring24–26 and a MoS2/carbon hybrid system.27–31 In addition, carbon-based materials (e.g., graphene) as metal-free catalysts32–40 have also been considered as innovative alternatives to Pt catalyst due to their abundance, variable structures, and tunable composition. Chemical doping of graphene with heteroatoms is an effective way to manipulate its electronic structure and electrochemical properties.41–44 Both experimental and theoretical research reveal that the effects of various dopants can improve the electrocatalytic activities of graphene. In these graphene-based metal-free electrocatalysts, the chemical dopants and geometric lattice defects of graphene play an important role in the superior HER catalysis. However, not only the fundamental effect of dopants but also tuning the electronic properties of doped graphene is still being studied.
In this work, we describe a structure–activity relationship in graphene-based electrocatalysts for HER based on a thorough understanding of the effects of dopants, such as second row elements (B and N) and third row elements (Si, P and S). We explore the causes of variation in HER performance with respect to the type of dopant by comparing geometric and electronic structures of heteroatom-doped graphenes (GXs, where G and X represent graphene and the heteroatom dopant). In addition, we have investigated the effect of doping level by systematically changing primitive cell size, where we uncovered an interesting regularity in HER activity attributed to periodic lattice defects.
Computational details
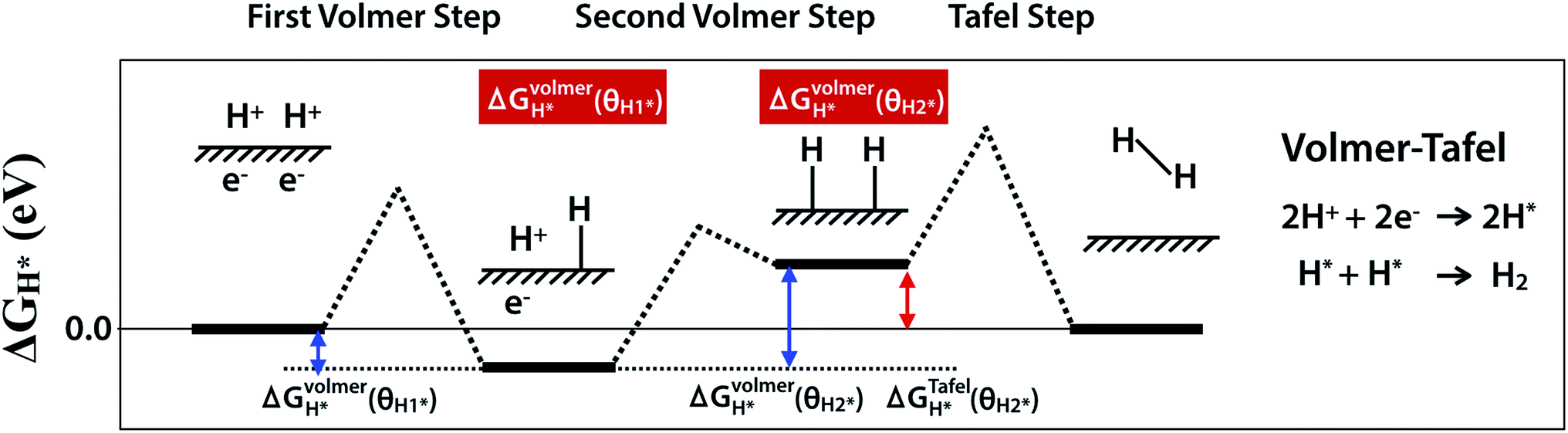
HER is a multi-step process that takes place on the surface of catalyst and there are two proposed mechanisms, Volmer–Tafel and Volmer–Heyrovsky. Both Volmer–Tafel and Volmer–Heyrovsky mechanisms describe the hydrogen atom adsorption and hydrogen molecule desorption reactions among (1) an initial state 2H+ + 2e−, (2) an intermediate adsorbed state H* + H+ + e− or 2H*, and (3) a final product state H2, where the * and H* denote the active site and adsorbed hydrogen atom on the surface of the catalyst, respectively.45,46 Because the initial and final states are equivalent at equilibrium reduction potential, U = 0, the Gibbs free energy of the intermediate state, |ΔGH*|, has been considered as a major descriptor of the HER activity for a wide variety of catalysts. Therefore, the optimum value of |ΔGH*| should be zero for a spontaneous reaction without activation energy barrier. The Pt catalyst facilitates HER with a low activation energy, |ΔGH*|∼0.09 eV.45 In this work, we have considered the Volmer–Tafel mechanism to calculate the Gibbs free energy of the intermediate state, ΔGVolmerH*(θH1*), ΔGVolmerH*(θH2*), and, ΔGTafelH*(θH2*) with different hydrogen coverage at the active sites, θH1* and, θH2*, as shown in Fig. 1. The Gibbs free energy46 of the adsorbed hydrogen is calculated as:| | |
ΔGH* = ΔEH* + ΔEZPE − TΔSH
| (1) |
where ΔEH* is hydrogen adsorption energy and ΔEZPE is the difference in zero point energy (ZPE) between the adsorbed state and the gas phase. ΔSH refers to the entropy of adsorption of 1/2H2, which is 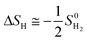 , where
, where 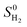 is the entropy of H2 in the gas phase at STP. ZPE and entropic corrections, ΔEZPE − TΔSH, are listed in Table S1.† The hydrogen adsorption energy ΔEH* can be defined in two ways: the integral and differential H adsorption energy as a function of the H coverage in eqn (2) and (3), respectively:
is the entropy of H2 in the gas phase at STP. ZPE and entropic corrections, ΔEZPE − TΔSH, are listed in Table S1.† The hydrogen adsorption energy ΔEH* can be defined in two ways: the integral and differential H adsorption energy as a function of the H coverage in eqn (2) and (3), respectively:| |
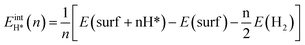 | (2) |
| | |
EdiffH*(θH*) = ΔEintH*(n)/δn = (EintH*(n) − EintH*(n − 1))/Δn
| (3) |
where n, H* and θH* refer to the number of hydrogen atoms, adsorbed hydrogen on the surface, and hydrogen coverage, respectively.
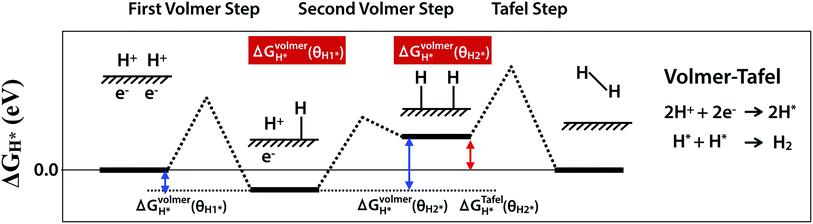 |
| | Fig. 1 Schematic of Volmer–Tafel mechanism. ΔGVolmerH*(θH1*), ΔGVolmerH*(θH2*) and ΔGTafelH*(θH2*) are free energies of the first- and the second-Volmer steps and Tafel step, where θH1* and θH2* indicate different hydrogen coverage of active sites. | |
In contrast to the single hydrogen reaction of the Volmer step, two hydrogen atoms mediate the Tafel step. Therefore, we obtain the Gibbs free energy of the intermediate state during the Volmer and Tafel steps with the following equations to determine the different hydrogen coverages of active sites.
| | |
ΔGVolmerH* = ΔEVolmerH* + ΔZPE − TΔS
| (4) |
| | |
ΔEVolmerH* = EdiffH*(θH*) = (EintH*(n) − EintH*(n − 1))/Δn, Δn = 1
| (5) |
| | |
ΔGTafelH* = ΔETafelH* + ΔZPE − TΔS
| (6) |
| | |
ΔETafelH* = EdiffH*(θH*) = (EintH*(n − 2) − EintH*(n))/Δn, Δn = 2
| (7) |
All calculations were performed using the Vienna Ab initio Simulation Package (VASP 5.3.5).47–50 Ab initio calculations were carried out using the projector augmented wave (PAW) method51,52 with a generalized gradient approximation based on the Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional.53,54 Integration in the Brillouin zone was performed on the basis of the Monkhorst–Pack scheme using a Γ centered 10 × 10 × 1 k-point mesh in each primitive lattice vector of the reciprocal space for geometry optimization and density of states (DOS). A plane-wave cutoff energy of 500 eV was used. 20 k-points along each high symmetry line in the Brillouin zone had been used for band structure. Lattice constants and internal atomic positions were optimized until the residual forces became less than 0.04 eV A−1. The vacuum slab space of a unit cell in the Z direction was 20 Å, to avoid interaction between layers.
Results and discussion
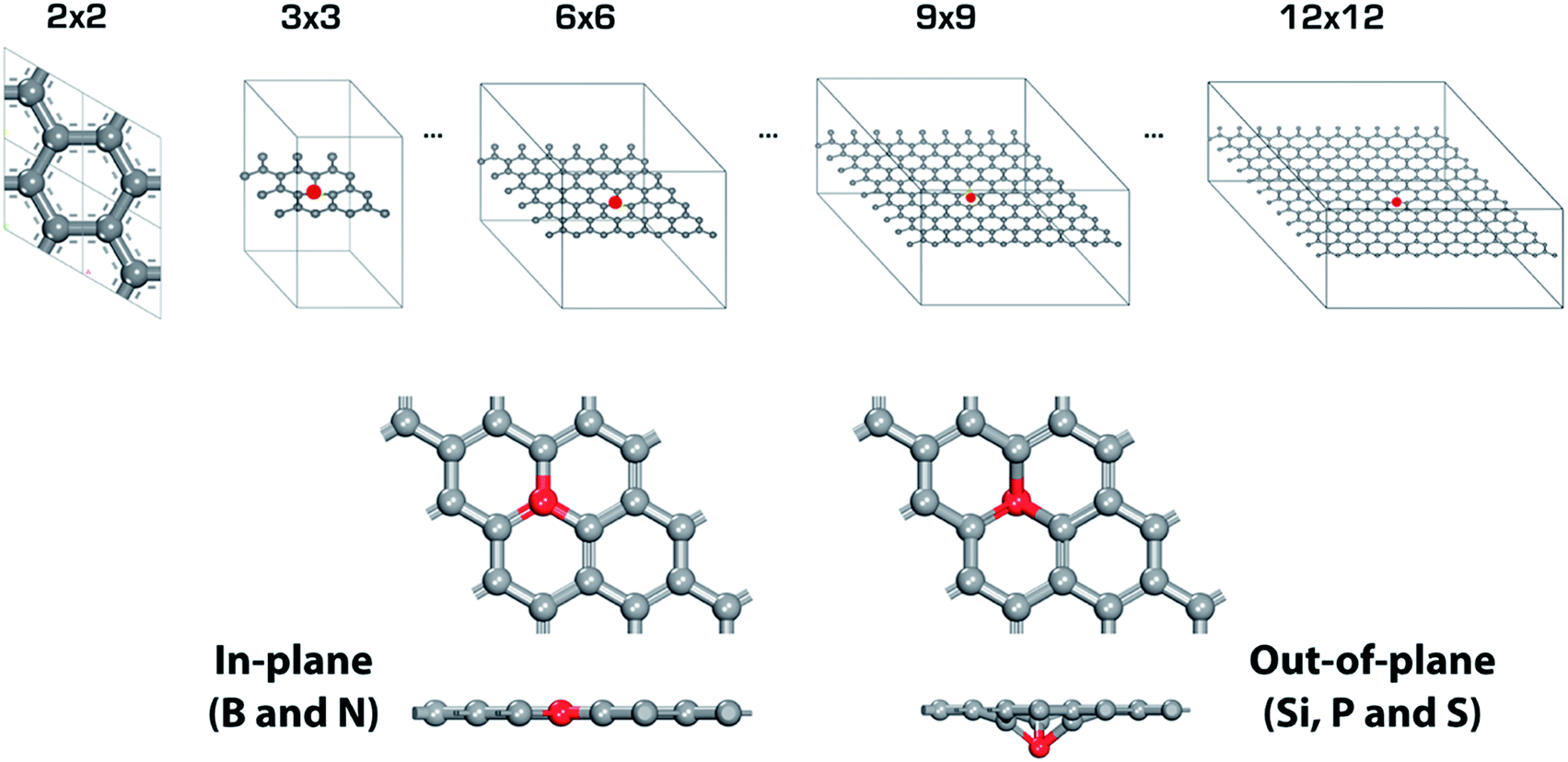
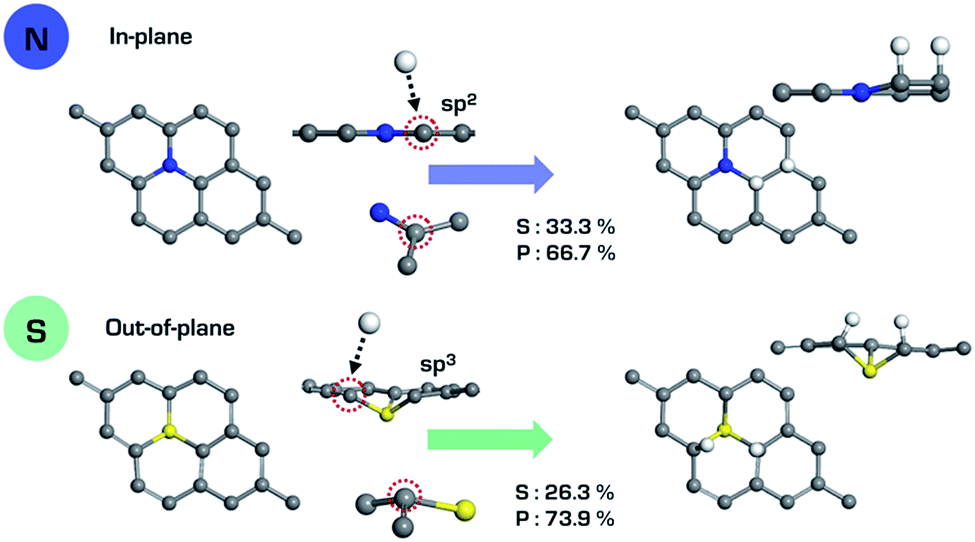
Fig. 2 shows a schematic of the heteroatom doped-graphene (GX, where G means graphene and X is B, N, Si P and S dopant) structures. We have used the second row elements (B and N) and the third row elements (Si, P and S) in the periodic table in order to investigate the structural and electronic doping effects on HER activity, because the third row elements are relatively larger than the second row elements and p- and n-type doping effects can be expected from the electron deficient B and electron rich N, P and S elements. As shown in Fig. 2, the second and third row elements give in-plane and out-of-plane structures, respectively, due to the size of the dopants.36. In addition, we have considered doping level by controlling the primitive cell size of heteroatom doped-graphene structures, having only one dopant of size N × N (where N is an integer from 3 to 12) which is a supercell of the pure graphene unit cell. Therefore, we can also expect an interesting feature of HER activity to be attributed to periodic lattice defects. Using these model structures, we have systematically evaluated the relationship between structure and HER activity of the GXs.
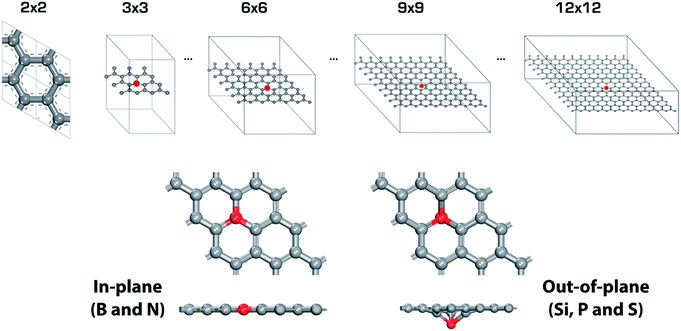 |
| | Fig. 2 Primitive cells of single heteroatom doped-graphene (GX, where G means graphene and X is B, N, Si P and S dopant). Red mark indicates dopant. There are two different type of structures depending on dopant, in-plane for GB and GN, and out-of-plane for GSi, GP and GS. | |
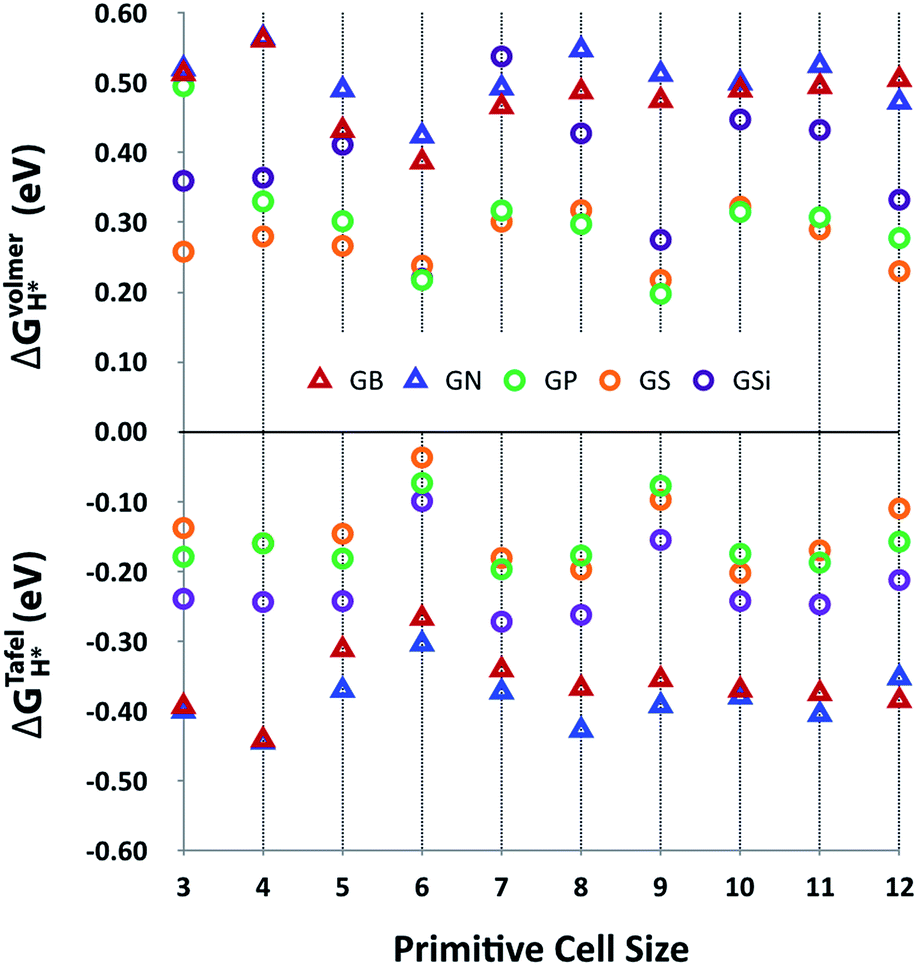
Fig. 3 shows the Gibbs free energy of the intermediate state of the Volmer and Tafel steps, ΔGVolmerH* and ΔGTafelH*, of GX as a function of primitive cell size. This plot indicates that the adsorption of the hydrogen atom in the Volmer step is unfavorable with positive ΔGVolmerH* values, but molecular hydrogen is likely to desorb at the Tafel step with negative ΔGTafelH* values, even though the ideal catalyst requires a near zero value of ΔGH*. However, it is worth noticing that there are two remarkable features of HER activity with respect to the type of dopant and the size of the primitive cell. At first, it seems that the third row element doped out-of-plane graphene structures (GSi, GP and GS) have better HER activity than the second row element doped in-plane graphene structures (GB and GN) with lower values of |ΔGH*|. In addition, |ΔGH*| has physical regularity attributed to periodic lattice defects as a function of primitive cell size, especially in the third row element doped graphene structures, where every 3N × 3N (N is integral) primitive cell has outstanding HER activity.
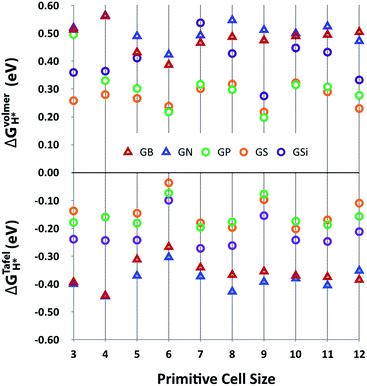 |
| | Fig. 3 HER activities, ΔGVolmerH* and ΔGTafelH*, of heteroatom doped-graphenes (GB, GN, GP, GS, and GSi) as a function of primitive cell size. ΔGVolmerH* is denoted as the average value of ΔGVolmerH*(θH1*) and ΔGVolmerH*(θH2*) for clear comparison between ΔGVolmerH* and ΔGTafelH*. | |
A. Structural and electronic doping effects on HER activity
The relatively large atomic size of the third row elements causes structural deformation of graphene, which improves HER activity by shifting ΔGVolmerH* and ΔGTafelH* to zero, as shown in Fig. 3. This means that the out-of-plane deformation enhances the hydrogen binding energy, because the hydrogen adsorption (Volmer step) and desorption (Tafel step) reactions, respectively, are endothermic and exothermic on the surface of GXs. Looking at the active sites on the surface of GXs, in-plane and out-of-plane GXs have different active sites, as shown in Fig. 4. Although the first hydrogen adsorption site is near the first neighboring carbons of the dopant regardless of the GX structure, the second hydrogen adsorption site is different. In contrast, the second preferable hydrogen adsorption site of out-of-plane GXs is the same as the first hydrogen adsorption site, in-plane GXs have the second neighboring carbons of the dopant as the second hydrogen adsorption site. On the atomic orbital hybridization characters of adjacent carbon atoms of dopant, the natural bond orbital (NBO) analysis55 show an increased p orbital contribution from sp2 to sp3 hybridization of carbons adjacent to the dopant due to structural deformation from in-plane to out-of-plane. Compared to sp2 hybridized carbon, sp3 hybridized carbons more readily form an extra a hydrogen atom without additional structural change. Therefore, in out-of-plane structures, the subsequent two hydrogen atoms prefer to bind to only sp3 hybridized carbons adjacent to the dopant. However, in the case of in-plane structures having only sp2 hybridized carbons, the first hydrogen atom should result in structural deformation to form sp3 hybridized carbons. Therefore, the first hydrogen adsorption on the in-plane structure is less favorable than the reaction on out-of-plane structures. And the second hydrogen atom can favorably bind to sp3 hybridized second neighboring carbons of dopant (see the adsorption energies of hydrogen atoms in subsequent Volmer step considering possible binding sites around the dopant in Table S3†). Consequently, structural deformation with dopants that are third row elements is associated with improved HER activity due to atomic orbital hybridization.
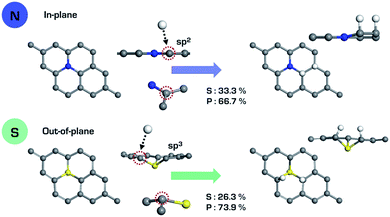 |
| | Fig. 4 Hybridization characters of carbons adjacent to the dopant on the in-plane (GN) and out-of-plane (GS) structures. | |
In the same out-of-plane structures, the stability of the intermediate state (H*) also depends on the charge of the carbons around the active site with ionic bond characters. Therefore, the negativity of carbon charges around active sites by hydrogen binding can improve HER activity. The order of negativity of the carbon charges around active site is well agreement with the order of HER activities of the out-of-plane structures, GS > GP > GSi (see the discussion in Fig. S1†).
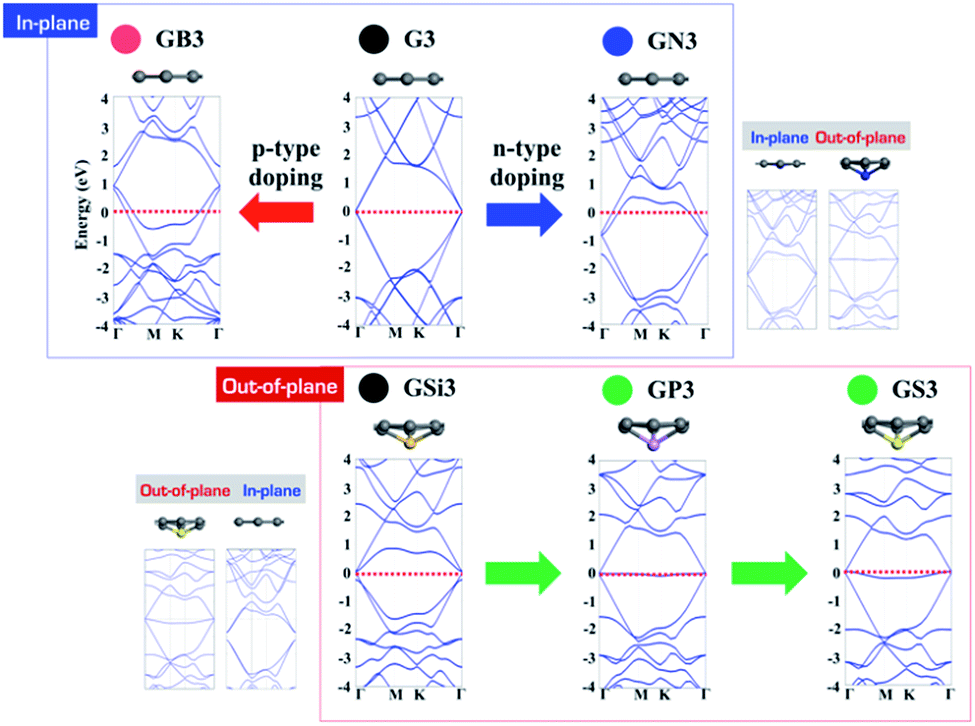
Looking at the electronic structures of GXs, p- and n-type doping effects are also expected from electron deficient and rich elements, as shown in Fig. 5. In the case of in-plane GXs, electron deficient boron shifts its band structure up by withdrawing an electron from graphene, and electron rich nitrogen shifts its band structure down by donating an electron to graphene. Interestingly, in the case of out-of-plane GXs, electron rich phosphorous and sulfur dopant have no associated band shift. The origin of the flat band can be understood based on the localization of an extra electron onto the dopant site. In order to verify the relationship between geometric and electronic structures of GXs, we have systematically changed the structures of GXs from in-plane to out-of-plane structures and vice versa. These calculations clearly show that dopant can produce n- and p-type doping states as well as a localized state depending on the structure of GXs induced by the type of dopants. When the out-of-plane (in-plane) deformation is applied in the in-plane (out-of-plane) GXs, band structures change from the p-type doping state (localized state) to the localized state (p-type doping state). Therefore, it is worth mentioning that the localized electronic states can be associated with physical regularity of HER activities on the out-of-plane GXs attributed to periodic lattice defects as shown in Fig. 3.
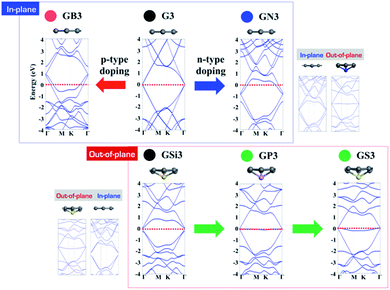 |
| | Fig. 5 Band structures of B, N, Si, P and S doped graphenes (GB, GN, GSi, GP and GS) with 3 × 3 cells. | |
B. Effects of periodic lattice defect on HER activity
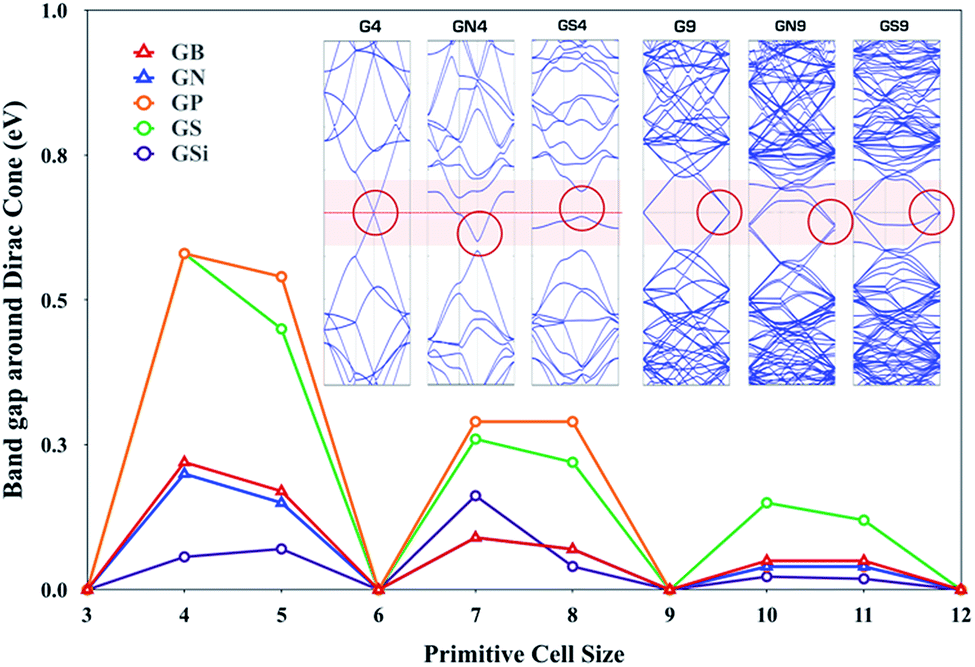
The third row elements, i.e., doped GXs (GSi, GP and GS), show an interesting physical regularity described by a simple 3N rule: GXs give outstanding HER activity whenever its primitive cell size has a 3N × 3N (N is integral) supercell size of pure graphene. In order to account for this tendency, we have preferentially studied electron band structures of GXs as a function of the primitive cell size and compared the behavior of the band gap around the Dirac point, as shown in Fig. 6, where GXs can either have a gap or remains gapless at the Dirac point, which is dependent on the size of the primitive cell.
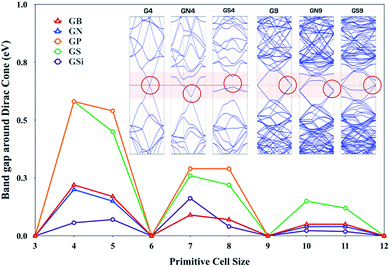 |
| | Fig. 6 The gaps around the Dirac cones of the B, N, Si, P and S doped graphenes (GB, GN, GSi, GP and GS) with N × N primitive cell sizes. Insects are band structure of pure graphene with 4 × 4 primitive cell size. | |
Interestingly, there is close correlation between the electronic properties of GXs and their HER activities; whenever the primitive cell size has 3N × 3N (N is integral), GXs have zero band gap around the Dirac point as well as enhanced HER activity.
A perfect graphene is known to be semi-metallic, with a Fermi level (the Fermi level is at 0 eV.) that is always aligned with the Dirac point. This 3N rule can be explained by band folding, as shown in Fig. S2.†56 For a 3N × 3N supercell, the Dirac points are moved to the Γ point via a translational symmetry operation, hence the Dirac points are folded to the Γ points, which produces two bands near the Γ points. Dopants and their types can distort these two degenerate bands at the Dirac point. Looking at the inset band structures of Fig. 6, compared to G9 of two gapless degenerated bands at the Dirac point, GN9 and GS9 show one split band at the Dirac point. Therefore, 3N × 3N primitive cells of GN and GS can preserve a gapless Dirac cone even though one of two degenerate bands is opened, which can be understood by the Bloch states analysis in Fig. S3.† However, because GN4 and GS4 have a single band at the Dirac point, a band gap opening is shown at the Dirac point. In addition, GN9 and GS9 give different behaviors due to different electronic states induced by the dopant, which are associated with the doping state of the GN9 in-plane structure and the localized state of the GS9 out-of-plane deformed structure. Note that GS4 has more pronounced semiconducting properties than the metallic features of all other systems because the out-of-plane structure has a localized state without a band shift.
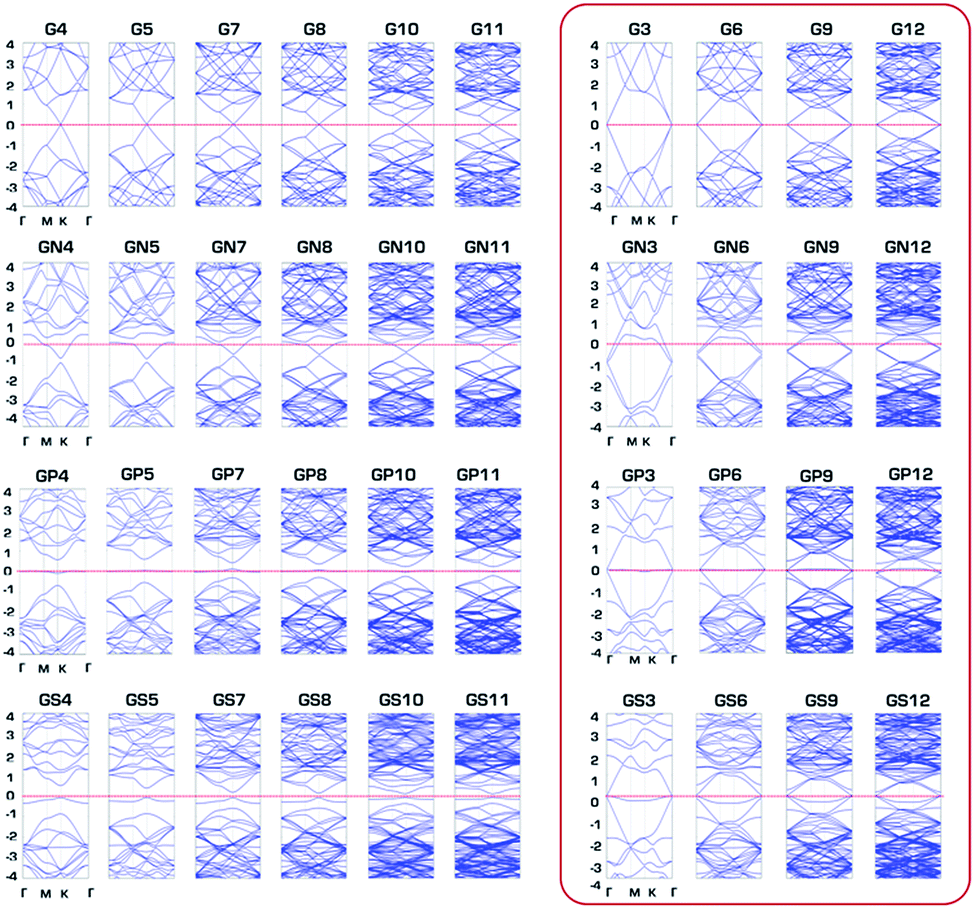
The overall semiconducting band gap is clearly seen in Fig. 7, which shows band structures of G, GN, GP and GS with N × N primitive cell sizes (band structures of GB and GSi are depicted in Fig. S4†). Only out-of-plane structures of GP and GS have a semiconductor band gap as semiconductor at none 3N × 3N primitive cell sizes. Based on the charge transfer from GXs to the approaching proton for effective hydrogen atom binding at the Volmer step, it seems reasonable that semiconducting none 3N × 3N primitive cells cannot facilitate the hydrogen binding necessary for HER activity compared to metallic 3N × 3N primitive cells. Consequently, physical regularity in the HER activity of the third row element-doped GXs (GSi, GP and GS) is attributed to the changes in band gap associated with periodic lattice defects, where HER catalytic activity is enhanced for every metallic 3N × 3N primitive cell.
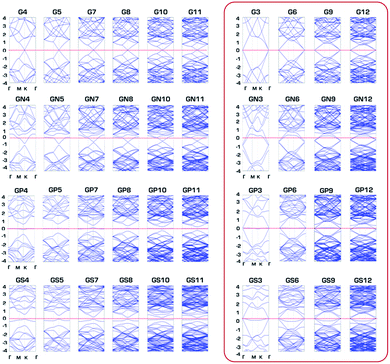 |
| | Fig. 7 Band structures of pure graphene (G) and N, P and S doped graphenes (GN, GP and GS) with N × N primitive cell sizes. | |
Conclusions
We have systematically investigated the structure–activity relationship of the hydrogen evolution reaction (HER) for heteroatom-doped graphenes (GXs, where G and X represent graphene and the heteroatom dopant). In this work, we demonstrated that the HER activity of GXs can be modulated by structural factors, including structural deformation and periodic lattice defects. Both structural factors enhance hydrogen atom binding during the Volmer step in HER by generating sp3 hybridized carbons and facilitating charge transfer between hydrogen atoms and GXs as metallic properties. Therefore, third row element-doped GXs have improved HER activity compared to second row element-doped GXs. In addition, physical regularity of HER activity described by the 3N rule appears due to periodic lattice defects, where the HER activity is improved at every metallic 3N × 3N primitive cell. Especially, the localized electronic state of the out-of-plane GXs was the origin of physical regularity of HER activity. This understanding of the structure–activity relationship can facilitate development of new electrocatalytic materials.
Acknowledgements
This research was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (NRF-2015R1C1A1A02036670).
References
- J. O. M. Bockris, Int. J. Hydrogen Energy, 2002, 27, 731–740 CrossRef CAS.
- J. R. Rostrup-Nielsen and R. Nielsen, Catal. Rev., 2004, 46, 247–270 CAS.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS PubMed.
- B. E. Conway and B. V. Tilak, Electrochim. Acta, 2002, 47, 3571–3594 CrossRef CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed. Engl., 2015, 54, 52–65 CrossRef CAS PubMed.
- F. Safizadeh, E. Ghali and G. Houlachi, Int. J. Hydrogen Energy, 2015, 40, 256–274 CrossRef CAS.
- C. G. Morales-Guio, L. A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- J. Q. Zhuo, T. Y. Wang, G. Zhang, L. Liu, L. B. Gan and M. X. Li, Angew. Chem., Int. Ed., 2013, 52, 10867–10870 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- I. E. Stephens and I. Chorkendorff, Angew. Chem., Int. Ed. Engl., 2011, 50, 1476–1477 CrossRef CAS PubMed.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Metaye, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS PubMed.
- M. Gong, D. Y. Wang, C. C. Chen, B. J. Hwang and H. J. Dai, Nano Res., 2016, 9, 28–46 CrossRef CAS.
- W. J. Zhou, J. Zhou, Y. C. Zhou, J. Lu, K. Zhou, L. J. Yang, Z. H. Tang, L. G. Li and S. W. Chen, Chem. Mater., 2015, 27, 2026–2032 CrossRef CAS.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- W. F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed. Engl., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef CAS PubMed.
- T. F. Jaramillo, J. Bonde, J. D. Zhang, B. L. Ooi, K. Andersson, J. Ulstrup and I. Chorkendorff, J. Phys. Chem. C, 2008, 112, 17492–17498 CAS.
- J. Kubisztal, A. Budniok and A. Lasia, Int. J. Hydrogen Energy, 2007, 32, 1211–1218 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Norskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jorgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Norskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- D. S. Kong, H. T. Wang, J. J. Cha, M. Pasta, K. J. Koski, J. Yao and Y. Cui, Nano Lett., 2013, 13, 1341–1347 CrossRef CAS PubMed.
- H. T. Wang, D. S. Kong, P. Johanes, J. J. Cha, G. Y. Zheng, K. Yan, N. A. Liu and Y. Cui, Nano Lett., 2013, 13, 3426–3433 CrossRef CAS PubMed.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- A. Behranginia, M. Asadi, C. Liu, P. Yasaei, B. Kumar, P. Phillips, T. Foroozan, J. C. Waranius, K. Kim, J. Abiade, R. F. Klie, L. A. Curtiss and A. Salehi-Khojin, Chem. Mater., 2016, 28, 549–555 CrossRef CAS.
- Y. G. Li, H. L. Wang, L. M. Xie, Y. Y. Liang, G. S. Hong and H. J. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- M. R. Gao, J. X. Liang, Y. R. Zheng, Y. F. Xu, J. Jiang, Q. Gao, J. Li and S. H. Yu, Nat. Commun., 2015, 6, 5982 CrossRef CAS PubMed.
- J. J. Duan, S. Chen, B. A. Chambers, G. G. Andersson and S. Z. Qiao, Adv. Mater., 2015, 27, 4234–4241 CrossRef CAS PubMed.
- D. Y. Wang, M. Gong, H. L. Chou, C. J. Pan, H. A. Chen, Y. Wu, M. C. Lin, M. Guan, J. Yang, C. W. Chen, Y. L. Wang, B. J. Hwang, C. C. Chen and H. Dai, J. Am. Chem. Soc., 2015, 137, 1587–1592 CrossRef CAS PubMed.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed. Engl., 2015, 54, 2100–2104 CrossRef CAS PubMed.
- Y. Ito, W. Cong, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed. Engl., 2015, 54, 2131–2136 CrossRef CAS PubMed.
- L. K. Putri, B. J. Ng, W. J. Ong, H. W. Lee, W. S. Chang and S. P. Chai, ACS Appl. Mater. Interfaces, 2017, 9, 4558–4569 CAS.
- H. J. Cui, Z. Zhou and D. Z. Jia, Mater. Horiz., 2017, 4, 7–19 RSC.
- Y. Jiao, Y. Zheng, K. Davey and S. Z. Qiao, Nat. Energy, 2016, 1, 16130 CrossRef CAS.
- S. Gadipelli and Z. X. Guo, Prog. Mater. Sci., 2015, 69, 1–60 CrossRef CAS.
- J. J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Catal., 2015, 5, 5207–5234 CrossRef CAS.
- Y. Zheng, Y. Jiao, Y. H. Zhu, L. H. Li, Y. Han, Y. Chen, A. J. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 Search PubMed.
- Y. Zheng, Y. Jiao, L. H. Li, T. Xing, Y. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2014, 8, 5290–5296 CrossRef CAS PubMed.
- B. R. Sathe, X. X. Zou and T. Asefa, Catal. Sci. Technol., 2014, 4, 2023–2030 Search PubMed.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 Search PubMed.
- Y. Shim, J. Han, Y. J. Sa, S. Lee, K. Choi, J. Oh, S. Kim, S. H. Joo and S. Park, J. Ind. Eng. Chem., 2016, 42, 149–156 CrossRef CAS.
- L. K. Putri, W. J. Ong, W. S. Chang and S. P. Chai, Appl. Surf. Sci., 2015, 358, 2–14 CrossRef CAS.
- X. W. Wang, G. Z. Sun, P. Routh, D. H. Kim, W. Huang and P. Chen, Chem. Soc. Rev., 2014, 43, 7067–7098 RSC.
- L. Song, Z. Liu, A. L. M. Reddy, N. T. Narayanan, J. Taha-Tijerina, J. Peng, G. H. Gao, J. Lou, R. Vajtai and P. M. Ajayan, Adv. Mater., 2012, 24, 4878–4895 CrossRef CAS PubMed.
- J. K. Norskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and J. K. Norskov, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef CAS.
- E. Skulason, V. Tripkovic, M. E. Bjorketun, S. Gudmundsdottir, G. Karlberg, J. Rossmeisl, T. Bligaard, H. Jonsson and J. K. Norskov, J. Phys. Chem. C, 2010, 114, 18182–18197 CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- Y. C. Zhou, H. L. Zhang and W. Q. Deng, Nanotechnology, 2013, 24, 225705 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04115b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab
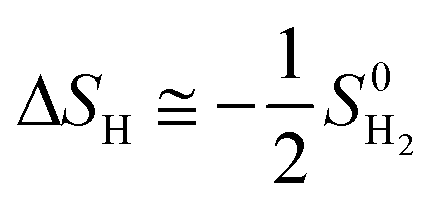 , where
, where 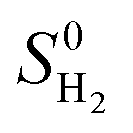 is the entropy of H2 in the gas phase at STP. ZPE and entropic corrections, ΔEZPE − TΔSH, are listed in Table S1.† The hydrogen adsorption energy ΔEH* can be defined in two ways: the integral and differential H adsorption energy as a function of the H coverage in eqn (2) and (3), respectively:
is the entropy of H2 in the gas phase at STP. ZPE and entropic corrections, ΔEZPE − TΔSH, are listed in Table S1.† The hydrogen adsorption energy ΔEH* can be defined in two ways: the integral and differential H adsorption energy as a function of the H coverage in eqn (2) and (3), respectively: