
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new guanylhydrazone derivative as a potential acetylcholinesterase inhibitor for Alzheimer's disease: synthesis, molecular docking, biological evaluation and kinetic studies by nuclear magnetic resonance†
Denise Cristian Ferreira Neto a,
Marcelle de Souza Ferreiraa,
Elaine da Conceição Petronilhoa,
Josélia Alencar Limaa,
Sirlene Oliveira Francisco de Azeredoa,
Juliana de Oliveira Carneiro Bruma,
Claudia Jorge do Nascimentob and
José Daniel Figueroa Villar*a
a,
Marcelle de Souza Ferreiraa,
Elaine da Conceição Petronilhoa,
Josélia Alencar Limaa,
Sirlene Oliveira Francisco de Azeredoa,
Juliana de Oliveira Carneiro Bruma,
Claudia Jorge do Nascimentob and
José Daniel Figueroa Villar*a
aChemistry Department, Military Institute of Engineering, Praça General Tibúrcio 80, 22290-270, Praia Vermelha, Rio de Janeiro, Brazil. E-mail: jdfv2009@gmail.com; Tel: +55 21 2546 7057
bInstitute of Biosciences – Federal University of the State of Rio de Janeiro, Urca, Rio de Janeiro, Brazil
First published on 5th July 2017
Abstract
The development of pharmacological agents for the treatment of Alzheimer's Disease (AD) is very relevant since this is the most common type of dementia. The inhibition of acetylcholinesterase (AChE) is important to increase the low levels of acetylcholine (ACh) neurotransmitter observed in sick people, which is associated with the memory loss. In this work, a new guanylhydrazone was designed and synthesized as an AChE inhibitor. This new compound was compared to tacrine and other guanylhydrazones. All of them were studied by molecular docking and tested in vitro as AChE inhibitors by Ellman's test and Fig-NMR method. A high inhibition of AChE by the new compound was observed, showing that this compound has great potential for the treatment of AD.
Introduction
Alzheimer's Disease (AD) is characterized by the loss of cholinergic function and a chronic and progressive neurodegenerative disorder affecting memory, cognition and behavior.1,2 It is the most common cause of dementia in old people, it being estimated that there were around 46.8 million cases worldwide in 2015.3,4Due to the increase in life expectancy, a significant increase in the number of cases is expected for the next years. This number will almost double every 20 years, reaching 74.7 million in 2030 and 131.5 million cases in 2050.3,4 Usually, AD takes about 8 to 10 years from the first symptoms to death, its diagnosis being mainly done through predetermined clinical criteria with the exclusion of other causes of dementia.5–7
Currently, the most accepted hypothesis is that some of the symptoms of AD result from changes due to the deficiency of neurotransmitters that are responsible for the transmission of nerve signals from one neuron to another. The progression of AD occurs through the decline in the level of neurotransmitters including the cholinergic neurons and the forebrain.8,9 Patients with AD have a remarkable alteration of the cholinergic system, with decreasing activity of the choline acetyltransferase, mainly in the cerebral cortex and hippocampus. Also the activity of acetyl coenzyme A is decreased which leads to a very important decrease in the acetylcholine (ACh) levels. This is known as cholinergic hypothesis and reveals that a possible treatment, which has been quite effective in reducing the symptoms, is the use of inhibitors of acetylcholinesterase (AChE) to prevent the breakdown of acetylcholine (ACh).10–12
The current therapy of AD is based on three pillars: improving cognition, slowing the progression of the disease and relieving the symptoms and behavioral changes.13–15 Currently, six drugs are approved by the Food and Drug Administration (FDA). Four of them are AChE inhibitors: donepezil, galantamine, rivastigmine, and tacrine.16–18 Tacrine was the first commercial drug approved by the FDA, in 1993, for the treatment of AD.17,18 However, its use is associated with increased risk of hepatotoxicity observed in 30% to 50% of the cases. Nowadays it is used only when other drugs do not show good results in treatment. Its structure, however, has been widely used for the design of many other bioactive analogs with different levels of selectivity and lower toxicity17–27 and has motivated the development of analogues to improve the pharmacokinetic profile.22,24
In this work, we synthesized a new guanylhydrazone (2) that showed high inhibition of AChE. This new compound was compared to tacrine and other guanylhydrazones previously synthesized by our group28–30 (Fig. 1). The guanylhydrazones represent a class of compounds that presents a great variety of biological activity, and have been successfully used as antihypertensive,31 trypanocidal,32,33 antineoplastic,34 antibacterial35 and more recently as very effectives inhibitors of HuAChE for AD.36
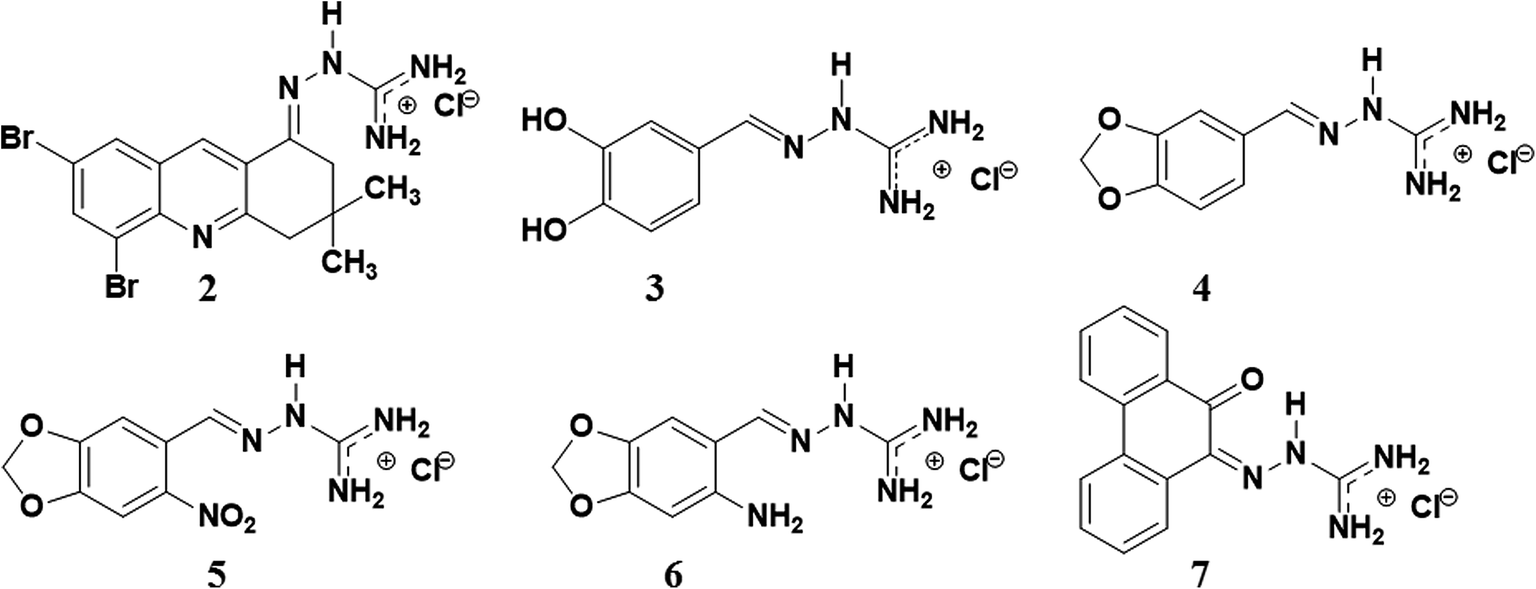 | ||
| Fig. 1 The structure of guanylhydrazones 2, 3, 4, 5, 6 and 7 designed for the treatment of Alzheimer's disease. | ||
These synthesized compounds were tested as inhibitors of the AChE enzyme by NMR test (Fig-NMR)37 and the results were compared to the Ellman's test.38
Results and discussion
Chemistry
In order to interact with the active site of the AChE ligands should have groups with positive charge. In previous studies by our research group using molecular modeling and inhibition tests it has been found that the cationic site of guanylhydrazones are appropriate to interact with the anionic region of the AChE active site and compete with acetylcholine.39,40 For this reason, a new guanylhydrazone (2) having some structural similarity with tacrine was prepared. Its guanidine group, a cationic group, can interact with the anionic site of the active site of the enzyme, and NMR results showed that this new guanylhydrazone is a more efficient inhibitor than tacrine.The synthesis of compound 2 involved two steps: (i) the reaction of 2-amino-3,5-dibromobenzaldehyde with dimedone to obtain compound 1 (5,7-dibromo-3,4-dihydro-3,3-dimethylacridin-1(2H)-one) (Scheme 1); (ii) reaction between (1) and aminoguanidine hydrochloride (H2N–NH–C(NH2)·HCl) giving compound 2 (Scheme 1) with yield of 80%. For this reaction, there are two different step mechanisms. In the first one, the dimedone reacts with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the benzaldehyde; it follows the reaction of the –NH2 group with the CO of dimedone. The second one would be the reaction between the –NH2 group from the benzaldehyde and one of the carbonyl groups of dimedone, followed by the reaction of the –CH2 group of dimedone with the CO group of the benzaldehyde. This mechanism is illustrated in the ESI (Scheme S.1†).
O of the benzaldehyde; it follows the reaction of the –NH2 group with the CO of dimedone. The second one would be the reaction between the –NH2 group from the benzaldehyde and one of the carbonyl groups of dimedone, followed by the reaction of the –CH2 group of dimedone with the CO group of the benzaldehyde. This mechanism is illustrated in the ESI (Scheme S.1†).
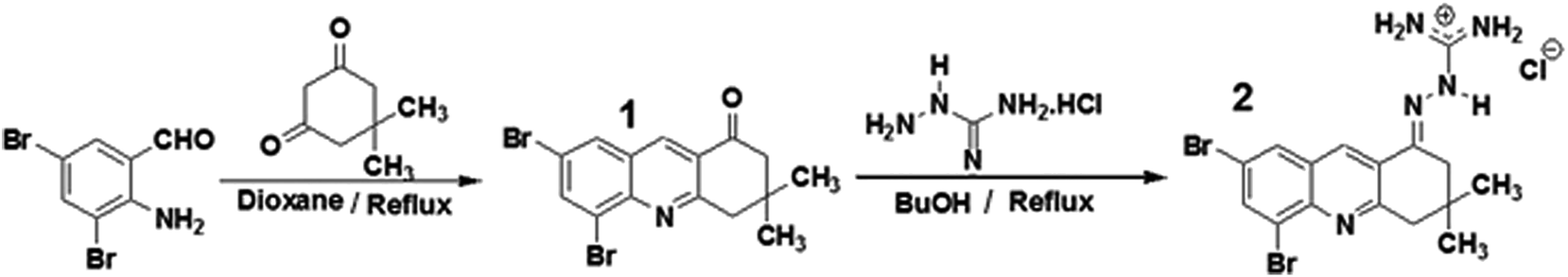 | ||
| Scheme 1 Synthesis of compounds 1 and 2 from the reaction between 2-amino-3,5-dibromobenzaldehyde and dimedone. | ||
The other guanylhydrazones (3, 4, 5, 6 and 7) were prepared by the reaction of aminoguanidine hydrochloride with some aldehydes (see ESI, Scheme S.2†), as previously described.28–30 Although guanylhydrazones 3–7 have already been reported in the literature, they had not been tested yet as AChE inhibitors.
The NMR data and IR spectra for compounds 2–7 are given in the ESI (Fig. S.1–S.21†).
In silico bioactivity study
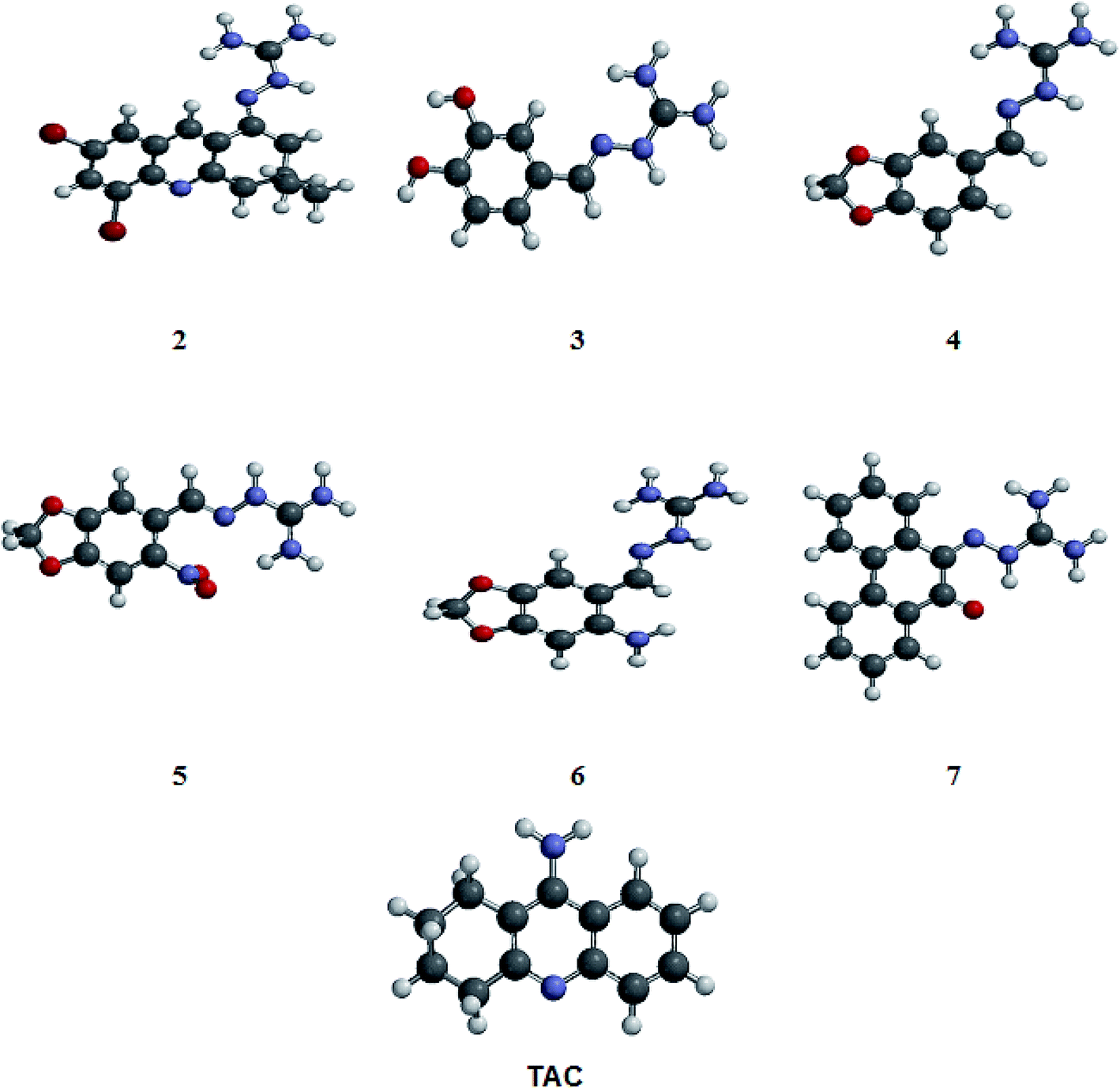 | ||
| Fig. 2 Optimized structures of compounds 2–7 (showed in Fig. 1) and tacrine (TAC) obtained from Spartan'10 program. The structures were designed and submitted to the conformational analysis using MMFF force field, to explore the energetically favorable forms of the molecules. The most stable conformation showed in this figure was selected for the docking studies. | ||
For development of AChE inhibitors with enhanced pharmacological profile, the prediction of physicochemical properties is the most emphasizing parameter. Molecular descriptors and drug likeliness properties of compounds were analyzed using the tool Molinspiration and Osiris Property explorer, based on the Lipinski Rules of five42,43 (see ESI, Table S.1†). The solubility (clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) S) of synthesized compounds was found in an acceptable range (<4). All compounds showed lipophilicity (clogp) <5, indicating that they have a good drug-likeness profile.43,44 They also showed a low molecular weight (<500) which means that drug molecules can be easily transported, diffused and absorbed compared to heavy molecules.42,43 The topological polar surface area (TPSA) is the sum of the van der Waals surface areas of the polar atoms (oxygen and nitrogen) and it is related to the evaluation of cellular permeability and the in vivo bioavailability of a therapeutic agent. The synthesized compounds 2 and 7 showed TPSA < 90 Å (see ESI, Table S.1†), indicating that they have good bioavailability by oral route.42,43 In this study, drug-likeliness property44–46 and toxicity were studied using Osiris tools. Results indicated no toxicity risks (mutagenicity, tumorogenicity, irritation, reproduction), and revealed a good score when compared to the control tacrine (see ESI, Table S.1†).
S) of synthesized compounds was found in an acceptable range (<4). All compounds showed lipophilicity (clogp) <5, indicating that they have a good drug-likeness profile.43,44 They also showed a low molecular weight (<500) which means that drug molecules can be easily transported, diffused and absorbed compared to heavy molecules.42,43 The topological polar surface area (TPSA) is the sum of the van der Waals surface areas of the polar atoms (oxygen and nitrogen) and it is related to the evaluation of cellular permeability and the in vivo bioavailability of a therapeutic agent. The synthesized compounds 2 and 7 showed TPSA < 90 Å (see ESI, Table S.1†), indicating that they have good bioavailability by oral route.42,43 In this study, drug-likeliness property44–46 and toxicity were studied using Osiris tools. Results indicated no toxicity risks (mutagenicity, tumorogenicity, irritation, reproduction), and revealed a good score when compared to the control tacrine (see ESI, Table S.1†).
Molecular docking studies
For the study of the interaction between the guanylhydrazones and the enzyme AChE by molecular docking, AutoDock 4.2 software was used. The results were compared to the molecular docking of tacrine (used as a reference). To perform molecular docking, it was necessary to validate the Grid Box. This was done through molecular redocking with the enzyme TcAChE and tacrine, since the three-dimensional structure of that protein (cod: 1ACJ, PDB) was co-crystallized with this ligand (Fig. 3).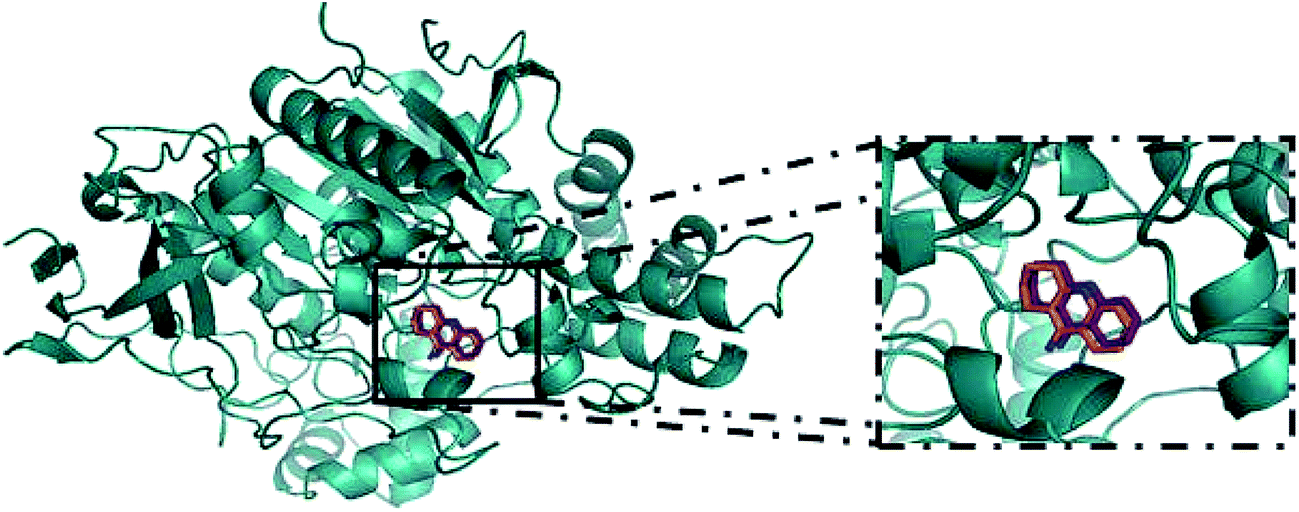 | ||
| Fig. 3 Redocking of tacrine in Torpedo californica AChE (cod: 1ACJ, in PDB) was done in the AutoDock 4.2 program for validation of the Grid Box.; in orange, tacrine from the crystal and in magenta, tacrine generated by redocking.47 | ||
The RMSD obtained for the redocking of tacrine inside of the 3D structure of TcAChE was 0.61, confirming the effectiveness of AutoDock 4.2. After that, docking for compounds 2–7 and tacrine (TAC) was performed. Results (Table 1) show the possible interactions between the enzyme and the binder, such as binding and intermolecular energies and inhibition constant. Negative values of free energy and intermolecular energy are observed for all compounds, indicating they all have affinity for the active site of the enzyme. Guanylhydrazones 2 and 7 form the most stable complexes, showing these compounds should have a higher affinity with the enzyme.
| Compound | Binding energy (kcal mol−1) | Intermolecular energy (kcal mol−1) |
|---|---|---|
| 2 | −10.44 | −10.74 |
| 3 | −7.06 | −8.26 |
| 4 | −7.93 | −8.52 |
| 5 | −6.96 | −7.85 |
| 6 | −7.74 | −8.63 |
| 7 | −10.45 | −10.75 |
| TAC | −8.01 | −8.01 |
The results obtained for possible hydrogen bonds and van der Walls interactions are listed in Table S.2 (ESI†) and in Fig. 4 showing the docking model for compounds 2–7 and tacrine.
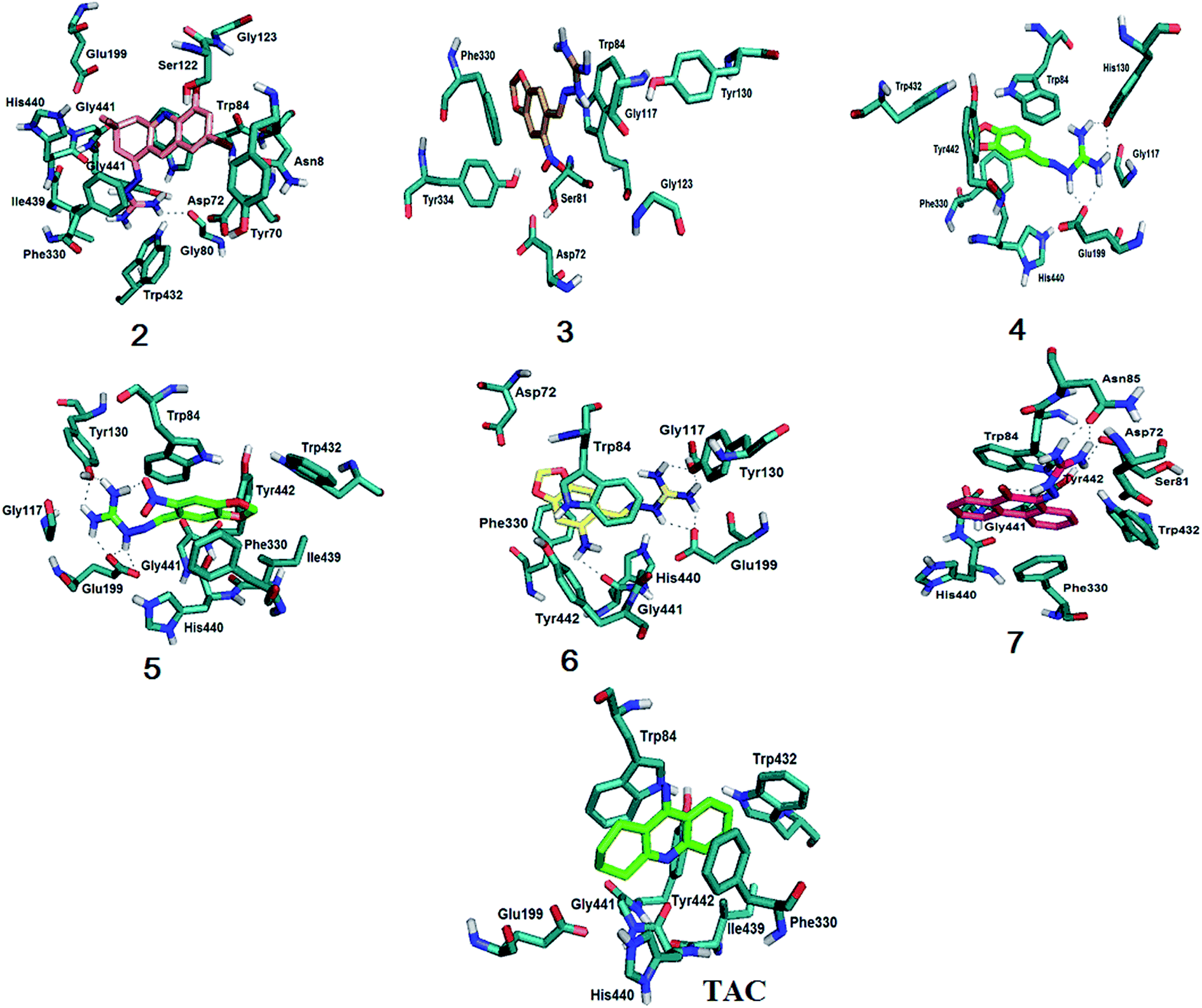 | ||
| Fig. 4 Molecular docking results for compounds 2–7 and tacrine with EeAChE. All compounds interact with Phe330 and Trp84, important residues for the stabilization in the active site. Compound 3 is the only one that doesn't interact with the catalytic triad (Ser200–His440–Glu327). Compounds 2 and 7 show pi stacking when interacting with the enzyme. For more details, see text and ESI, Fig. S.22–S.28†. | ||
The coupling results (Fig. 4) showed that, like tacrine, guanylhydrazone 2 interacts with His440 that is located in the catalytic triad at the entrance of the active site. Interaction with Asp72, Ser122 and Phe330 residues of the peripheral anion site (PAS) was also observed. The Phe330 residue is directly involved in the recognition of quaternary linkers.46–48 This residue is very flexible and adopts different conformations through π–π stacking or cation π interaction with the ligands that bind to it. The PAS ligand that is stacked between the side chains of the Phe330 residue clogs the active site cavity. Thus, binding of a ligand at this position blocks the substrate inflow and outlet of the active site base, having a great functional importance.49 Guanylhydrazone 2 has a cationic group (the guanidine group) that showed interaction with the residues Trp84, Glu199 and Gly441 at the anionic site of the active site of the enzyme.
Compounds 4–7 also showed interaction with His440, being this interaction crucial for the hydrolysis of the ACh substrate. Exception is observed for compound 3, that can indicate this compound would not be a good AChE inhibitor. It is also observed for all compounds an interaction involving Phe330 and Trp84. Silva et al.46 showed the importance of this binding because Phe330 is responsible for the stabilization of tacrine in the active site, which also enables the interaction with Trp84. This amino acid is located in the catalytic cavity, close to the catalytic triad (Ser200–His440–Glu327).48 It has been used as an important marker for successful molecular docking studies due to the cation–pi interaction between its indole ring and the quaternary salt group from acetylcholine.50
According to the docking results, the interactions observed for compounds 2 and 7 are very important and could guarantee the good degree of inhibition indicating that they are promising inhibitors in the treatment of AD.
Biological evaluation and kinetic study by Ellman's test and NMR methods
To evaluate the inhibition potential of the compounds, two different methodologies were used: the NMR method (Fig-NMR)37 and Ellman's test38 using tacrine as reference. The AChE from E. electricus (EeAChE) was chosen as it displays an identical active site and a very similar activity to the human enzyme.51,52 Typically, enzyme inhibition studies are conducted using UV-visible spectroscopy, which is fast, very cheap and has high sensitivity. However it has some disadvantages. As it is a colorimetric method, analysis using colored compounds (this is the case of the synthesized compounds) can lead to conflicting results. In other cases, in order of observing the results, UV analysis requires the use of a second enzyme, which is used to convert the substrate of the primary enzyme into a secondary one. This is the case of Ellman's test, commonly used for kinetic studies of AChE, which uses acetylthiocholine instead of acetylcholine (ACh) as substrate for the test.38For these reasons, it is important to perform other methods of analysis in parallel. In this work, in addition to the traditional Ellman's test, we also used the NMR kinetics test37 for the inhibition studies of the compounds with AChE. This method is simple and can be applied using acetylcholine as substrate to test all kinds of inhibitor compounds, including the colored compounds of this work (ESI, Fig. S.39†). Knowing that in the hydrolysis of acetylcholine by AChE two compounds are formed, the acetic acid (or acetate, due to the aqueous medium) and choline, the methyl groups of those two compounds were monitored. This is because these signals absorb as distinct singlets in the 1H-NMR spectrum. Because of the stoichiometric ratio (1:1), for each mole of acetylcholine that is consumed in the reaction, one mole of acetate is formed. So direct integration of the two signals can be directly used in the analysis.
Table 2 shows an important agreement between the results for both methods, confirming that NMR is a very good and useful method for the biological evaluation of acetylcholinesterase inhibitors. NMR also allowed the kinetic study and the determination of the inhibition percentage for all compounds (see Fig. S.29–S.35 in ESI†). By comparing the results between the methods, it is interesting to observe that by NMR compound 2 is a better inhibitor when compared to tacrine.
| Compounds | Ellman's test EeAChE inhibition (%) | NMR test EeAChE inhibition (%) |
|---|---|---|
| 2 | 93.49 ± 0.52 | 94.31 ± 0.60 |
| 3 | 14.93 ± 2.66 | 29.63 ± 2.91 |
| 4 | 40.12 ± 1.22 | 47.08 ± 1.12 |
| 5 | 89.77 ± 0.45 | 81.76 ± 1.07 |
| 6 | 37.50 ± 2.56 | 38.81 ± 3.73 |
| 7 | 88.77 ± 1.35 | 92.55 ± 1.97 |
| TAC | 100.00 ± 0.42 | 93.50 ± 5.42 |
Guanylhydrazones 3 and 4 showed inhibition of 29.63% and 47.08% respectively, by the NMR method (Table 2). These compounds are not good EeAChe enzyme inhibitors, as expected from the energy values from docking results (Table 1).
Also by NMR, the guanylhydrazone 5 showed 81.76% inhibition (Table 3 and Fig. S.32 in ESI†) and the guanylhydrazone 6, 38.81% (Table 2 and Fig. S.33 in ESI†).
|
||
|---|---|---|
| Atoms | Nuclear charge of 5 | Nuclear charge of 6 |
| 2 | −0.034 | 0.310 |
| 4 | −0.206 | −0.615 |
| 7 | −0.200 | −0.604 |
| 13 | 0.544 | 0.876 |
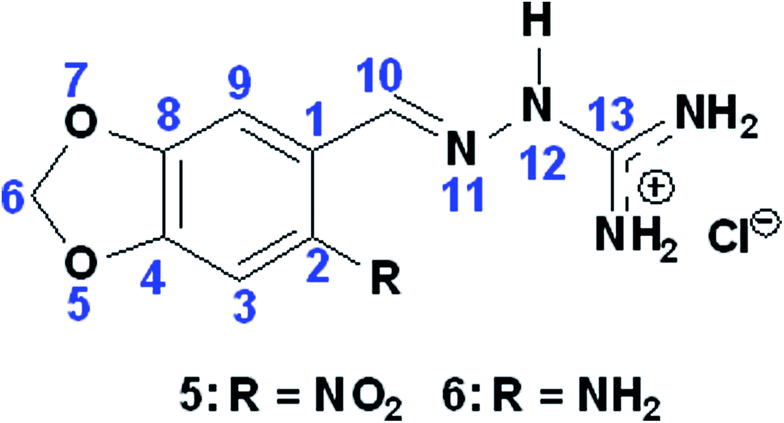
Docking results demonstrated that both compounds exhibited the same interactions with those residues (see ESI, Table S.2†). However, also from docking results, the obtained energies (Table 1) suggested that compound 5 should not be a better inhibitor than compound 6. This can be explained because sometimes docking studies (theoretical) show different results from in vitro experiments due to program limitations. Despite of these compounds (5 and 6) having a similar structures (by changing only the nitro substituent by the amino group in the molecule), in vitro results suggest that an electronegative substituent, such as the nitro group of compound 5, may favor the inhibition process. This happens by shifting the electron cloud and leaving the oxygen of the molecule less polarized and consequently the molecule has a stronger positive charge. This could induce an increasing interaction with the amino acid residues of the active site. To confirm this statement, using Spartan'10 program, we measured the values of the natural atomic charge of some atoms of the two molecules (Table 3). The results indicate that the oxygen in compound 5 has a lower electron density than the oxygen in compound 6, thus confirming that the density in that region decreased with the presence of the –NO2 group. These observations are important for the development of new and more effective compounds for inhibiting the enzyme AChE.
The IC50 for the compounds that showed better inhibition of the EeAChE enzyme (Table 2) by both methods (NMR and Ellman) was calculated. They were 1.97 ± 0.28 μM (compound 2), 6.54 ± 0.61 μM (compound 7) and 7.77 ± 0.73 μM (compound 5). The inhibition curves are available in the ESI (Fig. S.36–S.38†). According to these results, guanylhydrazones 2, 5 and 7 are good inhibitors, while the others do not show a good inhibition.
Looking at the NMR results, the new guanylhydrazone 2 inhibited about 94.31% (Fig. 5) showing greater inhibition than tacrine (93.50%). It is the most active guanylhydrazone in this study (IC50 1.97 ± 0.28 μM). This novel guanylhydrazone has a cationic group, which is the guanidine group that interacts with the residues Trp84, Glu199 and Gly441 at the anionic site of the active site of the enzyme. All those interactions have been confirmed by molecular docking and are very important as they can guarantee the good inhibition of the compound, indicating that it is a promising inhibitor in the treatment of AD.
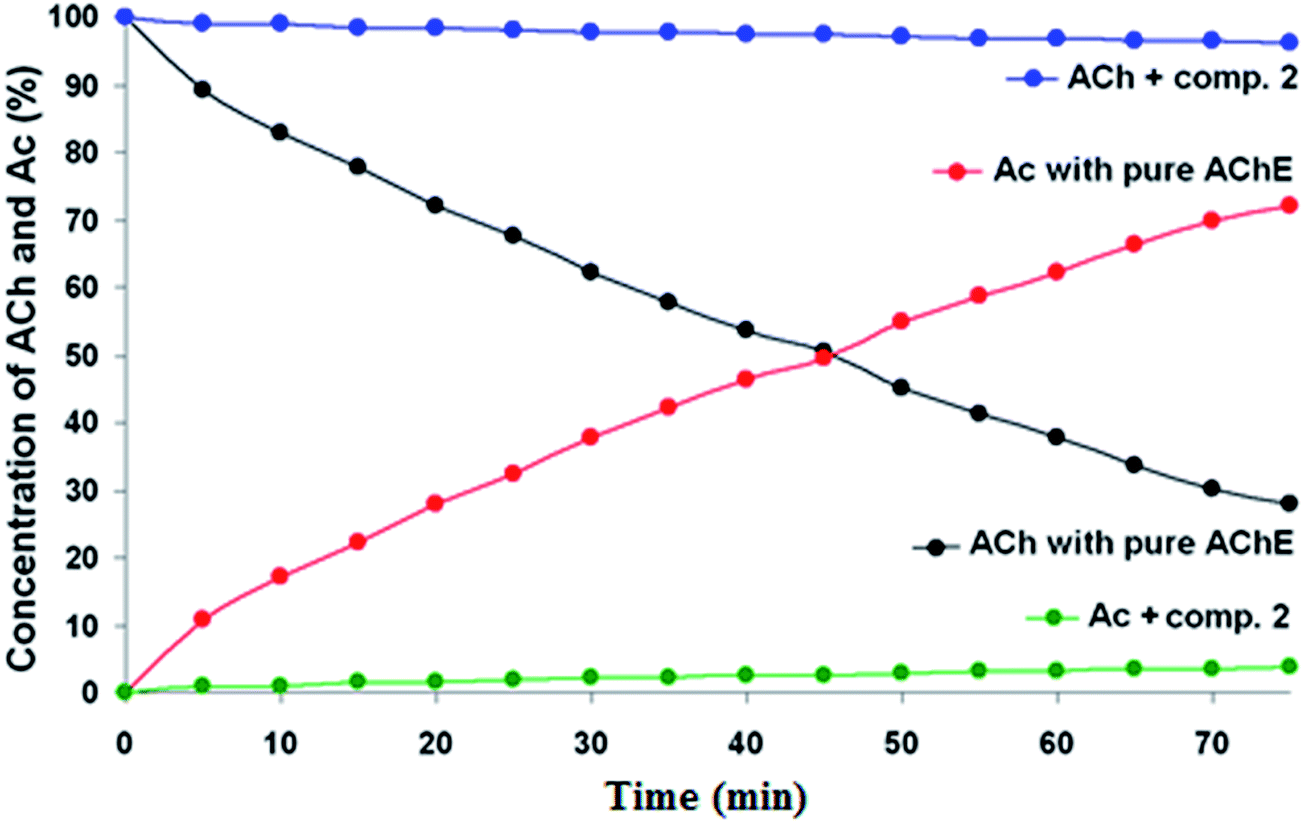 | ||
| Fig. 5 AChE inhibition by compound 2 using NMR test. The concentrations were determined from direct integration of the methyl peaks of ACh and Ac (see text for more information). As ACh is consumed (black), Ac is formed (red). However, in the presence of the good inhibitor (compound 2), the reaction doesn't take place: ACh is not consumed (blue) and, consequently, no Ac is formed (green). | ||
Conclusion
In this work some guanylhydrazones were synthesized and tested as AChE inhibitors. Docking studies and kinetic tests by Ellman's and NMR test were performed. Both in vitro tests showed similar results, proving that NMR method is appropriate for inhibition studies. Also the structure–activity relationship (SAR) of all compounds was studied. All results revealed that compounds 2 and 7 are very promising drugs. These compounds show interaction with the amino acid His440 (catalytic triad), important for avoiding the hydrolysis of ACh. It was also observed interaction with Trp84 (catalytic cavity), which has been widely used as an important marker for successful molecular docking studies with AChE. The results presented here indicated that some guanylhydrazones can very effective drugs for the treatment of Alzheimer's disease and can be a promising drug class for AD. The new guanylhydrazone from 5,7-dibromo-3,4-dihydro-3,3-dimethylacridin-1(2H)-one, compound 2, was proven to be a very good inhibitor of EeAChE. Despite of having similar inhibitory activity, SAR studies showed compound 2 is better than tacrine, but biological experiments should be performed for confirming the theoretical results.Materials and methods
Chemistry
Solvents (methyl alcohol, ethyl alcohol 95%, butyl alcohol, dioxane) were purchased from VETEC (Brazil) and used with further purification. All other reagents were purchased from Merck and Sigma-Aldrich (Brazil) and used without further purification. Reactions were monitored by TLC using DCAlufolien Kieselgel 60 F254 (Merck, Darmstadt, Germany). All NMR measurements were performed at 25 °C on a Varian Premium COMPACT™ 600 MHz (software VNMRJ version 3.2) spectrometer using a 5 mm NMR probe and dimethyl sulfoxide-d6 (DMSO-d6) and deuterium oxide (D2O). Chemical shifts are given in ppm (d) with TMS as an internal standard. J values were given in Hertz. Abbreviations for 1H NMR data quoted are as follows: s (singlet); d, (doublet); t, (triplet); q, (quartet); m, (multiplet); bs, (broad singlet). IR spectra of the compounds were recorded on a Spectrum 100 spectrometer. All IR and NMR spectra are available in the ESI.†Synthesis of the compounds
General procedure for guanylhydrazones (4–6)
1 mmol of aminoguanidine hydrochloride was dissolved in 15 mL of 95% ethanol; the corresponding aldehyde (1 mmol) and 8 drops of HCl (6 M) were added to a 50 mL round-bottom flask. The solution was stirred and heated under reflux for 72 h. The solid obtained after eliminating the solvent under vacuum was recrystallized from ethanol.In silico bioactivity study
Oral bioavailability, physicochemical properties and toxicity activity were evaluated for all synthesized compounds and tacrine. Lipophilicity (c-logp), water solubility (c-logS), molecular weight (MW), polar surface area (PSA), drug likeness and toxicity were calculated using online Molinspiration Cheminformatics (http://www.molinspiration.com/cgi-bin/properties) property calculation toolkit and online OSIRIS Property explorer (http://www.organic-chemistry.org/prog/peo/). The bioactivity score and drug likeness properties of the tested compounds were compared to tacrine.
Molecular docking studies
The three-dimensional structures of the ligands were built using Spartan'10 program, and the conformer distribution with molecular mechanics using 100 conformers was examined. The partial atomic charges were calculated using the RM1 semi-empirical method, and the molecular energies were calculated using Hartree–Fock 6-31G*. The ligands' rotatable bonds and atomic charges were defined. As the EeAchE is not reported in the literature involving complexes ligands/enzymes, the TcAchE (Torpedo californica AChE, PDB Code: 1ACJ) was used for docking studies. To validate the docking protocol, it was first performed the docking simulation of tacrine against the active site of TcAchE and then the result was compared to the crystallographic structure. The interactions between ligands and the active site of TcAChE were performed using AutoDock 4.2, using each ligand with 50 poses and grid points of 48 × 38 × 42 with 0.375 Å spacing. The figures were generated using a PyMOL Molecular Graphics System and AutoDock Tools.Biological evaluation
Accession codes
Ordinate and structure factors were deposited to the PDB code for Torpedo californica AChE: 1ACJ.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
We would like to thank the financial support afforded by CAPES, CNPq and INBEB.References
- R. Annicchiarico, A. Federici, C. Pettenati and C. Caltagirone, Ther. Clin. Risk Manage., 2007, 3, 1113 CAS.
- B. Abdalla, B. Bisharat, M. Abir, M. Yassin and Z. Armaly, Traditional and Modern Medicine Harmonizing the Two Approaches in the Treatment of Neurodegeneration (Alzheimer's Disease—AD), in Complementary Therapies for the Contemporary Healthcare, ed. S. Marcelo and R. de Medeiros, INTECH Open Access Publisher, 2012 Search PubMed.
- M. Prince, M. Guerchet and M. Prina, CurrentState and Future Trends, World health Organization, Geneva, 2015 Search PubMed.
- M. Prince, M. Guerchet and M. Prina, World Alzheimer Report 2013: Journey of Caring – An Analysis of Long-Term Care for Dementia, Alzheimer’s Disease International, London, 2013 Search PubMed.
- R. N. Kalaria, G. E Maestre, R. Arizaga, R. P. Friedland, D. Galasko, K. Hall and M. Prince, Lancet Neurol., 2008, 7, 812 CrossRef PubMed.
- D. M. Holtzman, J. C. Morris and A. M. Goate, Sci. Transl. Med., 2011, 3, 77 Search PubMed.
- A. Alzheimer's, Alzheimer's Dementia, 2015, 11, 332 CrossRef.
- D. S. Auld, T. J. Kornecook, S. Bastianetto and R. Quirion, Prog. Neurobiol., 2002, 68, 209 CrossRef CAS PubMed.
- E. J. Mufson, S. E. Counts, S. E. Perez and S. D. Ginsberg, Expert Rev. Neurother., 2008, 8, 1703 CrossRef CAS PubMed.
- S. Paul, W. K. Jeon, J. L. Bizon and J. S. Han, Front. Aging Neurosci., 2015, 7, 43 Search PubMed.
- E. L. Konrath, B. M. Neves, P. S. Lunardi, C. dos Santos Passos, A. Simões-Pires, M. G. Ortega and A. T. Henriques, J. Ethnopharmacol., 2012, 139, 58 CrossRef PubMed.
- I. Aprahamian, J. E. Martinelli and M. S. Yassuda, Revista da Sociedade Brasileira de Clínica Médica, 2009, 7, 27 Search PubMed.
- D. M. Bowen, C. B. Smith, P. White and A. N. Davison, Brain, 1976, 99, 459 CrossRef CAS PubMed.
- P. Davies and A. J. F. Maloney, Lancet, 1976, 308, 1403 CrossRef.
- E. K. Perry, B. E. Tomlinson, G. Blessed, K. Bergmann, P. H. Gibson and R. H. Perry, Br. Med. J., 1978, 2, 1457 CrossRef CAS PubMed.
- P. J. Whitehouse, D. L. Price, R. G. Struble, A. W. Clark, J. T. Coyle and M. R. Delon, Science, 1982, 215, 1237 CAS.
- R. T. Bartus, R. Dean, B. Beer and A. S. Lippa, Science, 1982, 217, 408 CAS.
- J. T. Coyle, D. L. Price and M. R. De Long, Science, 1983, 219, 1184 CAS.
- A. V. Savonenko, T. Melnikova, A. Hiatt, T. Li, P. F. Worley, J. C. Troncoso and D. L. Price, Neuropsychopharmacology, 2012, 37, 261 CrossRef CAS PubMed.
- K. R. Scott and A. M. Barrett, Expert Rev. Neurother., 2007, 7, 407 CrossRef PubMed.
- K. L. Lanctôt, R. D. Rajaram and N. Herrmann, Ther. Adv. Neurol. Disord., 2009, 2, 163 CrossRef PubMed.
- D. A. Casey, D. Antimisiaris and J. O'Brien, P&T, 2010, 35, 208 Search PubMed.
- M. B. Colovic, D. Z. Krstic, T. D. Lazarevic-Pasti, A. M. Bondzic and V. M. Vasic, Curr. Neuropharmacol., 2013, 11, 315 CrossRef CAS PubMed.
- M. L. Crismon, Ann. Pharmacother., 1994, 28, 744 CrossRef CAS PubMed.
- J. Viegas, V. D. S. Bolzani, E. J. Barreiro and C. A. Manssour Fraga, Mini-Rev. Med. Chem., 2005, 5, 915 CrossRef PubMed.
- M. Mehta, A. Adem and M. Sabbagh, Int. J. Alzheimer's Dis., 2012, 2012, 1 CrossRef PubMed.
- D. K. Lahiri, M. R. Farlow, N. H. Greig and K. Sambamurti, Drug Dev. Res., 2002, 56, 267 CrossRef CAS.
- J. C. Messeder, L. W. Tinoco, J. D. Figueroa-Villar, E. M. Souza, R. Santa Rita and S. L. De Castro, Bioorg. Med. Chem. Lett., 1995, 5, 3079 CrossRef CAS.
- T. L. Martins, T. C. França, T. C. Ramalho and J. D. Figueroa-Villar, Synth. Commun., 2004, 34, 3891 CrossRef CAS.
- S. O. F. De Azeredo, E. M. Sales and J. D. Figueroa-Villar, J. Braz. Chem. Soc., 2016, 27, 10 Search PubMed.
- W. O. Foye, B. Almassian, M. S. Eisenberg and T. J. Maher, J. Pharm. Sci., 1990, 79, 527 CrossRef CAS PubMed.
- R. J. Sundberg, D. J. Dahlhausen, G. Mannikumar, B. Mavunkel, A. Biwas, V. Srinivasan, H. A. Mussallam, W. A. Reid and A. L. Ager, J. Med. Chem., 1990, 33, 298 CrossRef CAS PubMed.
- J. C. Messeder, L. W. Tinoco and J. D. Figueroa-Villar, Bioorg. Med. Chem. Lett., 1995, 5, 3079 CrossRef CAS.
- A. Andreani, A. Leoni, A. Locatelli, R. Morigi, M. Rambaldi, M. Recantini and V. Garaliene, Bioorg. Med. Chem., 2000, 8, 2359 CrossRef CAS PubMed.
- A. K. Gadad, C. S. Mahahjanshetti, S. Nimbalkar and A. Raichurkar, Eur. J. Med. Chem., 2000, 35, 853 CrossRef CAS PubMed.
- J. D. Figueroa-Villar, RSC Adv., 2017, 7, 23457 RSC.
- S. F. D. C. X. Soares, A. A. Vieira, R. T. Delfino and J. D. Figueroa-Villar, Bioorg. Med. Chem., 2013, 21, 5923 CrossRef PubMed.
- G. L. Ellman, K. D. Courtney, V. Andres and R. M. Featherstone, A new and rapid colorimetric determination of acetylcholinesterase activity, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
- E. da C. Petronilho, M. do N. Rennó, N. G. Castro, F. M. Da Silva, A. da C. Pinto and J. D. Figueroa-Villar, J. Enzyme Inhib. Med. Chem., 2016, 31, 1–10 Search PubMed.
- R. T. Delfino and J. D. Figueroa-Villar, J. Phys. Chem. B, 2009, 113, 8402 CrossRef CAS PubMed.
- G. B. Rocha, R. O. Freire, A. M. Simas and J. J. Stewart, J. Comput. Chem., 2006, 27, 11 CrossRef PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3 CrossRef CAS PubMed.
- C. A. Lipinski and A. Christopher, Drug Discovery Today: Technol., 2004, 1, 337 CrossRef CAS PubMed.
- W. P. Walters and M. A. Murcko, Adv. Drug Delivery Rev., 2002, 54, 255 CrossRef CAS PubMed; V. V. Zernov, K. V. Balakin, A. A. Ivaschenko, N. P. Savchuk and I. V. Pletnev, J. Chem. Inf. Comput. Sci., 2003, 43, 2048 CrossRef PubMed.
- C. H. Da Silva, V. L. Campo, I. Carvalho and C. A. Taft, J. Mol. Graphics Modell., 2006, 2, 169 CrossRef PubMed.
- L. G. De Souza, Dissertation (Master in Chemistry), Military Institute of Engineering, 2016.
- M. Harel, I. Schalk, L. Ehret-Sabatier, F. Bouet, M. Goeldner, C. Hirth and J. Sussman, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 9031 CrossRef CAS.
- H. Dvir, I. Silman, M. Harel, T. L. Rosenberry and J. L. Sussman, Chem.-Biol. Interact., 2010, 187, 10 CrossRef CAS PubMed.
- S. D. Bembenek, J. M. Keith, M. A. Letavic, R. Apodaca and A. J. Barbier, Bioorg. Med. Chem., 2008, 16, 2968 CrossRef CAS PubMed.
- K. Takeuchi and G. Wagner, Curr. Opin. Struct. Biol., 2006, 16, 109 CrossRef CAS PubMed.
- M. Goldflam, T. Tarragó, M. Gairí and E. Giralt, Methods Mol. Biol., 2012, 831, 233 CAS.
- J. A. Lima, R. S. Costa, R. A. Epifânio, N. G. Castro, M. S. Rocha and A. C. Pinto, Pharmacol., Biochem. Behav., 2009, 92, 508 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures, tables, graphs, IR and NMR spectra are available for a better illustration and comprehension of the results. See DOI: 10.1039/c7ra04180b |
| This journal is © The Royal Society of Chemistry 2017 |