
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A multianalyte fluorescent carbon dots sensing system constructed based on specific recognition of Fe(III) ions†
Pei Song a,
Lisha Zhanga,
Hao Longa,
Meng Menga,
Ting Liub,
Yongmei Yin*a and
Rimo Xi*a
a,
Lisha Zhanga,
Hao Longa,
Meng Menga,
Ting Liub,
Yongmei Yin*a and
Rimo Xi*a
aState Key Laboratory of Medicinal Chemical Biology, College of Pharmacy, Tianjin Key Laboratory of Molecular Drug Research, Nankai University, Tianjin 300353, China. E-mail: xirimo@nankai.edu.cn; yinyongmei@nankai.edu.cn
bTianjin Medical University Eye Hospital, Tianjin 300384, China
First published on 31st May 2017
Abstract
In this research, we developed a multianalyte fluorescence sensing system through a carbon dots (CDs)-based fluorescent probe that can specifically recognize Fe(III) by fluorescence quenching. The CDs prepared using black tea by a hydrothermal method show outstanding properties like low cytotoxicity, high photostability, excellent biocompatibility, and high sensitivity. It was found that the fluorescence of CDs can be quenched by micromolar concentrations of Fe(III) in both aqueous solutions as well as living cells. It is well known that glucose can be oxidized by glucose oxidase (GOx) to release H2O2, which, in turn, can oxidize Fe(II) to Fe(III). Based on this consideration, a multianalyte sensing system was established. Therefore the quantitative analysis of Fe(III), H2O2, and glucose with detection limits of 0.25 μM, 0.82 μM, and 1.71 μM, respectively, was achieved by the simple and cost-effective multianalyte CDs sensing system constructed. The sensing system showed high photostability and negligible cytotoxicity toward HeLa cells, which enables it to be applied in the visualization of Fe(III) or H2O2 in living cells. The system was further applied in the detection of Fe(III) or glucose in human serum, and satisfactory results were obtained.
Introduction
As the most abundant transition metal ion in cellular systems, ferric ions perform crucial roles in biological systems involved in enzyme catalysis, cellular metabolism, oxygen transport in hemoglobin, and as a cofactor in enzyme-based reactions.1–5 Iron overload and deficiency can disturb cellular homeostasis and give rise to various diseases resulting from biological disorders, such as anemia,6 dysfunction of kidney7,8 and liver,9,10 arthritis, Alzheimer's disease,11,12 heart failure, diabetes, and cancer.13,14 Therefore, the determination of Fe(III) is of great significance for early diagnoses of these diseases. Among the various strategies for Fe(III) sensing, fluorescence assays have emerged as a rational option for highly sensitive and fast analyses. So far, many fluorescent probes including semiconductor quantum dots, fluorescent metal nanoclusters, and fluorescent metal organic frameworks have been developed for the fluorescence detection of Fe(III).15–17 However, the preparation of these materials needs specialized synthetic skills and complicated purification procedures. Additionally, because of the toxicity and water-insolubility, the application of these chemosensors has been greatly limited, especially in living cells.As newcomers to the carbon nanomaterials family, carbon dots (CDs) have attracted tremendous attention owing to their captivating properties such as wavelength-tuneable emission, low cytotoxicity, excellent photostability, aqueous solubility, and favorable biocompatibility.18,19 As a consequence, CDs have been widely used in fluorescence sensing, bioimaging, drug delivery, etc.20–22 For chemical sensing applications, CDs-based Fe(III) sensors have been developed.23–30 For specifically sensing Fe, the challenge is to avoid the interference from Fe(II). However, it is not so easy to set up a CDs-based sensing system to detect Fe(III) without interruptions from Fe(II).23,25 Even if such sensor could be constructed with acceptable discrimination of Fe(III) and Fe(II), few could respond well to Fe(III) over a wide range of pH values.24
In this research, we prepared the CDs through a simple one-pot hydrothermal method from used black tea, and the CDs can be efficiently quenched by Fe(III). The prepared CDs not only respond well to Fe(III) in a buffer over a range of pH from 4.5 to 9.0, but also sensitively respond to Fe(III) in water with negligible interference from Fe(II). Hence, a multianalyte sensing system was constructed to determine Fe(III), H2O2, and glucose, since H2O2 can oxidize Fe(II) to Fe(III) and glucose can be oxidized to yield H2O2 (Scheme 1). The sensing system was evaluated in human serum and living cells.
 | ||
| Scheme 1 A multianalyte fluorescent CDs-sensing system for sensing of Fe(III), H2O2, and glucose. | ||
Experimental section
Materials
Jinjunmei black tea was bought from the local tea supermarket. FeCl3, FeCl2, Pb(NO3)2, ZnCl2, CuSO4, AlCl3, Hg(NO3)2, CdCl2, MgCl2, CaCl2, BaCl2, MnCl2, CoCl2, NiCl2, CrCl3, AgNO3, glycine, cysteine, H2O2, glucose, and glucose oxidase were purchased from Aladdin Chemistry Co., Ltd. (Shanghai, China). Other chemicals were of analytical grade. Human serum was purchased from Beijing Solarbio Science & Technology Co., Ltd. Purified water was prepared through a Millipore system and used throughout the experiment.Instruments
UV-vis absorption spectra were recorded on a Shimadzu UV-1750 spectrophotometer with one pair of 10 mm quartz cell. Fluorescence spectral recording and emission measurements were conducted on a Hitachi F-4600 FL spectrophotometer using 5/5 nm slit width and equipped with a 10 mm quartz cell. Fourier transform infrared spectra (FTIR) were acquired on a Bio-Rad FTS 6000 spectrometer using KBr pellets. The transmission electron microscopic (TEM) image was acquired on a FEI Tecnai G2 F20 microscopy operating at 200 kV. The particle size distribution was measured by using a Nicomp 380 submicron particle sizer. The X-ray photoelectron spectra (XPS) were carried out on an AXIS Ultra DLD X-ray photoelectron spectrometer. Fluorescence imaging experiments were performed on a Zeiss LSM710 confocal laser scanning microscope with an objective lens (×40).Preparation of fluorescent CDs
A hydrothermal method was used to prepare the CDs. Briefly, 1.8 g drying used black tea was added into 10 mL ultrapure water; then, the mixture was transferred into a 50 mL Teflon-lined stainless steel autoclave. The mixture was heated at 200 °C for 5 h and then cooled to room temperature. The CDs solution was collected after removing the carbide slag through centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. The supernatant was purified by filtration through a 0.22 μm filter membrane and then diluted with ultrapure water for further use.
000 rpm for 10 min. The supernatant was purified by filtration through a 0.22 μm filter membrane and then diluted with ultrapure water for further use.
General procedure for fluorescence measurements
The selectivity of CDs for sensing Fe(III) was evaluated by the interfered metal ions (Fe2+, Cu2+, Cd2+, Cr3+, Zn2+, Mn2+, Ni2+, Co2+, Al3+, Pb2+, Ag+, Hg2+, Mg2+, Ba2+, Ca2+, Na+, and K+) under the same conditions. Besides, 100 μM common anions and organic compounds (PO43−, SO42−, CO32−, Cl−, cysteine, dopamine, glycine, and ascorbic acid) were analyzed by the sensor with the same procedures as indicated above.
Additionally, the feasibility of the proposed method for the analysis of Fe(III) in human serum was investigated. After the samples of human serum were centrifuged at 10000 rpm for 10 min and the precipitation was removed, the supernatant was kept for further use. Thus, different concentrations of Fe(III) ions (10 μL) and supernatant (10 μL) were added in the CDs solution (980 μL). After incubation for 10 min at room temperature, the fluorescence emission spectra were recorded.
For the analysis of glucose, appropriate concentrations of GOx (20 μg mL−1) and Fe(II) (120 μM) were added into the CDs solution, followed by the addition of different concentrations of glucose (0, 0.1, 0.5, 2.5, 5, 10, 20, 30, 40, 50, 80, and 100 μM). The fluorescence emission spectra of the mixture were recorded after the reaction for 30 min. For all the fluorescence measurements, the excitation wavelength was set at 320 nm, and the emission was monitored at 400 nm.
Cytotoxicity investigation
The cell viability was measured using the MTT assay. HeLa cells were propagated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Briefly, the cells were seeded into 96-well plates at a density of 8 × 103 cells in each well. After incubating for 24 h at 37 °C in a 5% CO2 humidified atmosphere, the cells were treated with various concentrations (0, 20, 40, 80, 100, 200, 400, and 800 μg mL−1) of CDs solution and cultured for another 24 h. At the end of the treatment, 20 μL of MTT (5 mg mL−1) was added to each well, followed by additional incubation for 4 h. The culture medium was then removed, and 150 μL of DMSO was then added to dissolve the formazan crystals. The optical density (OD) of each well was measured at 570 nm with a microplate reader. The cell viability was estimated according to the following equation:| Cell viability (%) = (ODtreated/ODcontrol) × 100% |
Fluorescence imaging of living cells
HeLa cells were seeded in a glass-bottom cell culture dish at 37 °C in 5% CO2 for 24 h. The growth medium was removed and replaced by the culture medium containing CDs (12 μg mL−1). After incubation for 24 h, the cells were washed for three times with PBS. For monitoring endogenous H2O2, cells were pretreated with 1 μg mL−1 phorbol myristate acetate (PMA) or 1 mM N-acetylcysteine (NAC) for 1 h. Similarly, for imaging of exogenous H2O2, cells were treated with 100 μM H2O2 for 1 h. After incubation, cells were washed thoroughly three times and kept in PBS for the fluorescence imaging. Thereafter, 200 μM Fe(III) or 120 μM Fe(II) was added and incubated for 30 min. The fluorescence signal was recorded with the excitation of 405 nm.Results and discussion
Characterization of CDs
The CDs were prepared by a simple one-pot hydrothermal method from used black tea. Fig. 1 shows the representative TEM image and size distribution histogram of the products, revealing that the average size of the monodispersed nanoparticles was around 4.6 nm. FTIR spectrum was obtained to identify the surface functional groups presented on the as-prepared CDs. As shown in Fig. S1,† the characteristic absorption band at 3286 cm−1 was assigned to the stretching vibration of O–H and N–H, while C–H stretching vibration was observed at 2962 cm−1. The peaks at 1666 and 1450 cm−1 could be identified as the asymmetric and symmetric vibrations of COO−, respectively.31 The absorption band of –NH2 stretching vibrations at 1543 cm−1 was also observed, and the bands in the range of 1083–1323 cm−1 were attributed to the large amount of C–O groups.32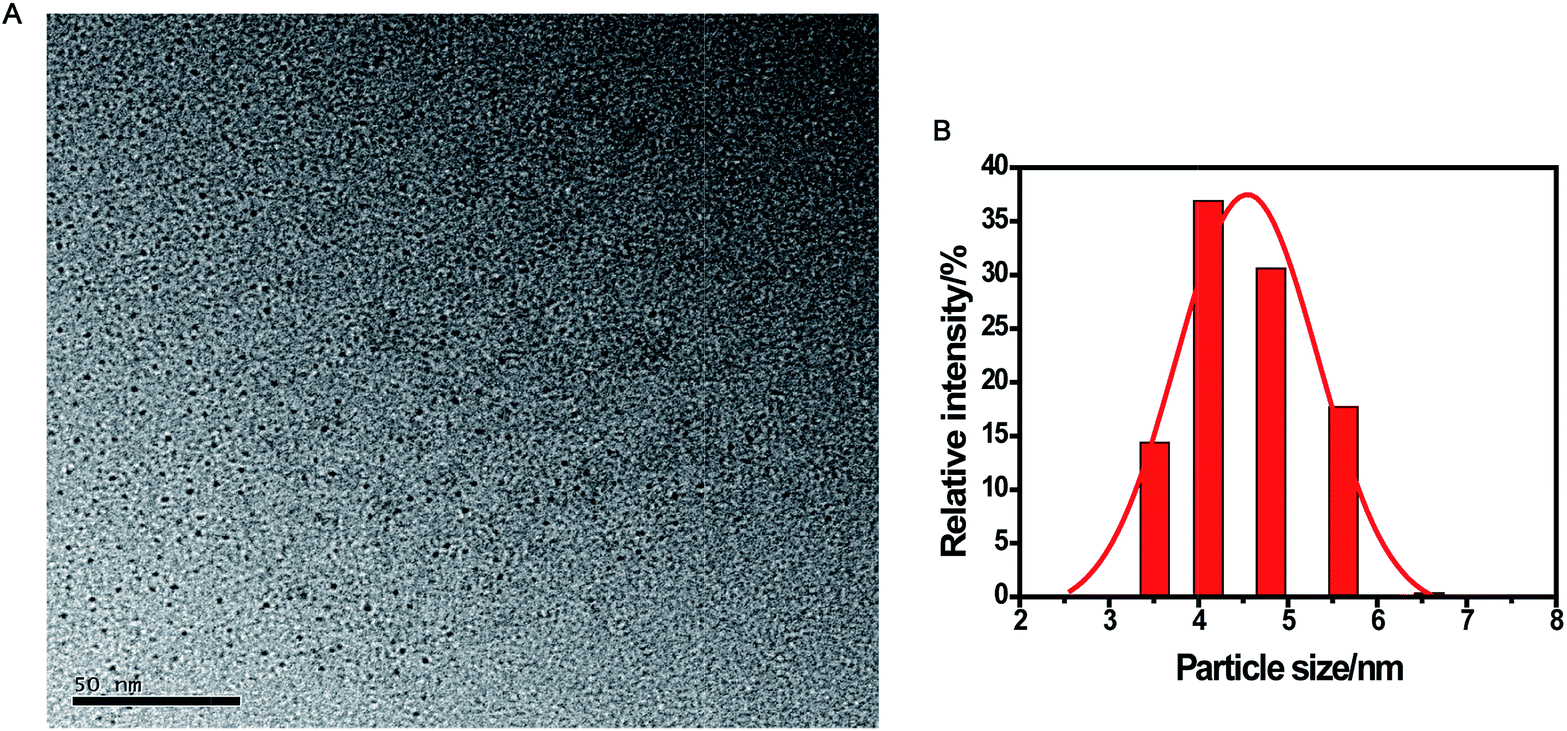 | ||
| Fig. 1 TEM image (A) and the corresponding size distribution histograms (B) of the CDs. | ||
The surface composition and elemental analysis for the resultant nanoparticles were characterized by XPS. The XPS survey spectrum (Fig. 2A) revealed that the CDs were mainly composed of carbon (68.68%), oxygen (23.42%), and nitrogen (7.9%). The high-resolution XPS spectrum of C 1s (Fig. 2B) showed three peaks at 284.6, 286.0, and 287.8 eV, which were assigned to C–C, C–N/C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively.33 The two peaks at 531.3 and 532.1 eV in the O 1s spectrum (Fig. 2C) were associated with the CO and C–OH/C–O–C bands, respectively. Additionally, the N 1s spectrum (Fig. 2D) showed two peaks at 399.7 and 401.4 eV, which were attributed to C–N–C and N–H, respectively.34 The surface components of the resultant CDs determined by XPS were consistent with the FTIR results. These data demonstrated that the as-prepared CDs function with hydroxyl and carboxylic/carbonyl moieties that may originate from the carbohydrates in black tea.
O, respectively.33 The two peaks at 531.3 and 532.1 eV in the O 1s spectrum (Fig. 2C) were associated with the CO and C–OH/C–O–C bands, respectively. Additionally, the N 1s spectrum (Fig. 2D) showed two peaks at 399.7 and 401.4 eV, which were attributed to C–N–C and N–H, respectively.34 The surface components of the resultant CDs determined by XPS were consistent with the FTIR results. These data demonstrated that the as-prepared CDs function with hydroxyl and carboxylic/carbonyl moieties that may originate from the carbohydrates in black tea.
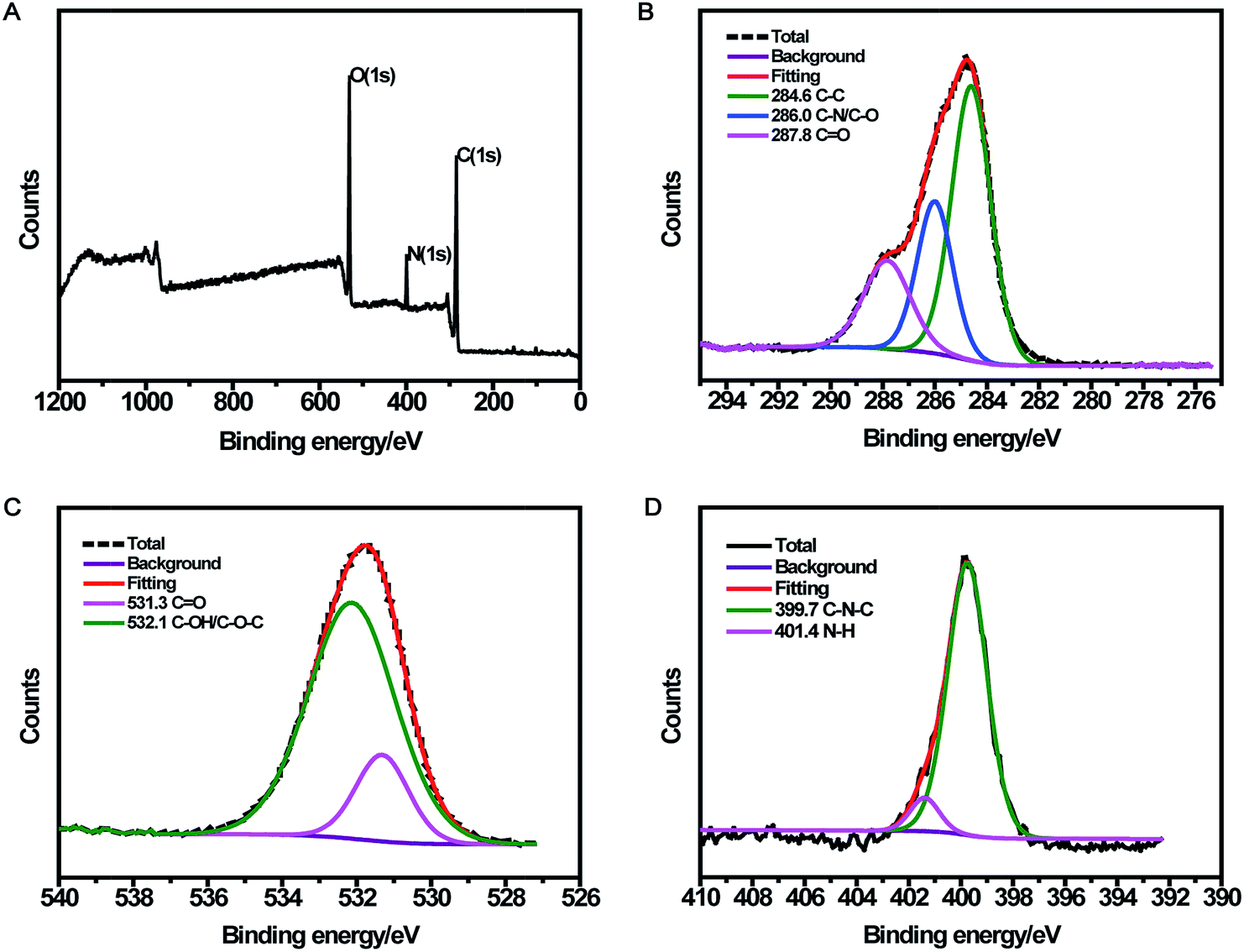 | ||
| Fig. 2 Survey (A) and high-resolution C 1s (B), O 1s (C), and N 1s (D) XPS spectra of CDs. | ||
Optical properties of CDs
The remarkable optical properties of the synthesized CDs were confirmed by the UV-vis absorption and fluorescence spectra of the aqueous dispersion of the nanoparticles. As illustrated in Fig. 3, the UV-vis spectrum shows two absorption peaks at 270 nm and 320 nm, which were attributed to the π–π* transition of CC bonds and n–π* transition of CO bonds in CDs, respectively.25,35 The emission spectra showed a strong emission with the maximum emission wavelength at 400 nm under excitation at 320 nm. A bright blue luminescence of CDs was observed under UV light (365 nm) (Fig. 3, inset), revealing their blue photoluminescence property. Additionally, with an increase in excitation wavelength from 290 nm to 420 nm, the maximum emission shifted from 398 nm to 490 nm along with variation in the emission intensity (Fig. S2†). The strongest emission spectrum was observed at the excitation wavelength of 320 nm. The excitation-dependent fluorescence behavior of CDs resulted from the different sizes and surface states of nanoparticles,36 as in the case of most of the luminescent CDs and graphene quantum dots.37–39
 | ||
| Fig. 3 UV-vis absorption spectra and emission spectra of CDs at the excitation of 320 nm. Inset: photographs of CDs under visible light (left) and a UV beam of 365 nm (right). | ||
Stability of CDs
To investigate the stability of CDs, fluorescence intensity toward various pH values, high ionic strength, and continuous UV irradiation were measured. Fig. S3A† presents the emission spectrum of CDs at different pH values. Evidently, CDs exhibited strong and stable fluorescence in the range of pH from 4.5 to 9. The stable fluorescence intensities could be understood in terms of the change in surface charge owing to protonation-deprotonation.36 As shown in Fig. S3B,† the fluorescence intensity changed slightly with varying NaCl concentrations, which resulted from no ionization of groups located on the CDs surface.40 Moreover, the fluorescence intensity did not significantly change after continuous Xe lamp (365 nm) irradiation for 60 min (Fig. S3C†), demonstrating excellent photostability of the CDs. Considering the results above, it was confirmed that the CDs were highly stable under different conditions.Fluorescence response of CDs toward Fe(III)
As displayed in Fig. 4, the CDs showed strong fluorescence with the excitation wavelength of 320 nm and the fluorescence was quenched dramatically upon the addition of 100 μM Fe(III). According to the XPS data, oxygen was the main component of CDs, and the –OH/–O–C groups make up about 81% of the entire oxygen-containing groups. Meanwhile, the FTIR results demonstrated that plentiful hydroxyl groups exist on the surface of the CDs. Furthermore, the electronegativity of oxygen-containing groups was higher than that of nitrogen-containing groups.41 Thus, we suggested that the quenching of fluorescence was mostly attributed to the binding affinity of hydroxyl groups on the surface of CDs to Fe(III). Fe(III) could react with the hydroxyl groups of CDs to form the complex by a special coordination interaction. In order to further explore the mechanism of fluorescence quenching by Fe(III), fluorescence decay curves of CDs in the absence and presence of Fe(III) were examined. As displayed in Fig. S4,† two fluorescence decay curves were completely overlapped and had no significant change. Moreover, the average fluorescence lifetime was calculated to be 5.53 ns for CDs and 5.50 ns for CDs–Fe(III) complex. This is because static quenching arising from the formation of a ground-state complex between the fluorescence material and quencher could not lead to the change of lifetime. However, dynamic quenching could result in a decrease of lifetime.20,42,43 Our result showed no significant change in the lifetime, indicating the occurrence of static quenching and the formation of a complex.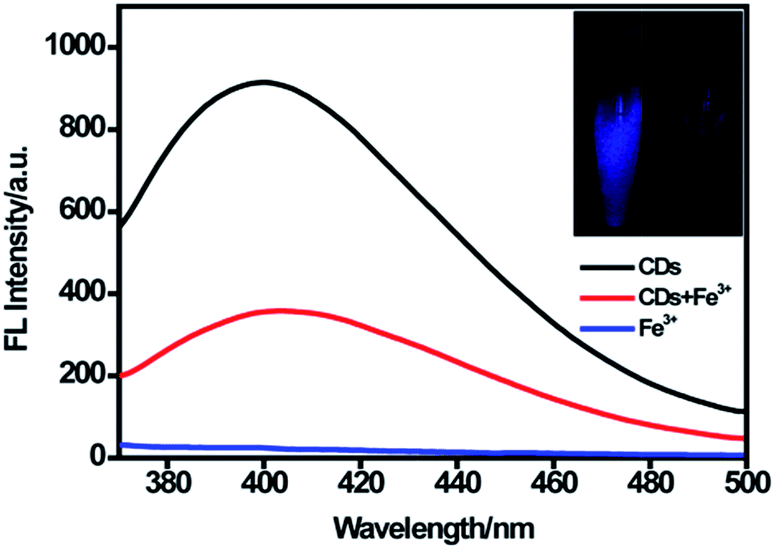 | ||
| Fig. 4 Fluorescence spectra of CDs in the absence and presence of 100 μM Fe(III) at the excitation of 320 nm. Insets showed the photographs of the CDs solutions in the absence and presence of Fe(III) under UV light (365 nm). | ||
Fluorescence sensing for Fe(III)
For better performance in sensing Fe(III), the effects of reaction time and medium were evaluated. Fig. S5A† shows the fluorescence response of CDs after adding different concentrations of Fe(III) (10 μM and 100 μM). It was found that the fluorescence response became stable in 10 min. In addition, the fluorescence response toward Fe(III) in ultrapure water and buffer with different pH values is shown in Fig. S5B.† The results indicated that CDs could respond well to Fe(III) in ultrapure water and buffer over the range of pH from 4.5 to 9.0. The wide pH range for sensing Fe(III) could promote the application of CDs at various pH conditions, especially in cells. Some studies did not investigate the pH range for sensing Fe(III). Shi and co-workers developed the CDs-based sensor for Fe(III) over the range of pH in 6.0–8.0.23 Sachdev et al.25 did obtain a wide pH range; however, the results of linear range with 0–6 μM and F/F0 value of 0.8 for Fe(II) were not satisfactory (F0 and F are fluorescence intensities at 400 nm in the absence and presence of ions, respectively).Fig. S6† presented a gradual decrease in the fluorescence intensity with increasing Fe(III) concentration, revealing a response of CDs to Fe(III). In the presence of 600 μM Fe(III), the quenching efficiency could reach up to 89% (Fig. 5A). (F0/F) showed a perfect linear relationship (R2 = 0.999) vs. the concentration of Fe(III) in the range of 0.25–60 μM (Fig. 5B). Likewise, the limit of detection (LOD) of Fe(III) was estimated to be 0.25 μM (according to a signal-to-noise ratio of 3), which was comparable to those detection limits obtained by other fluorescent probes for Fe(III) in Table 1.25,44–47
 | ||
| Fig. 5 (A) The variation of the quenching efficiency of CDs in the presence of different concentrations of Fe(III) from 0 to 600 μM. (B) Fluorescence emission spectra of CDs upon addition of various concentrations of Fe(III) (from top to bottom: 0, 0.25, 2.5, 5, 10, 20, 30, 40, 50, 60, 80, and 100 μM). Inset is the linear calibration plot for F0/F versus different concentrations of Fe(III) from 0.25 μM to 60 μM. | ||
| Fluorescence probes | Carbon source | Detection limit (μM) | Linear range (μM) | Refs |
|---|---|---|---|---|
| Graphene quantum dots | Aspartic acid | 0.26 | 0–50 | 44 |
| Carbon dots | DL-Malic acid | 0.8 | 0–200 | 45 |
| Carbon dots | Chitosan, acetic acid and ethylenediamine | 5.3 | 0.17–32 | 46 |
| Carbon dots | Citric acid and tris | 1.3 | 2–50 | 47 |
| Carbon dots | Coriander leaves | 0.4 | 0–6 | 25 |
| Multianalyte sensing system | Used black tea | 0.25 | 0.25–60 | This work |
Selectivity is another important parameter to evaluate the performance of the sensing system. Therefore, the fluorescence intensity of CDs in the presence of different metals ions was monitored (Fig. 6). A much lower fluorescence intensity was observed for CDs upon the addition of Fe(III). However, the fluorescence intensity did not tremendously decrease after adding Fe(II) into the CDs dispersion with a F/F0 value of 0.96, which is larger than those of carbon nanomaterials reported in the literature. For example, Sachdev et al.25 reported a F/F0 value of 0.8 for Fe(II) using CDs from coriander leaves. Zhang et al.44 and Chen et al.48 reported F/F0 values of 0.45 and 0.89, respectively. Although a negligible interference from Fe(II) to the CDs in sensing Fe(III) was observed, the efficiency of Fe(III) sensing greatly relied on the pH of the sample solution.24 The prepared CDs in our study showed more specific recognition of Fe(III) from Fe(II) and responded well to Fe(III) in a wide range of pH (4.5 to 9.0), indicating the satisfactory tolerance of this sensing system. The discrimination effect for Fe(III) ions might originate from the exceptional coordination between Fe(III) ions and hydroxyl groups of CDs.46 Besides metal ions, various anions and organic molecules that may co-exist with Fe(III) were observed under the same conditions. As shown in Fig. S7,† in the presence of these interfering species, the fluorescence intensity had no significant change. The results confirmed the good selectivity of the proposed method for Fe(III) sensing.
 | ||
| Fig. 6 Fluorescence response of CDs in the presence of different metal ions. The concentration of metal ions was 100 μM. | ||
Multianalyte sensing system for H2O2 and glucose
A multianalyte fluorescent CDs sensing platform was designed for the qualitative and quantitative analysis of H2O2 and glucose, which depended on the stoichiometric relationship in the biochemical reaction.In the presence of H2O2 (Fig. 7A), Fe(II) was oxidized into Fe(III), resulting in the fluorescence quenching of CDs. To demonstrate the feasibility of the multianalyte sensing system for H2O2 detection, we evaluated the quenching phenomena of CDs caused by H2O2 (Fig. S8†). In order to establish the assay for detecting H2O2, the reaction time and concentration of Fe(II) for sensing H2O2 were optimized. As illustrated in Fig. S9A,† the relative fluorescence intensities had no obvious change when the concentration of Fe(II) was higher than 120 μM. The fluorescence was gradually stable within 10 min, showing that the reaction was complete (Fig. S9B†). Thus, 120 μM Fe(II) and 10 min incubation time were chosen for the quantitative determination of H2O2. Under the optimum experimental conditions, the sensitivity, linear response range, and detection limit of the multianalyte sensing system for H2O2 were measured. (F0 − F)/F0 vs. concentration of H2O2 from 0.1 to 100 μM was observed (Fig. S10†). As shown in Fig. 7B, there was a good linear correlation between (F0 − F)/F0 and the concentration of H2O2 in the range from 0.1 μM to 40 μM, providing a detection limit of 0.82 μM.
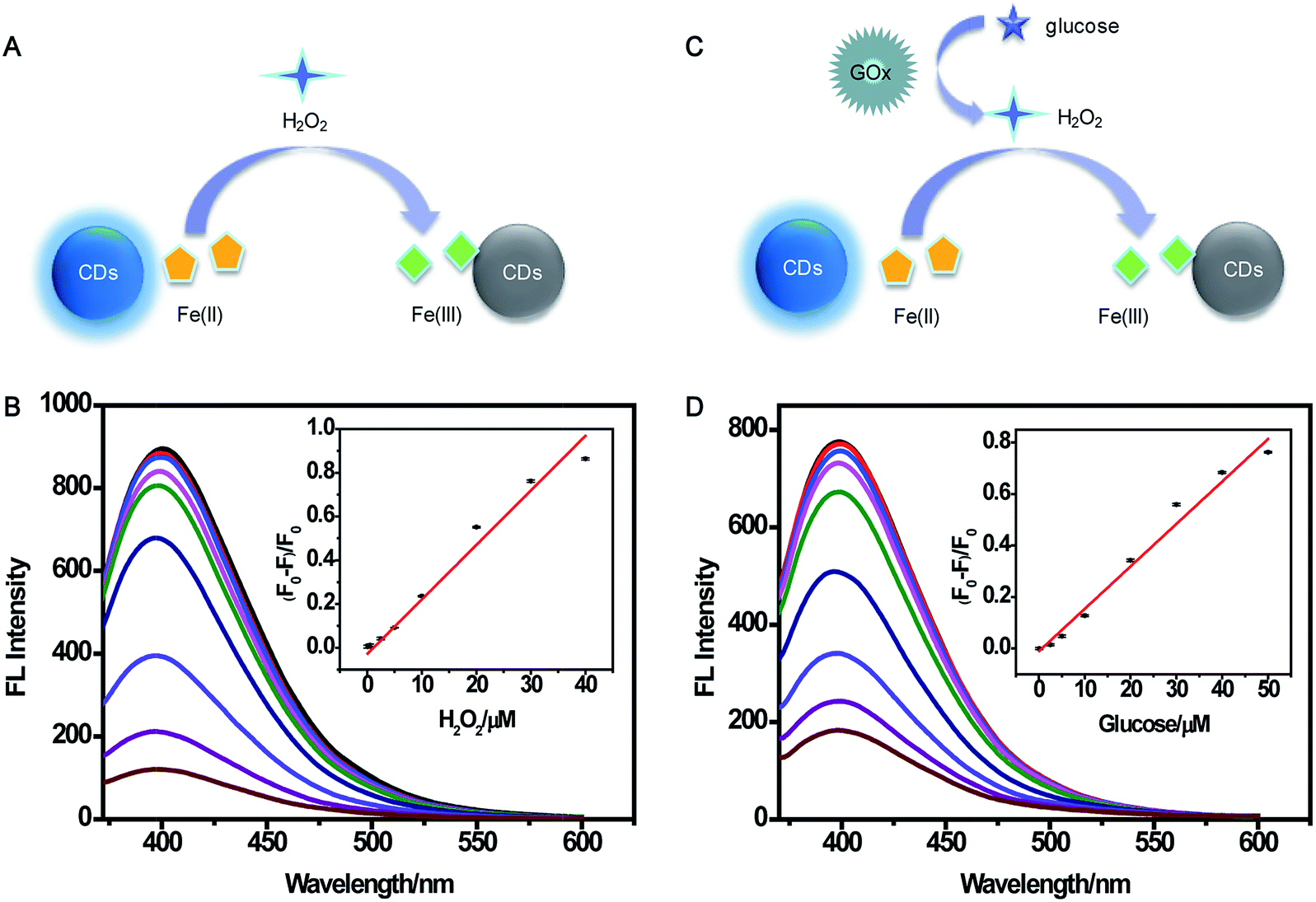 | ||
| Fig. 7 Schematic illustration of H2O2 (A) and glucose (C) detection mechanism using CDs. (B) Fluorescence emission spectra for the determination of H2O2 (0.1–40 μM). Inset: (F0 − F)/F0 vs. [H2O2]. (D) Fluorescence emission spectra for the determination of glucose (0.1–50 μM). Inset: (F0 − F)/F0 vs. [glucose]. | ||
As illustrated in Fig. 7C, H2O2 was produced in the oxidization reaction of glucose in the presence of GOx. Under the existence of the H2O2, Fe(II) was oxidized to Fe(III), and Fe(III) could yield the fluorescence quenching of CDs. Based on this consideration, indirect detection of glucose can be performed. A comparison of the relative fluorescence intensity changes of CDs after the addition of glucose, GOx, and “glucose + GOx + Fe(II)” is shown in Fig. S11,† suggesting the feasibility of the multianalyte sensing system for sensing glucose. Then, the concentration of GOx and reaction time were optimized. When the concentration of GOx was higher than 20 μg mL−1, the fluorescence intensity had no significant change (Fig. S12A†). Fig. S12B† shows that the reaction was complete within 30 min. Thus, 20 μg mL−1 GOx and 30 min incubation time were chosen as the optimal conditions for glucose assay. Then, the sensitive determination of glucose was performed by adding different concentrations of glucose into the sensing system of CDs, Fe(II), and GOx under the optimum experimental conditions (Fig. S13†). The (F0 − F)/F0 value indicated that the linear relationship was good in relation to glucose concentration ranging from 0.1 μM to 50 μM (Fig. 7D). The detection limit was estimated to be 1.71 μM. This demonstrated the sensing system could be used for glucose detection with satisfactory sensitivity.
Intracellular imaging of Fe(III) and H2O2
For further biological applications, MTT assays were performed to evaluate the cytotoxicity of the CDs using cervical cancer HeLa cells. As expected, Fig. S14† showed that the cell viability was estimated to be greater than 90% upon the addition of CDs over a wide concentration range of 20 to 800 μg mL−1. High cell viability confirmed the low cytotoxicity and excellent biocompatibility of the as-prepared CDs, which indicated the great potential of CDs for monitoring Fe(III) and H2O2 in living cells.Taken together, the CDs gave advantages of small size, high selectivity, good photostability, and, especially, biocompatibility, substantially making them superior in potential bioimaging applications. Accordingly, confocal fluorescence images were obtained to further demonstrate the availability of CDs in intracellular Fe(III) and H2O2 imaging for HeLa cells. As illustrated in Fig. 8A, the fluorescence can be observed in the CDs-loaded cell membrane and the cytoplasmic area, while the fluorescence signal was quite weak in the cell nucleus. The fluorescence intensity decreased distinctly upon the addition of 200 μM Fe(III) and incubation of 30 min at room temperature, and the quenching efficiency was calculated to be about 74%. Furthermore, the bright-field measurements indicated that the cells are viable throughout the imaging experiments. All the observations indicated that the proposed multianalyte CDs could be applied for the effective semi-quantitative imaging of Fe(III) in living cells. Further, the sensing system for monitoring H2O2 in living biological systems was conducted. Fig. 8B shows that HeLa cells loaded with 12 μg mL−1 CDs showed bright fluorescence. The treatment of CDs-loaded cells with 100 μM H2O2 for 60 min caused no change in the fluorescence intensity. A striking fluorescence decrease was observed upon the addition of 120 μM Fe(II) into the growth medium and incubation of 30 min.
 | ||
| Fig. 8 Confocal fluorescence images of living HeLa cells labeled with CDs (12 μg mL−1) under different conditions. Bright-field image (1, A–E) and fluorescence image (2, A–E) of HeLa cells labeled with CDs. (A) Representative images of HeLa cells treated with CDs and Fe(III): (A3) only CDs; (A4) CDs + 200 μM Fe(III); (A5) the relative fluorescence intensities of CDs in the absence and presence of Fe(III). (B) Representative images of HeLa cells treated with CDs, H2O2, and Fe(II): (B3) CDs + 100 μM H2O2; (B4) CDs + 100 μM H2O2 + 120 μM Fe(II); (B5) the relative fluorescence intensities of CDs in the absence and presence of H2O2 and Fe(II). (C) Representative images of HeLa cells treated with CDs, PMA, and Fe(II): (C3) CDs + 1 μg mL−1 PMA; (C4) CDs + 1 μg mL−1 PMA + 120 μM Fe(II); (C5) the relative fluorescence intensities of CDs in the absence and presence of PMA and Fe(II). (D) Representative images of HeLa cells treated with CDs, NAC, and Fe(II): (D3) CDs + 1 mM NAC; (D4) CDs + 1 mM NAC + 120 μM Fe(II); (D5) the relative fluorescence intensities of CDs in the absence and presence of NAC and Fe(II). (E) Representative images of HeLa cells treated with CDs and Fe(II): (E3) only CDs; (E4) CDs + 120 μM Fe(II); (E5) the relative fluorescence intensities of CDs in the absence and presence of Fe(II). | ||
To prove further that the generation of fluorescence signal was dependent on H2O2, PMA that can induce H2O2 generation49 and NAC that can scavenge free-radicals in cancer cells50 were used as the modulators of intracellular H2O2 levels. PMA (1 μg mL−1) or NAC (1 mM) was added to test the ability of the sensing system to monitor the endogenous bursts of H2O2 produced within living cells. After the addition of Fe(II), the fluorescence intensity in the PMA-treated cells in Fig. 8C was significantly decreased, similar to that of H2O2-treated cells, while the fluorescence intensity in the NAC-treated cells had a negligible change (Fig. 8D).
In addition, we investigated the effect of the native H2O2 level of tumor cells on fluorescence imaging. The CDs-loaded cells were incubated with Fe(II) for 30 min; then, the fluorescence signals within the cells were analyzed. As shown in Fig. 8E, a decrease in fluorescence intensity was observed, which could be attributed to the H2O2 existing in the tumor cells.51 Consequently, all the results demonstrated that the multianalyte CDs sensing system has good potential for the imaging of intracellular H2O2.
Detection of Fe(III) and glucose in human serum samples
In order to evaluate the efficiency of the multianalyte sensing system in detecting Fe(III) or glucose, human serum samples were selected. Human serum samples were spiked with different concentrations of Fe(III)/glucose and measured with the proposed methods: high analytical precision and good recoveries were obtained (Table S1†). These results further justified the reliability and feasibility of the multianalyte sensing system for monitoring Fe(III) or glucose in biological samples.Conclusions
A high-performance multianalyte fluorescent CDs sensing platform based on the specific recognition of Fe(III) from Fe(II) has been successfully developed. With the property of specific response to Fe(III), the system was applied to detect not only Fe(III) but also H2O2 and glucose. The sensitive assays for Fe(III), H2O2, and glucose analyses were established with LOD values of 0.25 μM, 0.82 μM, and 1.71 μM, respectively. The application of the system in sensing Fe(III) and glucose in human serum showed excellent sensitivity and accuracy. Further, the system can be used to monitor Fe(III) and H2O2 in living cells. These results demonstrated that the system was promising as an efficient platform for sensing these analytes for clinical diagnosis.Acknowledgements
This work was supported by the financial support of the International Science & Technology Cooperation Program of China (No. 2015DFR40460), National Natural Science Foundation under Grant (No. 81402896, No. 81573390), the National High-Tech Research and Development Program of China (863 Program) under Grant (No. 2014AA022303).Notes and references
- T. Walczyk and F. Von Blanckenburg, Science, 2002, 295, 2065–2066 CrossRef CAS PubMed.
- T. A. Rouault, Nat. Chem. Biol., 2006, 2, 406–414 CrossRef CAS PubMed.
- M. W. Hentze, M. U. Muckenthaler, B. Galy and C. Camaschella, Cell, 2010, 142, 24–38 CrossRef CAS PubMed.
- B. D'Autreaux, N. P. Tucker, R. Dixon and S. Spiro, Nature, 2005, 437, 769–772 CrossRef PubMed.
- R. S. Eisenstein, Annu. Rev. Nutr., 2000, 20, 627–662 CrossRef CAS PubMed.
- D. A. Weinstein, C. N. Roy, M. D. Fleming, M. F. Loda, J. I. Wolfsdorf and N. C. Andrews, Blood, 2002, 100, 3776–3781 CrossRef CAS PubMed.
- W. H. Horl, J. Am. Soc. Nephrol., 2007, 18, 382–393 CrossRef PubMed.
- R. Agarwal, N. Vasavada, N. G. Sachs and S. Chase, Kidney Int., 2004, 65, 2279–2289 CrossRef CAS PubMed.
- G. T. Sucak, Z. A. Yegin, Z. N. Ozkurt, S. Z. Aki, T. Karakan and G. Akyol, Bone Marrow Transplant., 2008, 42, 461–467 CrossRef CAS PubMed.
- J. C. Duvigneau, C. Piskernik, S. Haindl, B. Kloesch, R. T. Hartl, M. Huttemann, I. Lee, T. Ebel, R. Moldzio, M. Gemeiner, H. Redl and A. V. Kozlov, Lab. Invest., 2008, 88, 70–77 CrossRef CAS PubMed.
- S. Altamura and M. U. Muckenthaler, J. Alzheimer's Dis., 2009, 16, 879–895 CrossRef PubMed.
- J. A. Duce, A. Tsatsanis, M. A. Cater, S. A. James, E. Robb, K. Wikhe, S. L. Leong, K. Perez, T. Johanssen, M. A. Greenough, H. H. Cho, D. Galatis, R. D. Moir, C. L. Masters, C. McLean, R. E. Tanzi, R. Cappai, K. J. Barnham, G. D. Ciccotosto, J. T. Rogers and A. I. Bush, Cell, 2010, 142, 857–867 CrossRef CAS PubMed.
- S. Zhang, J. Li, M. Zeng, J. Xu, X. Wang and W. Hu, Nanoscale, 2014, 6, 4157–4162 RSC.
- N. Narayanaswamy and T. Govindaraju, Sens. Actuators, B, 2012, 161, 304–310 CrossRef CAS.
- C. X. Yang, H. B. Ren and X. P. Yan, Anal. Chem., 2013, 85, 7441–7446 CrossRef CAS PubMed.
- X. Y. Qu, Q. Liu, X. N. Ji, H. C. Chen, Z. K. Zhou and Z. Shen, Chem. Commun., 2012, 48, 4600–4602 RSC.
- J. A. Ho, H. C. Chang and W. T. Su, Anal. Chem., 2012, 84, 3246–3253 CrossRef PubMed.
- J. Zong, X. L. Yang, A. Trinchi, S. Hardin, I. Cole, Y. H. Zhu, C. Z. Li, T. Muster and G. Wei, Biosens. Bioelectron., 2014, 51, 330–335 CrossRef CAS PubMed.
- H. T. Li, Z. H. Kang, Y. Liu and S. T. Lee, J. Mater. Chem., 2012, 22, 24230–24253 RSC.
- S. J. Zhu, Q. N. Meng, L. Wang, J. H. Zhang, Y. B. Song, H. Jin, K. Zhang, H. C. Sun, H. Y. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- X. T. Zheng, A. Than, A. Ananthanaraya, D. H. Kim and P. Chen, ACS Nano, 2013, 7, 6278–6286 CrossRef CAS PubMed.
- Y. Li, Y. Zhao, H. H. Cheng, Y. Hu, G. Q. Shi, L. M. Dai and L. T. Qu, J. Am. Chem. Soc., 2012, 134, 15–18 CrossRef CAS PubMed.
- B. Shi, Y. Su, L. Zhang, M. Huang, R. Liu and S. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 10717–10725 CAS.
- Y. Chen, Y. Wu, B. Weng, B. Wang and C. Li, Sens. Actuators, B, 2016, 223, 689–696 CrossRef CAS.
- A. Sachdev and P. Gopinath, Analyst, 2015, 140, 4260–4269 RSC.
- P. Li, X. Y. Sun, J. S. Shen and B. Liu, RSC Adv., 2016, 6, 61891–61896 RSC.
- G. M. Li, N. Lv, J. L. Zhang and J. Z. Ni, RSC Adv., 2017, 7, 16423–16427 RSC.
- X. Wang, X. Shen, B. Z. Li, G. Y. Jiang, X. M. Zhou and H. J. Jiang, RSC Adv., 2016, 6, 18326–18332 RSC.
- J. Gong, X. Lu and X. Q. An, RSC Adv., 2015, 5, 8533–8536 RSC.
- L. Wang, H. R. Zhang, X. H. Zhou, Y. L. Liu and B. F. Lei, RSC Adv., 2016, 6, 98554–98562 RSC.
- M. J. Bojdys, J. O. Muller, M. Antonietti and A. Thomas, Chemistry, 2008, 14, 8177–8182 CrossRef CAS PubMed.
- J. Yu, N. Song, Y.-K. Zhang, S.-X. Zhong, A.-J. Wang and J. Chen, Sens. Actuators, B, 2015, 214, 29–35 CrossRef CAS.
- S. Liu, J. Tian, L. Wang, Y. Luo, J. Zhai and X. Sun, J. Mater. Chem., 2011, 21, 11726–11729 RSC.
- S. Liu, J. Tian, L. Wang, Y. Zhang, X. Qin, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Adv. Mater., 2012, 24, 2037–2041 CrossRef CAS PubMed.
- A. Sachdev, I. Matai and P. Gopinath, RSC Adv., 2014, 4, 20915–20921 RSC.
- Y. Q. Dong, H. C. Pang, H. B. Yang, C. X. Guo, J. W. Shao, Y. W. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS PubMed.
- A. Jaiswal, S. S. Ghosh and A. Chattopadhyay, Chem. Commun., 2012, 48, 407–409 RSC.
- D. Y. Pan, J. C. Zhang, Z. Li and M. H. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef CAS PubMed.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- H. J. Zhang, Y. L. Chen, M. J. Liang, L. F. Xu, S. D. Qi, H. L. Chen and X. G. Chen, Anal. Chem., 2014, 86, 9846–9852 CrossRef CAS PubMed.
- J. Mullay, J. Am. Chem. Soc., 1985, 107, 7271–7275 CrossRef CAS.
- X. J. Gong, W. J. Lu, M. C. Paau, Q. Hu, X. Wu, S. M. Shuang, C. Dong and M. M. F. Choi, Anal. Chim. Acta, 2015, 861, 74–84 CrossRef CAS PubMed.
- Y. L. Zhai, Z. J. Zhu, C. Z. Zhu, J. T. Ren, E. K. Wang and S. J. Dong, J. Mater. Chem. B, 2014, 2, 6995–6999 RSC.
- C. Zhang, Y. Cui, L. Song, X. Liu and Z. Hu, Talanta, 2016, 150, 54–60 CrossRef CAS PubMed.
- W. J. Lu, X. J. Gong, M. Nan, Y. Liu, S. M. Shuang and C. Dong, Anal. Chim. Acta, 2015, 898, 116–127 CrossRef CAS PubMed.
- X. Gong, W. Lu, M. C. Paau, Q. Hu, X. Wu, S. Shuang, C. Dong and M. M. Choi, Anal. Chim. Acta, 2015, 861, 74–84 CrossRef CAS PubMed.
- M. Zhou, Z. Zhou, A. Gong, Y. Zhang and Q. Li, Talanta, 2015, 143, 107–113 CrossRef CAS PubMed.
- L. Chen, C. Wu, P. Du, X. Feng, P. Wu and C. Cai, Talanta, 2017, 164, 100–109 CrossRef CAS PubMed.
- S. R. Tyagi, M. Tamura, D. N. Burnham and J. D. Lambeth, J. Biol. Chem., 1988, 263, 13191–13198 CAS.
- K. Ishikawa, K. Takenaga, M. Akimoto, N. Koshikawa, A. Yamaguchi, H. Imanishi, K. Nakada, Y. Honma and J. Hayashi, Science, 2008, 320, 661–664 CrossRef CAS PubMed.
- S. J. McQuaker, C. L. Quinlan, S. T. Caldwell, M. D. Brand and R. C. Hartley, ChemBioChem, 2013, 14, 993–1000 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04122e |
| This journal is © The Royal Society of Chemistry 2017 |