
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ construction of polymer-encapsulated Au nanoparticle dimers based on a C–C coupling reaction
Qi Jin,
Chenjie Zhang,
Jing Zhang,
Yaxian Yuan*,
Minmin Xu and
Jianlin Yao *
*
College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, China. E-mail: yuanyaxian@suda.edu.cn; jlyao@suda.edu.cn
First published on 16th May 2017
Abstract
The assembly of simple nanostructures has already attracted significant interest in the field of plasmonic devices and other relevant areas. From the viewpoint of theoretical simulation and practical application, the precise control of nanoparticles still remains a significant challenge. Herein, a strategy was successfully developed to fabricate in situ a polymer-encapsulated Au nanoparticle dimer based on the C–C coupling reaction of p-aminophenylacetylene (p-APAC). The balance between the polymerization processes and the coupling reaction resulted in Au nanoparticle assemblies with different configurations, such as monomer, dimer, and multimer, depending on the concentration of p-APAC. The gap distance of about 1.8 nm was well consistent with the length of the coupling products of p-APAC, i.e. the gap distance was about double the length of a single p-APAC molecule. The observation of a longitudinal peak in the UV-vis spectrum demonstrated that the aspect ratio of the Au nanoparticles was about 2.5, indicating the formation of Au dimers with reasonable yield. Moreover, the thickness of the polymer shell was well-controlled via changing the concentration of p-APAC. The gap of the dimer resulted in a very large coupling effect of the localized surface plasmon resonance (LSPR), and the surface enhanced Raman spectroscopy (SERS) signal of the molecules was accordingly enhanced in the gap areas, which served as the hot spots. Based on the characteristic spectral feature of the coupling products, the single Au nanoparticle dimer was positioned via SERS mapping. The large LSPR coupling effect in the gap area allowed the conversion of p-nitrothiophenol (PNTP) to dimercaptoazobenzene (DMAB) with high efficiency. Thus, it was confirmed that the SPR-catalyzed coupling reaction preferentially occurred on a hot spot area. The proposed approach is expected to be developed into a promising tool for precisely controlling the gap distance of a nanoparticle assembly, and it may serve as a simple model for theoretical consideration in understanding the SERS mechanism(s).
Introduction
Surface-enhanced Raman spectroscopy (SERS) has rapidly developed in the last few decades due to the contribution of the hot spot effect generated in adjacent plasmonic nanoparticles.1–3 Normally, the incident light was harvested around the nanostructure, resulting in a large enhancement of the electromagnetic field. It was well-accepted as the main mechanism of enhancement by the SERS community,4 and it efficiently contributed to signal enhancement even up to single molecular detection. Therefore, the huge enhancement effect from the hot spots has been widely explored for the purpose of the practical application of SERS.5,6 In addition, the well-structured assembly of the hot spots improved the SERS performance in quantitative detection.7 On account of its great importance, significant efforts have been made to construct and control the assembly of plasmonic nanoparticles into discrete clusters,8–11 especially dimers, which are the simplest nanoparticle aggregates with only one gap and suitable as a model for theoretical simulations.12During the last few decades, a number of reports have demonstrated the fabrication of plasmonic nanoparticle dimers. Pioneering methods reported by Alivisatos et al.13 and Feldheim et al.14,15 included generation of Au dimers and trimers through DNA and organic molecules as linkers, respectively. These were the initial attempts to assemble discrete groups of plasmonic nanoparticles. After this, scientists are focused on how to increase the yield of dimers towards either fundamental research in plasmonics or applications in spectroscopy. To date, plasmonic dimers created via methods including organic molecule or DNA-assisted linkage,13–15 electrostatic assembly,16 polymer-encapsulation assembly,17,18 chain reorganization of the amphiphilic polymer brushes,19 and lithography20–22 have been well developed. Chen et al. reported the PS154-b-PAA60 encapsulated Au dimers obtained by the addition of HCl, and this encapsulation could be removed via oxidation. The dimers were enriched to 60% via utilizing different centrifugal forces.17 However, the Au nanoparticles were too small to provide detectable SERS signals. To improve the SERS sensitivity, Au@Ag core–shell nanoparticles were substituted by small Au nanoparticles. Via the same approach, high-purity dimers and trimers enriched to 85% and 70%, respectively, were separated.18 Recently, depending on the sophisticated technology of lithographic fabrication, it was possible to extensively generate dimers and structure them on a very large scale.23 Crozier and his coworkers fabricated dimers in a two-dimensional array with hundreds nanometer unit cell and micrometer array via lithography.20 However, high-cost is the key obstacle to the development of the abovementioned method. Based on the key issues of SERS, the plasmonic dimers are essential to fulfill the following requirements: the generality for different metal nanoparticles, the anchoring of probe molecules in the gap region, easy fabrication, and tunable gap size.24 Well-assembled metal dimers are explored for a wide range of SERS applications such as investigations on the quantitative relationship between the nanogap and plasmonic coupling, ultrasensitive detection, and biosensors for tumors.25–27 For example, using a high-yield method for preparing the SERS-active dimeric gold–silver core–shell nanodumbbells, Lee and coworkers found a detectable single-molecule SERS signal in a gap with an appropriate size.25 Xu and coworkers developed an ultrasensitive and highly selective approach for dopamine detection based on Ag-coated aptamer-mediated dimers.26 Recently, highly SERS-active metal dimers have played an important role in biomedicine, especially in specific tumor detection. Fabris and coworkers fabricated spherical Au nanoparticle dimers using a rigid dithiolate linkage. The cell-internalized Au nanoparticle dimers with highly specific cellular recognition and excellent stability could serve as novel candidates for tumor phenotype detection.27 Thus, the controllable fabrication of a metal dimer is highly desired for both theoretical simulations and practical application. Although researchers have exploited various methods as abovementioned, a lack of balance between flexibility, high yield, and low-cost has resulted in the preparation of only a single-functionality of the dimers.
Herein, a facile approach combining the functional synthesis and flexibility was proposed to extend the applications of dimers; this approach was mainly based on the precise positioning of individual coupling systems and investigation of the coupling reaction in the finite region. Based on the C–C coupling reaction,28 Au nanoparticles modified with p-aminophenylacetylene (p-APAC) were dimerized through the linkage of the coupling products on two Au nanoparticles, along with the encapsulation of the polymer shells during dimerization, which were spontaneously polymerized by p-APAC. The polymer shell was designed to play an important role in isolating the dimers from each other. This process is beneficial for the simplification of the fabrication procedure, as well as precise control of the aggregation via the balances between the polymerization and C–C coupling reactions. The great coupling effect in the dimers gap contributed to the strong SERS effect and also produced the SPR around the nanoparticles for the relevant catalysis reaction. Thus, the SERS effect and SPR-driven catalytic reaction were investigated using the polymer-encapsulated Au nanoparticle dimers.
Experimental
Chemicals
Chloroauric acid tetrahydrate (HAuCl4·4H2O), trisodium citrate dihydrate (Na3C6H5O7·2H2O), hydroxylamine hydrochloride (NH2OH·HCl), and cuprous iodide (CuI) were purchased from Sinopharm Chemical Reagent Co. (China). p-Aminophenylacetylene (p-APAC) and diethyl azodicarboxylate (DEAD) were obtained from J&K. 4-Nitrothiophenol (PNTP, 98%) was purchased from Matrix Scientific Trade Co. 1-Methylimidazole (C4H6N2) was purchased from Medipharm Co. (China), which was purified before use. The process of purification was as follows: 1-methylimidazole was stirred with the addition of NaOH and then distilled. The distillate obtained at 198 °C was used.Characterizations
Transmission electron microscopy (TEM) measurements were carried out using a TecnaiG220 microscope at 200 kV operating voltage. Scanning electron microscopy (SEM) was performed using a Hitachi S-4700 microscope operated at 15 kV. Raman spectra were obtained using a fully automatic microprobe Raman system (XploRA PLUS, Horiba Jobin Yvon). The sizes of the slit and pinhole were 100 and 300 μm, respectively. The excitation laser wavelength was 638 nm.Preparation of the Au nanoparticles
Au nanoparticles with a diameter of 30 nm were synthesized via a two-step method. The Au seed solution was first prepared through the standard citrate reduction method.29 In brief, 1 mL of HAuCl4·4H2O solution (1% w/v) in 100 mL of water was heated to boiling under vigorous stirring, and then, 2 mL of trisodium citrate solution (1% w/v) was quickly added. The solution was allowed to boil for 15 min, then cooled down to room temperature, and used as the seed solution. The diameter of Au seed nanoparticles was about 15 nm. Then, 25 mL of Au seed solution was stirred at room temperature followed by the sequential addition of 1 mL of trisodium citrate solution (1% w/v) and 20 mL of 25 mM NH2OH·HCl aqueous solution. To perform the growth of the Au seeds, 20 mL of HAuCl4·4H2O solution (0.1% w/v) was injected to the abovementioned solution under mild stirring through a peristaltic pump at an injection rate of 1 mL min−1. The mixture was constantly stirred for 1 h before use.Formation of the Au dimers
The 30 nm Au nanoparticles were concentrated via centrifugation and then mixed with 10 mL ethanol solution of 15 mM p-APAC for 0.5 h at a stirring rate of 1000 rpm for the alkynylation of Au nanoparticle surface via multiple bonds between the Au and the amine group. The molar ratio of p-APAC to Au nanoparticles was adjusted to 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 40:1, 60:1, 100:1, 140:1, and 200:1 to obtain the aggregates containing different numbers of Au nanoparticles. Subsequently, 1.4 × 10−3 g CuI, 9 μL 1-methylimidazole, and 14.4 μL DEAD, which acted as catalysts, alkali, and hydrogen acceptors, respectively, were sequentially added to trigger the coupling reaction. The mixture was stirred at 1000 rpm for 2 h, with the color change from reddish brown to dark brown. To increase the yield of the dimers, the as-prepared colloidal solution was centrifuged at 2000 rpm to remove the large-sized nanoparticle aggregates and then enriched via centrifugation at 5000 rpm.
:1, 40:1, 60:1, 100:1, 140:1, and 200:1 to obtain the aggregates containing different numbers of Au nanoparticles. Subsequently, 1.4 × 10−3 g CuI, 9 μL 1-methylimidazole, and 14.4 μL DEAD, which acted as catalysts, alkali, and hydrogen acceptors, respectively, were sequentially added to trigger the coupling reaction. The mixture was stirred at 1000 rpm for 2 h, with the color change from reddish brown to dark brown. To increase the yield of the dimers, the as-prepared colloidal solution was centrifuged at 2000 rpm to remove the large-sized nanoparticle aggregates and then enriched via centrifugation at 5000 rpm.
SERS mapping measurements
The distinctive marks on a clean Si wafer were made via a diamond cutter. For SERS mapping, the square area measuring 6 μm × 6 μm was selected and observed using a 100× objective lens, which showed the dimer and monomer along with the SEM image. Taking 1 μm as a step, SERS spectra of 6 × 6 points were accumulated at an integration time of 10 s and integrated to the SERS mapping.Results and discussion
The principal procedure for fabricating the Au dimer
Scheme 1 demonstrates the schematic of the fabrication of Au dimers through an organic coupling reaction. In the present strategy, p-APAC was immobilized onto the Au nanoparticle surface through the amine group and severed as the linker for the formation of the dimer after the acetylene coupling reaction. Thus, the inter-nanoparticle gap was occupied by the coupling product p-APAC-CAPA-p molecules. Normally, the continuous C–C coupling of the terminal groups on adjacent Au nanoparticles allow the cross-connection of the nanoparticles to generate network structures. However, under controlled conditions, the spontaneous polymerization of p-APAC occurred and produced the shell on the Au nanoparticle dimer surface. Hence, the continuous coupling reaction of p-APAC was hindered. As a consequence, the Au dimer dominated in the final products. However, the coverage of p-APAC on Au nanoparticles was crucial for the formation of different configurations such as monomers, dimers, or multimers. Moreover, the following polymerization processes resulted in the formation of a continuous shell on the Au nanoparticle dimer. This shell isolated the dimers from each other to avoid further aggregation. Thus, the monodispersed Au dimer covered with a polymer shell was obtained, and the probe molecules were anchored onto two Au nanoparticles. Therefore, it was beneficial to focus the SERS investigation on the molecules localized in the nanoparticle gap area.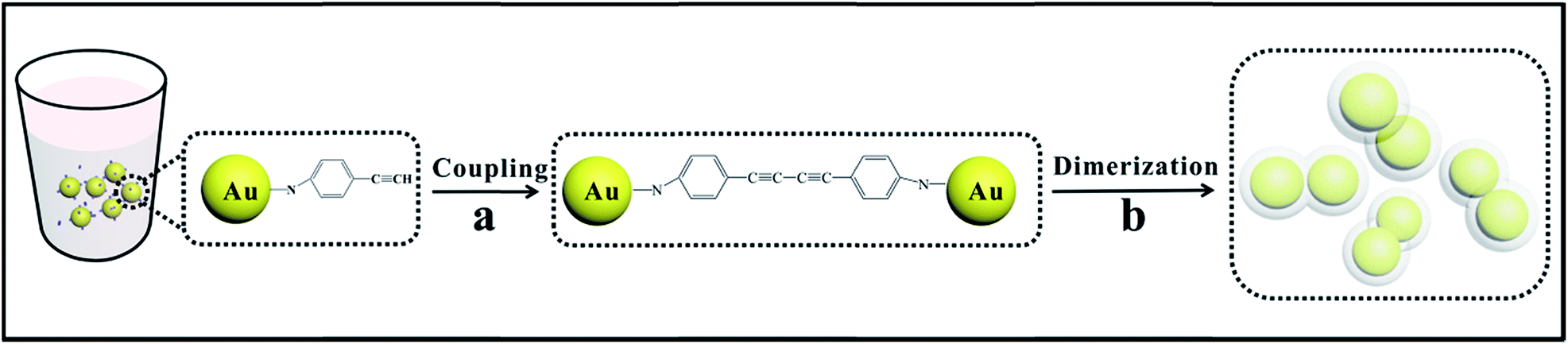 | ||
| Scheme 1 Schematic for the fabrication procedure of Au dimers. (a): Au nanoparticles absorbing p-APAC were dimerized via a C–C coupling reaction, (b): polymer shells were formed by spontaneous polymerization of excess p-APAC during dimerization to maintain stability and modification. | ||
The morphology of the Au dimers
Fig. 1 presents the TEM images of Au dimers assembled by controlling the optimal reaction conditions, where the molar ratio of p-APAC to Au nanoparticles was 60:1. Large amount of p-APAC was used to improve the coupling efficiency for constructing Au dimers. As the description shown in Scheme 1, the polymerization was immediately followed by the coupling reaction of p-APAC. It created the polymer shell on the Au dimers, which prevented the nanoparticles from further linkage and aggregation, resulting in the high yield of the Au dimers. A gap distance of about 1.8 nm was unambiguously observed (as shown in the inset TEM image). Actually, the molecular length of the product obtained by the coupling reaction was about 1.9 nm for the rigid structure. Therefore, it was reasonable to speculate that the formation of the Au dimer was mainly contributed by the coupling reaction of two p-APAC molecules anchored onto different Au nanoparticles. To verify the role of the coupling reaction, a similar protocol was performed in the absence of the catalyst (CuI). Normally, the catalyst plays an essential role in the C–C coupling reaction of p-APAC. Therefore, it can be expected that in the absence of a C–C coupling reaction, the Au dimers would not be formed. Although there were barely any Au dimers, the polymerization of p-APAC still occurred and the single Au nanoparticle was then encapsulated by the polymer shell. Based on the abovementioned experimental evidence, the Au nanoparticle dimers were successfully constructed through the C–C coupling reaction of p-APAC with the gap distance of about 1.8 nm, and the formation of the polymer shell allowed the monodispersity of Au dimers. The linkage of two Au nanoparticles by the in situ coupling reaction provided a promising strategy in the function-directed assembly of nanostructures for both theoretical simulation and practical applications.
 | ||
| Fig. 1 TEM image of the Au dimers assembled in the solution with the molar ratio of 60:1 of p-APAC to Au nanoparticles. Upper left inset: the corresponding TEM image of the dimer with a gap distance of ∼1.8 nm, and lower left inset: Au nanoparticle multimer assembled using the solution with a molar ratio of 200:1. | ||
Generally, the extensive coupling reaction among Au nanoparticles result in a complex network structure rather than a controllable dimer configuration. In the present study, the occurrence of a C–C coupling reaction between two adjacent Au nanoparticles became the critical requirement for constructing the Au dimers. Moreover, simultaneous polymerization around the Au nanoparticles was beneficial for inhibiting the continuous linkage of the third or more Au nanoparticles. Therefore, the final configuration was critically dependent on the balance between the coupling reaction and the polymerization. The larger amount of p-APAC improved the efficiency of the polymerization and coupling reaction, and more Au nanoparticles were linked via a coupling reaction and covered by the polymer shell. However, the lower dosage resulted in a low efficiency of the coupling reaction, and it was difficult to link the nanoparticles together. Thus, it is reasonable to speculate that three possible nanostructures will be formed: monomer, dimer, and multimer. The good balance between the polymerization and coupling reaction allowed the simultaneous occurrence of the abovementioned two procedures, resulting in the formation of Au dimers for a broad range of molar ratios of p-APAC to Au nanoparticles. The relatively fast coupling reaction originated from the high dosage of p-APAC and was beneficial to link more Au nanoparticles together to form multimers, and the polymerization was also accordingly improved, which blocked, to some extent, the coupling reaction by encapsulating the nanostructures in a polymer shell. As a consequence, the Au multimers configuration containing more than ten Au nanoparticles was dominant in the solution with a high concentration of p-APAC (as shown in the inset of Fig. 1). The formation of dimer and multimer was undoubtedly confirmed by the abovementioned experiment at the molar ratios of 60:1 and 200:1 of p-APAC to Au nanoparticles (as shown in Fig. 1). Via reducing the dosage of p-APAC, the surface coverage of p-APAC was dramatically decreased and the efficiency of the coupling reaction accordingly decreased; this resulted in difficulties in linking Au nanoparticles together, and the monomer became the dominant product. Therefore, it was certainly important to reduce the surface coverage of p-APAC to verify the abovementioned hypothesis. After decreasing the molar ratio of p-APAC to Au nanoparticles to 12:1 (5 times lower than that required for the coverage of the dimers), the Au nanoparticle monomers were fabricated with a 4–5 nm thick polymer shell (as shown in Fig. 2). By carefully comparing the UV-vis spectra of the monomers and the original Au nanoparticles (as shown in Fig. 2B), it was found that the surface plasmon resonance (SPR) peak at 535 nm contributed by the Au nanoparticles gradually red-shifted after the continuous polymer shell was covered.
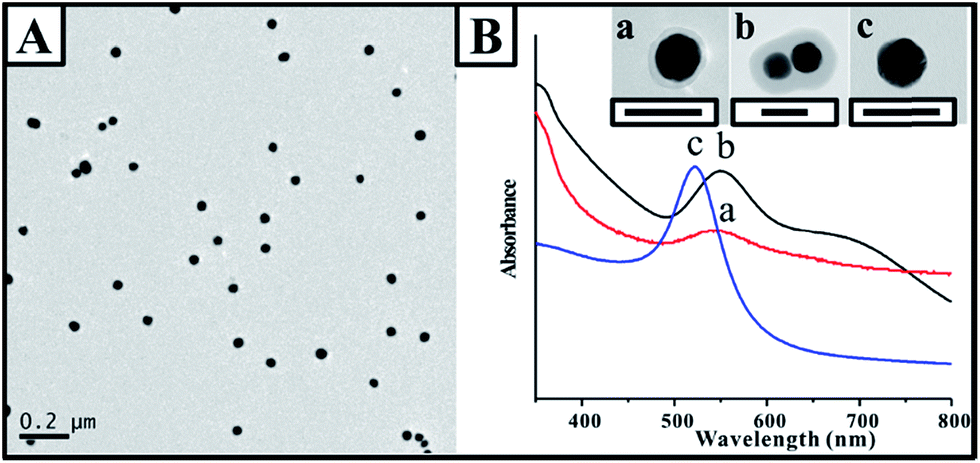 | ||
| Fig. 2 (A) TEM image of the Au clusters under the conditions of the 12:1 molar ratio of p-APAC to Au NPs. (B) UV-vis spectra of monomers (a), dimers (b), and original Au nanoparticles (c), scale bar: 50 nm. | ||
Note that a shoulder peak appeared at ∼675 nm in addition to that at 550 nm, which was consistent with the polymer-encapsulated monomer. It was in agreement with the characteristic feature of the anisotropic gold nanorods.19 Pileni and coworkers simulated the absorption efficiency using the discrete dipole approximation (DDA) method; an equation was developed for estimating the longitudinal peak location of the Au nanorod with different aspect ratios.30 In our present study, based on the equation, the maximum position of the longitudinal peak at about 660 nm demonstrated an aspect ratio of about 2.5, suggesting dimerization and strong coupling within the dimers.31 The longitudinal peak of the dimer was comparable to that of the nanorod with an aspect ratio of 2. Note that the small deviation in the aspect ratio was mainly due to the ellipsoidal shape of the Au nanoparticle and the difference in the shape of the dimer and nanorod. The longitudinal peak of the nanoparticle dimer became broader with a weak intensity, which was mainly contributed by the presence of an interparticle gap and the continuous polymer shell.
Tuning the thickness of the polymer shell
As abovementioned, the polymer shell was formed by the spontaneous polymerization of p-APAC in the solution. Consequently, the thickness of the polymer shell could be tuned via the change in the concentration of p-APAC. The shell thickness at different concentration ratios is summarized in Fig. 3. Note that the yield of the dimer was critically dependent on the dosage of p-APAC in the solution. The low concentration of p-APAC produced isolated Au nanoparticles covered with a thin polymer shell. The high dosage of p-APAC allowed a significant decrease in the yield of the dimer and an increase in the yield of the multimer. However, the dimer still existed in the solution with a high concentration of p-APAC. In the present case, the thickness of the outer polymer on the dimer was explored to demonstrate the effect of the dosage on the polymer thickness. For the same reaction duration, shell thickness ranging from 4 nm to 50 nm could be obtained with the molar ratio of p-APAC to Au nanoparticles increasing from 12 to 200 (as shown in Fig. 3). Actually, the coupling reaction between two nanoparticles was quickly blocked via polymerization around the nanostructures, followed by the further growth of the polymer shell. Therefore, the thicker shell was obtained in the solution with a high concentration of p-APAC.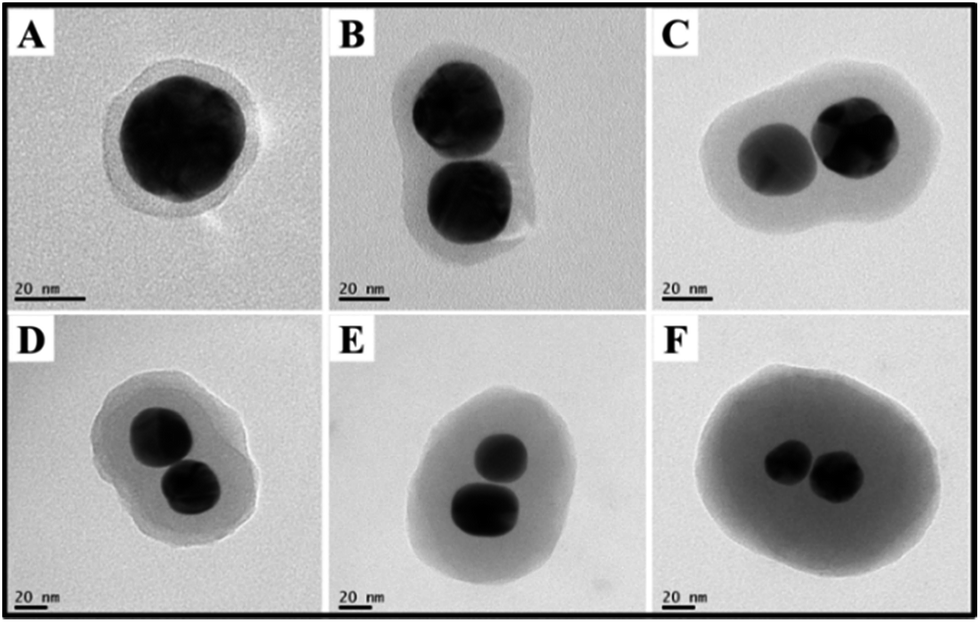 | ||
| Fig. 3 TEM image of the shell thickness at different molar ratios of p-APAC to Au NPs: (A) 1:12, (B) 1:40, (C) 1:60, (D) 1:100, (E) 1:140, and (F) 1:200. | ||
SERS investigation of the Au dimer
An additional advantage of the present approach was that the gap region of the dimer was filled with linkers, which could serve as probes for investigating the unique optical properties in the dimer gap. For example, due to the strong coupling effect of SPR from Au nanoparticles, the large localized SPR contributed the extremely strong electromagnetic field, resulting in the strong SERS signal of the probe molecules in the gap. To confirm the SERS signal of the probe on the surface and in the gap, the adsorption of p-APAC and its coupling products were investigated by SERS on the Au nanoparticle film surface, together with the normal Raman spectrum obtained from the theoretical simulation based on the density functional theory (DFT) at B3LYP/6-311+G** (as shown in Fig. 4). Herein, two main bands were observed, at 1180 and 1600 cm−1, and these were assigned to the phenyl ring vibrational mode (9a and 8a vibrational mode).32 Note that a new band at 2205 cm−1 appeared as p-APAC coupled. The theoretical simulation of the coupling product presented in Fig. 4B was used for comparison, and a strong band at 2224 cm−1 was assigned to the conjugated C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C symmetrical stretching vibrational mode.33 Moreover, the difference in the spectral features of SERS and Raman spectra in the region from 1000 cm−1 to 1700 cm−1 could be observed. It was mainly due to the change in the symmetry after the probe was adsorbed onto the Au surface. Therefore, it was reasonable to assign the new band at 2205 cm−1 to the CC symmetrical stretching vibrational mode after the coupling reaction of p-APAC. Furthermore, this band was also appropriate as a characteristic feature for confirming the formation of the Au dimer and could serve as the marker for screening the position of the dimer.
C symmetrical stretching vibrational mode.33 Moreover, the difference in the spectral features of SERS and Raman spectra in the region from 1000 cm−1 to 1700 cm−1 could be observed. It was mainly due to the change in the symmetry after the probe was adsorbed onto the Au surface. Therefore, it was reasonable to assign the new band at 2205 cm−1 to the CC symmetrical stretching vibrational mode after the coupling reaction of p-APAC. Furthermore, this band was also appropriate as a characteristic feature for confirming the formation of the Au dimer and could serve as the marker for screening the position of the dimer.
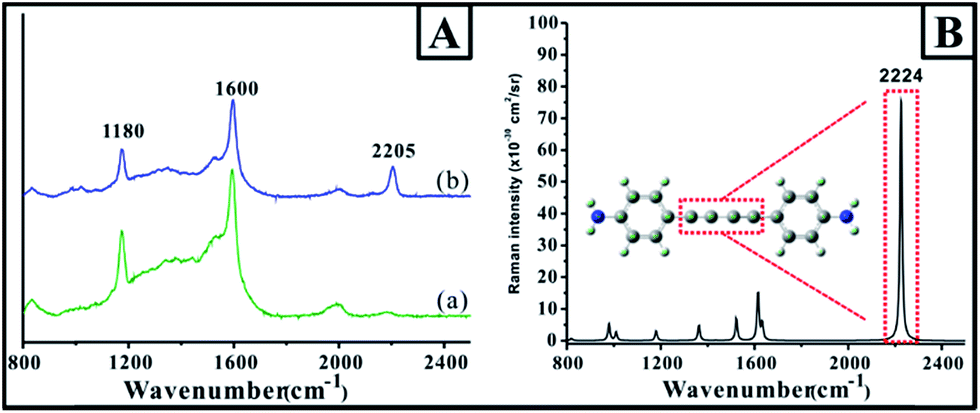 | ||
| Fig. 4 (A) The SERS spectra of the molecules absorbed on the Au film. (a) p-APAC and (b) coupling product of p-APAC. (B) The theoretical Raman spectra of the coupling product. (Calculation parameters: Gaussian 09 B01, B3LYP/6-311+G**). | ||
Further characterization via a combination of SEM and SERS proved the feasibility of the abovementioned concept for analyzing the Au nanoparticle dimers. The as-prepared solution containing dimers was diluted to improve the monodispersity of the dimers. The diluted solution was then dispersed onto a clean Si wafer with a distinctive mark in micrometer dimensions, which made the observation of the position easy using an optical microscope. As observed from Fig. 5A and B, an area containing only a dimer was carefully selected. A monomer away from the dimer with a distance of about 4.7 μm was selected for comparison. Generally, based on the confocal Raman spectroscopy, the size of the laser spot was less than 2 μm. To avoid the overlap of the laser spot, it was preferred to select the dimer and monomer with a separation distance larger than the size of the laser spot. Using the distinctive mark on the Si wafer, it was easy to obtain the image via an optical microscope attached to a Raman spectrometer, and the image was well consistent with the SEM image. The result of SERS mapping based on the intensity of the band at 2205 cm−1 in the marked area is presented in Fig. 5E, and all the SERS spectra are presented in Fig. 5D. It was demonstrated that the SERS signal assigned to the coupling products was observed around area a, and the signal to noise ratio was quite low beyond this area. The SERS mapping also indicated a similar feature, in which the positive signal was present in area a. whereas no signal was present in area b. Actually, the areas of a and b were corresponding to the location of the dimer and monomer, respectively. The distance between the monomer and dimer was estimated to be about 4.9 μm from the SERS mapping, which was close to the real distance determined by the SEM image. The typical SERS spectra for areas a and b were obtained from the mapping image and are shown in the inset of Fig. 5E. The spectral feature contributed by the area a was very similar to that where the coupling products were adsorbed onto the Au nanoparticle film surface (as shown in Fig. 4). Therefore, all the abovementioned experimental evidence revealed that the weak band at 2205 cm−1 was contributed by the coupling products in the gap of the dimer. The orange region suggested the location of the dimer, whereas the monomer area location displayed no signal because of the low SERS activity of the isolated single nanoparticles. The inset spectra in Fig. 5E revealed the concrete intensity with approximately 10 cps, an extremely weak signal due to only one hot spot between two Au nanoparticles.
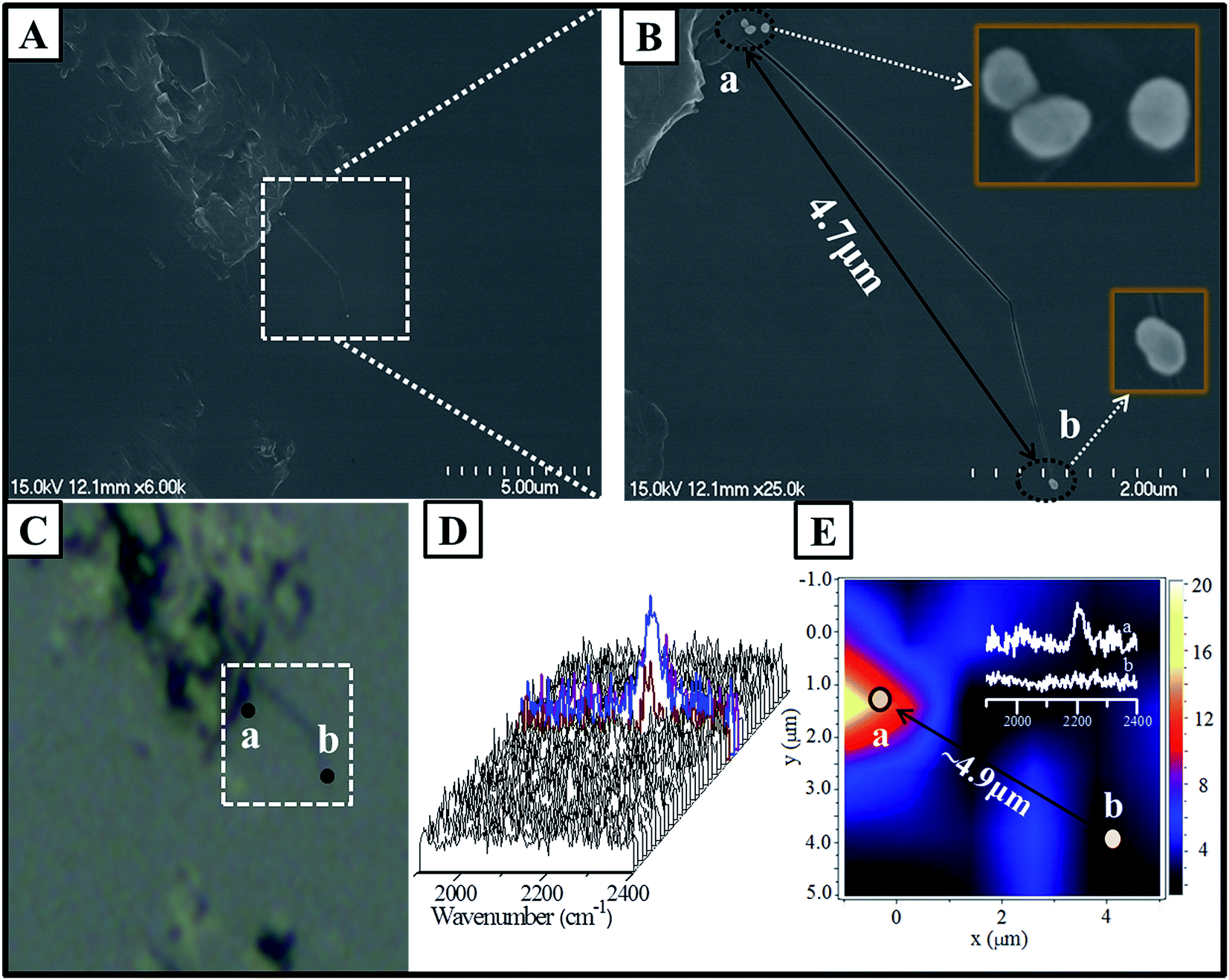 | ||
| Fig. 5 SEM image of the Si wafer with the dimer and the monomer in the marked region (A and B). The corresponding image obtained via an optical microscope attached to a Raman spectrometer (C). All the SERS spectra were obtained via mapping (D). SERS mapping of the area corresponding to the dotted box (E), inset: SERS spectra extracted from the position of the dimer (a) and monomer (b). | ||
Although large fraction of the surface of Au nanoparticles was modified with p-APAC, the SERS signal of the terminal CC group was absent due to the poor electromagnetic enhancement on the monomer owing to the lack of any coupling effect. For the dimer, due to the very large coupling effect of SPR in the gap area, the SERS signal was still detected although the amount of the coupling products was extremely low. However, it was difficult to precisely measure the amount of the coupling products linked to two Au nanoparticles (inside of the gap); thus, the surface enhanced factor from one hot spot of the dimer was not estimated in the present study. However, it can be speculated that the coupling products connecting two Au nanoparticles achieved maximum enhancement. However, successful identification of the Au dimers coated with the polymer shell via SERS would provide a great opportunity for the deeper understanding of the formation and application of Au dimers, as well as the complex SERS enhancement mechanism(s).
SPR-catalyzed reaction of PNTP on the Au dimer gap
As is well known, the localized SPR is dramatically enhanced as two Au nanoparticles come infinitely close to each other. Generally, the very large SPR effect would drive the relevant catalytic reaction; for example, the coupling of PNTP to form dimercaptoazobenzene (DMAB). Herein, the distinctive gap distance of the fabricated dimers as compared to those of the monomer and Au nanoparticle film, which possessed tiny gap distances, allowed us to experimentally investigate the efficiency of the SPR-catalyzed coupling reaction of PNTP. Fig. 6 presents the time-dependent SERS spectra of PNTP on the dimer and the Au nanoparticle monolayer film.7 PNTP was pre-absorbed on the core of the p-APAC-modified Au nanoparticles before the formation of the dimer, and it was directly immobilized onto the Au nanoparticle monolayer film. In the present case, the SERS of PNTP from the dimer and Au nanoparticle films was carefully compared. The strong bands at 1180 and 1600 cm−1 originated from p-APAC and the corresponding coupling products, respectively. The band at 1330 cm−1 was assigned to PNTP, whereas the bands at 1140, 1388, and 1430 cm−1 were due to the formation of DMAB, which represented the SPR-catalyzed coupling products of PNTP.34 Moreover, the intensity of the band at 2205 cm−1 dramatically decreased due to the introduction of PNTP. This was mainly due to the competitive adsorption of PNTP and the steric hindrance effect, which contributed to the decrease of the number of products p-APAC coupled in the gap region. As is well-known, the absolute intensity of the SERS signal is critically dependent on the number of nanoparticles; thus, it was not suitable to be used as an indicator for exploring the progress of the catalytic reaction. As a consequence, the relative intensity of 1140 cm−1 to 1330 cm−1 was selected for estimating the efficiency of the SPR-catalyzed coupling reaction of PNTP. In our previous reports, the assembly of two-dimensional Au nanoparticles produced numerous hot spots for enhancing the SERS signal of the probe molecules, and the conversion of PNTP to DMAB occurred in the initial exposure to laser with an appropriate power.35 Herein, the absence of the SERS signal of DMAB indicated that the SPR-catalyzed conversion of PNTP to DMAB was not achieved on the Au nanoparticle monolayer films. However, on using the same laser power, the SPR-catalyzed conversion was unambiguously observed (as shown in Fig. 6a) in the Au dimers. Herein, two spectral features were observed as follows: (i) the relative intensity of 1330 cm−1 significantly decreased, and it was weaker than that of DMAB; (ii) after 5 seconds exposure to laser, the relative intensity of PNTP to DMAB remained constant. For the dimer structure, PNTP was adsorbed onto the Au nanoparticles together with p-APAC, and the contribution of PNTP adsorbed on the area beyond the gap was negligible due to the lack of LSPR coupling effect. Therefore, the weak SERS signal originated from PNTP located in the hot spot (gap) area, which did not undergo the conversion reaction. However, the low relative intensity of 1330 cm−1 was associated with the low coverage of PNTP, indicating that a large fraction of PNTP in the hot spot area underwent the SPR-catalyzed conversion reaction to produce DMAB. Numerous previous studies, including those performed by our group, have reported that this conversion reaction occurs with low efficiency, and only the molecules in the very hot area are transferred to DMAB. Therefore, the final SERS spectra were contributed by PNTP and DMAB together, in which the band at 1330 cm−1 was still dominant. Fig. 6b demonstrated that the efficiency of the SPR coupling reaction on the dimer revealed a sharp upward jump and then maintained stability during the first 20 s laser exposure. For the Au nanoparticle film, although strong signal intensity was obtained due to a large number of hot spots, the unchanged SERS spectral feature suggested that no coupling reaction occurred (as shown in Fig. 6c). This was mainly due to the following two reasons: (i) to make a comparison of the hot spot between the dimer and Au nanoparticles film, lower laser power was used to inhibit the conversion reaction on the Au nanoparticle monolayer film; (ii) considering that the molecular length of DMAB was about 1.9 nm, the tiny gap distances on the Au nanoparticle monolayer film prevented PNTP coupling as a result of the steric hindrance effect. Therefore, our present results suggest that the SPR-catalyzed conversion reaction preferentially occurred in the hot spots with an appropriate gap distance.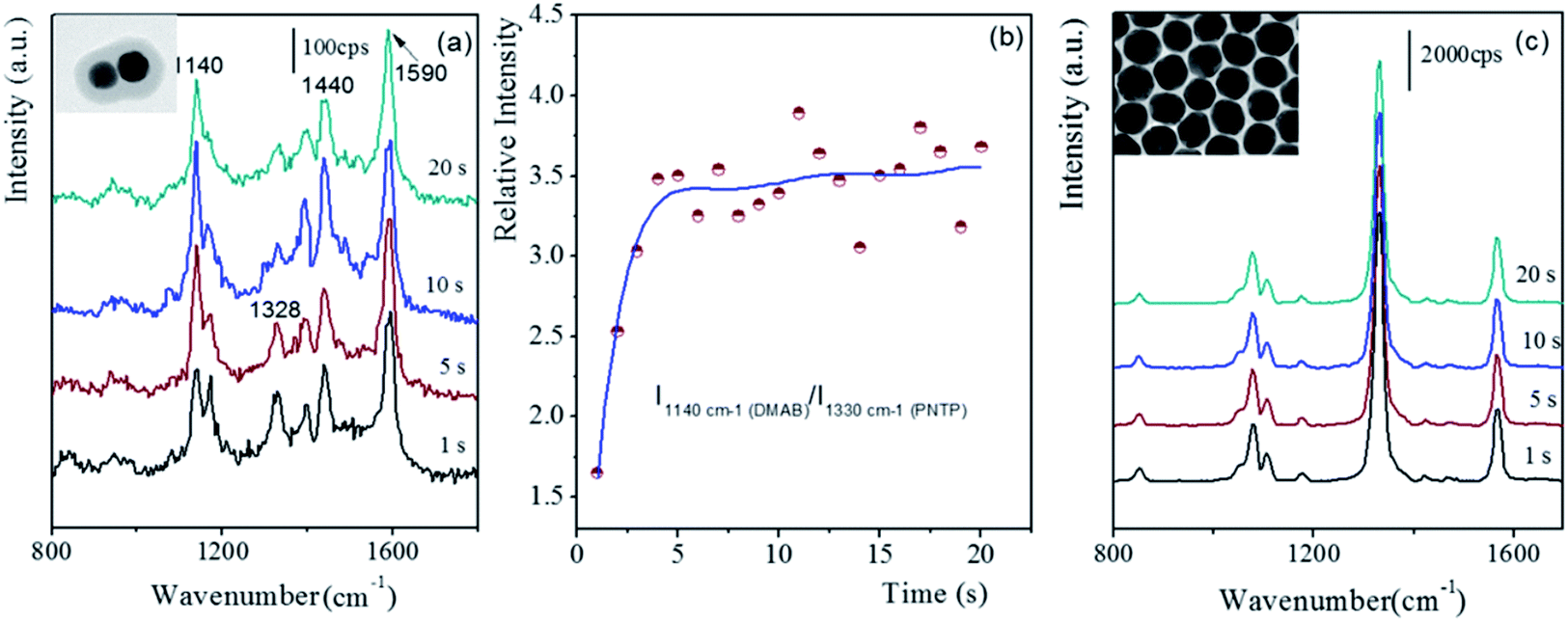 | ||
| Fig. 6 The time-dependent SERS spectra of PNTP adsorbed on different substrates with the initial 20 seconds exposure to laser. (a) Dimer, (b) relative intensity–time profile, and (c) on Au nanoparticles monolayer film.7 | ||
Conclusions
A facile approach for the assembly of polymer-encapsulated Au dimers with fixed gap distances was successfully developed based on the organic C–C coupling reaction between two molecules anchored onto two Au nanoparticles. The fabrication process combined both dimerization and modification, designed for maintaining stability during preparation, which greatly simplified the entire procedure but remained adjustable and flexible. Based on the coupling products located at the gap region, the positions of the dimers were identified by integrating SERS mapping and SEM images, providing potential applications for track monitoring. The polymerization processes and coupling reaction of p-APAC occurred together, and the balance of these two procedures resulted in different configurations of the Au nanoparticle assembly. The low amount of p-APAC resulted in the polymer-encapsulated Au monomer, whereas a broad range of p-APAC concentration led to dimers, and a high dosage of p-APAC produced multimers. The gap distance of the dimer of about 1.8 nm was well consistent with the length of the coupling products of p-APAC, i.e. the gap distance was about double the length of a single p-APAC molecule. Moreover, the obtained Au nanoparticle assembly exhibited high monodispersity with a tunable shell thickness. The very large coupling effect of LSPR was achieved in the gap area of the dimer, which accordingly enhanced the SERS signal. By avoiding the influence of the molecules beyond the gap area, the conversion of PNTP to DMAB was achieved with high efficiency. It confirmed that the SPR-catalyzed coupling reaction preferentially occurred in the hot spot area. The proposed approach is expected to be developed into a promising tool for connecting nanostructures with functional organic molecules and precisely controlling the gap distance of the nanoparticle assembly. It may serve as a simple model for obtaining a deeper understanding of the SERS and the SPR catalytic reaction mechanism(s).Acknowledgements
The authors gratefully acknowledge the financial support received from the Natural Science Foundation of China (21673152 and 21473128) and the National Instrumentation Program (2011YQ031240402). The partial financial support received from a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) is gratefully acknowledged.Notes and references
- S. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- A. M. Michaels, J. Jiang and L. Brus, J. Phys. Chem. B, 2000, 104, 11965–11971 CrossRef CAS.
- E. Hao and G. C. Schatz, J. Chem. Phys., 2004, 120, 357–366 CrossRef CAS.
- J. P. Camden, J. A. Dieringer, Y. Wang, D. J. Masiello, L. D. Marks, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2008, 130, 12616–12617 CrossRef CAS PubMed.
- D. K. Lim, K. S. Jeon, H. M. Kim, J. M. Nam and Y. D. Suh, Nat. Mater., 2010, 9, 60–67 CrossRef CAS PubMed.
- Q. H. Guo, M. M. Xu, Y. X. Yuan, R. A. Gu and J. L. Yao, Langmuir, 2016, 32, 4530–4537 CrossRef CAS PubMed.
- R. Klajn, K. J. Bishop, M. Fialkowski, M. Paszewski, C. J. Campbell, T. P. Gray and B. A. Grzybowski, Science, 2007, 316, 261–264 CrossRef CAS PubMed.
- K. L. Wustholz, A. I. Henry, J. M. Mcmahon, R. G. Freeman, N. Valley, M. E. Piotti, M. J. Natan, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2010, 132, 10903–10910 CrossRef CAS PubMed.
- F. S. Ou, M. Hu, I. Naumov, A. Kim, W. Wu, A. M. Bratkovsky, X. M. Li, R. S. Williams and Z. Y. Li, Nano Lett., 2011, 11, 2538–2542 CrossRef CAS PubMed.
- N. Vilar-Vidal, S. Bonhommeau, D. Talaga and S. Ravaine, New J. Chem., 2016, 40, 7299–7302 RSC.
- N. J. Halas, S. Lal, W. S. Chang, S. Link and P. Nordlander, Chem. Rev., 2011, 111, 3913–3961 CrossRef CAS PubMed.
- C. J. Loweth, W. B. Caldwell, X. Peng, A. P. Alivisatos and P. G. Schultz, Angew. Chem., Int. Ed., 1999, 38, 1808–1812 CrossRef CAS.
- L. C. Brousseau Iii, J. P. Novak, S. M. Marinakos and D. L. Feldheim, Adv. Mater., 1999, 11, 447–449 CrossRef.
- J. P. Novak and D. L. Feldheim, J. Am. Chem. Soc., 2000, 122, 3979–3980 CrossRef CAS.
- N. Zohar and G. Haran, Langmuir, 2014, 30, 7919–7927 CrossRef CAS PubMed.
- X. J. Wang, G. P. Li, T. Chen, M. X. Yang, Z. Zhang, T. Wu and H. Y. Chen, Nano Lett., 2008, 8, 2643–2647 CrossRef CAS PubMed.
- G. Chen, Y. Wang, M. X. Yang, J. Xu, S. J. Goh, M. Pan and H. Y. Chen, J. Am. Chem. Soc., 2010, 132, 3644–3645 CrossRef CAS PubMed.
- L. Cheng, J. B. Song, J. Yin and H. W. Duan, J. Phys. Chem. Lett., 2011, 2, 2258–2262 CrossRef CAS.
- W. Q. Zhu, M. G. Banaee, D. X. Wang, Y. Z. Chu and K. B. Crozier, Small, 2011, 7, 1761–1766 CrossRef CAS PubMed.
- R. Near, C. Tabor, J. Duan, R. Pachter and M. El-Sayed, Nano Lett., 2012, 12, 2158–2164 CrossRef CAS PubMed.
- M. Chirumamilla, A. Toma, A. Gopalakrishnan, G. Das, R. P. Zaccaria, R. Krahne, E. Rondanina, M. Leoncini, C. Liberale, F. De Angelis and E. Di Fabrizio, Adv. Mater., 2014, 26, 2353–2358 CrossRef CAS PubMed.
- S. L. Kleinman, R. R. Frontiera, A. I. Henry, J. A. Dieringer and R. P. Van Duyne, Phys. Chem. Chem. Phys., 2013, 15, 21–36 RSC.
- N. Zohar, L. Chuntonov and G. Haran, J. Photochem. Photobiol., C, 2014, 21, 26–39 CrossRef CAS.
- J. H. Lee, J. M. Nam, K. S. Jeon, D. K. Lim, H. Kim, S. Kwon, H. Lee and Y. D. Suh, ACS Nano, 2012, 6, 9574–9584 CrossRef CAS PubMed.
- L. J. Tang, S. Li, F. Han, L. Q. Liu, L. G. Xu, W. Ma, H. Kuang, A. K. Li, L. B. Wang and C. L. Xu, Biosens. Bioelectron., 2015, 71, 7–12 CrossRef CAS PubMed.
- A. S. D. S. Indrasekara, B. J. Paladini, D. J. Naczynski, V. Starovoytov, P. V. Moghe and L. Fabris, Adv. Healthcare Mater., 2013, 2, 1370–1376 CrossRef CAS PubMed.
- Z. P. Yang, B. N. Wang, X. L. Xu, H. Wang and X. N. Li, Chin. J. Org. Chem., 2015, 35, 207 CrossRef CAS.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- A. Brioude, X. C. Jiang and M. P. Pileni, J. Phys. Chem. B, 2005, 109, 13138–13142 CrossRef CAS PubMed.
- W. Y. Li, P. H. Camargo, X. M. Lu and Y. N. Xia, Nano Lett., 2009, 9, 485–490 CrossRef CAS PubMed.
- M. Osawa, N. Matsuda, K. Yoshii and I. Uchida, J. Phys. Chem., 1994, 98, 12702–12707 CrossRef CAS.
- A. G. Meister and F. F. Cleveland, J. Chem. Phys., 1944, 12, 393–398 CrossRef CAS.
- B. Dong, Y. R. Fang, L. X. Xia, H. X. Xu and M. T. Sun, J. Raman Spectrosc., 2011, 42, 1205–1206 CrossRef CAS.
- H. Y. Weng, Q. H. Guo, X. R. Wang, M. M. Xu, Y. X. Yuan, R. A. Gu and J. L. Yao, Spectrochim. Acta, Part A, 2015, 150, 331–338 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |