DOI:
10.1039/C7RA03180G
(Paper)
RSC Adv., 2017,
7, 26161-26169
Loading docetaxel in β-cyclodextrin-based micelles for enhanced oral chemotherapy through inhibition of P-glycoprotein mediated efflux transport†
Received
17th March 2017
, Accepted 11th May 2017
First published on 16th May 2017
Abstract
Oral administration of BCS Class IV drugs still faces great challenges owing to their poor dissolubility in the gastro-intestinal (GI) tract and low permeation capacity across the GI membrane. Here, we constructed a β-cyclodextrin-based polymeric micelle (PELC) to effectively deliver docetaxel (DTX), a typical BCS Class IV drug, by oral administration. Cellular uptake and bidirectional transport studies of PELC/DTX on MDCK-MDR1 cells revealed significantly enhanced absorption of DTX through inhibition of P-glycoprotein mediated efflux. In vivo pharmacokinetic studies showed that the relative oral bioavailability of PELC/DTX was 4.6-fold higher than free DTX. Correspondingly, orally administered PELC/DTX achieved superior anti-cancer efficacy against mouse sarcoma 180 tumor in vivo than free DTX with decreased subacute toxicity. Furthermore, we confirmed the vital effect of β-cyclodextrin on various functions of PELC throughout this study. These findings indicate that PELC copolymer displays great potential for the effective oral delivery of DTX.
1. Introduction
Drugs categorized as Class IV drugs according to the Biopharmaceutics Classification System (BCS)1 always face much more severe challenges with limited oral bioavailability than other drugs, not only because of their poor dissolubility in the aqueous environment of the gastrointestinal (GI) tract but also low permeation capability to cross the GI membrane.2 So far, however, few available measures have been taken to enhance the oral absorption of such drugs.3 Docetaxel (DTX), a first-line anti-mitotic chemotherapy medication,4 belongs to Class IV drugs5 and plays an important role in cancer therapy.6 The currently marketed DTX formulation Taxotere® uses polysorbate 80 and ethanol as cosolvents to improve DTX solubility for intravenous infusion, which could cause untoward hypersensitivity reactions, febrile neutropenia, cumulative fluid retention and nail changes.4,5,7 In order to reduce the adverse effects, non-injection and polysorbate 80-free formulations are particularly desired. Oral administration is the most convenient route for drug delivery. However, its oral absorption rate is quite low (<5%).8 It has been confirmed that the main reason for the low oral bioavailability of DTX is closely related to the membrane transporter P-glycoprotein (P-gp),9,10 which is a typical ATP-binding cassette transporter protein. In the GI tract, P-gp is present on the villus tip of the apical brush border membrane of gut enterocytes and actively initiates the efflux of drugs from gut epithelial cells back into the intestinal lumen.11
Recently, increased investigations have been carried out to explore possible solutions to improve the bioavailability of DTX. Applying P-gp inhibitors such as cyclosporine A, valspodar, and elacridar has been the most popular idea. However, these P-gp inhibitors are still unacceptable in clinics owing to their toxicity, interference of drug metabolism and immune suppression.12 On the other hand, nanotechnology displays potentials to improve the oral administration of drugs by solubilizing poorly water-soluble drugs, protecting drugs from the harsh environment of the GI tract, or prolonging the drug residence time in the gut by mucoadhesion.13 In addition, nanotechnology could enhance the intracellular uptake of epithelial cells by means of optimal particle size and modification of active targeting groups.14 Therefore, nanotechnology-based drug delivery systems such as micelles, solid lipid nanoparticles and nanoemulsions could benefit the oral administration of DTX to some extent. Among them, the most attractive carrier is D-α-tocopherol polyethylene glycol 1000 succinate (TPGS), a kind of water-soluble derivative of natural vitamin E. TPGS can act as an excellent emulsifier to solubilize hydrophobic drugs in the form of micelles. In addition, TPGS has been proved to improve drug permeability across biological membranes by inhibiting P-gp pumps, thereby increasing the drug absorption. Nevertheless, TPGS micelles would easily dissociate in an aqueous solution since the CMC (0.02% w/w) of TPGS is relatively high.15 Therefore, TPGS is often mixed with other amphiphilic materials to form composite micelles or is linked to certain polymers to construct novel micelles to improve its stability and drug encapsulation capability.16–19
Nanoparticles such as micelles have been widely used in cancer therapy study and achieved some promising results.20–22 In our previous study,23 we synthesized an amphiphilic copolymer PELC by sequentially linking poly(ethylene glycol), poly(D,L-lactide) and β-cyclodextrin and found that this micellar carrier could reverse doxorubicin (DOX) resistance to DOX resistant human breast carcinoma MCF-7/ADR cells. Based on a series of molecular biology tests on MCF-7/ADR cells and P-gp overexpressed zebrafish model, it has been verified that PELC could inhibit the efflux function of P-gp by the several mechanisms including intracellular ATP depletion, P-gp expression decrease, the increase of P-gp ATPase activity and the inhibition of DOX efflux as a competitive P-gp substrate.
Based on the above results, we aim to make use of PELC micelles with excellent physiochemical properties (particle size, stability, drug loading, biodegradability and biocompatibility) as nano-carriers to achieve high bioavailability of DTX via oral delivery and further verify the inhibition effect of PELC on P-gp function. In this study, we analyzed in vitro DTX transport across both Madin–Darby canine kidney cells (MDCK) and MDR1-transfected MDCK cells (MDCK-MDR1) with the PELC micelles. We also compared the data from rat in situ single pass intestinal perfusion (SPIP) models and mouse in vivo pharmacokinetics and pharmacodynamics studies to characterize the intestinal absorption behavior and to evaluate the potential of using PELC micelles in the oral delivery of DTX. More specially, we emphasized the effect of β-cyclodextrin on various functions of PELC throughout this study.
2. Materials and methods
2.1 Materials, cells and animals
D,L-Lactide (LA) and monomethoxy poly(ethylene glycol) (mPEG, Mn: 5000 Da) was purchased from Dai Gang Biotechnology Co. Ltd (Jinan, China). Stannous octoate (Sn(Oct)2) was provided by Sigma Chemical (Steinheim, Germany). β-Cyclodextrin (β-CD) was obtained from Shanghai Chemical Reagents Co. Ltd. (Shang Hai, China) and was recrystallized before use. Docetaxel (DTX, HPLC purity: 98.5%) was kindly supplied by Hisun pharmaceutical Co. Ltd. (Tai Zhou, China). Rhodamine 123 (R123) and N,N-carbonyldiimidazole (CDI) were purchased from Sigma-Aldrich (St. Louis, Mo, USA). Chlorpromazine hydrochloride, amiloridehydrochloride and nystatin were obtained from Shanghai Chemical Reagents Co. Ltd. (Shang Hai, China). All other reagents were commercially purchasable. Madin–Darby canine kidney (MDCK) cell lines and stable MDCK-MDR1 cell lines were obtained from Genscript company (Jiangsu, China). Cells were cultured in MEM medium (10% FBS, 1% penicillin–streptomycin) and humidified atmosphere at 37 °C, 5% CO2. The mice sarcoma 180 (S180) cells were purchased from KeyGen Biotechnology Co., LTD (Nanjing, China), cultured in RPMI-1640 medium (10% FBS, 1% penicillin–streptomycin) and humidified atmosphere at 37 °C, 5% CO2. Female ICR mice (4–6 weeks old, weighing 20 ± 2 g) were purchased from Experimental Animal Center, Zhejiang Chinese Medical University (Hangzhou, China). All animal studies were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals with the approval of the Scientific Investigation Board of Zhejiang University (Hangzhou, China).
2.2 Synthesis and characterization of PELC copolymer
The synthesis of β-CD based amphiphilic copolymer (PELC) followed a three-step reaction. Methoxy poly(ethylene glycol)–polylactide block copolymer (PEL) was synthesized by ring-opening polymerization. After carboxylated by succinic anhydride, the carboxylated PEL was conjugated with β-CD activated by CDI. More details were clarified in the ESI.†
2.3 Preparation and characterization of DTX-loaded micelles
The DTX-loaded micelles were prepared using thin film hydration method. Briefly, DTX (1.0 mg) and PELC or PEL (20.0 mg) were dissolved at a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 in acetonitrile. The organic solvent was evaporated at 50 °C by rotary evaporation to form a thin film. Then the dried film was hydrated with the pre-warmed distilled water for 5 min and filtered through a 0.45 μm filter to remove the free DTX. After lyophilization, DTX loaded PELC (PELC/DTX) or DTX loaded PEL (PEL/DTX) micelles were obtained. More details were clarified in the ESI.† The drug loading content (LC) and the encapsulation efficiency (EE) were calculated from the following equations:
:20 in acetonitrile. The organic solvent was evaporated at 50 °C by rotary evaporation to form a thin film. Then the dried film was hydrated with the pre-warmed distilled water for 5 min and filtered through a 0.45 μm filter to remove the free DTX. After lyophilization, DTX loaded PELC (PELC/DTX) or DTX loaded PEL (PEL/DTX) micelles were obtained. More details were clarified in the ESI.† The drug loading content (LC) and the encapsulation efficiency (EE) were calculated from the following equations:| | |
LC (%) = (weight of DTX in micelle)/(weight of DTX loaded micelle) × 100%
| (1) |
| | |
EE (%) = (weight of DTX in micelle)/(weight of the feeding DTX) × 100%
| (2) |
2.4 Cell cytotoxicity assay
MDCK and MDCK-MDR1 cells were seeded in 96-well culture plates (5000 cells per well) and grown for 24 h. Then free DTX and PELC/DTX micelle were added to medium in the DTX concentration range of 0–200 μg mL−1 for 2 h. Then, 31.5 μL of MTT solution (5 mg mL−1 in PBS) was added into each well followed by 4 h incubation at 37 °C in darkness. Finally, the medium was removed and 200 μL DMSO was added into each well. The absorbance intensity was tested using Multiskan MK3 (Thermo, USA) at 570 nm. Cell viability of the control group was regarded as 100%. Relative viability was determined by comparing the absorbance intensity of groups treated with different concentration of DTX with the control group.
2.5 Characterization of MDCK-MDR1 and parent MDCK cells
To study P-gp expression in MDCK-MDR1 and parent cells, samples were analyzed by western blots. Subsequently, the cells were harvested, washed and lysed. The concentrations of the proteins in the supernatants were quantified by BCA Protein Assay Kit (Biyuntian, China). They were then diluted into the same protein concentration. Equal micrograms of the supernatant of each sample were electrophoresed by SDS-PAGE and then transferred to nitrocellulose membrane. After blocked with 5% fat-free milk, the monoclonal antibodies P-gp (1:1000) were incubated overnight. Goat anti-mouse IRDye-680CW secondary antibodies (1:10000) was incubated at room temperature for 1 h and the blots were visualized on the Odyssey® scanner.
2.6 Cellular uptake investigation
HPLC analysis of DTX. For quantitative study, MDCK and MDCK-MDR1 cells were seeded into a 12-well culture plate (1 × 105 cells per well) and allowed to attach for 24 h. The medium was then removed and treated with fresh MEM containing free DTX and PELC or PEL (20, 50, 100 μg mL−1) at 37 °C for 2 h. After incubation, the cells were washed with PBS, lysed with 10% sodium lauryl sulfate for 20 min, and extracted by equivalent acetonitrile. DTX amount was detected by HPLC and normalized by cellular protein levels as determined by BCA protein assay kit.
Confocal analysis of R123. The uptake of free R123 by MDCK and MDCK-MDR1 cells were visualized by a confocal laser scanning microscope (CLSM, Nikon A1, Japan). MDCK or MDCK-MDR1 cells were cultured in sterile cover slips. 5 μM fresh MEM dispersion of R123 and 10, 50, 100 μg mL−1 PELC was added and incubated for 2 h, respectively. In the end of incubation, cells were washed with cold PBS, fixed by 4% paraformaldehyde, treated by Hoechst 33258 and observed by CLSM.
2.7 Bidirectional transport studies
Transport studies across MDCK and MDCK-MDR1 cell monolayer. The transport experiments of free DTX, PEL/DTX micelles, PELC/DTX micelles were carried out on MDCK or MDCK-MDR1 cells, more details were clarified in the ESI.† For the direct imaging of the transport for drug-loaded micelles across the monolayer, both apical and basolateral medium were collected after the transport studies with PELC/DTX at 37 °C for 2 h. Then the medium were placed onto copper grids and viewed under TEM.
Endocytosis pathways of PELC/DTX in transport assay. To identify the effect of the polymer vehicle on the transcellular transport of DTX by endocytosis, the absorptive transport experiment was performed with various inhibitors of different endocytosis pathways and PELC/DTX. Briefly, MDCK or MDCK-MDR1 cell monolayers were treated with PELC/DTX in HBSS containing chlorpromazine (20 μg mL−1), amiloride (171 μg mL−1) or nystatin (15 μg mL−1). The media in the basolateral side was replaced by fresh HBSS containing the corresponding inhibitor at a specified time interval. The apparent permeability coefficients (Papp) for the formulations were calculated according to the following formulation:where Papp (10−6 cm s−1) is the permeability, dQ/dt is the apparent appearance rate of drug in the receiver side calculated using linear regression of amounts in receiver chamber versus time, C is the drug concentration in the donor chamber and S is the surface area of the monolayer (S = 1.13 cm2).The efflux ratio (ER) of drugs was calculated from the bidirectional Papp values in the BL–AP and AP–BL directions.
| | |
ER = (Papp B–A)/(Papp A–B)
| (4) |
where
Papp B–A is the
Papp value measured from BL to AP and the
Papp A–B is the
Papp value measured from AP to BL. The net efflux ratio (NER) calculated by dividing the ER in MDCK-MDR1 monolayer by ER in MDCK monolayer.
| | |
NER = ERMDCK-MDR1/ERMDCK
| (5) |
2.8 In situ single pass intestinal perfusion (SPIP)
The test solutions were obtained by adding DTX, PEL/DTX micelles or PELC/DTX micelles into the blank HBSS perfusion solution (concentration of DTX was 20 μg mL−1), respectively. The phenol red (20 μg mL−1) was used as a non-absorbable marker for correcting the change of DTX concentration. The SPIP studies were performed using methods as reported.24–26 More details were clarified in the ESI.† The effective permeability (Peff, cm s−1) was calculated by the following equation.| | |
Peff = (lnCin/Cout(corr))/2πrL
| (6) |
where L is the length of the intestinal segment, r is the radius of the segment lumen, Qin is the flow rate of inlet perfusate, Cin and Cout(corr) are the DTX concentration of inlet solution and the concentration of DTX in the outlet solution corrected for water flux, respectively. The Cout(corr) can be calculated by the following equation.| | |
Cout(corr) = CoutCp(in)/Cp(out)
| (7) |
| | |
Ka = (1 − Cout(corr)/Cin)Qin/πr2L
| (8) |
where Cout is the drug concentration of outlet solution, Cp(in) and Cp(out) are the concentration of phenol red in the inlet solution and the concentration of phenol red in the outlet solution, Ka is absorption rate constant, respectively.
2.9 Pharmacokinetic studies
The mice were fasted 12 h before experiment with free access to water and were divided randomly into three groups: group 1, free DTX solution (2 mg kg−1) via intravenous injection (i.v.); group 2, oral gavage with free DTX-water suspension (20 mg kg−1); group 3, oral gavage with PEL/DTX (20 mg kg−1); group 4, oral gavage with PELC/DTX (20 mg kg−1). More details were clarified in the ESI.† The relative bioavailability (FR) and absolute bioavailability (FA) of formulations was calculated as following:| | |
FR% = (AUC0–∞, PELC/DTXp.o.)/(AUC0–∞, free DTXp.o.) × 100%
| (9) |
| | |
FA% = (AUC0–∞, PELC/DTXp.o. × dosei.v.)/(AUC0–∞, free DTXi.v. × dosep.o.) × 100%
| (10) |
where AUC0–∞, PELC/DTXp.o., AUC0–∞, free DTXp.o., AUC0–∞, free DTXi.v. present the total area under the plasma concentration versus time curves of PELC/DTX micelle or free DTX following oral or intravenous administration.
2.10 In vivo anti-tumor activity and toxicity study
The in vivo inhibitory efficacy of free DTX, PELC/DTX micelle and PELC/DTX micelle via different administration was evaluated against S180 solid tumor. After S180 cells being subcutaneously implanted into the armpits, the tumor bearing mice were randomly divided into six groups (5 mice per group): group 1, control (normal saline); group 2, intravenous injection with free DTX (10 mg kg−1); group 3, oral gavage with free DTX (20 mg kg−1); group 4, oral gavage with free DTX (30 mg kg−1); group 5, oral gavage with PELC/DTX micelle (20 mg kg−1); group 6, oral gavage with PELC/DTX micelle (30 mg kg−1); group 7, oral gavage with PEL/DTX micelle (30 mg kg−1). More details were clarified in the ESI.† Tumor volume and inhibitory of rate of tumor (IRT) were calculated as the following:| | |
V = [length × (width)2]/2
| (11) |
| | |
IRT = (Vcontrol − Vtreated)/Vcontrol × 100%
| (12) |
where the Vcontrol and Vtreated present the average volume of tumor of control group and different formulation treated groups, respectively.
2.11 Statistical analysis
Data were showed as mean ± SD and using the one way analysis of variance (ANOVA). A significant difference was considered as p < 0.05. Very significant difference was regarded as p < 0.01.
3. Results and discussion
3.1 Preparation and characterization of PELC copolymer and PELC/DTX micelles
The chemical structure of PELC was identified by 1H-NMR assay. As shown in Fig. 1A(a). The weight ratio of mPEG to PLA was calculated to be 5000/4582 by comparing the area of the –CH peak (5.18 ppm) in PLA and the –OCH3 peak (3.38 ppm) in mPEG, consistent with the feed ratio of mPEG to LA. The conjugation ratio of PEL to β-CD was 1.3 by calculating the peak area ratio of –OCH3 (3.38 ppm) in mPEG to the C1 hydrogen in β-CD (4.8 ppm) and the molecular weight of the resultant copolymer was 13592. The GPC result showed that the number-average molecular weight (Mn) of PELC was 12003, which was similar to the molecular weight determined by 1H-NMR, with a narrow polydispersity of 1.17. The CMC of PEL was calculated to be 0.0025 mg mL−1. The CMC of PELC was calculated to be 0.00123 mg mL−1, more than 16-fold lower than that of TPGS and 2-fold lower than that of PEL. In the D2O-solvent 1H-NMR spectrum, the typical PLA resonance peaks were hardly observed (Fig. 1A(b)), owing to the restricted motions of these protons in the micelle core, further suggesting PELC formed micelles in an aqueous solution. In fact, the TEM image (Fig. 1B(a)) shows the particles of PELC to be spherical with uniform distribution, and the size was determined to be 134 ± 21.3 nm by DLS.
 |
| | Fig. 1 (A) 1H NMR spectra of PELC in (a) DMSO-d6 and (b) D2O (*solvent peak), and (B) TEM image of (a) blank PELC micelles, scale bar = 0.5 μm and (b) DTX-loaded PELC micelles, scale bar = 0.2 μm. | |
The drug loading content (LC) and encapsulation efficiency (EE) of PELC/DTX reached 12 ± 3.6% and 92.2 ± 10.2%, respectively, which displayed the superior loading capability than PEL (LC 11 ± 2.4%, EE 67.2 ± 8.9%) due to the combination effect of hydrophobic inner cavity of β-CD and hydrophobic poly(D,L-lactide) blocks in PELC on DTX payload.27 The size of PELC/DTX was determined to be 87.2 ± 10.6 nm by DLS with spherical morphology as shown in Fig. 1B(b). PEL/DTX micelle displayed the similar shape with the size about 60 nm.
3.2 Cytotoxicity studies
In order to evaluate the cytotoxicity of materials, the MDCK and MDCK-MDR1 cells were treated with several concentrations of free DTX or PELC/DTX micelles. No significant toxicity toward both cell lines was observed in MTT assay when DTX concentration was increased from 2 μg mL−1 to 66.6 μg mL−1 where the relative cell viability exceeded 95% (Fig. S1†). Therefore, PELC/DTX micelle and free DTX at 60 μg mL−1 of DTX were chosen as the test concentration in next studies.
3.3 Cellular uptake of DTX and R123
Before the cellular uptake study of DTX on MDCK cells and MDCK-MDR1 cells, P-gp overexpression in MDCK-MDR1 cells was confirmed by western blot analysis. As shown in Fig. 2A, the significant amounts of P-gp were present in MDCK-MDR1 cells whereas parent MDCK cells had no trace of P-gp. As displayed in Fig. 2B and C, the uptake of free DTX in MDCK cells was 84.6-fold higher than that in MDCK-MDR1 cells. The micelles induced a small accumulation of cellular DTX in MDCK cells. On the contrary, in MDCK-MDR1 cells, the cellular uptake of DTX with PELC at 20 50, 100 μg mL−1 concentrations significantly improved, which was 10.8-fold, 12.1-fold, and 13.6-fold higher than the control, respectively (P < 0.01). In the meantime, we observed the significantly weaker cellular uptake by the PEL group at the corresponding concentrations as compared with PELC (Fig. 2D), which is corresponding with our previous study in MCF-7/ADR cells.23
 |
| | Fig. 2 (A) P-gp expression in parent MDCK and MDR1-MDCK cells. (B) MDCK cells uptake of DTX with different concentration of PELC (20, 50, 100 μg mL−1). (C) MDCK-MDR1 cells uptake of DTX with different concentration of PELC (20, 50, 100 μg mL−1). (D) MDCK-MDR1 cells uptake of DTX with different concentration of PEL (20, 50, 100 μg mL−1), n = 3, P < 0.01 when compared with control (free DTX). | |
To further confirm the positive effect of PELC on the cellular uptake of P-gp substrate, we performed an accumulation study of R123. R123 is a classic substrate of P-gp with poor permeability according to the Papp values,28 and is usually used as a marker in cellular uptake studies. CLSM was used to observe the internalization of fluorescent R123 into MDCK and MDCK-MDR1 cells after exposure to free R123 or to R123 with 10, 50, 100 μg mL−1 PELC micelles at 37 °C for 2 h. Fig. 3 shows the intracellular distribution of R123 was quite different in the two cell lines. Strong fluorescence was observed in the cytoplasm of MDCK cells. However, R123 fluorescence was quite weak in MDCK-MDR1 cells while the micelles could significantly enhance the cellular uptake at any polymer concentration.
 |
| | Fig. 3 CLSM images of MDCK cells and MDCK-MDR1 cells after 2 h exposure of 5 μM R123 (red fluorescence) or various concentration of PELC with 5 μM R123. Nucleus was stained by Hoechst 33258. | |
3.4 Bidirectional transport studies
Validation of P-gp activity in MDCK and MDCK-MDR1 cells. Higher values of permeability coefficients in the absorptive direction (Papp A–B) and lower values of those in the secretory direction (Papp B–A) would indicate a great permeability across the intestinal barrier and optimal oral bioavailability. P-gp activity was tested in MDR1-MDCK cells and parent MDCK cells using the transport assay with the substrate R123. Involvement of a P-gp-mediated efflux mechanism could be concluded if the net efflux ratio (NER) was above 2.29 In Table 1, R123 exhibited a strong polarized efflux in MDCK-MDR1 cells (Papp B–A ≫ Papp A–B), and the efflux ratio (ER) of R123 in MDCK-MDR1 cells was 4.641. As expected, the polarized efflux of R123 measured by ER was much higher in MDCK-MDR1 cell monolayers than that in MDCK parent cell monolayers (4.641 vs. 1.508). The NER was 2.97 indicating the R123 was a P-gp substrate.
Table 1 Transport and permeability of R123 across MDCK and MDCK-MDR1 cells at 37 °C
| Drug |
Cells |
Papp (10−6 cm s−1) |
ER |
NER |
| R123 |
MDCK |
A–B |
3.265 ± 1.046 |
1.508 |
2.97 |
| B–A |
4.925 ± 0.300 |
| MDCK-MDR1 |
A–B |
0.920 ± 0.036 |
4.641 |
| B–A |
4.267 ± 0.047 |
ATP-dependent transport. It has been reported that in many cases drug transport is very sensitive to ATP depletion and requires an ATP regenerating system for maximal transport.30–32 As for MDCK-MDR1 cells, the drug-efflux process is heavily ATP-dependent. Depletion of ATP in cell results in inhibition of active mechanisms. It is necessary to compare the transport of DTX or DTX-loaded micelles at 4 °C and at 37 °C, since ATP-dependent processes of cells are inhibited at 4 °C. As shown in Table 2, lowering the temperature from 37 °C to 4 °C significantly reduced the Papp B–A values of DTX in MDCK-MDR1 monolayers, while the ER decreased from 6.873 to 1.339, indicating that a large extent of free DTX was transported via P-gp efflux. On the other hand, there was no significant difference in Papp B–A values between PELC/DTX micelle groups.
Table 2 Transport and permeability of DTX and PELC/DTX across MDCK-MDR1 cells at 4 °C or 37 °C
| Drug or micelle |
Temperature |
Papp (10−6 cm s−1) |
ER |
| DTX |
4 °C |
A–B |
0.401 ± 0.033 |
1.339 |
| B–A |
0.537 ± 0.042 |
| 37 °C |
A–B |
0.340 ± 0.020 |
6.873 |
| B–A |
2.336 ± 0.274 |
| PELC/DTX |
4 °C |
A–B |
1.578 ± 0.685 |
0.539 |
| B–A |
0.850 ± 0.049 |
| 37 °C |
A–B |
1.584 ± 0.257 |
0.567 |
| B–A |
0.899 ± 0.518 |
Transport permeability influence comparison among PELC micelles and verapamil using MDCK and MDCK-MDR1 monolayers. To assess the capability of PELC micelles in enhancing DTX transport, MDCK and MDCK-MDR1 monolayers were treated with free DTX, DTX plus verapamil, DTX plus PELC, and PELC/DTX micelles at a concentration of 60 μg mL−1 at 37 °C. In Table 3, a significant efflux of free DTX groups was found in the MDCK-MDR1 cell monolayer. The ER of 60 μg mL−1 DTX was 6.873 in the MDCK-MDR1 cell monolayer, which was much higher than that in the MDCK cell monolayer (1.664), increasing the NER to 4.13. So DTX was confirmed to be subjected to efflux by P-gp. The presence of the P-gp inhibitor verapamil caused a 3.4-fold increase of the Papp A–B value in the MDCK-MDR1 cell monolayer and a 1.6-fold decrease of the Papp B–A value. Therefore, the ER decreased from 6.873 to 1.247 compared with free DTX, and the NER significantly decreased from 4.13 to 1.72. Similarly, when DTX was administered with PELC micelles, the ER in the MDCK-MDR1 cell monolayers also decreased to 2.362 with a low NER of 1.46. Most strikingly, the Papp A–B value of PELC/DTX in the MDCK-MDR1 cell monolayer was 4.7-fold higher and the Papp B–A value was 2.6-fold lower than in the free DTX group. The ER of PELC/DTX micelles with same concentration of DTX in the MDCK-MDR1 cell monolayers decreased from 6.873 to 0.567 and achieved the lowest NER of 0.26. In addition, the presence of PEL/DTX increased the ER from 0.576 to 2.072 (Table S1†) compared with PELC/DTX micelles in the MDCK-MDR1 cell monolayer, indicating that the modification of β-CD in the polymers was vital to inhibit the efflux of DTX, which was consistent with the results from the DTX cellular accumulation assay.
Table 3 DTX transport and permeability of various groups across MDCK and MDCK-MDR1 cells at 37 °C
| Drugs |
Cells |
Papp (10−6 cm s−1) |
ER |
NER |
| DTX |
MDCK |
A–B |
2.132 ± 0.500 |
1.664 |
4.13 |
| B–A |
3.548 ± 0.482 |
| MDCK-MDR1 |
A–B |
0.340 ± 0.020 |
6.873 |
| B–A |
2.336 ± 0.274 |
| DTX + verapamil |
MDCK |
A–B |
1.496 ± 0.438 |
0.727 |
1.72 |
| B–A |
1.087 ± 0.423 |
| MDCK-MDR1 |
A–B |
1.163 ± 0.397 |
1.247 |
| B–A |
1.450 ± 0.105 |
| DTX + PELC |
MDCK |
A–B |
1.221 ± 0.621 |
1.618 |
1.46 |
| B–A |
1.975 ± 0.259 |
| MDCK-MDR1 |
A–B |
0.740 ± 0.059 |
2.362 |
| B–A |
1.748 ± 0.301 |
| PELC/DTX micelles |
MDCK |
A–B |
1.008 ± 0.097 |
2.182 |
0.26 |
| B–A |
2.200 ± 0.330 |
| MDCK-MDR1 |
A–B |
1.584 ± 0.257 |
0.567 |
| B–A |
0.899 ± 0.518 |
3.5 Endocytosis pathways of PELC/DTX in transport assay
Transcellular transportation is generally considered the main route for most nanocarriers.33–35 Nanocarriers can employ multiple transcellular pathways, including macropinocytosis, clathrin-mediated endocytosis, caveolae-mediated endocytosis, and caveolae- and clathrin-independent endocytosis.36 Based on the demonstration that drug-loaded micelles had the ability to enhance drug permeability through MDCK-MDR1 monolayers with decreased secretory efflux, the possible transport routes of PELC/DTX micelles were investigated at 37 °C. In order to identify whether the enhancement on MDCK-MDR1 cell permeability by DTX was associated with the cellular transport of PELC/DTX micelles and to determine the endocytosis pathways of PELC/DTX micelles, inhibitors for clathrin-mediated endocytosis (chlorpromazine), caveolae-mediated endocytosis (nystatin) and macropinocytosis (amiloride) were used in the 2 h transport studies. The data in Fig. 4A indicated that the most effective inhibition in cell endocytosis of PELC/DTX micelles was by nystatin (60.5% decrease), followed by amiloride (39.5% decrease). Chlorpromazine did not lead to a reduction in Papp A–B values compared with the control group without inhibitors added. Therefore, the study demonstrated the involvement of caveolae- and macropinocytosis-dependent and clathrin-independent endocytosis of the PELC micelles in MDCK-MDR1 monolayers, which is desirable for achieving endosomal escape into the cytoplasm since the lysosome has a highly acidic environment (pH 4.5–5.5) and numerous enzymes can be detrimental to the integrity of the cargo.37
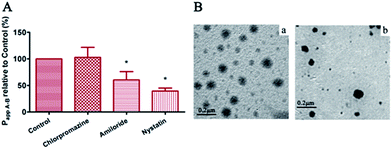 |
| | Fig. 4 (A) Relative transport permeability (Papp A–B) of PELC/DTX with 60 mg mL−1 DTX in absence or presence of various endocytosis inhibitors in MDCK-MDR1 cell monolayers (37 °C, n = 3, *P < 0.01 vs. control). (B) TEM images of (a) apical and (b) basolateral medium after incubation of Transwell filter grown MDCK-MDR1 cell monolayer with PELC/DTX at 37 °C for 2 h. Bar = 0.2 μm. | |
The TEM images of apical and basolateral medium after incubation with PELC/DTX micelles at 37 °C for 2 h are illustrated in Fig. 4B. The size and morphology of the particles in the basolateral medium were almost the same as those in the apical medium despite declining concentrations. This result again confirmed that PELC/DTX micelles underwent clathrin-independent endocytosis to avoid lysosomal fusion and then were effluxed on the basolateral side of MDCK-MDR1 cells.
3.6 In situ single pass intestinal perfusion
In situ single pass intestinal perfusion (SPIP) is a commonly used technique to assess the permeability of drugs through the intestine, and is a robust tool for predicting in vivo drug fate following oral administration.38,39 In the next experiment, the effective permeability (Peff) values of PEL/DTX micelles, PELC/DTX micelles and free DTX were compared. The Peff value of PELC/DTX micelles (0.887 ± 0.066 × 10−4 cm s−1) was nearly 2.6-fold higher than that of free DTX (0.347 ± 0.099 × 10−4 cm s−1, p < 0.01) and was nearly 1.4-fold higher than that of PEL/DTX micelles (0.614 ± 0.076 × 10−4 cm s−1, p < 0.05). The absorption rate constant (Ka) of PELC/DTX micelle (0.0088 ± 0.0024) is higher than that of free DTX (0.0044 ± 0.0010, p < 0.05) and is also higher than that of PEL/DTX micelles (0.0068 ± 0.0009).
3.7 Pharmacokinetic studies
Fig. 5 and Table 4 display the plasma concentration–time curves of free DTX and PELC/DTX micelles via different administration routes and the corresponding pharmacokinetic parameters. PELC/DTX micelles yielded much higher plasma maximum concentrations (Cmax) than free DTX did (with 292.130 ng mL−1 vs. 27.078 ng mL−1). The area under the time–concentration curve (AUC0–∞) of PELC/DTX was 506.529 ng L−1 h−1 while that of DTX solution was 110.105 ng L−1 h−1. Moreover, the t1/2 of DTX-loaded micelles was 1.25-fold longer than that of free DTX, and the body clearance CLz/F was 5.9-fold lower. These results indicate that the drug absorption of PELC/DTX micelles was enhanced and the plasma levels was remained high and steady for a certain period, which was attributed to the improved cellular uptake and permeability of DTX via PELC micelles. The pharmacokinetic study of free DTX solution via a single intravenous administration at 2 mg kg−1 was conducted to calculate the absolute bioavailability (FA) of oral formulations. The oral absorption of free DTX was extremely limited with the FA of 2.56%, which was in agreement with previous reports.18 The oral absorption of PEL/DTX was improved by 7.36%. However, in the case of PELC/DTX micelle, the FA was increased to 11.78%. As a result, the relative bioavailability (FR) of PELC/DTX micelle reached 461%, which was 1.6-fold larger than that of PEL/DTX (FR = 288%). The oral bioavailability of DTX was greatly improved via PELC because of the following reasons. First, the hydrophobic core of PELC micelles can improve the solubility of DTX. Second, the β-CD unit facilitated the inhibition effect of PELC on P-gp efflux according to the transport study.
 |
| | Fig. 5 Plasma concentration vs. time profile of free DTX, PEL/DTX micelle and PELC/DTX micelle following single treatment of i.v. (2 mg kg−1) or oral administration (20 mg kg−1). All data represent as mean ± SD (n = 3). | |
Table 4 Pharmacokinetic parameters of various DTX formulations after single treatment of i.v. (2 mg kg−1) or oral administration (20 mg kg−1) in mice (n = 3)
| Formulation |
DTX solution (p.o.) |
PEL/DTX (p.o.) |
PELC/DTX (p.o.) |
Free DTX (i.v.) |
| Cmax (ng mL−1) |
27.08 ± 7.84 |
190.30 ± 76.60 |
292.10 ± 42.50 |
— |
| Tmax (h) |
0.75 |
0.26 |
0.25 |
— |
| t1/2 (h) |
1.89 ± 1.16 |
1.96 ± 0.44 |
2.36 ± 1.09 |
1.85 ± 0.14 |
| AUC0–∞ (ng mL−1 h−1) |
110.11 ± 74.72 |
316.09 ± 64.70 |
506.53 ± 91.00 |
451.60 ± 33.51 |
| CLz/F (L h−1 kg−1) |
0.24 ± 0.16 |
0.53 ± 0.12 |
0.04 ± 0.007 |
4.44 ± 0.33 |
| FA |
2.56% |
7.36% |
11.78% |
— |
| FR |
— |
288% |
461% |
— |
3.8 In vivo anti-tumor activity and toxicity study
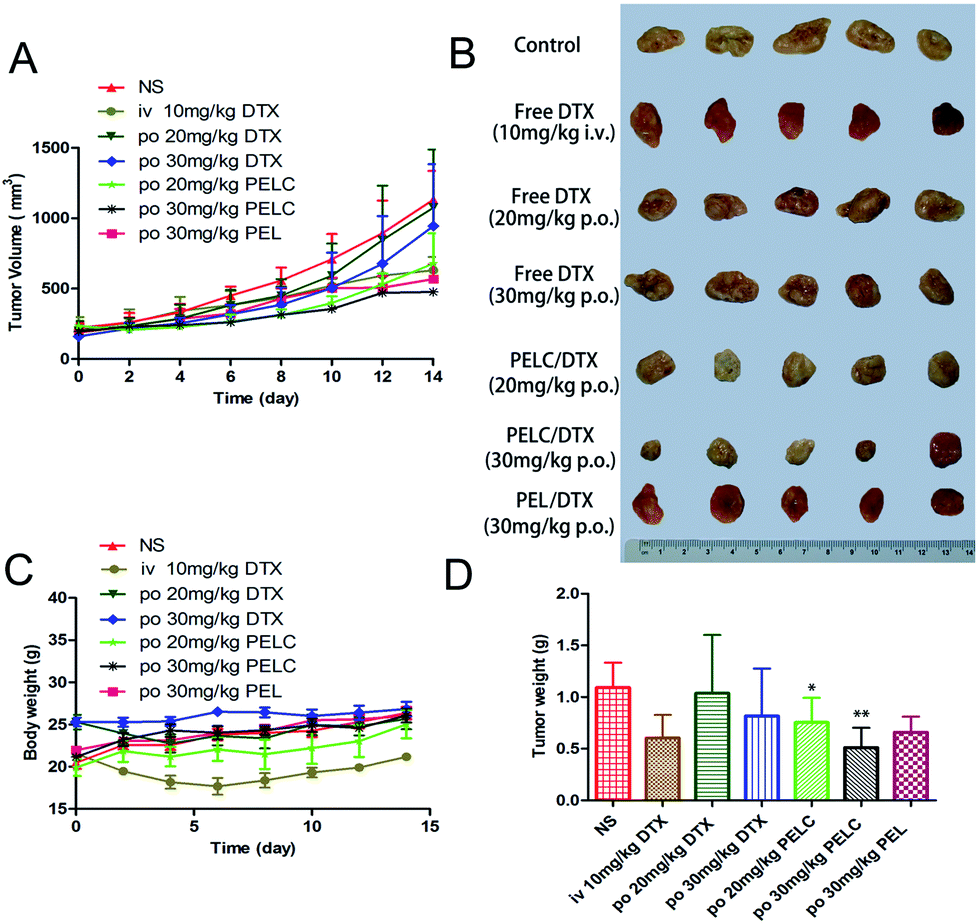
The antitumor ability was studied in S180 tumor-bearing mice. Because tumor growth sometimes could not be inhibited effectively by a single dose, multiple doses were carried out in this study to enhance the anti-tumor effect.40 The changes in tumor volume and body weight are showed in Fig. 6, and time-related changes in tumor volume were observed between the different treatment groups. Tumor volume increased rapidly without significant difference in mice orally treated with saline and free DTX at 20 mg kg−1 and 30 mg kg−1, indicating that free DTX had little effect on tumor growth due to poor oral bioavailability. However, the inhibitory of rate of tumor (IRT) of mice treated with PELC/DTX at 20 mg kg−1 and 30 mg kg−1 reached 40.14% and 57.82%, respectively, and the inhibitory effect increased at increasing doses. In addition, the IRT of mice treated with PEL/DTX at 30 mg kg−1 reached 49.8%, which was lower than PELC/DTX at 30 mg kg−1 (IRT = 57.82%), which indicating the stronger anti-tumor activity of PELC/DTX.
 |
| | Fig. 6 In vivo anti-tumor activity in S180 bearing mice. (A) Tumor progressions after oral administration of free DTX, PEL/DTX and PELC/DTX. (B) Photos of the tumors collected from different groups of mice. (C) Body weights of mice after various treatments. All data represent as mean ± SD (n = 5). (D) Average tumor weights in each treatment group on day 14. **p < 0.01, *p < 0.05, vs. Saline group. | |
To further investigate the in vivo effects of free DTX, PEL/DTX and PELC/DTX micelles via oral administration, S180 tumors from each group were analyzed with H&E staining (Fig. 7A). Apoptosis occurred in S180 tumor slices treated with PELC/DTX at both 20 mg kg−1 and 30 mg kg−1 dosage. Severe necrosis and definitive changes in the cytoarchitecture were observed in the group PELC/DTX at 30 mg kg−1, leading to tumor regression, which is consistent with the results of tumor growth profile and inhibitory rate. In contrast, tumors with free DTX displayed a much lower necrotic level with a large amount of living tumor cells existing.
 |
| | Fig. 7 (A) Histology of S180 tumor, stomach and intestine tissues (scale bar is 50 or 100 μm) and (B) hematological toxicity of free DTX and PELC/DTX (20 mg kg−1 and 30 mg kg−1) on mice on the 15th day after 4 times oral administration. | |
The major target of anti-cancer drugs is the rapidly proliferating cells of the body, in particular the GI, bone marrow, hair follicle cells, causing severe side effects such as nausea, vomiting, stomatitis, diarrhea, severe myelosuppression and neutropenia, and weight loss.41,42 As shown in Fig. 6C, no noticeable change in body weight after both PELC/DTX and free DTX administration was observed since the sudden high concentration of DTX was prevented. However, the body weight decreased when treated with intravenous DTX. The onset of neutropenia was quite rapid, occurring between day 5 and day 8 after DTX administration finished, but a very rapid recovery appeared after that.42 Therefore, cumulative effect on neutropenia from DTX could be avoided. To evaluate systemic toxicity of PELC/DTX formulation in vivo, hematological parameters, including number counts of red blood cells (RBC), white blood cells (WBC), and neutrophils (NEUT), were measured on day 14 (Fig. 7B). RBC values did not change in all the treatments. Although the level of the two markers in the PELC/DTX groups decreased slightly compared with that of the control group, more significant decreases in counts of WBC and NEUT were found in the free DTX group. The results indicate that the PELC/DTX showed lower toxicity than free DTX at the equivalent dosage.
Among the non-hematological side effects, GI toxicities, such as nausea, diarrhea and vomiting, are the predominant adverse effects, therefore, it is necessary to evaluate the damages of intestinal mucosa induced by oral administration. In order to identify the extent of disruption to the GI tract after oral administration, stomach and intestine tissues were analyzed using H&E staining (Fig. 7A). Epithelial cell shedding and intense parietal cell loss were observed in groups orally administered with free DTX, suggesting that repeated administration of free DTX could induce toxicity to gastric tissues. However, it is clear that stomach tissues from the groups orally administered with PELC/DTX micelle displayed normal histological structures with intact epithelial tissues as well as evenly distributed parietal and chief cells arranged orderly and tightly. This result indicated that PELC micelles were stable enough to hold DTX in the GI tract and protect the tissues from damage induced by free DTX, and thus ensure the safe use of DTX via oral delivery.
4. Conclusion
In summary, we constructed a novel DTX loaded micelle system based on an amphiphilic β-CD modified polymer PELC for the oral delivery of DTX and reported the transport features across MDCK-MDR1 monolayers, pharmacokinetic behaviors, and the therapeutic outcome of S180 tumor-bearing mice. Indeed, PELC/DTX micelles exhibited higher oral absorption and better antitumor efficacy than PEL/DTX micelles and free DTX did, due to the improved stability and absorbed by clathrin-independent endocytosis instead of excreted by P-gp. Moreover, compared with free DTX orally administered, the subacute toxicity in terms of body weight loss and hematological toxicity markedly decreased after treatment with PELC/DTX micelles, indicating their potential for further clinical applications. Taken together, the PELC micelle system is a promising oral delivery platform for Class IV drugs.
Acknowledgements
This work was supported by National Natural Science Foundation (81473173) and National Natural Science Funds for Excellent Young Scholar (81222047).
References
- Y. Kawabata, K. Wada, M. Nakatani, S. Yamada and S. Onoue, Int. J. Pharm., 2011, 420, 1–10 CrossRef CAS PubMed.
- M. S. Ku, AAPS Journal, 2008, 10, 208–212 CrossRef CAS PubMed.
- C. Luo, J. Sun, Y. Q. Du and Z. G. He, J. Controlled Release, 2014, 176, 94–103 CrossRef CAS PubMed.
- F. K. Engels, R. A. Mathot and J. Verweij, Anti-Cancer Drugs, 2007, 18, 95–103 CrossRef CAS PubMed.
- B. Nuijen, M. Bouma, J. H. Schellens and J. H. Beijnen, Investig. New Drugs., 2001, 19, 143–153 CrossRef CAS.
- W. Li, J. Peng, L. Tan, J. Wu, K. Shi, Y. Qu, X. Wei and Z. Qian, Biomaterials, 2016, 106, 119–133 CrossRef CAS PubMed.
- H. Song, H. Q. Geng, J. Ruan, K. Wang, C. C. Bao, J. Wang, X. Peng, X. Q. Zhang and D. X. Cui, Nanoscale Research Letters, 2011, 6, 1–12 Search PubMed.
- H. Zhang, J. Dou, Y. Zhai, A. Liu and G. Zhai, J. Drug Target, 2014, 22, 87–94 CrossRef CAS PubMed.
- E. M. Kemper, T. Buckle, W. Leenders, W. Boogerd, J. H. Beijnen and O. Van Tellingen, Proceedings of the American Association for Cancer Research Annual Meeting, 2005, vol. 46, p. 970 Search PubMed.
- E. J. Wang, C. N. Casciano, R. P. Clement and W. W. Johnson, Cancer Res., 2001, 61, 7525–7529 CAS.
- L. Benet, C. Cummins and C. Wu, Int. J. Pharm., 2004, 277, 3–9 CrossRef CAS PubMed.
- P. Breedveld, J. H. Beijnen and J. H. Schellens, Trends Pharmacol. Sci., 2006, 27, 17–24 CrossRef CAS PubMed.
- L. Plapied, N. Duhem, A. des Rieux and V. Préat, Curr. Opin. Colloid Interface Sci., 2011, 16, 228–237 CrossRef CAS.
- Z. Khatun, M. Nurunnabi, K. J. Cho, Y. Byun, Y. H. Bae and Y. K. Lee, J. Controlled Release, 2014, 177, 64–73 CrossRef CAS PubMed.
- N. Duhem, F. Danhier and V. Préat, J. Controlled Release, 2014, 182, 33–44 CrossRef CAS PubMed.
- Y. Mi, Y. Liu and S.-S. Feng, Biomaterials, 2011, 32, 4058–4066 CrossRef CAS PubMed.
- S.-S. Feng, L. Mei, P. Anitha, C. W. Gan and W. Zhou, Biomaterials, 2009, 30, 3297–3306 CrossRef CAS PubMed.
- J. Dou, H. Zhang, X. Liu, M. Zhang and G. Zhai, Colloids Surf., B, 2014, 114, 20–27 CrossRef CAS PubMed.
- K. K. Gill, A. Kaddoumi and S. Nazzal, Eur. J. Pharm. Sci., 2012, 46, 64–71 CrossRef CAS PubMed.
- L. Zhang, L. Tan, L. Chen, X. Chen, C. Long, J. Peng and Z. Qian, Sci. Rep., 2016, 6, 36957 CrossRef CAS PubMed.
- C. H. Lin, X. X. Lin, L. Lin, J. M. Wang, Z. X. Lin and J. M. Lin, Chin. Chem. Lett., 2015, 26, 1225–1230 CrossRef CAS.
- Y. L. Wang, H. Zhao, J. R. Peng, L. J. Chen, L. W. Tan, Y. X. Huang and Z. Y. Qian, J. Biomed. Nanotechnol., 2016, 12, 2097–2111 CrossRef CAS.
- L. Zhang, J. Lu and L. Qiu, Int. J. Nanomed., 2016, 11, 5205–5220 CrossRef PubMed.
- B. X. Xiao, Q. Wang, L. Q. Fan, L. T. Kong, S. R. Guo and Q. Chang, J. Ethnopharmacol., 2014, 151, 846–851 CrossRef CAS PubMed.
- F. Cao, J. Jia, Z. Yin, Y. Gao, L. Sha, Y. Lai, Q. Ping and Y. Zhang, Mol. Pharm., 2012, 9, 2127–2135 CAS.
- F. Cao, Y. Gao, M. Wang, L. Fang and Q. Ping, Mol. Pharm., 2013, 10, 1378–1387 CrossRef CAS PubMed.
- L. Y. Qiu, R. J. Wang, C. Zheng, Y. Jin and Q. Jin le, Nanomedicine, 2010, 5, 193–208 CrossRef CAS PubMed.
- M. D. Troutman and D. R. Thakker, Pharm. Res., 2003, 20, 1210–1224 CrossRef CAS.
- O. N. K. Martey, X. He, H. Xing, F. Deng, S. Feng, C. Li and X. Shi, Biopharm. Drug Dispos., 2014, 35, 195–206 CrossRef CAS PubMed.
- D. Y. Alakhova, N. Y. Rapoport, E. V. Batrakova, A. A. Timoshin, S. Li, D. Nicholls, V. Y. Alakhov and A. V. Kabanov, J. Controlled Release, 2010, 142, 89–100 CrossRef CAS PubMed.
- C. A. Doige and F. J. Sharom, Biochim. Biophys. Acta, 1992, 1109, 161–171 CrossRef CAS.
- H. Katzir, H. D. Yeheskely, R. Regev and G. D. Eytan, FEBS J., 2010, 277, 1234–1244 CrossRef CAS PubMed.
- P. Oh, P. Borgström, H. Witkiewicz, Y. Li, B. J. Borgström, A. Chrastina, K. Iwata, K. R. Zinn, R. Baldwin and J. E. Testa, Nat. Biotechnol., 2007, 25, 327–337 CrossRef CAS PubMed.
- J. Panyam and V. Labhasetwar, Pharm. Res., 2003, 20, 212–220 CrossRef CAS.
- G. Sahay, J. O. Kim, A. V. Kabanov and T. K. Bronich, Biomaterials, 2010, 31, 923–933 CrossRef CAS PubMed.
- S. D. Conner and S. L. Schmid, Nature, 2003, 422, 37–44 CrossRef CAS PubMed.
- S. Trapp, G. R. Rosania, R. W. Horobin and J. Kornhuber, Eur. Biophys. J., 2008, 37, 1317–1328 CrossRef CAS PubMed.
- A. H. Eriksson, M. V. Varma, E. J. Perkins and C. L. Zimmerman, J. Pharm. Sci., 2010, 99, 1574–1581 CrossRef CAS PubMed.
- M. V. Varma and R. Panchagnula, J. Pharm. Sci., 2005, 94, 1694–1704 CrossRef CAS PubMed.
- Z. Xu, L. Chen, W. Gu, Y. Gao, L. Lin, Z. Zhang, Y. Xi and Y. Li, Biomaterials, 2009, 30, 226–232 CrossRef CAS PubMed.
- V. Valero, F. A. Holmes, R. S. Walters, R. L. Theriault, L. Esparza, G. Fraschini, G. A. Fonseca, R. E. Bellet, A. U. Buzdar and G. N. Hortobagyi, J. Clin. Oncol., 1995, 13, 2886–2894 CrossRef CAS PubMed.
- G. Catimel, J. Verweij, V. Mattijssen, A. Hanauske, M. Piccart, J. Wanders, H. Franklin, N. L. Bail, M. Clavel and S. B. Kaye, Ann. Oncol., 1994, 533–537 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03180g |
| ‡ These two authors equally contributed to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a