
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct conversion of carbohydrates to diol by the combination of niobic acid and a hydrophobic ruthenium catalyst†
Ying Duanab,
Jun Zhanga,
Dongmi Lia,
Dongsheng Denga,
Lu-Fang Maa and
Yanliang Yang *a
*a
aHenan Key Laboratory of Function-Oriented Porous Material, College of Chemistry and Chemical Engineering, Luoyang Normal University, Luoyang 471934, P. R. China. E-mail: yangyl0410@126.com
bCollege of Food and Drug, Luoyang Normal University, Luoyang 471934, China
First published on 17th May 2017
Abstract
Tetrahydro-2,5-furandimethanol (THFDM) was obtained directly from a wide variety of carbohydrates by the combination of niobic acid and a hydrophobic ruthenium catalyst. Fructose, glucose, and polysaccharides consisting of fructose or glucose could be converted to THFDM in one-step. The selectivity to THFDM was kept around 60% while the glucose conversion varied from 9% to 49%. The as-synthesized niobic acid was characterized by TEM, N2 adsorption/desorption, XRD, NH3-TPD and FT-IR spectra of adsorpted pyridine. The niobic acid was proved to have medium and strong acid sites with a high Brönsted/Lewis ratio, which played a great role for keeping high THFDM selectivity using glucose as a substrate.
Introduction
Production of chemicals and fuels from carbohydrates is an attractive way for alleviating the shortage of non-renewable fossil resources.1–5 One of the main strategies for the valorization of carbohydrates is the synthesis of platform compounds and their high value transformation. The conversion of carbohydrates to platform compounds such as 5-(hydroxymethyl)furfural (HMF),6–10 furfural,11–13 levulic acid (LA)14,15 and the utilization of platform compounds have both been extensively researched. Furfuryl alcohol,16–20 tetrahydrofurfuryl alcohol,21,22 dimethylfuran,23 2,5-furandicarboxylic acid,24 diformylfuran,25–31 cyclopentanone,32,33 γ-valerolactone,14,34–36 and tetrahydro-2,5-furandimethanol37,38 are the typical target products.Considering that the platform compounds such as HMF are not stable and not easy to be separated, purified and stored, the conversion of carbohydrates to target products in one pot has attracted attention increasingly.39–47 A large number of works have been reported on the direct carbohydrate conversion. Sen and his co-workers gave reports on the direct conversion of fructose to 2,5-dimethyltetrahydrofuran or 5-methylfurfural by using RhCl3 as the hydrogenation catalyst and HI as acid promoter.45 Sels et al. obtained high yields of liquid straight-chain alkanes directly from cellulosic feedstock in an one-pot biphasic catalytic system.46
Alcohols, including monobasic alcohols and diols, are important chemical products from fossil resources. The carbohydrates are ideal renewable raw materials for the production of monobasic alcohols and diols as they contain a large amount of alcohol hydroxyl.48–56 A lot of research had been reported on this topic. For example, hexanols could be produced in high yield by conversion of cellulose over Ir–ReOx/SiO2 catalyst in biphasic reaction system.56 And 1,5-pentanediol could be obtained in high yield from biomass via furfural and tetrahydrofurfuryl alcohol.55
Tetrahydro-2,5-furandimethanol (THFDM) is a valuable diol that can be used as solvent or monomer57 and also could be converted to 1,6-hexanediol,58 an important monomer in the plastics industry, in very high yield. Recently, we achieved the direct conversion of fructose to tetrahydro-2,5-furandimethanol (THFDM) by the combination of Amberlyst-15 and hydrophobic Ru/SiO2-TM in a water/cyclohexane biphasic system.7 However, the reported procedure was only effective for the conversion of fructose, a monosaccharide not common in nature.
In this article, we reported a catalytic system which was able to provide THFDM directly from glucose, the monomer of most of the carbohydrates rich in nature. Based on this, we proved that this catalytic system was able to directly convert many kinds of carbohydrates including sucrose and starch to THFDM. The conversion of carbohydrates was illuminated in Scheme 1. The carbohydrates were first dehydrated to HMF in water phase on acids catalyst and then the HMF was hydrogenated to THFDM in oil phase on hydrophobic ruthenium catalyst. The hydrophobicity of ruthenium catalyst kept the ruthenium catalyst in oil phase. As a result the hydrogenation of carbohydrates under H2 and the degradation of HMF were both avoided.
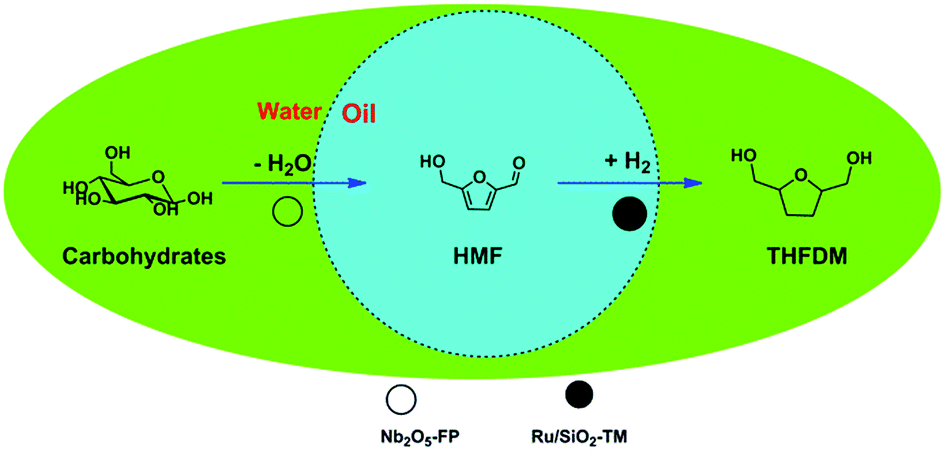 | ||
| Scheme 1 Conversion of carbohydrates into THFDM in the water/cyclohexane biphasic system. | ||
Material and methods
Materials
All chemicals were of analytical grade and used as received unless otherwise stated. Inulin, sucrose, maltose, cellobiose, starch (soluble) and zirconium oxychloride were obtained from Aladdin Chemistry Co. Ltd. Fructose, glucose, and HF were purchased from Tianjin Kemiou Chemical Reagent Co. Ltd. Amberlyst-70 was obtained from Dow Chemical Shanghai Co. Ltd. RuCl3 (Ru 37%) was purchased from Shenyang Research Institute of Nonferrous Metal. HY-zeolite (Si/Al = 5) was purchased from Nankai University Catalyst Co. Ltd. China. Niobium oxide was obtained from UCB Pharma. Pyridine and toluene were redistilled before used. All other reagents were commercially available.Preparation of catalysts
The SiO2 (4.00 g) was dispersed in the solution of RuCl3 (RuCl3: 542.1 mg, 2.00 mmol, H2O: 24.00 g). The mixture was stirred and kept at room temperature for 12 h and then dried at 393 K for 6 h. Before used, the catalyst was reduced at 673 K by H2 for 2 h and then passivated in 1% O2/N2 at room temperature to afford Ru/SiO2. The Ru/SiO2-TM catalyst was prepared by CH3-functionalization of Ru/SiO2. Ru/SiO2 (2.00 g) was transferred into a 100 mL round flask to which toluene (40 mL), then trimethylchlorosilane (10 mL) and pyridine (10 mL) were introduced. The mixture refluxed under N2 for 24 h, and then filtered and washed with alcohol for five times. The resulted solid were dried at 323 K in vacuum overnight.Nb2O5 (1.00 g) was added into H3PO4 aqueous solution (30 mL, 0.5 M) and stirred for 8 h at 343 K. Then the precipitation was washed with water (50 mL × 3 times) and dried overnight at 393 K. The resulted white powder was calcined in air at 573 K for 3 h to afford the H3PO4-treated niobic acid (Nb2O5-P).
Nb2O5 (10.00 g) was added in HF aqueous solution (45 mL, 40 wt%). The mixture was stirred at 353 K for 24 h to afford a clear solution. Then concentrated ammonia water was added drop wise to adjust the pH 8–9 under drastic stirring. The precipitation was centrifuged and washed with ammonia water for 6 times. The solid was dried at 393 K to afford niobic acid. The H3PO4-treated procedure was the same as the preparation of Nb2O5-P to afford the Nb2O5-FP.
ZrOCl·8H2O (16.20 g) was dissolved in water (100 mL). Then concentrated ammonia water was added drop wise to adjust the pH 8–9 under drastic stirring. After kept stand for 24 h at room temperature, the precipitation was centrifuged and washed with water until no Cl− was detected by AgNO3. The precipitation was dried at 373 K to afford zirconium hydroxide solid. Then the zirconium hydroxide was impregnated by H2SO4 (1 mol L−1) under vigorous stirring at room temperature for 4 h. After centrifuged, the precipitation was dried at 393 K overnight and calcined at 773 K for 4 h to afford the SO42−/ZrO2.
Characterization of catalysts
Fourier transform infrared (FT-IR) spectra were collected on a Bruker Tensor 27 FT-IR spectrometer in KBr media. Samples were thoroughly dried before measured.The X-ray powder diffraction (XRD) patterns were obtained using Rigaku D/Max 2500/PC powder diffractometer with Cu Kα radiation (λ = 0.15418 nm) at 40 kV and 200 mA in a scanning rate of 5° min−1.
The transmission electron microscopy (TEM) was conducted on a JEOLJEM-2000EX electron microscopy.
N2 physical adsorption/desorption measurement was carried out at liquid nitrogen temperature on an Autosorb-1 Quantachrome instrument. Samples were pre-degassed at 300 °C for about 10 h to remove water and other physical adsorbed species.
Temperature-programmed desorption of ammonia (NH3-TPD) was performed on a Micromeritics AutoChem II 2920 Instrument with a thermal conductivity detector (TCD). The catalyst was degassed at 673 K in an atmosphere of Ar.
The FT-IR spectra for pyridine adsorption of the catalyst were conducted on a Bruker Tensor 27 FT-IR spectrometer. Prior to pyridine adsorption, the catalyst was evacuated in vacuum at 523 K. After that, the sample was used for the background reference collected. Subsequently, pulses of pyridine were introduced in an in situ cell for adsorption. Then the catalyst was heated to 523 K and evacuated for 30 min. The spectra of the catalysts were collected after the reaction cell was cooled to room temperature.
Catalytic reactions
The catalytic reactions were performed in a 60 mL stainless steel autoclave equipped with a magnetic stirrer, a pressure gauge, and automatic temperature control apparatus. The reactor was connected to a hydrogen cylinder for reaction pressure. In a typical experiment, an aqueous solution of carbohydrates (3 mL, 1.00 mmol by monosaccharide), cyclohexane (6 mL), and catalysts were put into the reactor. After sealing and purging with H2 for 4 times to exclude air, the autoclave was heated to the desired temperature, and then H2 (4 MPa) was charged into the reactor. The magnetic agitator was set to 1200 rpm to start the reaction. After reaction, the autoclave was cooled. The aqueous phase and oil phase were sampled and analyzed respectively. After sampling ethanol was added to make the acid and Ru/SiO2-TM in one phase. The catalysts were centrifuged and washed with 50 mL ethanol for 3 times. After dried at 373 K under vacuum overnight, the catalyst was used for the next run.Products analysis
The products were identified by Agilent 6890N GC/5973MS as well as by comparison with the retention times to corresponding standards in GC and HPLC traces. The amounts of products were determined based on GC and HPLC data. Gas chromatography measurements were conducted on Agilent 6890A GC. HP-5 capillary column (30 m × 530 μm × 1.5 μm) was used for separation of reaction mixtures. 1,2,4,5-Tetramethylbenzene (TMB) was used as the internal standard. HPLC analysis was carried out using a Waters e2695 HPLC system equipped with a 2414 refractive index detector maintained at 303 K. The aqueous sample was separated using a Waters Sugar-Pak 1 column (6.5 mm × 30 cm) at 358 K, using ethylenediaminetetraacetic acid calcium disodium salt hydrate (EDTACa) aqueous solution (50 mg L−1) as the mobile phase at a flow rate of 0.55 mL min−1. The conversion and selectivity of main products were evaluated as below: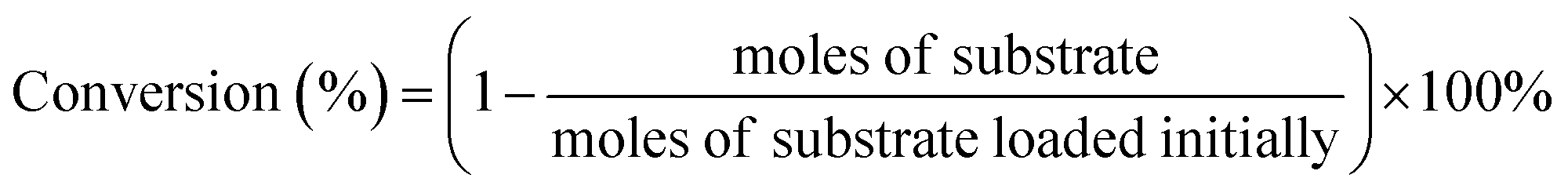
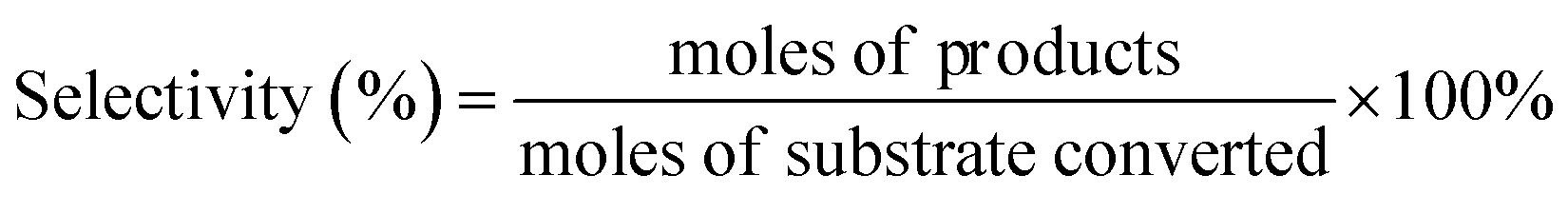
Results and discussion
We focused our attentions on the selection of different kinds of acid using glucose as the starting material. The HY, A-70, H2SO4, SO42−/ZrO2 and Nb2O5 were selected as acids. The result was exhibited in Fig. 1. The acids were first used in the conversion of glucose to HMF (Fig. 1a). All the acids except Nb2O5-FP gave poor selectivity to HMF while the Nb2O5-FP had 78% selectivity to HMF after 1 h. And the selectivity of HMF was 56% after 4 h with 27% conversion of glucose. The results showed that the Nb2O5-PF was an effective solid acid for the conversion of glucose to HMF as reported in the literature.59 When the acids were used in the conversion of glucose to THFDM, HY, A-70, H2SO4, SO42−/ZrO2 resulted poor selectivity of THFDM (no more than 20% Fig. 1b) in the experiment range. This was attributed to the poor selectivity of HMF from glucose when those acids were used as the catalyst (Fig. 1a). The selectivity of THFDM is positively correlated to the selectivity of HMF on the specific acid. The selectivity of THFDM increased to 31% with 35% glucose conversion when H3PO4-treated niobic acid (Nb2O5-P) was employed. Surprisingly, the selectivity of THFDM increased significantly to 64% when H3PO4-treated fresh niobic acid (Nb2O5-PF) was used, in spite of the conversion of glucose was only 9% after 1 h. When the reaction time was extended to 4 h, the selectivity of THFDM was 63% at 32% glucose conversion. The fresh niobic acid was prepared by dissolution of niobium oxide and then precipitated by aqueous ammonia. And the Nb2O5-FP was used in subsequence reactions.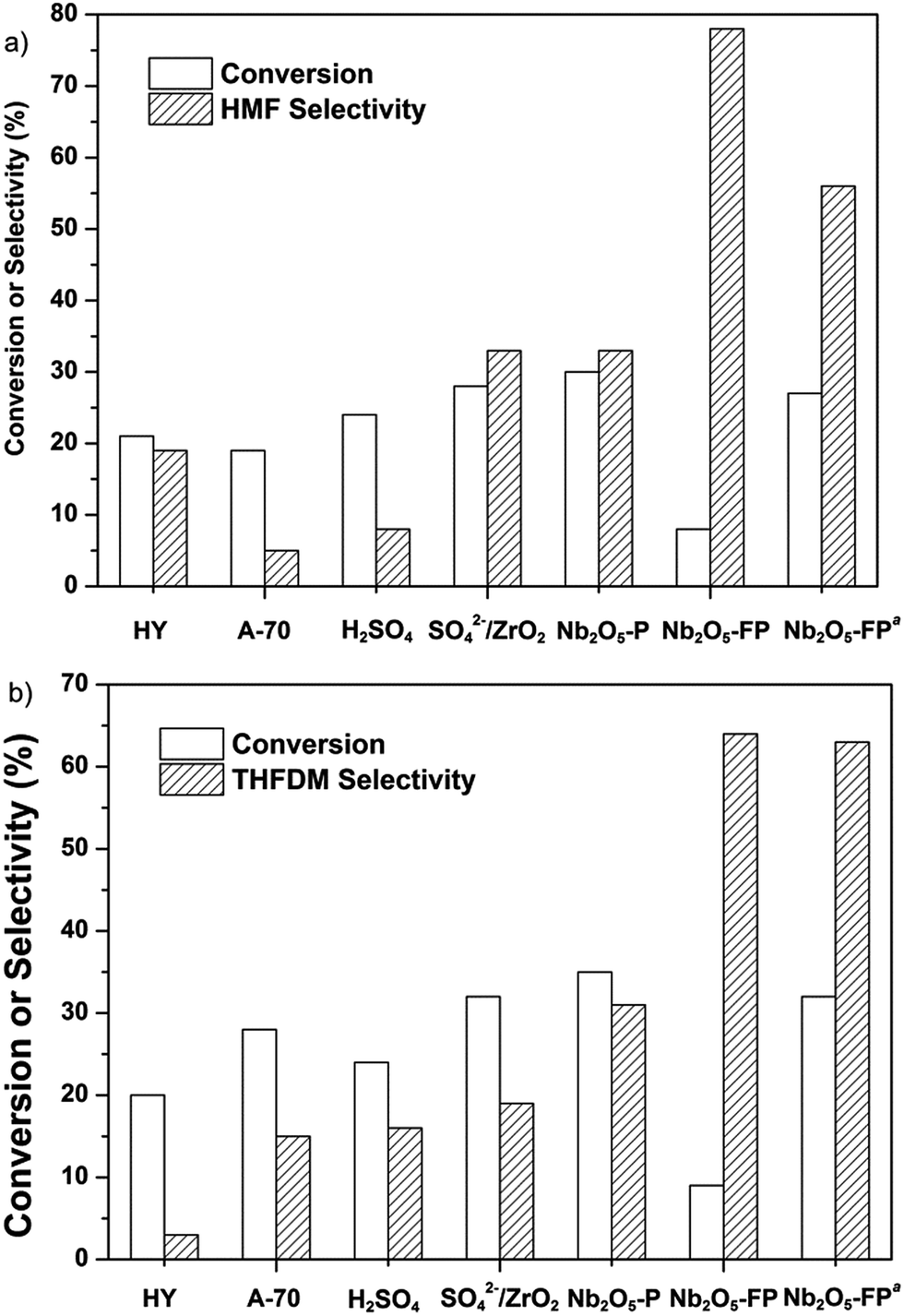 | ||
| Fig. 1 The conversion of glucose by (a) acids (b) Ru/SiO2-TM and acids. Reaction conditions: acids (40 mg), glucose aqueous solution (3 mL, glucose 1 mmol), cyclohexane (6 mL), H2 (4 MPa), 433 K, 1 h. Ru/SiO2-TM (30 mg) for (b). aReaction time 4 h. HY: HY-zeolite (Si/Al = 5), A-70: Amberlyst-70. | ||
The performance of Nb2O5-FP was further studied by carrying out the reaction in two cases. The case I was the conversion of glucose to HMF under H2 atmosphere by just using the Nb2O5-FP as the catalyst. And the combination of Nb2O5-FP and Ru/SiO2-TM was used for the other case named case II. The conversion–time profiles of both cases were almost the same as shown in Fig. 2. This should be ascribed to the hydrophobicity of the Ru/SiO2-TM. The Ru/SiO2-TM was kept in the oil phase where there had no glucose. Thus, the conversion of glucose was catalyzed by Nb2O5-FP in both cases which led to the same conversion of glucose at the same reaction time.
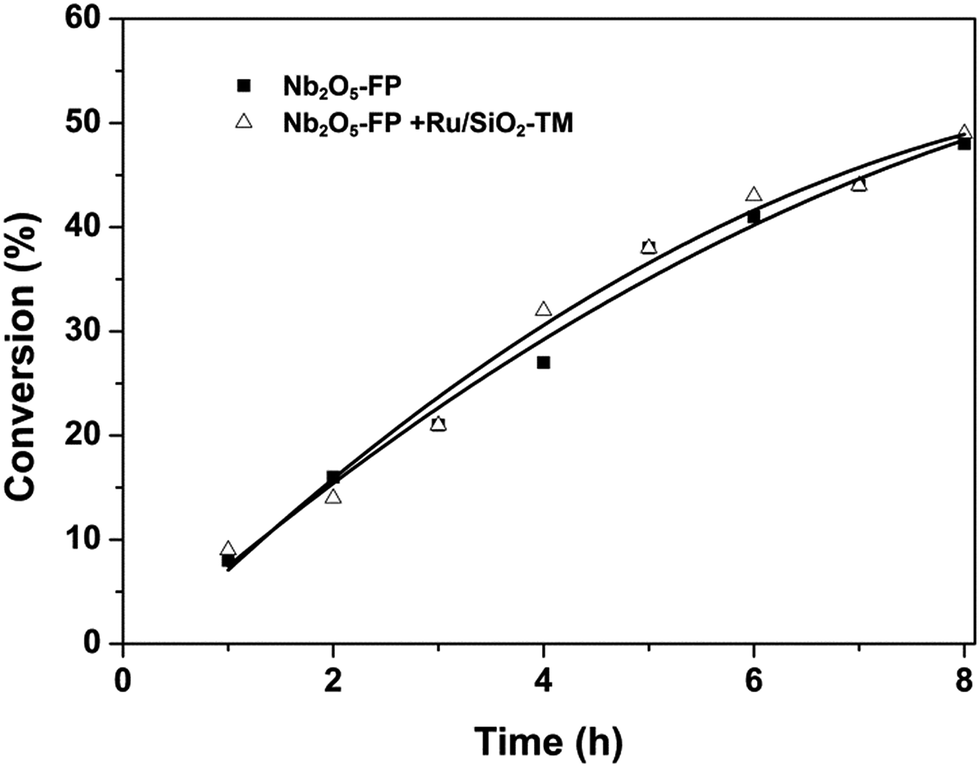 | ||
| Fig. 2 Conversion–time profiles of glucose dehydration to HMF and glucose conversion to THFDM. Reaction conditions: Nb2O5-FP (40 mg), glucose aqueous solution (3 mL, glucose 1 mmol), cyclohexane (6 mL), H2 (4 MPa), 433 K. Δ: Ru/SiO2-TM (30 mg). | ||
The products selectivity versus glucose conversion was shown in Fig. 3. Very high selectivity of HMF was obtained at the initial stage for the case I (Fig. 3b). Meanwhile, 24% selectivity of fructose was gained in this case. It showed that the Nb2O5-PF was able to convert glucose to fructose. With the time prolonged the selectivity of fructose decreased to 3%. This was caused by the conversion of fructose to HMF. However, the selectivity of HMF decreased sharply from 76% to 27%, revealing that the HMF was not stable and was easy to degrade under this condition. This was in accordance with the results we obtained previously.7 The case II was the conversion of glucose to THFDM by the combination of Nb2O5-FP and Ru/SiO2-TM. The results at the initial stage were analogous to that of the case I (Fig. 3a). High selectivity of THFDM with quite a number of fructose was obtained. To our delight, the selectivity of THFDM kept around at 60% with no obviously decrease as the time prolonged (Fig. 3a). This phenomenon was quite different from the selectivity of HMF in the case I. The reason was that the THFDM was much more stable than the HMF. The HMF was converted to THFDM once generated in the case II. As a result, the degradation of HMF was inhibited and the selectivity of THFDM was well kept.
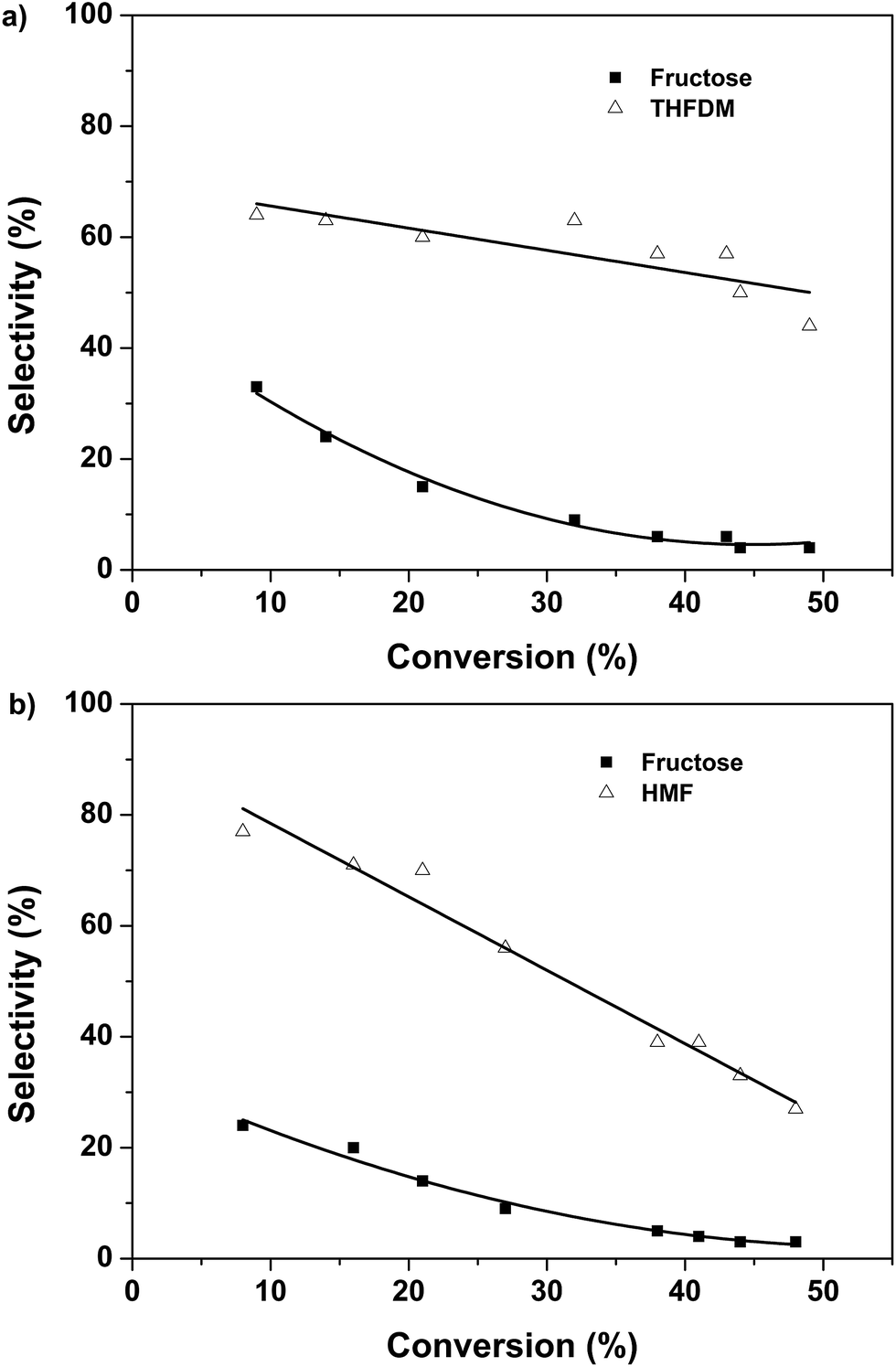 | ||
| Fig. 3 Products selectivity versus glucose conversion over (a) Nb2O5-FP and Ru/SiO2-TM, (b) Nb2O5-FP. Reaction conditions: Nb2O5-FP (40 mg), glucose aqueous solution (3 mL, glucose 1 mmol), cyclohexane (6 mL), H2 (4 MPa), 433 K. Ru/SiO2-TM (30 mg) for (a). | ||
The effect of temperature played an important role in the conversion of glucose and the results were shown in Fig. 4. The conversion of glucose increased as the temperature increased from 413 K to 453 K as expected. The selectivity of THFDM kept around 60% when the temperature was not higher than 433 K. However, once the temperature exceeded 433 K the selectivity of THFDM decreased drastically. The best result was obtained at a temperature of 433 K. And the reaction was repeated at this temperature. Similar conversion of glucose and selectivity of THFDM was acquired showing the repeatability of the catalytic system.
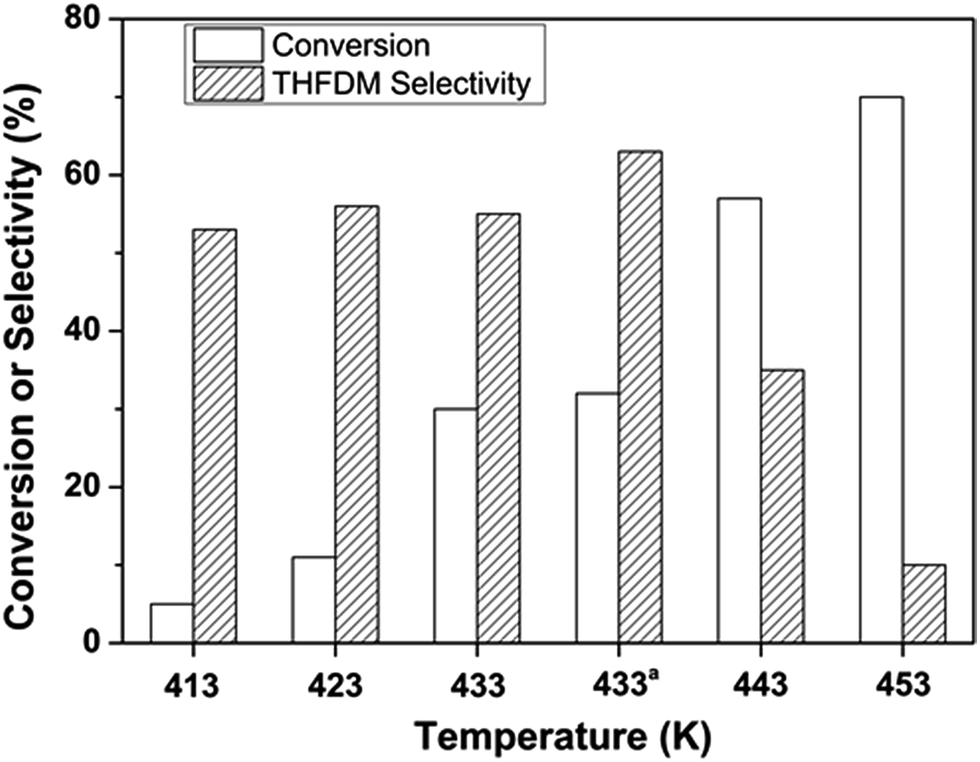 | ||
| Fig. 4 Effect of temperature on the conversion of glucose to THFDM. aRepeated test for the temperature at 433 K. Reaction conditions: Nb2O5-FP (40 mg), Ru/SiO2-TM (30 mg), glucose aqueous solution (3 mL, glucose 1 mmol), cyclohexane (6 mL), H2 (4 MPa), 433 K, 4 h. | ||
Fig. 5 showed the recyclability of the catalytic system. After the analysis, ethanol was added into the system to obtain one liquid phase. The catalyst was then centrifuged and fully washed with ethanol after each cycle. After dried at 373 K under vacuum, the catalyst was used for the next run. It could be seen from Fig. 5 that there had a small gradual decrease in conversion value. This should be ascribed to the incomplete recovery of catalyst after each reaction. The selectivity of THFDM kept around 60% in all the four consecutive runs. This illustrated the catalyst was stable under the reaction conditions.
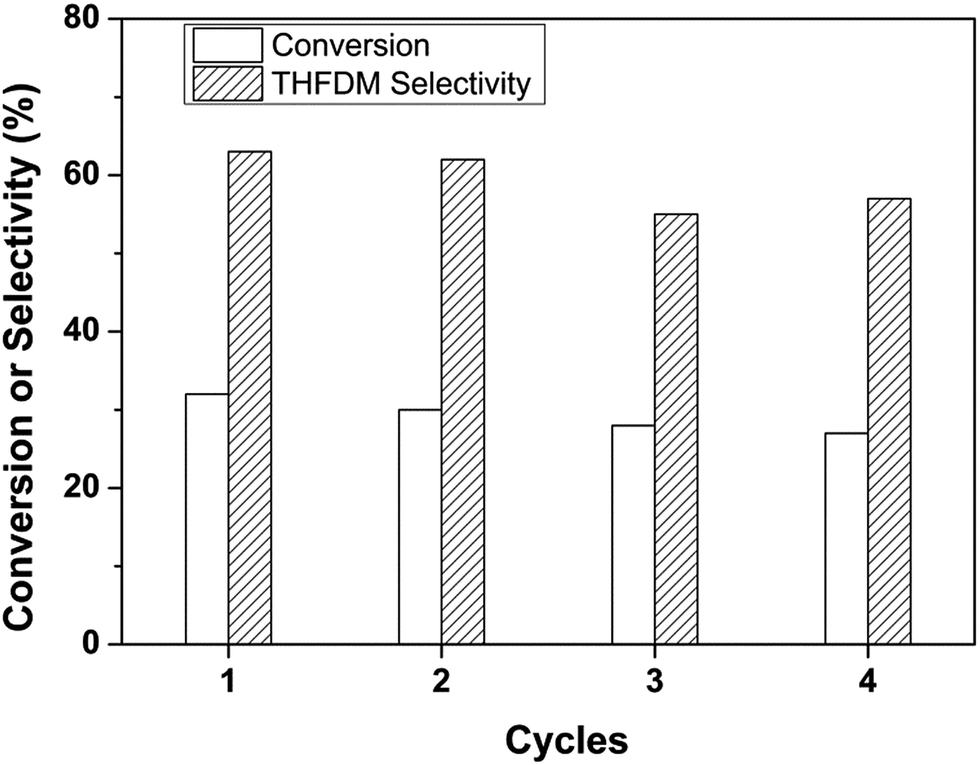 | ||
| Fig. 5 Cycle experiments of the conversion of glucose to THFDM. Reaction conditions: Nb2O5-FP (40 mg), Ru/SiO2-TM (30 mg), glucose aqueous solution (3 mL, glucose 1 mmol), cyclohexane (6 mL), H2 (4 MPa), 433 K, 4 h. | ||
We attempted to obtain THFDM directly from carbohydrates, containing glucose or fructose units, by the combination of Nb2O5-FP and Ru/SiO2-TM. The fructose was almost completely converted under the reaction condition (Table 1, entry 2). However, the selectivity of THFDM was only 41% showed that the conditions adopted here were not appropriate for the conversion of fructose. As a result, the inulin, a polymer consisting of chain-terminating glucosyl moieties and a repetitive fructosyl moiety, gave 33% selectivity of THFDM although with 96% of conversion (Table 1, entry 3). The conversion of sucrose was higher than that of the other two kinds of disaccharides maltose and cellobiose (Table 1, entries 4–6). The reason was that the sucrose was a disaccharide comprised of glucose and fructose while the maltose and cellobiose were comprised of glucose only. The selectivity of THFDM for the three disaccharides was around 40%. The starch, the polysaccharide of glucose, was also able to be converted to THFDM in one-step by this catalytic system. A conversion of 26% with 34% selectivity of THFDM was obtained under the same reaction conditions (Table 1, entry 7).
| Entry | Substrate | Conv.b (%) | Selectivity (%) | |||
|---|---|---|---|---|---|---|
| THFDM | Fructose | Othersc | U. D.d | |||
| a Reaction conditions: Nb2O5-FP (40 mg), Ru/SiO2-TM (30 mg), carbohydrate aqueous solution (3 mL, 1 mmol by monosaccharide), cyclohexane (6 mL), H2 (4 MPa), 433 K, 4 h.b Conversion was calculated based on the amounts of monosaccharide.c Others referred to furfuryl alcohol, methyl furfuryl alcohol, γ-valerolactone, 1,2-pentanediol, 1,2-hexanediol, 1,2,5-hexanetriol, 1,2,6-hexanetriol.d Unidentified products. | ||||||
| 1 | Glucose | 32 | 63 | 9 | 15 | 13 |
| 2 | Fructose | 98 | 41 | 1 | 27 | 31 |
| 3 | Inulin | 96 | 33 | 4 | 31 | 32 |
| 4 | Sucrose | 61 | 42 | 4 | 25 | 29 |
| 5 | Maltose | 37 | 42 | 7 | 22 | 29 |
| 6 | Cellobiose | 33 | 32 | 8 | 19 | 41 |
| 7 | Starch | 26 | 34 | 11 | 19 | 36 |
The physical properties of the Nb2O5-FP were characterized by BET and TEM. The results were shown in Fig. 6 and S3.† The BET surface of as-synthesized Nb2O5-FP was 62 m2 g−1. According to the IUPAC classifications, the sample had the characteristics of typical type IV physisorption isotherms, indicating the presence of mesoporous structure. There had a significantly increase in adsorbed volume at relatively high pressure which was the characteristic of the H3 type hysteresis loop indicating the existence of slit-shaped pores with a large pore size which was in accordance with the results of TEM. The distribution of pore size calculated by the non-local density functional theory (NLDFT) model showed there had very broad pore size distribution in the mesoporous range (2–50 nm) and macropore were also presented in the Nb2O5-FP.
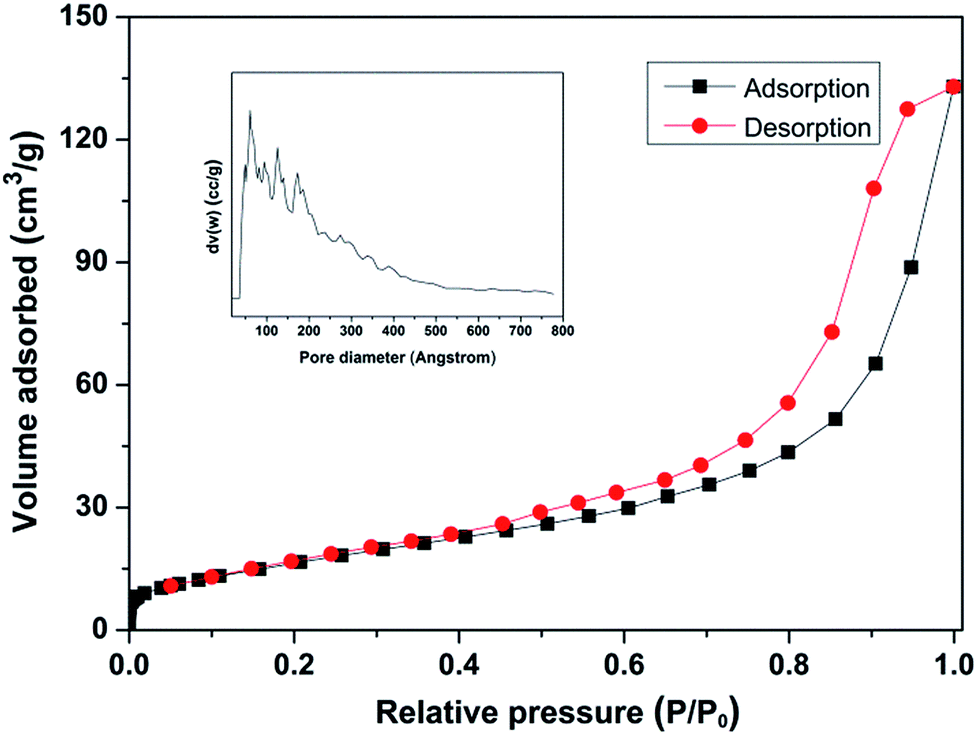 | ||
| Fig. 6 N2 adsorption/desorption isotherms of Nb2O5-FP. | ||
The acid property of the Nb2O5-FP was characterized by temperature-programmed desorption of ammonia (NH3-TPD) (Fig. 7) as well as Fourier transform infrared (FT-IR) spectroscopy of adsorbed pyridine (Fig. 8). There were two desorption peaks appeared in the NH3-TPD profile. The weak desorption peak at low temperature (400–500 K) should be ascribed to the weakly held ammonia, probably the hydrogen-bonded ammonia according to the literature.60 The strong broad desorption peak between 600 and 800 K corresponded to large amount of the medium and the strong acid sites in the sample. The FT-IR spectroscopy of adsorbed pyridine revealed the type of the acid sites.61,62 The medium peak at 1544 cm−1 was attributed to pyridine protonated on Brönsted sites, and the weak peak at 1450 cm−1 indicated the presence of Lewis acid sites. The peak intensity at 1544 cm−1 and 1450 cm−1 elucidated that the sample had a very high Brönsted/Lewis ratio. And the high Brönsted/Lewis ratio was favourable for high selectivity of HMF from glucose in the aqueous phase.63
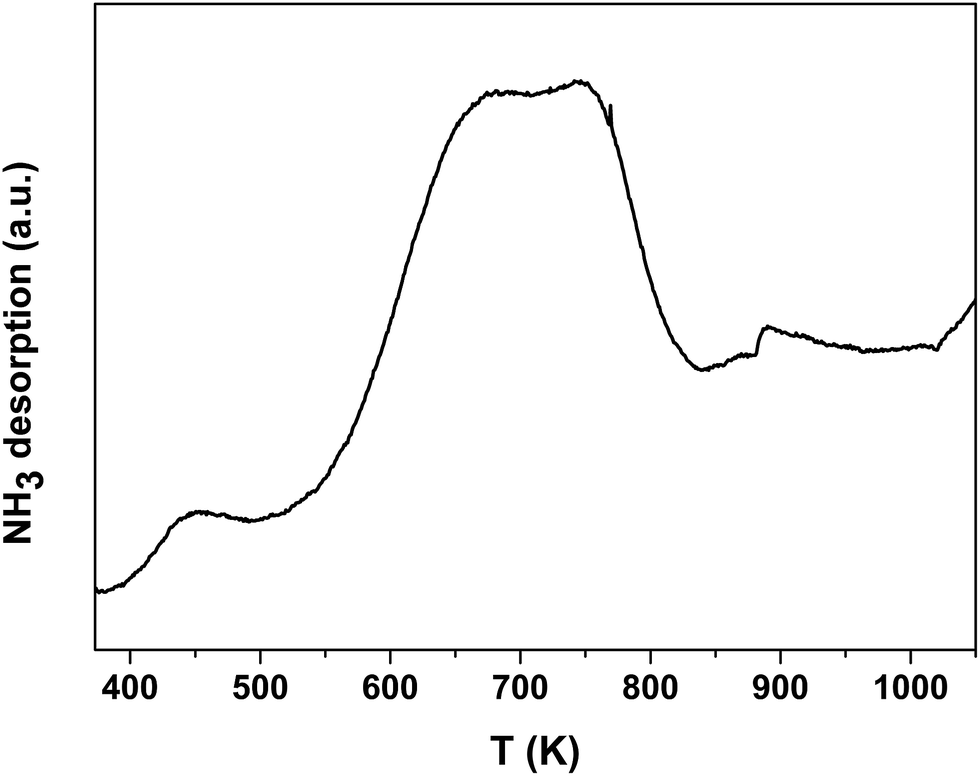 | ||
| Fig. 7 NH3-TPD profiles of Nb2O5-FP. | ||
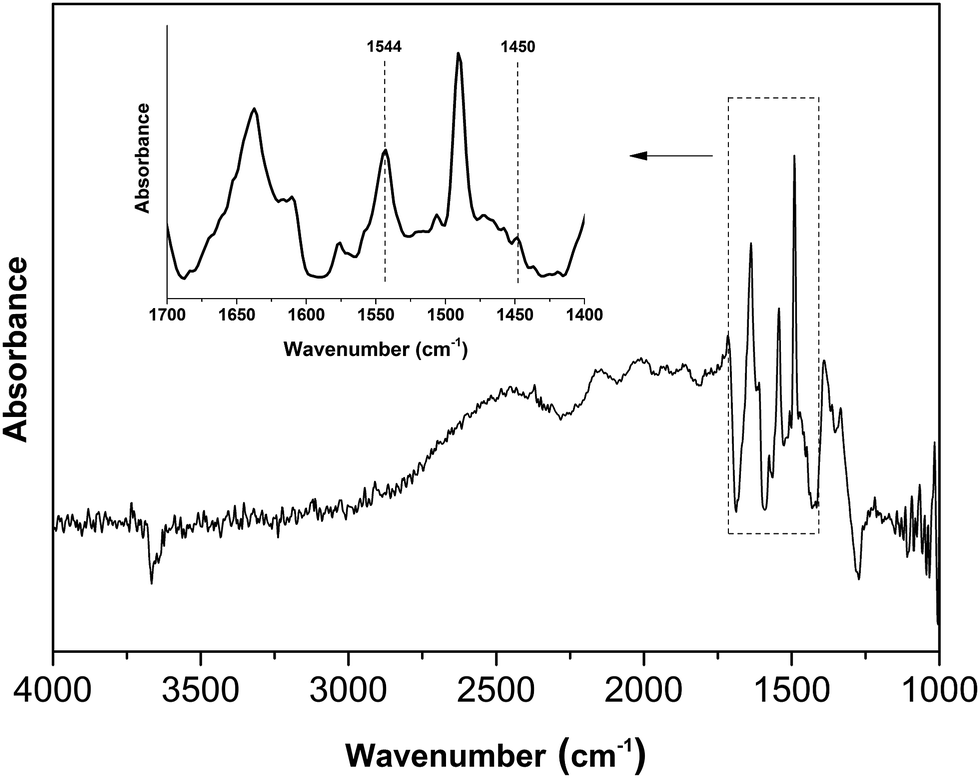 | ||
| Fig. 8 FT-IR spectrum of Nb2O5-FP after pyridine adsorption and subsequent evacuation, the sample was treated in vacuum to remove adsorbed water. | ||
Conclusions
In conclusion, the direct conversion of a wide variety of carbohydrates to THFDM was achieved by the combination of Nb2O5-FP and Ru/SiO2-TM in the water/cyclohexane biphasic system. The as-synthesized Nb2O5-FP, which was proved to have large amount of medium and strong acid sites with high Brönsted/Lewis ratio, was essential and effective for keeping high THFDM selectivity when glucose was used as substrate. This work provides a common strategy for the direct production of valuable chemicals from carbohydrates.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21272109 and 21371089) and the Intercollegiate Key Scientific Research Projects of Henan Province (16A150017). The authors express their gratitude for the help and guidance from Researcher Jie Xu at Dalian Institute of Chemical Physics, Chinese Academy of Sciences.References
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- D. Chen, F. Liang, D. Feng, F. Du, G. Zhao, H. Liu and M. Xian, Catal. Commun., 2016, 84, 159–162 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- J. Ma, W. Yu, M. Wang, X. Jia, F. Lu and J. Xu, Chin. J. Catal., 2013, 34, 492–507 CrossRef CAS.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- Y. Yang, Z. Du, J. Ma, F. Lu, J. Zhang and J. Xu, ChemSusChem, 2014, 7, 1352–1356 CrossRef CAS PubMed.
- Y.-R. Zhang, N. Li, M.-F. Li and Y.-M. Fan, RSC Adv., 2016, 6, 21347–21351 RSC.
- Y. Zhang, Y. Chen, Y. Shen, Y. Yan, J. Pan, W. Shi and L. Yu, ChemPlusChem, 2016, 81, 108–118 CrossRef CAS.
- P. Zhou and Z. Zhang, Catal. Sci. Technol., 2016, 6, 3694–3712 CAS.
- X. Hu, R. J. M. Westerhof, D. Dong, L. Wu and C.-Z. Li, ACS Sustainable Chem. Eng., 2014, 2, 2562–2575 CrossRef CAS.
- J. P. Lange, E. van der Heide, J. van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS PubMed.
- J. Zhang, J. Zhuang, L. Lin, S. Liu and Z. Zhang, Biomass Bioenergy, 2012, 39, 73–77 CrossRef CAS.
- D. Ding, J. Wang, J. Xi, X. Liu, G. Lu and Y. Wang, Green Chem., 2014, 16, 3846–3853 RSC.
- R. Weingarten, Y. T. Kim, G. A. Tompsett, A. Fernández, K. S. Han, E. W. Hagaman, W. C. Conner, J. A. Dumesic and G. W. Huber, J. Catal., 2013, 304, 123–134 CrossRef CAS.
- K. Fulajtárova, T. Soták, M. Hronec, I. Vávra, E. Dobročka and M. Omastová, Appl. Catal., A, 2015, 502, 78–85 CrossRef.
- A. B. Merlo, V. Vetere, J. F. Ruggera and M. L. Casella, Catal. Commun., 2009, 10, 1665–1669 CrossRef CAS.
- Q. Yuan, D. Zhang, L. V. Haandel, F. Ye, T. Xue, E. J. M. Hensen and Y. Guan, J. Mol. Catal. A: Chem., 2015, 406, 58–64 CrossRef CAS.
- M. Manikandan, A. K. Venugopal, A. S. Nagpure, S. Chilukuri and T. Raja, RSC Adv., 2016, 6, 3888–3898 RSC.
- F. Wang and Z. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 942–947 CrossRef CAS.
- Y. Nakagawa, H. Nakazawa, H. Watanabe and K. Tomishige, ChemCatChem, 2012, 4, 1791–1797 CrossRef CAS.
- Y. Yang, J. Ma, X. Jia, Z. Du, Y. Duan and J. Xu, RSC Adv., 2016, 6, 51221–51228 RSC.
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef CAS PubMed.
- S. Siankevich, G. Savoglidis, Z. Fei, G. Laurenczy, D. T. L. Alexander, N. Yan and P. J. Dyson, J. Catal., 2014, 315, 67–74 CrossRef CAS.
- Y. Liu, L. Zhu, J. Tang, M. Liu, R. Cheng and C. Hu, ChemSusChem, 2014, 7, 3541–3547 CrossRef CAS PubMed.
- J. Nie, J. Xie and H. Liu, J. Catal., 2013, 301, 83–91 CrossRef CAS.
- J. Chen, Y. Guo, J. Chen, L. Song and L. Chen, ChemCatChem, 2014, 6, 3174–3181 CrossRef CAS.
- R. Liu, J. Chen, L. Chen, Y. Guo and J. Zhong, ChemPlusChem, 2014, 79, 1448–1454 CrossRef CAS.
- N. Mittal, G. M. Nisola, L. B. Malihan, J. G. Seo, H. Kim, S.-P. Lee and W.-J. Chung, RSC Adv., 2016, 6, 25678–25688 RSC.
- M. Chatterjee, T. Ishizaka, A. Chatterjee and H. Kawanami, Green Chem., 2017, 19, 1315–1326 RSC.
- G. Li, Z. Sun, Y. Yan, Y. Zhang and Y. Tang, ChemSusChem, 2017, 10, 494–498 CrossRef CAS PubMed.
- Y. Yang, Z. Du, Y. Huang, F. Lu, F. Wang, J. Gao and J. Xu, Green Chem., 2013, 15, 1932–1940 RSC.
- G.-S. Zhang, M.-M. Zhu, Q. Zhang, Y.-M. Liu, H.-Y. He and Y. Cao, Green Chem., 2015, 18, 2155–2164 RSC.
- L. Deng, Y. Zhao, J. Li, Y. Fu, B. Liao and Q. X. Guo, ChemSusChem, 2010, 3, 1172–1175 CrossRef CAS PubMed.
- R. V. Maligal-Ganesh, C. Xiao, T. W. Goh, L.-L. Wang, J. Gustafson, Y. Pei, Z. Qi, D. D. Johnson, S. Zhang, F. Tao and W. Huang, ACS Catal., 2016, 6, 1754–1763 CrossRef CAS.
- V. V. Kumar, G. Naresh, M. Sudhakar, C. Anjaneyulu, S. K. Bhargava, J. Tardio, V. K. Reddy, A. H. Padmasri and A. Venugopal, RSC Adv., 2016, 6, 9872–9879 RSC.
- J. Chen, R. Liu, Y. Guo, L. Chen and H. Gao, ACS Catal., 2015, 5, 722–733 CrossRef CAS.
- Y. Guo and J. Chen, RSC Adv., 2016, 6, 101968–101973 RSC.
- K. Chen, M. Tamura, Z. Yuan, Y. Nakagawa and K. Tomishige, ChemSusChem, 2013, 6, 613–621 CrossRef CAS PubMed.
- S. Liu, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2014, 2, 1819–1827 CrossRef CAS.
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, Green Chem., 2016, 18, 165–175 RSC.
- Y. Weng, S. Qiu, C. Wang, L. Chen, Z. Yuan, M. Ding, Q. Zhang, L. Ma and T. Wang, Fuel, 2016, 170, 77–83 CrossRef CAS.
- Y. Liu, L. Chen, T. Wang, Q. Zhang, C. Wang, J. Yan and L. Ma, ACS Sustainable Chem. Eng., 2015, 3, 1745–1755 CrossRef CAS.
- Q. Xia, Z. Chen, Y. Shao, X. Gong, H. Wang, X. Liu, S. F. Parker, X. Han, S. Yang and Y. Wang, Nat. Commun., 2016, 7, 11162–11171 CrossRef PubMed.
- W. Yang and A. Sen, ChemSusChem, 2011, 4, 349–352 CrossRef CAS PubMed.
- B. Op de Beeck, M. Dusselier, J. Geboers, J. Holsbeek, E. Morré, S. Oswald, L. Giebeler and B. F. Sels, Energy Environ. Sci., 2015, 8, 230–240 CAS.
- B. Liu and Z. Zhang, ChemSusChem, 2016, 9, 2015–2036 CrossRef CAS PubMed.
- T. Buntara, S. Noel, P. H. Phua, I. Melian-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- B. Xiao, M. Zheng, X. Li, J. Pang, R. Sun, H. Wang, X. Pang, A. Wang, X. Wang and T. Zhang, Green Chem., 2016, 18, 2175–2184 RSC.
- W. Xu, H. Wang, X. Liu, J. Ren, Y. Wang and G. Lu, Chem. Commun., 2011, 47, 3924–3926 RSC.
- S. Koso, I. Furikado, A. Shimao, T. Miyazawa, K. Kunimori and K. Tomishige, Chem. Commun., 2009, 15, 2035–2037 RSC.
- S. P. Burt, K. J. Barnett, D. J. McClelland, P. Wolf, J. A. Dumesic, G. W. Huber and I. Hermans, Green Chem., 2017, 19, 1390–1398 RSC.
- Y. Nakagawa, M. Tamura and K. Tomishige, Catal. Surv. Asia, 2015, 19, 249–256 CrossRef CAS.
- S. Liu, Y. Amada, M. Tamura, Y. Nakagawa and K. Tomishige, Green Chem., 2014, 16, 617–626 RSC.
- Y. Nakagawa and K. Tomishige, Catal. Today, 2012, 195, 136–143 CrossRef CAS.
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, ChemSusChem, 2015, 8, 628–635 CrossRef CAS PubMed.
- C. Moreaua, M. N. Belgacemb and A. Gandini, Top. Catal., 2004, 35, 11–30 CrossRef.
- S. Koso, H. Watanabe, K. Okumura, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2012, 111–112, 27–37 CrossRef CAS.
- K. Nakajima, Y. Baba, R. Noma, M. Kitano, J. N. Kondo, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2011, 133, 4224–4227 CrossRef CAS PubMed.
- T. Miyamoto, N. Katada, J.-H. Kim and M. Niwa, J. Phys. Chem. B, 1998, 102, 6738–6745 CrossRef CAS.
- M. C. Kung and H. H. Kung, Catal. Rev., 1985, 27, 425–460 Search PubMed.
- B. A. Morrow and I. A. Cody, J. Phys. Chem., 1976, 80, 1995–1998 CrossRef CAS.
- V. V. Ordomsky, V. L. Sushkevich, J. C. Schouten, J. van der Schaaf and T. A. Nijhuis, J. Catal., 2013, 300, 37–46 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03939e |
| This journal is © The Royal Society of Chemistry 2017 |