DOI:
10.1039/C7RA04443G
(Paper)
RSC Adv., 2017,
7, 26478-26486
Three Pt(II) complexes based on thiosemicarbazone: synthesis, HSA interaction, cytotoxicity, apoptosis and cell cycle arrest†
Received
20th April 2017
, Accepted 8th May 2017
First published on 17th May 2017
Abstract
Three thiosemicarbazone-based platinum(II) complexes [Pt(MH-TSC)Cl] (1), [Pt(ME-TSC)Cl] (2) and [Pt(NH-TSC)2]Cl (3) (MH-TSC = (E)-2-(1-(pyridin-2-yl)ethylidene)hydrazinecarbothioamide, ME-TSC = (E)-N-ethyl-2-(1-(pyridin-2-yl)ethylidene) hydrazinecarbothioamide, NH-TSC = (Z)-2-(amino(pyridin-2-yl)methylene)hydrazinecarbothioamide) were synthesized and structurally characterized. X-ray analyses revealed that 1 and 2 possessed similar a neutral mononuclear unit in which one tridentate TSC ligand and one leaving group (Cl−) coordinated to Pt(II) ion, while 3 was cationic and formed by two NH-TSC ligands surrounding one Pt atom in a meridional arrangement. UV-visible and fluorescence spectra of human serum albumin (HSA) with the complexes displayed that the quenching mechanism of HSA by 1–3 might be a static binding mode. Moreover, synchronous fluorescence experiments proved that 1–3 affected the microenvironment of tryptophan residues of HSA. In addition, the antiproliferative activities against MCF-7 (human breast cancer lines), HepG-2 (human liver hepatocellular carcinoma cell line), NCI-H460 (non-small cell lung cancer lines) and HeLa (human epithelial cervical cancer cell line) were screened for 1–3. Inspiringly, their cytotoxic activity (IC50 = 1.7–9.6 μM) appeared much better than that of cisplatin (IC50 = 5.2–13.5 μM) against different cell lines, respectively. Among them, complex 3 exhibited the strongest inhibition on the viability of all tested cell lines with IC50 values of 1.7–2.2 μM. Inductively-coupled plasma mass spectrometry (ICP-MS) showed that 3 accumulated rapidly in cells and reached intracellular levels of up to 10-fold higher than those determined for 1 and 2. Furthermore, fluorescence microscopic observation and flow cytometric analysis revealed that 1–3 could effectively induce apoptosis of HeLa cells, which were arrested in the S phase after treatment with 1 (30.31%) and 3 (46.96%), and in G2 phase with 2 (20.2%). All the results mentioned above suggest that complexes 1–3 might be efficient antitumor agents.
Introduction
Cisplatin (cis-diamminedichloroplatinum(II)), capable of efficiently binding to and damaging DNA,1 has been widely used in the clinical treatment of various cancers since 1978. It functions in particular with respect to the therapy of head and neck, testicular, small lung cell and ovarian cancers.2–5 Unfortunately, cisplatin exhibits some drawbacks including severe side effects,6 acquired drug resistance7 and low water-solubility,8 which seriously limit its clinical applications. To combat those limitations, researchers have been exploring new Pt-compounds to improve antitumor efficiency.
Optimizing ligand structure is a usual strategy to design and construct new platinum antitumor agents. Therefore, a large number of so-called non-classical platinum complexes, which are structurally different from classical Pt drugs, have recently attracted researchers' attention.9 These complexes usually have monofunctional, cationic or high-valence Pt(IV) features, which might lead to a distinctly different manner of antitumor activity compared to current cisplatin-like drugs.10
Thiosemicarbazone (TSC) includes thousands of compounds, and many of them may possess various biological activities. It was reported that 2-formylpyridine TSC could have anti-leukemic activity in mice.11 Additionally, 3-AP (3-aminopyridine-2-carboxaldehyde thiosemicarbazone) is an inhibitor of ribonucleotide reductase (RNR) that catalyzes the reductive conversion of RNA building blocks into DNA building blocks for DNA replication and repair.12,13 So, 3-AP exhibites efficient antitumor activities and have undergone phase II clinical trials. Meanwhile, combination of 3-AP and cisplatin could enhance the clinical efficacy.14,15 Moreover, Dp44mT is a terminally dimethylated derivative of 3-AP, and it was shown to be 100-fold more potent than 3-AP with respect to cytotoxicity to tumor cells.16 Not only as antitumor agents, but also TSC could be applied in molecular imaging, metal-sensing, anti-viral, anti-microbial, and anti-inflammatory.17–19
Previously, our group synthesized a tetranuclear TSC-based Pt(II) complex in the laboratory.20 It showed intense nuclease activity that could degrade DNA completely. However, this complex was little cytotoxic to tumor cells, probably because of its low solubility and difficulty in the uptake by cells. In this work, we designed and synthesized three mononuclear complexes 1–3 based on ligands MH-TSC, ME-TSC, and NH-TSC, respectively, attempting to improve solubility and cytotoxicity, and explore the structure-activity relationship. The HSA-binding, cytotoxicity, and apoptotic induction and celluar uptake of the complexes were investigated.
Experimental
Materials and physical measurements
All chemicals were purchased from commercial sources and used without further purification. Tris–HCl buffer solution was prepared using triple-distilled water. Reactions were typically carried out in air, all chemical solvents were dried and distilled by standard methods before use (cautious: MeOH must dry over Mg and I2). IR spectra were recorded on a Nicolet 380 spectrometer as KBr pellets in the range 4000–375 cm−1 with the OMNIC software. NMR measurements were recorded on a Bruker AVANCE III 400 spectrometer with tetramethylsilane as the internal standard. Electrospray ionization mass spectrometry (ESI-MS) was analyzed using an Agilent MS instrument with an ion spray source using electrospray ionization in positive-ion mode. Single crystal data were collected on a Rigaku Saturn X-ray CCD diffractometer using MoKα radiation. UV spectra were measured using a JASCO V-770 spectrophotometer. Fluorescence spectral data were obtained using a Shimadzu RF-5301 fluorescence spectrophotometer at the approved temperature. 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT), Crystal Violet, Hoechst 33342, Propidium Iodide (PI), and HSA were purchased from Sigma-Aldrich Corporation. DMEM was obtained from Gbico and Fetal Bovine Serum (FBS) was from Hyclone. Annexin V-FITC/PI Apoptosis Detection Kit was purchased from MBL. Human cervical cancer cell lines (HeLa), human breast cancer lines (MCF-7), human hepatoma cell lines (HepG-2) and non-small cell lung cancer lines (NCI-H460) were obtained from American Type Culture Collection (Rockville, MD, USA). All the cells were cultured with DMEM which contained 10% FBS, 100 μg mL−1 penicillin and 100 μg mL−1 streptomycin in a humidified atmosphere including 5% CO2 at 37 °C.
Synthesis of MH-TSC and ME-TSC
The synthetic route of all ligands are based on literatures reported previously:21–23 2-acetylpyridine (6.65 g, 0.054 mol) was dissolved in a solution of sodium in MeOH which had been dried over CaSO4 under stirring for 0.5 h. The thiosemicarbazide (5 g, 0.054 mol) or N(4)-ethylthiosemicarbazide (6.43 g, 0.054 mol) was added in small portions to the above solution, then the mixture was refluxed for another 4 h. The solution was cooled to room temperature and filtered, after that, the yellowish residue was dissolved and recrystallized from methanol. MH-TSC: yield: 9.26 g (ca. 79.5%). IR, 1H and 13C NMR analysis data of MH-TSC were listed in ESI.† 1H NMR (DMSO-d6, δ/ppm): 10.30 (s, 1H), 8.57 (d, 1H), 8.41 (d, 2H), 8.17–8.06 (m, 1H), 7.78 (td, 1.5 Hz, 1H), 7.37 (dd, 1H), 2.38 (s, 3H). 13C NMR (DMSO-d6, δ/ppm): 179.07 (s), 154.62 (s), 148.37 (s), 148.21 (s), 136.34 (s), 123.90 (s), 120.84 (s), 12.08 (s). ME-TSC: yield: 10.73 g (ca. 82%). 1H NMR (DMSO-d6, δ/ppm): 10.27 (s, 1H), 8.70 (t, 1H), 8.59 (ddd, 1H), 8.42 (d, 1H), 7.83 (tt, 1H), 7.40 (ddd, 1H), 3.64 (m, 2H), 2.40 (s, 3H), 1.17 (t, 3H). 13C NMR (DMSO-d6, δ/ppm): 177.60 (s), 154.68 (s), 148.43 (s), 147.91 (s), 136.28 (s), 123.85 (s), 120.75 (s), 38.51 (s), 14.43 (s), 12.09 (s).
Synthesis of NH-TSC
This compound was synthesized in a similar way to that of MH-TSC, using 2-cyanopyridine (5.63 g, 0.054 mol) instead of 2-acetylpyridine (6.65 g, 0.054 mol). Yield: 6.38 g (ca. 60%). 1H NMR (d6-DMSO, δ/ppm): 10.06 (s, 1H), 8.55 (d, 1H), 8.44 (d, 1H), 7.95 (s, 1H), 7.86–7.75 (m, 2H), 7.43 (dd, 1H), 6.92 (s, 2H). 13C NMR (d6-DMSO, δ/ppm): 176.64 (s), 150.15 (s), 147.85 (s), 141.86–140.20 (m), 136.64 (s), 124.48 (s), 121.10 (s).
Synthesis of complexes
Synthesis of [Pt(MH-TSC)Cl] (1). A solution of K2PtCl4 (0.083 g, 0.2 mmol) in H2O (18 mL) was added into a white solution of MH-TSC (0.039 g, 0.2 mmol) in MeOH (12 mL) under stirring for 3 h. The yellow reaction mixture was filtered and left to stand at room temperature for slow evaporation, and the bolarious crystals suitable for X-ray analysis were collected after several days. Yield: 50 mg (ca. 41% based on Pt). Elemental analysis (%): calc. for 1: C, 22.65; H, 2.12; N, 13.21. Found: C, 22.64; H, 2.10; N, 13.27. Selected IR data (KBr, cm−1): 3423 (m), 3322 (s), 3184.1 (w), 1600.1 (s), 1500.2 (s), 1455 (vs), 1349.7 (w), 1313.2 (w), 1173.2 (m), 1126.6 (w), 1007.4 (w), 766.6 (m), 723.5 (w).
Synthesis of [Pt(ME-TSC)Cl] (2). Complex 2 was prepared according to the similar procedure of 1, using ME-TSC (0.044 g, 0.2 mmol) instead of MH-TSC (0.039 g, 0.2 mmol) to the reaction mixture. Yield: 51 mg (ca. 40% based on Pt). Elemental analysis (%): calc. for 2: C, 26.56; H, 2.88; N, 12.39. Found: C, 26.64; H, 2.85; N, 12.23. Selected IR data (KBr, cm−1): 3343.9 (s), 2973 (w), 1556.6 (w), 1513 (s), 1455.1 (s), 1373.9 (w), 1328.7 (w), 1170.5 (m), 1051 (w), 767.4 (m).
Synthesis of [Pt(NH-TSC)2]Cl (3). Complex 3 was prepared according to the similar procedure of 1, using NH-TSC (0.039 g, 0.2 mmol) instead of MH-TSC (0.039 g, 0.2 mmol) to the reaction mixture. Yield: 85 mg (ca. 53% based on Pt). Elemental analysis (%): calc. for 3: C, 26.33; H, 2.98; N, 21.94. Found: C, 26.40; H, 2.94; N, 21.98. Selected IR data (KBr, cm−1): 3380 (m), 3272.6 (m), 3165 (m), 2968 (w), 1647.7 (s), 1605.6 (m), 1550.9 (m), 1473 (s), 1413.9 (w), 1287.5 (w), 1185.4 (w), 1053 (w), 740.4 (w).
X-ray crystallographic studies
Crystals were picked up with a glass fiber and immediately mounted in a cold nitrogen stream to prevent loss of solvent. Intensity data were collected by a Rigaku Saturn CCD diffractometer using MoKα radiation (λ = 0.71073 Å) with the ω–2θ scan technique at 113(2) K. An empirical absorption correction was applied to the raw intensities. The structures were solved by direct methods SHELXS-97 and refined with full matrix least-squares technique on F2 using SHELXL-97.24 The hydrogen atoms were added theoretically, riding on the concerned atoms, and refined with fixed thermal factors. Structures were checked for higher symmetry using PLATON.25 Further crystallographic data and experimental details for structural analyses of 1–3 are summarized in Table 1. Selected bond lengths (Å) and bond angles (°) of 1–3 are listed in Table 2. Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, CCDC No. 1471505–1471507 for 1–3.†
Table 1 Summary of crystal data for 1–3
| Complex |
1 |
2 |
3 |
| Empirical formula |
C8H9ClPtN4S |
C10H13ClPtN4S |
C14H19ClPtN10S2O |
| Formula weight |
423.79 |
451.84 |
638.04 |
| Temperature/K |
113(2) |
113(2) |
113(2) |
| Wavelength/Å |
0.71073 |
0.71073 |
0.71073 |
| Space group |
P21/n |
C2/c |
P1 |
| a/Å |
7.4429(7) |
15.957(3) |
8.101(2) |
| b/Å |
13.5839(13) |
15.120(2) |
9.3191(19) |
| c/Å |
10.5375(10) |
12.362(2) |
14.094(3) |
| α/° |
90 |
90 |
92.2270(10) |
| β/° |
95.673(2) |
122.314(2) |
99.051(8) |
| γ/° |
90 |
90 |
104.437(8) |
| Volume/Å3 |
1060.16(17) |
2520.7(7) |
1014.2(4) |
| Z |
4 |
8 |
2 |
| ρ calcd/mg m−3 |
2.655 |
2.387 |
2.089 |
| F (000) |
784 |
1704 |
616 |
| Crystal size/mm |
0.15 × 0.12 × 0.10 |
0.20 × 0.15 × 0.12 |
0.20 × 0.18 × 0.15 |
| θ limits/° |
3.00–27.50 |
1.84–27.91 |
3.22–27.52 |
| Reflections collected |
13436 |
15956 |
13023 |
| Unique reflections |
2437 [R(int) = 0.0437] |
2885 [R(int) = 0.0276] |
4552 [R(int) = 0.0357] |
| Data/restraints/parameters |
2437/0/136 |
2885/3/166 |
4552/0/278 |
| Goodness of fit on F2 |
0.784 |
0.941 |
0.891 |
| R1(I > 2σ(I)) |
R1 = 0.0161 |
R1 = 0.0159 |
R1 = 0.0202, |
| wR2 (all data) |
wR2 = 0.0384 |
wR2 = 0.0326 |
wR2 = 0.0504 |
| Largest diff. peak and hole/e Å3 |
1.049 and −1.295 |
0.561 and −0.899 |
1.827 and −1.282 |
Table 2 Bond lengths [Å] and angles [°] for 1–3
| 1 |
| Pt(1)–N(2) |
1.951(2) |
N(2)–Pt(1)–N(1) |
80.77(9) |
| Pt(1)–N(1) |
2.041(2) |
N(2)–Pt(1)–S(1) |
85.27(7) |
| Pt(1)–S(1) |
2.2557(7) |
N(1)–Pt(1)–S(1) |
166.02(7) |
| Pt(1)–Cl(1) |
2.3233(7) |
N(2)–Pt(1)–Cl(1) |
178.59(7) |
| N(1)–Pt(1)–Cl(1) |
98.25(6) |
S(1)–Pt(1)–Cl(1) |
95.69(2) |
| C(8)–S(1)–Pt(1) |
94.14(9) |
C(6)–N(2)–Pt(1) |
117.8(2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| 2 |
| Pt(1)–N(2) |
1.959(2) |
N(2)–Pt(1)–N(1) |
80.79(10) |
| Pt(1)–N(1) |
2.039(2) |
N(2)–Pt(1)–S(1) |
85.24(7) |
| Pt(1)–S(1) |
2.2568(8) |
N(1)–Pt(1)–S(1) |
165.98(7) |
| Pt(1)–Cl(1) |
2.3145(7) |
N(2)–Pt(1)–Cl(1) |
177.77(7) |
| N(1)–Pt(1)–Cl(1) |
97.17(7) |
C(8)–S(1)–Pt(1) |
94.33(10) |
| S(1)–Pt(1)–Cl(1) |
96.82(3) |
C(1)–N(1)–Pt(1) |
129.0(2) |
|
| 3 |
| Pt(1)–N(3) |
1.971(2) |
N(3)–Pt(1)–N(1) |
80.43(10) |
| Pt(1)–N(1) |
2.059(2) |
N(3)–Pt(1)–S(1) |
84.54(7) |
| Pt(1)–S(1) |
2.2529(8) |
N(3)–Pt(1)–S(2) |
173.39(7) |
| Pt(1)–S(2) |
2.3049(8) |
N(1)–Pt(1)–S(1) |
164.96(7) |
| N(1)–Pt(1)–S(2) |
99.68(7) |
C(7)–S(1)–Pt(1) |
94.86(10) |
| S(1)–Pt(1)–S(2) |
95.31(3) |
C(14)–S(2)–Pt(1) |
108.46(10) |
HSA binding studies
Fluorescence quenching and UV spectrophotometry were used to carry out the protein (HSA) binding experiments. The protein was prepared in the phosphate buffer solution (containing 0.01 M NaH2PO4 and Na2HPO4 in aqueous medium at pH = 7.2). HSA solution concentration was controlled at 5 × 10−7 M (based on the molecular weight of HSA 66 kD), and then the solution was kept in the dark at 277 K overnight. Fluorescence quenching measurements were performed by keeping the concentration of HSA constant while gradually increasing the concentration of 1–3 at 25° (298 K), respectively. Fluorescence spectra were recorded at an excitation wavelength of 290 nm and emission wavelength range from 300 to 500 nm. UV spectrometry was performed using a JASCO V-770 spectrophotometer equipped with a 2 cm quartz cell. Absorption titration experiments were done by keeping the concentration of HSA constant (5 × 10−6 M) while adding the same concentration solution of 1–3, respectively. Synchronous fluorescence spectral experiments were operated with the same method as above while keeping the D values (Δλ) between excitation and emission wavelengths, which are stabilized at 15 and 60 nm respectively, at room temperature. All the data were obtained after each successive addition of HSA solution and equilibration (ca. 10 min).
Cytotoxicity evaluation
MTT assay. MTT assay was used to evaluate the cell viability effect of 1–3 with cisplatin as control. In brief, HeLa, MCF-7, HepG-2 and NCI-H460 cells were plated in 96 well-plate with three replica wells and incubated at 37 °C and 5% CO2 for 24 h. The concentration was 3000 cells per well. After the cells attached to wells, three complexes and cisplatin with various concentration (100, 50, 25, 12.5, 6.25, 3.12, 1.56, 0.78, 0.39 μM) were added and cultured for 48 h. After this period of time, 20 μL MTT (5 mg mL−1 in PBS) was added to each well and cells were incubated at 37 °C for 4 h. Finally, the medium was removed and 100 μL DMSO was added to dissolve the formed formazan crystals. Cell viability were determined by reading absorbance at 570 nm using an Enzyme-linked Immunosorbent Assay (ELISA) reader.
Time curve assay of three agents on HeLa cell line. Time curve assay was still evaluated by MTT assay. HeLa cells were treated with 1–3 and cisplatin with ranging from 100 to 0.39 μM for 12, 24 and 48 h, then incubated at 37 °C. The detection method was similar to the mentioned above.
Apoptosis evaluation
Hoechst 33342 staining. We adopted Hoechst 33342 staining to assess the inducing apoptosis ability of 1–3. HeLa cells, with a density of 1 × 105 cells per well, were seeded in 6 well-plate and incubated for 24 h at 37 °C. Then the cells were treated with fresh medium containing 10, 5, 2.5 μM three agents respectively. After 48 h, the cell was rinsed twice with PBS (pH = 7.4) to remove excess amount of agents and then stained with Hoechst 33342 (1 μg mL−1) for 15 min at room temperature. Finally, the cells were washed twice with PBS (pH = 7.4) to eliminate the surplus Hoechst 33342 interference and visualized with the aid of EPI fluorescence microscopy (Nikon eclipse E800).
Annexin V-FITC/PI double staining. For apoptosis quantitative analysis, 1 × 106 cells per well were plated in 6 well-plate and incubated at 37 °C for 24 h. Agents 1–3 at 2.5, 5, 10 μM were added to each well, respectively. After incubated for 48 h, cells were washed twice with PBS after the medium was removed and collected with trypsin (non-EDTA). Then the cells were centrifuged with 1200 rpm for 10 min and rinsed once with PBS. After centrifugation, supernatant was removed and pellet was resuspended with 1× binding buffer. Annexin V-FITC (10 μg mL−1) was added into the suspension and incubated for 15 min at room temperature and then stained with PI (5 μg mL−1) for 5 min at room temperature. The distribution of cell apoptosis was measured by a flow cytometer (BD FACS Calibur).
Cell proliferation assay
Cell cycle analysis. Cell cycle analysis was assessed by flow cytometry. Briefly, 1 × 106 cells were seeded in 6 well-plate and incubated for 24 h. After attaching to well, the HeLa cells were treated with 1–3 at 2.5, 5, 10 μM for 48 h. At the end of the culture period, the cells were rinsed twice with cold PBS (pH = 7.4), harvested with trypsin (containing EDTA) and centrifuged with 2000 rpm 10 min, then removed the supernatant and resuspended the pellets with 500 μL 70% cold ethanol at −20 °C over 48 h. After being centrifuged to remove the ethanol and washing once with cold PBS (pH = 7.4), the cells were digested with RNase A (100 μg mL−1) for 30 min at 37 °C to avoid RNA interference and stained with PI (50 μg mL−1) for 30 min at room temperature. Cell cycle distribution was analyzed immediately using a flow cytometer (BD FACS Calibur). The data was acquired and analyzed by ModFit 3.1 software.
Monoclonal formation assay
Cellular proliferation, an ability of cancer cells maintaining clonogenic capacity, was assessed by monoclonal formation assay. HeLa cells were plated with a density of 300 in 6 well-plate and incubated for 3 days until the cells attached to wells, complex 3 was added to the cells with various amounts (0, 2.5, 5, 10 μM) for 48 h, and then the medium containing 3 was changed with fresh medium and continuously incubated for two weeks. After this period of time, cells were washed twice with PBS (pH = 7.4) and fixed with methanol–acetic acid (3:1) solution for 15 min, stained with crystal violet solution (1% crystal violet, 20% ethanol) for 30 min, washed with PBS and examined with an inverted fluorescence microscope.
Platinum uptake assay
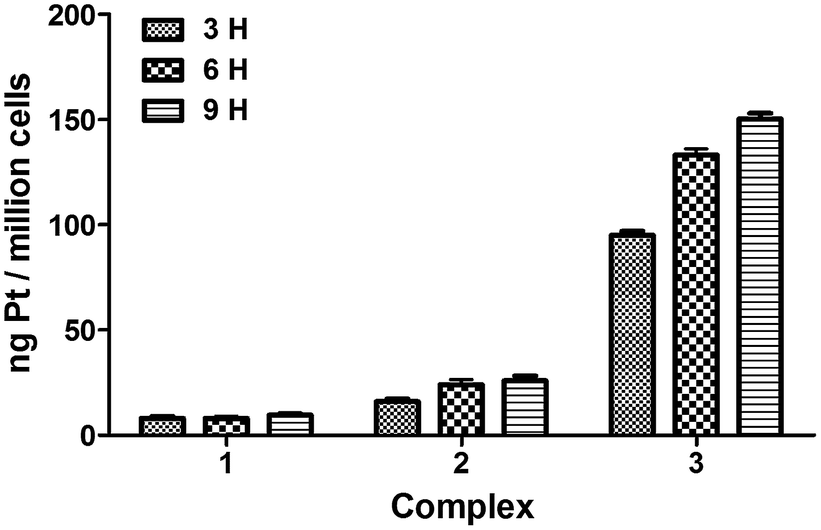
To assess the absorption capability of tumor cells to the three complexes, we examined the intracellular platinum content.26 In brief, cells (1 × 106 per well) were placed in 6-well plates and incubated for 24 h. Then, cells were treated with complexes (4 μM) at 37 °C for 3, 6 and 9 h, respectively. After treatment, the cells were washed three times with PBS, digested with trypsinization, collected with cold PBS and centrifuged at 1500 rpm for 5 min. Finally, supernatant was discarded and samples were lyophilized using a freeze dry system.
Prior to analysis, the lyophilized samples were digested with 0.2 mL highpurity nitric acid at room temperature for 4 h, diluted with 2 mL deionized water, centrifuged at 5000 rpm for 15 min, and transferred with an aliquot 0.2 mL to an ICP-MS autosampler tube for determining the Pt content. Thermo iCAP Q ICP-MS was calibrated with a Pt-containing acid matrix. The platinum content under each time point was expressed as ng Pt per million cells.
Results and discussion
X-ray structure characterization
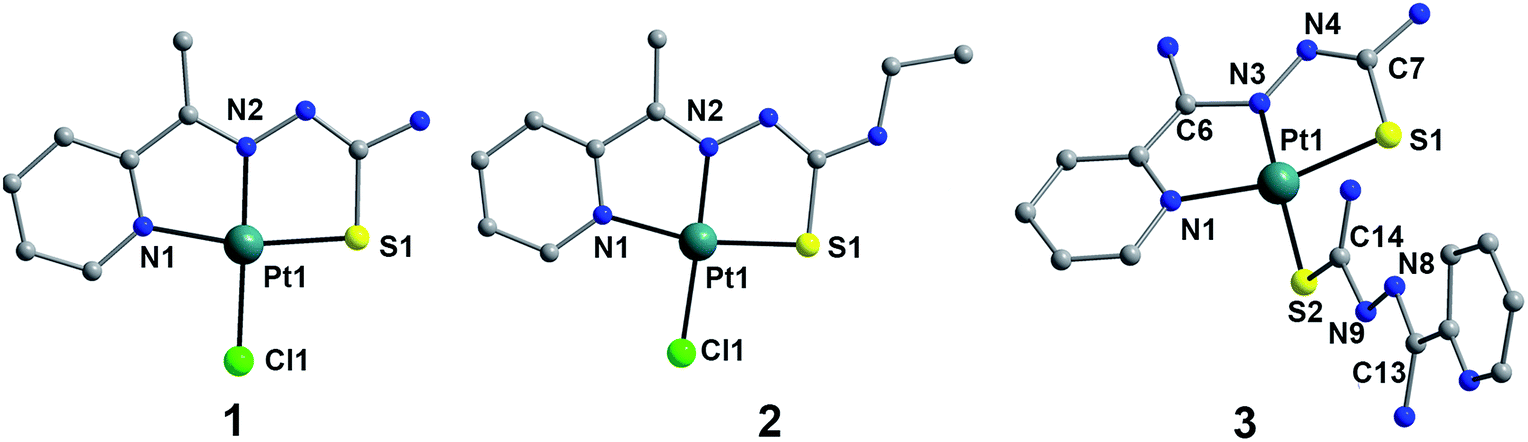
Complexes 1–3 have been structurally characterized by single crystal X-ray analysis. A labelled plot of 1–3 is shown in Fig. 1. Complex 1 crystallizes in the monoclinic system with space group P21/n and consists of neutral asymmetric mononuclear platinum(II) units without crystallization water. The structure of 1 consists of neutral asymmetric mononuclear unit, in which Pt(II) ion is tetracoordinated. The basal plane is formed by imine nitrogen N(1) [Pt(1)–N(1) = 2.041(2) Å], N(2) [Pt(1)–N(2) = 1.951(2) Å], thione S(1) [Pt(1)–S(1) = 2.2557(7) Å] from MH-TSC ligand and one chlorine Cl(1) [Pt(1)–Cl(1) = 2.3233(7) Å]. The donor atoms, N(1), N(2), S(1) and Cl(1), show a relatively good planarity, and Pt(1) displaced out of the basic plane towards the apex by 0.0151 Å. For 2, it crystallizes in monoclinic C2/c space group and presents a quite similar mononuclear Pt(II) structure to 1 owing to its slight ligand modification with ethyl instead of hydrogen in 1. As Fig. 1 shown, central Pt(II) ion in 2 is tetracoordinated, showing a good square planar geometry, surrounded by imine nitrogen N(1), N(2) and carboxylate thione S(1) from ME-TSC ligand and one chlorine Cl(1). Complex 3 crystallizes in the triclinic crystal system with P1 space group and consists of one [Pt(NH-TSC)2]+ cation, one non-coordinating chloride anion, and one water solvent molecule. Each Pt centre is coordinated with two NH-TSC ligands, by virtue of two nitrogen and one sulfur atoms from one ligand [Pt(1)–N(1) 2.059(2), Pt(1)–N(3) 1.971(2) and Pt(1)–S(1) 2.2529(8) Å], as well as one S atom from the other ligand [Pt(1)–S(2) 2.3049(8) Å] to build the square-planar coordination geometry. The bond distance Pt(1)–S(1) [2.2529(8) Å] is shorter than Pt(1)–S(2) [2.3049(8) Å]. Therefore, the two ligands do not display a similar coordinating pattern as common cases. The thiosemicarbazone moieties, C6–N3–N4–C7–S1 and C13–N8–N9–C14–S2 have mean plane deviations of 0.020(6) and 0.018(6) Å, respectively, and are at an angle to each other of 74.3°. Meanwhile, no interesting hydrogen bond and π–π stacking were found in these three complexes.
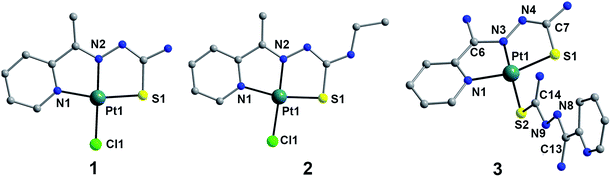 |
| | Fig. 1 DIAMOND views of 1–3 with the selected atom labeling scheme (hydrogen atoms omitted for clarity). | |
HSA binding experiments
Fluorescence spectroscopy of HSA. Qualitative analysis of binding of metal compounds to HSA was achieved by examining fluorescence spectra. The emission spectra of HSA in the presence of 1–3 were recorded in the wavelength range 300–500 nm by exciting the protein at 290 nm. A remarkable fluorescence decrease of HSA was observed with increasing concentration of 1–3, which indicated that complexes can bind to the proteins effectively. Generally, fluorescence quenching can be described by the following Stern–Volmer eqn (1).27| | |
F0/F = 1 + Kqt0[Q] = 1 + Ksv[Q]
| (1) |
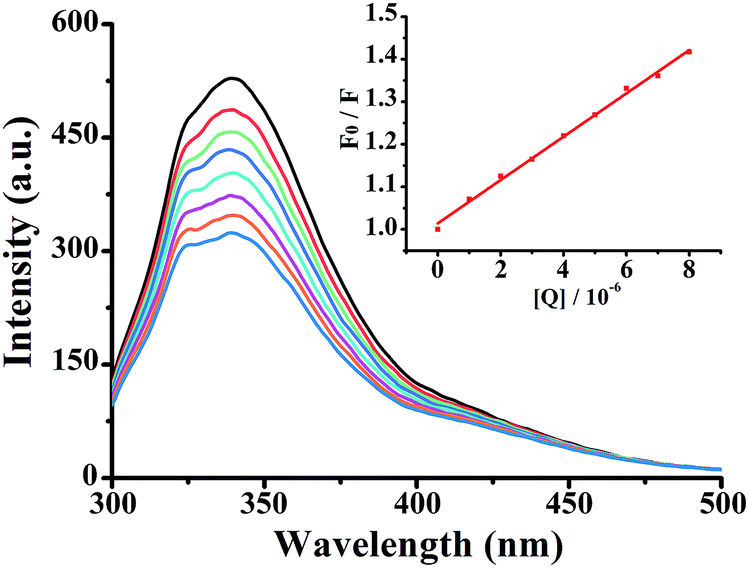
In this equation, F0 and F represent the fluorescence intensities in the absence and presence of the quencher, respectively. Kq is the biomolecular quenching rate constant, Ksv is the quenching constant, t0 is the average life-time of the biomolecule without the quencher (t0 = 6.2 ns) and [Q] is the concentration of quencher. The effect of complex 3 on HSA fluorescence intensity with Stern–Volmer plots are shown in Fig. 2. The corresponding Stern–Volmer quenching rate constant Kq of 1–3 were 4.25 × 1013, 7.09 × 1013 and 10.64 × 1013 L mol−1 s−1, respectively. The results showed that quenching constant Kq for 1–3 was on the order of 1013 L mol−1 s−1, which was 1000-fold higher than the maximum value possible for diffusion controlled quenching (i.e. 2.0 × 1010 L mol−1 s−1).28 This revealed that the quenching was not caused by dynamic collision but by a static one. The quenching mechanism should be further explored by UV-vis absorption spectrometry.
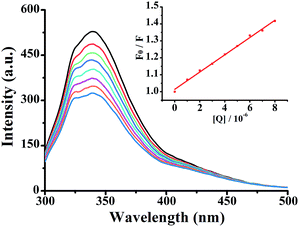 |
| | Fig. 2 Fluorescence spectra of HSA in the different concentrations of 3 at room temperature, [HSA] = 5 × 10−7 M, complex 3 (0–8 μM). Inset: plot of F0/F vs. [complex]. | |
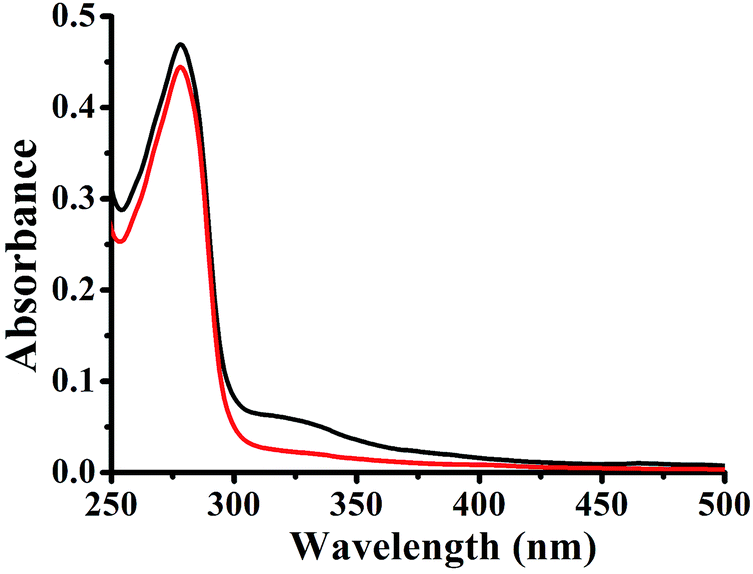
In a dynamic quenching mechanism, with the addition of a quencher, the absorption spectrum of the fluorescent substance is not changed, which due to the excited states of the fluorophore is not affected. While, the absorption spectrum will change in a static quenching, because new compound is formed between the quencher and HSA. The influences of the absorbance of HSA were observed by adding the same concentration of 1–3. From Fig. 3, the fluorescence of HSA caused by tryptophan, tyrosine, and phenylalanine residues exists.29 For 1–3, the absorption spectra decreased in the presence of the complex, indicating that the microenvironment of the three aromatic acid residues was changed, and this can prove that 1–3 formed a novel complex with HSA. These data suggested that the Pt(II) complex could interact with HSA through a static quenching mode.
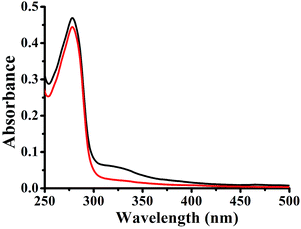 |
| | Fig. 3 UV-vis absorption spectra of HSA in the absence and presence of 3. Black line: the absorption spectrum of HSA. Red line: the absorption spectrum of HSA in the presence of 3 at the same concentration, [HSA] = [complex] = 5 × 10−6 M. | |
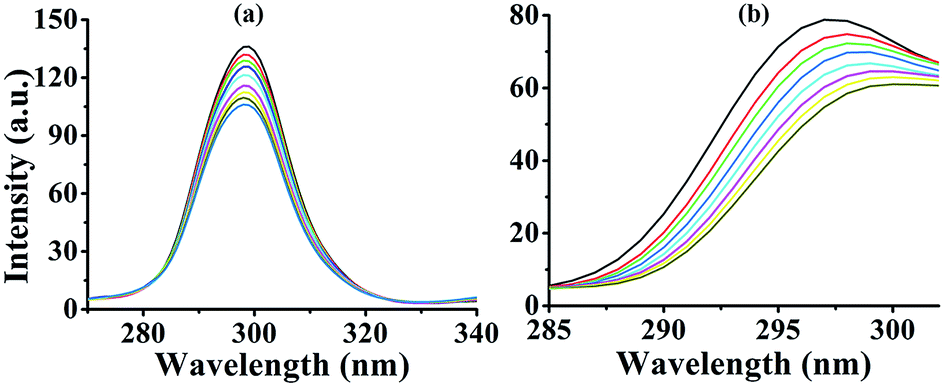
Synchronous fluorescence spectroscopy studies. It is reported that synchronous fluorescence spectroscopy is frequently used to characterize the interaction between the fluorescent probe and proteins,30 because it is of great importance for providing information about the molecular environment around the fluorophore molecules.31 The synchronous fluorescence spectroscopy performs the simultaneous scanning of excitation and emission spectra on a fluorimeter, and fixes the wavelength difference (Δλ) between them. When Δλ is stabilized at 15 nm, the synchronous fluorescence represents the state of tyrosine residues, whereas Δλ of 60 nm indicates that of tryptophan residues.32 The change of maximum emission wavelengths of tryptophan and tyrosine residues are related to the environmental change of their surroundings. From Fig. 4, it can be seen that upon addition of 1, the maximum emission wavelength showed no shift when Δλ was equal to 15 nm. However, the maximum emission wavelength displayed obviously red shift when Δλ was equal to 60 nm. The red shift indicated that the environment around HSA had changed with the transformation of conformation.33 Complexes 2 and 3 displayed similar manner to 1 (data were shown in ESI†).
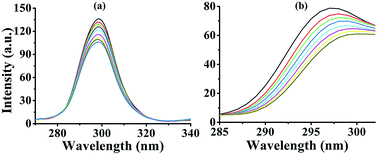 |
| | Fig. 4 Synchronous fluorescence spectra of HSA at room temperature upon addition of 3, Δλ = 15 nm (a); Δλ = 60 nm (b). Concentrations of complex is 0.0–8.0 μM. | |
MTT assay. MTT, the soluble yellow tetrazolium salt, is induced to purple formazan crystals by succinate dehydrogenase in the mitochondria of viable cell,34 while dead cells are dysfunction. Therefore, we adopted MTT assay to evaluate the cell viability of three complexes against HeLa, MCF-7, HepG-2 and NCI-H460 cells with cisplatin as control. The results were summarized in Table 3, complexes 1–3 and cisplatin all displayed antitumor effect against four cancer cells. The IC50 values of cisplatin ranged of 5.2–13.5 μM in accordance with the literature reported.35 Encouragingly, three complexes with IC50 values ranging of 1.7–9.6 μM showed obviously increased antitumor activity against human cancer cells, compared with cisplatin. Moreover, the antiproliferative activity of 3 was higher 7.9 and 6.2 times than cisplatin against MCF-7 and HeLa cells. Given that complex 3 is cation, it may take advantage of its cationic feature to pass through cells membrane easily.
Table 3 The IC50 values of three complexes in comparison with cisplatin against four cancer cell
| Cell |
Complex IC50 (μmol L−1) |
| 1 |
2 |
3 |
Cisplatin |
| MCF-7 |
6.6 ± 2.1 |
8.3 ± 3.3 |
1.7 ± 0.4 |
13.5 ± 4.3 |
| HepG-2 |
4.8 ± 1.6 |
7.2 ± 1.9 |
2.6 ± 0.6 |
9.71 ± 2.7 |
| NCI-H460 |
9.6 ± 3.1 |
6.6 ± 2.0 |
2.8 ± 1.1 |
5.2 ± 2.2 |
| HeLa |
7.4 ± 3.5 |
6.4 ± 2.4 |
2.2 ± 0.8 |
13.6 ± 4.6 |
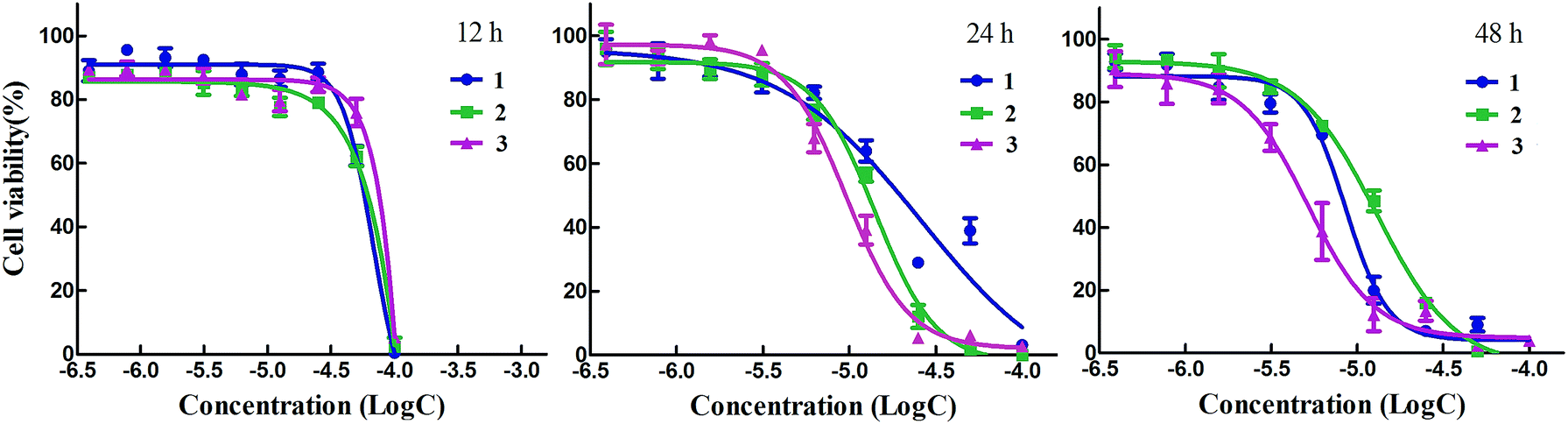
As three complexes exhibited strong antitumor effect, we determined to investigate the action manner of them by MTT assay. As shown in Fig. 5, complexes 1 and 3 exerted antitumor activity in a dose- and time-dependent behavior except 2. Along with the increasing concentration and time, the antitumor effect of 1–3 were strikingly enhanced. We did not expect that antitumor effect of 1 and 3 gradually reduced with the time extending while 2 exerted antiproliferation activity in a rapid manner. For example, when the HeLa cells were treated with 2 for 24 h, the IC50 values was lower 2 times than that of 1 and 3 with the same processing time. When the cells were incubated with 2 for 48 h, the IC50 values remained unchanged while that of 1 and 3 decreased two times than that of 2.
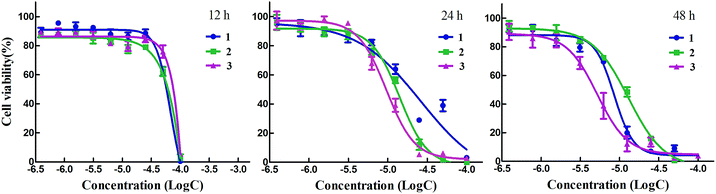 |
| | Fig. 5 The cell viability change of HeLa cells after complexes 1–3 treatment for 12, 24 and 48 h. | |
Apoptosis evaluation
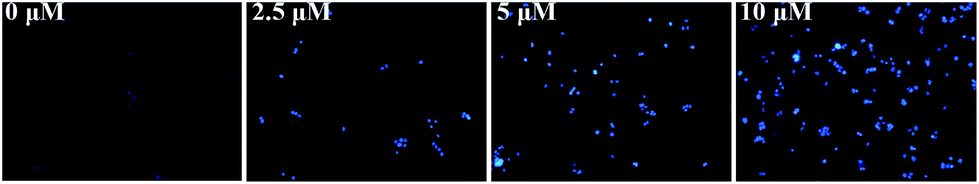
Hoechst 33342 staining. Hoechst 33342 dye could penetrate plasma membrane and selectively combine with DNA.36 The permeability of the apoptotic cells membrane were elaborated compared with normal membrane and more dyes gathered in nucleus. Meanwhile, obvious blue fluorescence could be observed in nucleus area. Therefore, we used this difference to distinguish the apoptotic cells and normal cells. HeLa cells were treated with 1–3 for 48 h and stained with Hoechst 33342, the results were shown in Fig. 6. As the concentration of complexes was increased, the intensity of bright vision was enhanced simultaneously, which indicated that 1–3 exactly caused cell apoptosis in HeLa cells.
 |
| | Fig. 6 Hoechst 33342 staining detected apoptosis in HeLa cells after treatment by 3 for 48 h at the concentrations of 0, 2.5, 5, and 10 μM. As concentration increased, the intensity of bright vision gradually enhanced. | |
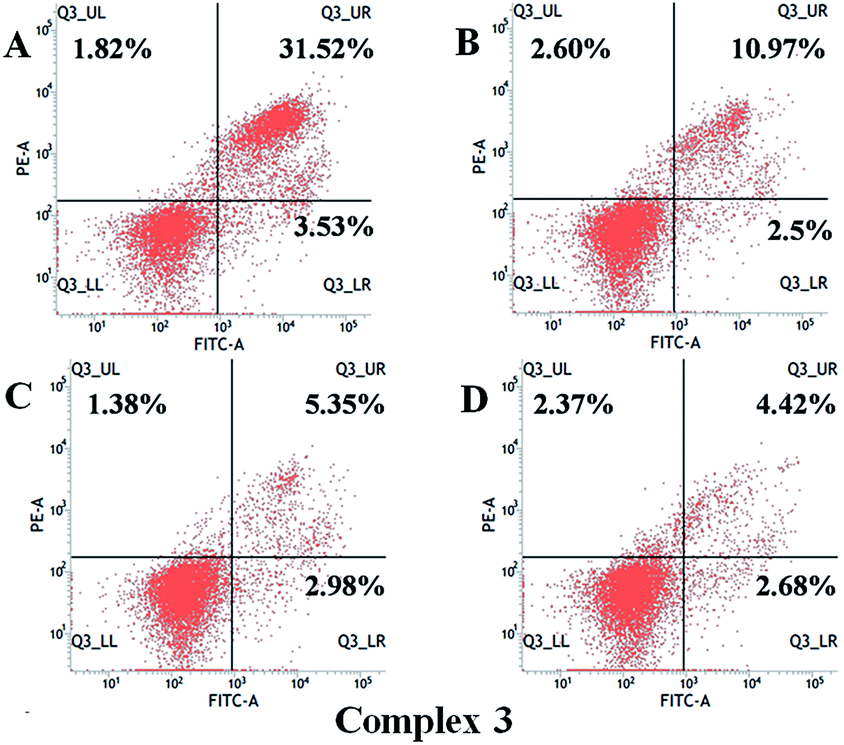
Annexin V-FITC/PI double staining. To quantitative analysis of the percentage of apoptosis, we adopted Annexin V-FITC/PI double staining to assess the apoptosis inducing ability. Briefly, the HeLa cells were stained by single Annexin V-FITC or single PI illustrating that the cancer cells occurred early apoptosis or necrosis. Annexin V-FITC/PI staining data was shown in Fig. 7, when concentration of 1–3 was elevated, the percentage of apoptosis was correspondingly enhanced especially for 3. The apoptosis inducing levels of three complexes were almost the same at 5 μM (19% for 1, 13% for 2 and 13% for 3). However, when the concentration was added to 10 μM, the inducing behavior of 3 (35%) was significantly stronger than that of 1 (19%) and 2 (25%). This result suggested that 1–3 exerted antitumor activities by inducing cell apoptosis and 3 would be the most promising antitumor agent.
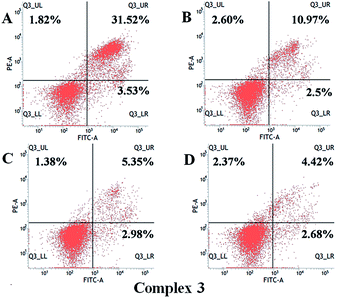 |
| | Fig. 7 The quantitative analysis of the proportion of viable, necrosis and apoptosis cells against HeLa cells after the treatment with 3 for 48 h at concentration of 0, 2.5, 5, and 10 μM. | |
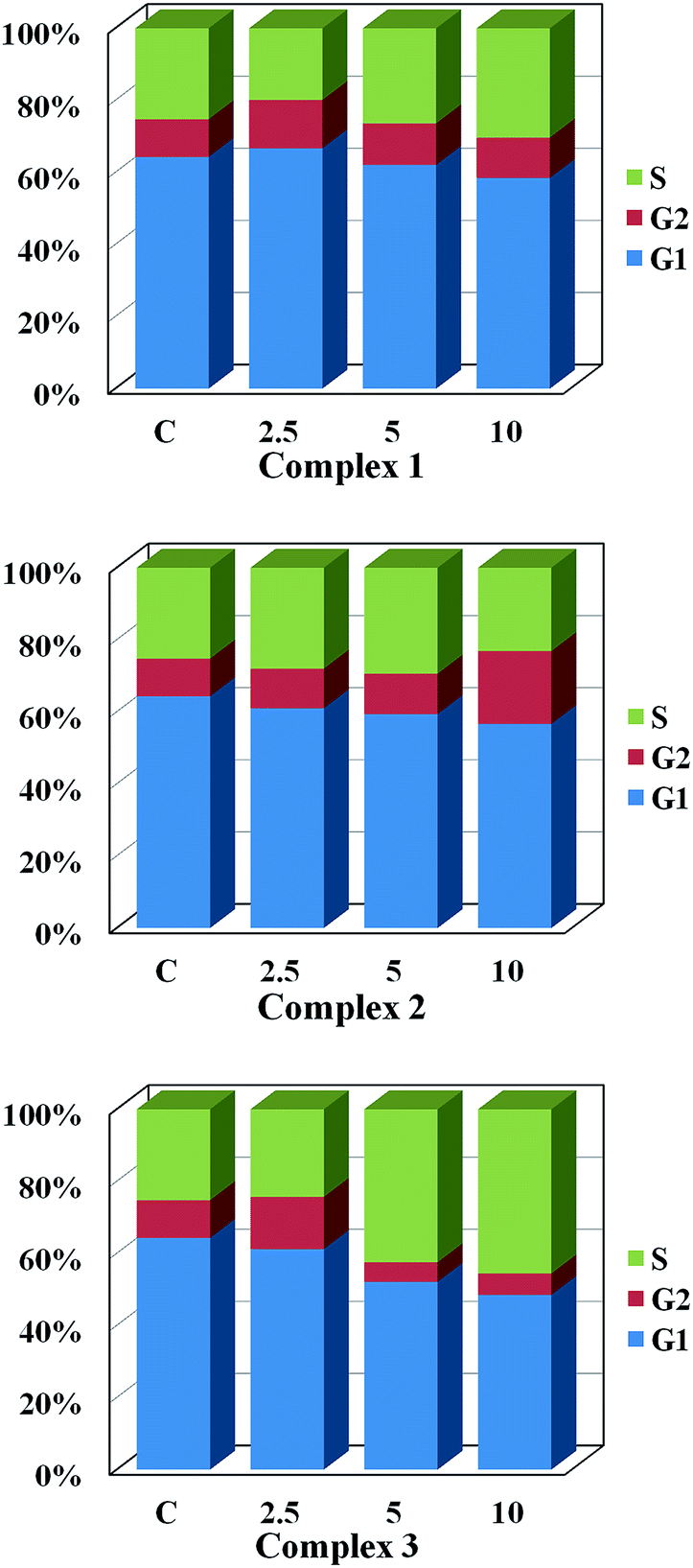
Cell cycle analysis. Cell cycle arrest played an important role in the development of anticancer agent. As three complexes were more efficient than cisplatin in terms of inducing apoptosis, we speculated that they may have influence on the cell cycle arrest. HeLa cells were treated with 1–3 and fixed for flow cytometry analysis. As shown in Fig. 8, all complexes exactly interfered cell cycle distribution and the effect displayed a concentration-dependent manner. Complex 2 could delay or inhibit cell cycle progression by arresting at G2 phase, while 1 and 3 induce cell cycle arrest at G2/M at low concentration. At high concentration, the complexes 1 and 3 induce cell cycle arrest at S phase, especially for 3. For example, compared with control group (25.4% in S phase, 10.44% in G2 phase), after the cells were treated with 10 μM complexes for 48 h, 46.96% of the cells treated with 3 and 30.31% of that treated with 1 were arrested in S phase, while 20.2% of the cells treated with 2 were stopped in G2 phase. Additionally, the monoclonal formation assay also confirmed that 3 had a major pact on cell proliferation (ESI†). These results demonstrated that three complexes would be the potential candidates for antitumor agents.
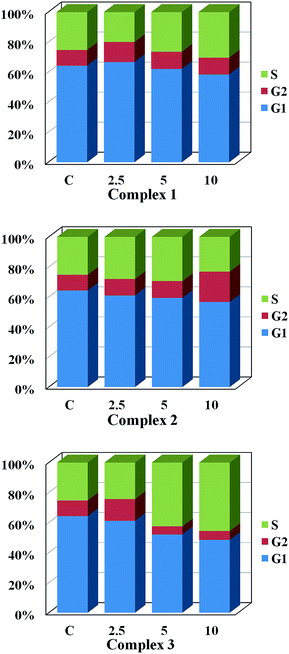 |
| | Fig. 8 Flow cytometric analysis of cell cycle distribution of HeLa cells treated with 1–3 for 48 h at the concentration of 0, 2.5, 5, 10 μM. | |
Platinum uptake assay. The major goal of the experiment described here was to test if the cytotoxic responses produced by 1–3 in cancer cells correlate with the platinum content in cells. As Fig. 9 showed that complex 3 rapidly accumulated in cancer cells with a maximum platinum level reached after a 9 h incubation period, at this time point, the content of the hybrid agent is approximately 10-fold higher than that determined for 1 and 2. As we expected, after tumor cells were treated with complexes, the content of 3 was strikingly higher than that of complexes 1 and 2 in cells, indicating that 3 was easily absorbed by tumor cells which may be related with its cationic feature. The results were in accordance with above biological activities.
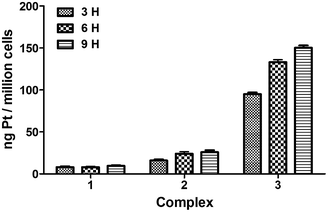 |
| | Fig. 9 Cellular platinum content in tumor cells treated with 4 μM complexes 1–3 for 3, 6, and 9 h, respectively. The data are calculated through 3 duplicated experiments. | |
Conclusions
In this work, three new platinum(II) complexes [Pt(MH-TSC)Cl] (1), [Pt(ME-TSC)Cl] (2) and [Pt(NH-TSC)2]Cl (3) were synthesized and characterized. According to the results obtained from the HSA interaction experiments, we concluded that the fluorescence quenching mechanism of HSA by 1–3 might be a static binding process. Complexes 1–3, with monofunctional or cationic features, showed higher cytotoxic activity towards four tumor cell lines than that of cisplatin under the same condition, and also superior to other thiosemicarbazone-based metal complexes with similar structure published in literatures.37–39 Previous reports displayed that bi-ligand complexes like 3, exhibited prominent anticancer activity with an extra ligand changing physicochemical features such as planarity, hydrophobicity and coordination geometry of metal complex.40–42 Consistently, cationic complex 3 had a distinct transport manner, resulted in higher ability for cellular uptake, which might explain its superior cytotoxic activity. Comparing the structures of 1 and 2, we see that there is one primary amine (–NH2) group for the ligand in 1, instead of secondary amine in 2, which might be available for hydrogen bonding with DNA base pairs and promising to improve the biological activity. So, it demonstrates that different transport manners or structural units will play a key role in the complexes' antiproliferative activity to cancer cells. In addition, the apoptotic study exhibited that complexes 1–3 could effectively induce apoptosis of HeLa cells. Meanwhile, cell proliferation assay demonstrated that treatments of HeLa cells with 1 and 3 hindered the cancer cells in S phase, while 2 processed a G2 phase arrest. These agents might trigger different signal pathways that eventually result in cell cycle arrest in different checkpoints. Further studies to elaborate its cytotoxicity mechanism are currently underway.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 21371135), Tianjin Municipal Natural Science Foundation (Nos 13JCZDJC28200 & 17JCZDJC33100).
Notes and references
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed
 .
. - D. Rades, D. Seidl, S. Janssen, A. Bajrovic, S. G. Hakim, B. Wollenberg and S. E. Schild, Oral Oncol., 2016, 57, 40–45 CrossRef CAS PubMed .
- M. Bandak, N. Jorgensen, A. Juul, J. Lauritsen, M. G. G. Kier, M. S. Mortensen and G. Daugaard, Eur. Urol. Focus, 2016 DOI:10.1016/j.euf.2016.11.018 .
- Z. Xiao, C. Wang, L. Chen, X. Tang, L. Li, N. Li, J. Li, Q. Gong, F. Tang and J. Feng, Oncotarget, 2017, 8, 1329–1342 Search PubMed .
- X. Zhu, H. Shen, X. Yin, L. Long, C. Xie, Y. Liu, L. Hui, X. Lin, Y. Fang and Y. Cao, Oncogene, 2016, 35, 323–332 CrossRef CAS PubMed .
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed .
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed .
- P. J. Loehrer, S. D. Williams and L. H. Einhorn, J. Natl. Cancer Inst., 1988, 80, 1373–1382 CrossRef PubMed .
- K. S. Lovejoy and S. J. Lippard, Dalton Trans., 2009, 10651–10659 RSC .
- J. Zhang, D. Liu, Y. Li, J. Sun, L. Wang and A. Zang, Mini-Rev. Med. Chem., 2009, 9, 1357–1366 CrossRef CAS PubMed .
- R. W. Brockman, J. R. Thomson, M. J. Bell and H. E. Skipper, Cancer Res., 1956, 16, 167–170 CAS .
- K. Agrawal and A. Sartorelli, Prog. Med. Chem., 1978, 15, 321–356 CrossRef CAS PubMed .
- M. Miller 3rd, C. Stineman, J. Vance, D. West and I. Hall, Anticancer Res., 1997, 18, 4131–4139 Search PubMed .
- C. A. Kunos, T. Radivoyevitch, S. Waggoner, R. Debernardo, K. Zanotti, K. Resnick, N. Fusco, R. Adams, R. Redline and P. Faulhaber, Gynecol. Oncol., 2013, 130, 75–80 CrossRef CAS PubMed .
- C. A. Kunos, T. Radivoyevitch, J. Pink, S. M. Chiu, T. Stefan, J. Jacobberger and T. J. Kinsella, Radiat. Res., 2010, 174, 574–581 CrossRef CAS PubMed .
- K. Ishiguro, Z. P. Lin, P. G. Penketh, K. Shyam, R. Zhu, R. P. Baumann, Y. L. Zhu, A. C. Sartorelli, T. J. Rutherford and E. S. Ratner, Biochem. Pharmacol., 2014, 91, 312–322 CrossRef CAS PubMed .
- A. E. Stacy, D. Palanimuthu, P. V. Bernhardt, D. S. Kalinowski, P. J. Jansson and D. R. Richardson, J. Med. Chem., 2016, 59(10), 4965–4984 CrossRef CAS PubMed .
- E. M. Jouad, G. Larcher, M. Allain, A. Riou, G. M. Bouet, M. A. Khan and X. Do Thanh, J. Inorg. Biochem., 2001, 86, 565–571 CrossRef CAS PubMed .
- B. S. Holla, K. Malini, B. S. Rao, B. Sarojini and N. S. Kumari, Eur. J. Med. Chem., 2003, 38, 313–318 CrossRef CAS PubMed .
- J. Shao, W. G. Bao, H. Tian, B. Li, X. F. Zhao, X. Qiao and J. Y. Xu, Dalton Trans., 2014, 43, 1663–1671 RSC .
- A. Castiñeiras, I. Garcia, E. Bermejo and D. X. West, Polyhedron, 2000, 19, 1873–1880 CrossRef .
- M. Carcelli, D. Delledonne, A. Fochi, G. Pelizzi, M. C. Rodríguez-Argüelles and U. Russo, J. Organomet. Chem., 1997, 544, 29–35 CrossRef CAS .
- D. X. West, G. A. Bain, R. J. Butcher, J. P. Jasinski, Y. Li, R. Y. Pozdniakiv, J. Valdés-Martínez, R. A. Toscano and S. Hernández-Ortega, Polyhedron, 1996, 15, 665–674 CrossRef CAS .
- G. Sheldrick, Acta Crystallogr., Sect. E: Struct. Rep. Online, 1997, 1600–5368 Search PubMed .
- A. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, c34–c34 Search PubMed .
- A. Ghezzi, M. Aceto, C. Cassino, E. Gabano and D. Osella, J. Inorg. Biochem., 2004, 98, 73–78 CrossRef CAS PubMed .
- J. Toneatto and G. A. Argüello, J. Inorg. Biochem., 2011, 105, 645–651 CrossRef CAS PubMed .
- P. Kandagal, S. Ashoka, J. Seetharamappa, V. Vani and S. Shaikh, J. Photochem. Photobiol., A, 2006, 179, 161–166 CrossRef CAS .
- F. Wang, W. Huang and Z. Dai, J. Mol. Struct., 2008, 875, 509–514 CrossRef CAS .
- L. N. Hordge, K. L. McDaniel, D. D. Jones and S. O. Fakayode, Talanta, 2016, 152, 401–409 CrossRef CAS PubMed .
- M. B. Bolattin, S. T. Nandibewoor, S. D. Joshi, S. R. Dixit and S. A. Chimatadar, RSC Adv., 2016, 6, 63463–63471 RSC .
- W. He, Y. Li, C. Xue, Z. Hu, X. Chen and F. Sheng, Bioorg. Med. Chem., 2005, 13, 1837–1845 CrossRef CAS PubMed .
- G. Zhang, Y. Wang, H. Zhang, S. Tang and W. Tao, Pestic. Biochem. Physiol., 2007, 87, 23–29 CrossRef CAS .
- J. Seo, B. Kim, D. N. Dhanasekaran, B. K. Tsang and Y. S. Song, Cancer Lett., 2016, 371, 30–37 CrossRef CAS PubMed .
- L. Sun and J. Fang, Cell Cycle, 2016, 15, 2647–2655 CrossRef CAS PubMed .
- G. Chandrasekher and D. Sailaja, Invest. Ophthalmol. Visual Sci., 2004, 45, 3577–3588 Search PubMed .
- M. Mohamed Subarkhan, R. N. Prabhu, R. Raj Kumar and R. Ramesh, RSC Adv., 2016, 6, 25082 RSC .
- B. Q. Zou, X. Lu, Q. P. Qin, Y. X. Bai, Y. Zhang, M. Wang, Y. C. Liu, Z. F. Chen and H. Liang, RSC Adv., 2017, 7, 17923 RSC .
- J. Shao, Z. Y. Ma, A. Li, Y. H. Liu, C. Z. Xie, Z. Y. Qiang and J. Y. Xu, J. Inorg. Biochem., 2014, 136, 13 CrossRef CAS PubMed .
- A. Li, Y. H. Liu, L. Z. Yuan, Z. Y. Ma, C. L. Zhao, C. Z. Xie, W. G. Bao and J. Y. Xu, J. Inorg. Biochem., 2015, 146, 52–60 CrossRef CAS PubMed .
- V. Gandin, M. Porchia, F. Tisato, A. Zanella, E. Severin, A. Dolmella and C. Marzano, J. Med. Chem., 2013, 56, 7416–7430 CrossRef CAS PubMed .
- C. Rajarajeswari, M. Ganeshpandian, M. Palaniandavar, A. Riyasdeen and M. A. Akbarsha, J. Inorg. Biochem., 2014, 140, 255–268 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR of ligand, corresponding figures of 1 and 2. CCDC 1471505–1471507 for 1–3. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra04443g |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ya-Hong Liu‡a,
Cheng-Zhi Xie
a,
Ya-Hong Liu‡a,
Cheng-Zhi Xie