
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benzene-glycol nucleic acid (BGNA)–DNA chimeras: synthesis, binding properties, and ability to elicit human RNase H activity†
Nazuki Niwaa,
Saki Shimizud,
Yusuke Maedaa,
Hidekazu Hiroakd and
Yoshihito Ueno*abc
aCourse of Applied Life Science, Faculty of Applied Biological Sciences, Gifu University, Japan. E-mail: uenoy@gifu-u.ac.jp; Fax: +81-58-293-2919; Tel: +81-58-293-2919
bUnited Graduate School of Agricultural Science, Gifu University, Japan
cCenter for Highly Advanced Integration of Nano and Life Sciences (G-CHAIN), Gifu University, 1-1 Yanagido, Gifu 501-1193, Japan
dDivision of Structural Biology, Graduate School of Pharmaceutical Sciences, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8601, Japan
First published on 11th May 2017
Abstract
This paper describes the synthesis and properties of benzene-glycol nucleic acid (BGNA)–DNA chimeras containing four nucleoside analogs – thymidine, cytidine, adenosine, and guanosine – with a base-benzene-glycol structure. We found that the BGNA–DNA chimeras are able to form thermally and thermodynamically stable duplexes with complementary RNAs, and have base-discriminating abilities. The BGNA–DNA chimeras were 20-fold more stable in a buffer containing 30% bovine serum than unmodified DNA. Furthermore, BGNA–DNA chimera/RNA duplexes were found to be good substrates for human RNase H. Thus, BGNA–DNA chimeras are good candidates for the development of therapeutic antisense molecules.
Introduction
Antisense oligonucleotides (AONs) are single-stranded DNAs consisting of 17–30 bases that can inhibit translation by binding to complementary target mRNAs.1 AONs have recently attracted attention as chemotherapeutic agents, since they can be rationally designed and synthesized given the sequences of disease-causing genes.2 Fomivirsen, a drug for cytomegalovirus (CMV) retinitis in patients with acquired immune deficiency syndrome (AIDS), is the first AON drug approved by the U.S. Food and Drug Administration (FDA).3 Mipomersen is an AON drug for patients with homozygous familial hypercholesterolemia. Eteplirsen, a drug used to treat patients with Duchenne muscular dystrophy, has also recently been approved by the FDA.4As phosphodiester linkages in DNA are easily hydrolyzed by the nucleases present inside and outside cells, chemical modifications are needed to impart nuclease-resistant properties to DNAs for them to function as antisense drugs. For example, fomivirsen is composed of phosphorothioate linkages instead of natural phosphate linkages.3 Mipomersen is partially composed of nucleoside analogs modified with methoxyethyl (MOE) groups at the 2′-hydroxyl positions. Eteplirsen has a backbone composed of methylenemorpholine rings and phosphorodiamidate linkages.
Recently, we reported the synthesis of two nucleoside analogs, a cytidine analog (1) and an adenosine analog (2), comprising a base-benzene-glycol structure (Fig. 1).5 We found that DNAs partially incorporating the benzene-glycol nucleic acids (BGNAs), termed BGNA–DNA chimeras, form thermally stable duplexes with complementary DNA and RNA, and these duplexes are more resistant to hydrolysis by a snake venom phosphodiesterase (a 3′-exonuclease) than unmodified DNA. As an extension of our study on the synthesis of BGNAs, we report in this paper the synthesis of a thymidine analog (3) and a guanosine analog (4), and properties of BGNA–DNA chimeras containing these four analogs as antisense molecules.
 | ||
| Fig. 1 Structures of the nucleoside analogs. | ||
Results and discussion
Synthesis
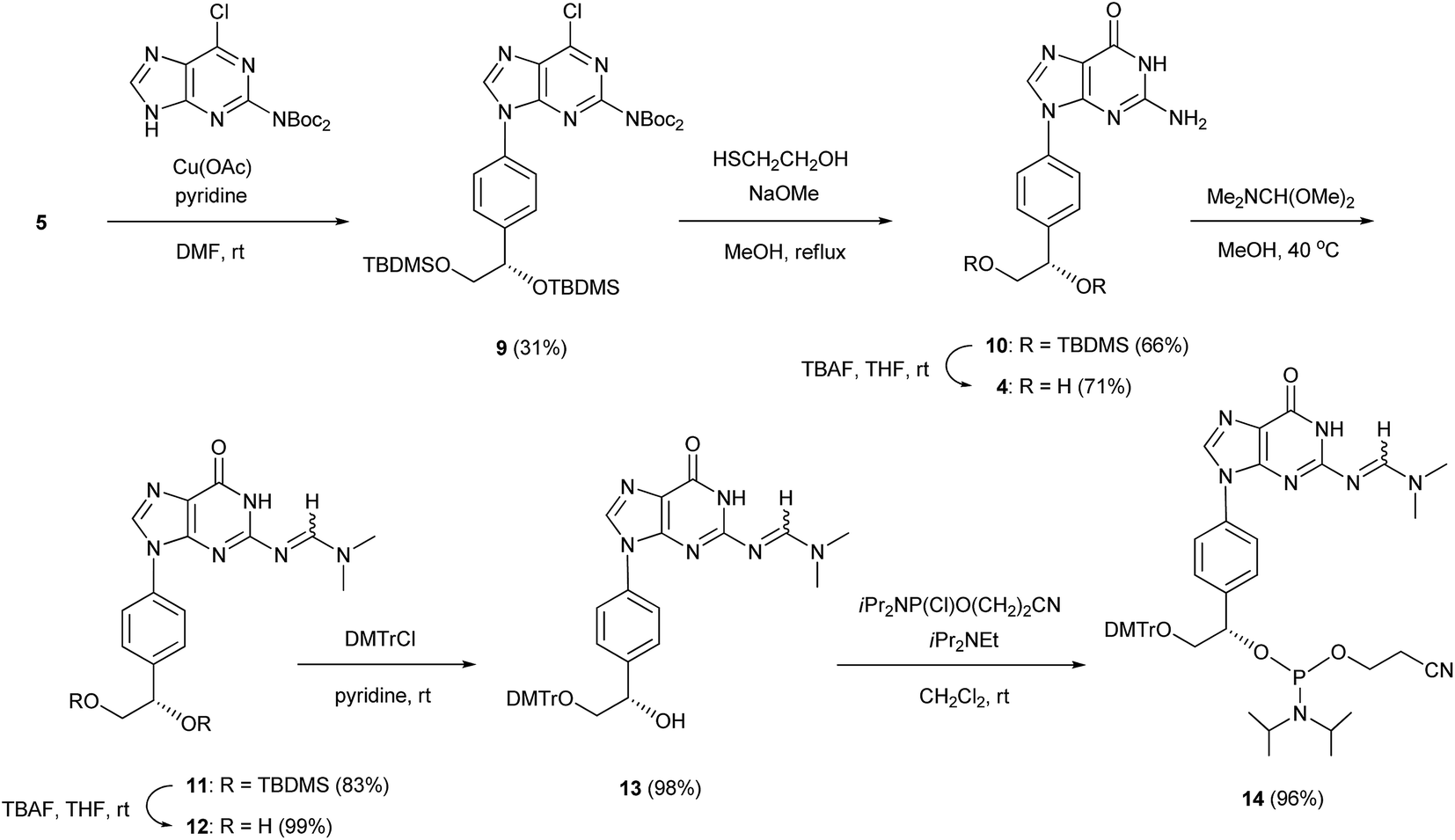
We previously reported the synthesis of the cytidine and adenosine analogs using the Chan–Lam–Evans reaction between an arylboronic acid and nucleobases.5–7 Synthesis of the thymidine and guanosine analogs was achieved using the same reaction. To prevent the side reaction between the N3 position of thymine and the arylboronic acid, we used an N3-protected thymine, N3-benzoylthymine, as a substrate for the synthesis of the thymidine analog. To solve the problem of the insolubility of guanine, a protected purine base, 2-bis(tert-butoxycarbonyl)amino-6-chloropurine, was used as a substrate for the synthesis of the guanosine analog.5b The synthetic routes to the thymidine and guanosine analogs, and the corresponding phosphoramidite units (8 and 14, respectively), are shown in Schemes 1 and 2.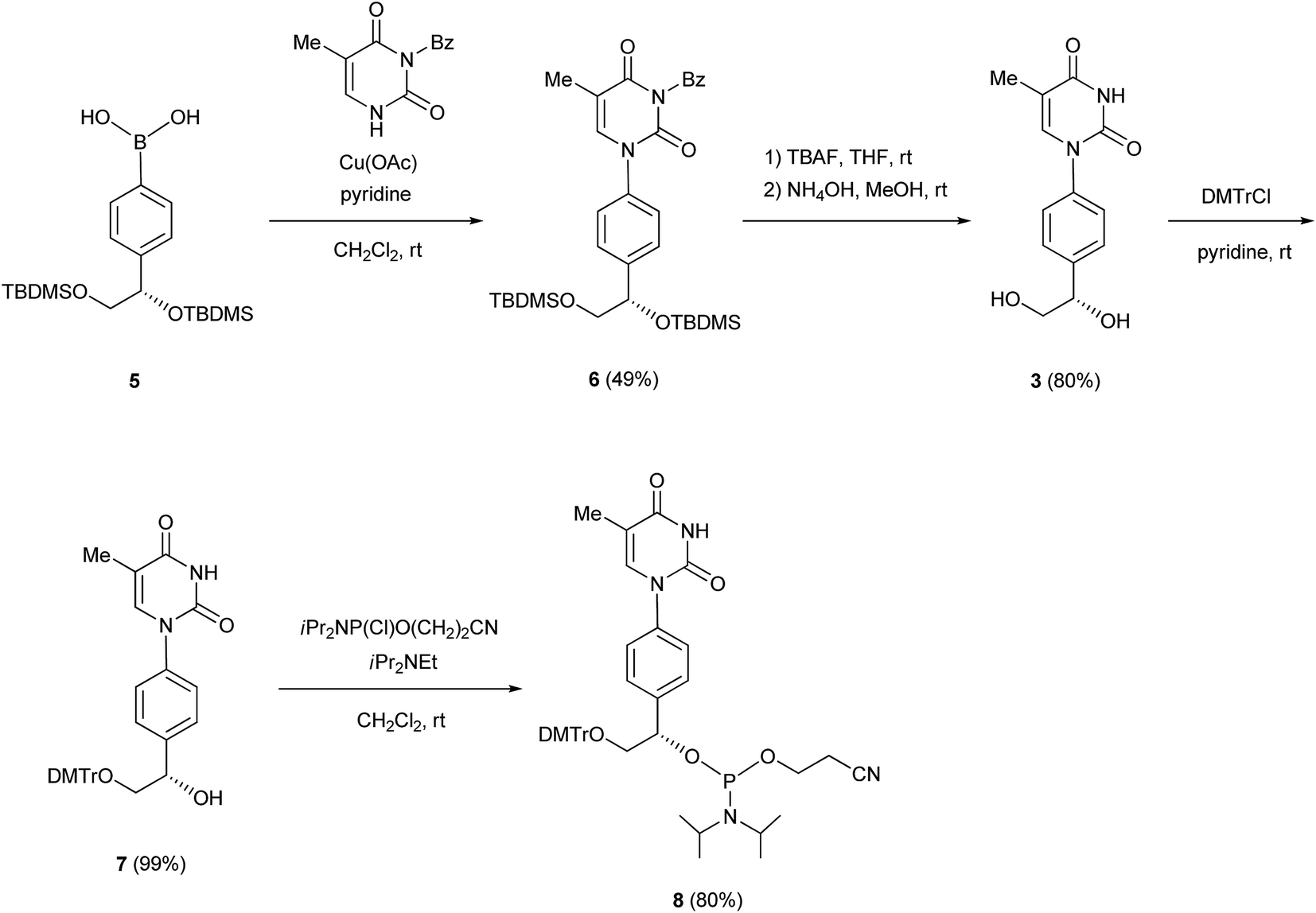 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
N3-Benzoylthymine was coupled with the arylboronic acid derivative 5 in the presence of Cu(OAc)2 and pyridine as a base in CH2Cl2 to give an N1-aryl thymine derivative (6) with a yield of 49%. Deprotection of the silyl and benzoyl groups of 6 with tetra-n-butylammonium fluoride (TBAF) and NH4OH produced the thymine analog 3 with a yield of 80%. The primary hydroxy group of 3 was protected with a 4,4′-dimethoxytrityl (DMTr) group to give a mono-DMTr derivative (7) with a yield of 99%. Compound 7 was phosphitylated using the standard procedure, leading to the formation of the corresponding phosphoramidite (8) with a yield of 80%.
In a similar manner, 2-bis(tert-butoxycarbonyl)amino-6-chloropurine was coupled with the arylboronic acid derivative 5 in the presence of Cu(OAc)2 and pyridine in DMF to form an N9-arylpurine derivative (9) with a yield of 31%. The treatment of 9 with 2-mercaptoethanol and NaOMe in MeOH gave an N9-arylguanine derivative (10) with a yield of 66%. The silyl groups of 10 were removed by treatment with TBAF to give the guanosine analog 4 with a yield of 71%. The exo-amino function of 10 was protected with a dimethylaminomethylene group to produce a fully protected guanosine derivative (11) with a yield of 83%. After deprotection of the silyl groups with TBAF, the primary hydroxy group of 12 was protected with a DMTr group to obtain a mono-DMTr derivative (13) with a yield of 98%. Compound 13 was phosphitylated by the standard procedure to lead to the formation of the corresponding phosphoramidite (14) with a yield of 96%.
All oligonucleotides (ONs) containing the analogs 1–4 were synthesized using the phosphoramidites 8 and 14, and the phosphoramidites of the cytidine and adenosine analogs, which were synthesized using the previously published procedure.5c The ON sequences used are given in Table 1. The ONs were analyzed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF/MS), and the observed molecular weights obtained from these analyses were consistent with the ON structures.
| Duplex | ON | Sequencesa | Tm (°C) | ΔTm (°C) | ΔH° (kcal mol−1) | ΔS° (cal mol−1 K−1) |
|
|---|---|---|---|---|---|---|---|
| a F indicates a fluorescein. | |||||||
| Duplex 1 | DNA 1 | 5′-d(AAAGTTCACTACTCAAA)-3′ | 52.3 | −98.4 | −275.2 | −13.1 | |
| RNA 1 | 3′-r(UUUCAAGUGAUGAGUUU)-5′ | ||||||
| Duplex 2 | ON 1 | 5′-d(AAbAbGTTCACTACTCAbAbAb)-3′ | 55.6 | +3.3 | −94.7 | −259.8 | −14.1 |
| RNA 1 | 3′-r(UUUCAAGUGAUGAGUUU)-5′ | ||||||
| Duplex 3 | DNA 2 | 5′-d(ACAGTTCACTACTCACA)-3′ | 58.0 | −144.1 | −406.6 | −18.0 | |
| RNA 2 | 3′-r(UGUCAAGUGAUGAGUGU)-5′ | ||||||
| Duplex 4 | ON 2 | 5′-d(ACbAbGTTCACTACTCAbCbAb)-3′ | 57.9 | −0.1 | −113.6 | −315.0 | −16.0 |
| RNA 2 | 3′-r(UGUCAAGUGAUGAGUGU)-5′ | ||||||
| Duplex 5 | DNA 3 | 5′-d(AGAGTTCACTACTCAGA)-3′ | 57.5 | −132.5 | −373.5 | −16.8 | |
| RNA 3 | 3′-r(UCUCAAGUGAUGAGUCU)-5′ | ||||||
| Duplex 6 | ON 3 | 5′-d(AGbAbGTTCACTACTCAbGbAb)-3′ | 56.4 | −1.1 | −112.3 | −313.0 | −15.3 |
| RNA 3 | 3′-r(UCUCAAGUGAUGAGUCU)-5′ | ||||||
| Duplex 7 | DNA 4 | 5′-d(ATAGTTCACTACTCATA)-3′ | 54.8 | −122.0 | −343.9 | −15.4 | |
| RNA 4 | 3′-r(UAUCAAGUGAUGAGUAU)-5′ | ||||||
| Duplex 8 | ON 4 | 5′-d(ATbAbGTTCACTACTCAbTbAb)-3′ | 56.0 | +1.2 | −117.6 | −329.6 | −15.5 |
| RNA 4 | 3′-r(UAUCAAGUGAUGAGUAU)-5′ | ||||||
| Duplex 9 | DNA 5 | 5′-d(AAATCACTACAAA)-3′ | 36.8 | −89.6 | −260.9 | −8.7 | |
| RNA 5 | 3′-r(UUUAGUGAUGUUU)-5′ | ||||||
| Duplex 10 | ON 5 | 5′-d(AAbAbTCACTACAbAbAb)-3′ | 35.0 | −1.8 | −66.0 | −186.1 | −8.4 |
| RNA 5 | 3′-r(UUUAGUGAUGUUU)-5′ | ||||||
| Duplex 11 | DNA 6 | 5′-d(ACATCACTACACA)-3′ | 47.7 | −108.6 | −310.5 | −12.4 | |
| RNA 6 | 3′-r(UGUAGUGAUGUGU)-5′ | ||||||
| Duplex 12 | ON 6 | 5′-d(ACbAbTCACTACAbCbAb)-3′ | 41.8 | −5.9 | −99.9 | −288.8 | −10.4 |
| RNA 6 | 3′-r(UGUAGUGAUGUGU)-5′ | ||||||
| DNA 7 | 5′-F-d(AAACACCGACGGCGAAA)-3′ | ||||||
| ON 7 | 5′-F-d(AAbAbCACCGACGGCGAbAbAb)-3′ | ||||||
| RNA 7 | 5′-F-r(UUUGAGUAGUGAACUUU)-3′ | ||||||
| RNA 8 | 5′-F-r(UUUGUAGUGAUUU)-3′ | ||||||
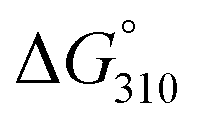
Thermal and thermodynamic stabilities of duplexes
The thermal stabilities of DNA/RNA and ON/RNA duplexes containing the analogs were compared by measuring their melting temperatures (Tms) in a 10 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl (Table 1 and Fig. S1†). Furthermore, to examine the stability of the duplexes in detail, we calculated the thermodynamic parameters of duplex formation based on the slope of the plot of 1/Tm vs. ln(CT/4), where CT is the total concentration of single-stranded ON and RNA (Table 1 and Fig. S3†). Duplexes 1–8 are composed of 17 base pairs, while duplexes 9–12 comprise 13 base pairs. All ON/RNA duplexes contain two and three analogs at the 5′- and 3′-regions of the ONs, respectively. Duplex 2 has five Ab:rU base pairs among its 17 base pairs, whereas duplexes 4, 6, and 8 contain two Cb:rG, Gb:rC, and Tb:rA base pairs in addition to the Ab:rU base pairs in the sequences, respectively. Duplex 10 has five Ab:rU base pairs among its 13 base pairs, while duplex 12 contains two Cb:rG base pairs in addition to the Ab:rU base pairs in the sequence.The thermal and thermodynamic stabilities of the duplexes containing the analogs were found to be dependent on their sequences. Duplexes 2 and 8, containing the Ab:rU and Tb:rA base pairs, were thermally and thermodynamically more stable than the corresponding DNA/RNA duplexes 1 and 7, respectively, while duplexes 4 and 6, containing the Cb:rG and Gb:rC base pairs, were thermally and thermodynamically less stable than the corresponding DNA/RNA duplexes 3 and 5, respectively. However, the differences in the thermal and thermodynamic stabilities between the duplexes containing the analogs and the unmodified DNA/RNA duplexes were not large (ΔTm = −1.1 ∼ +3.3 °C, 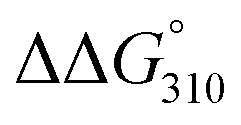 = +2.0 ∼ −1.0 kcal mol−1). On comparison of the thermodynamic parameters, it was found that duplex formation between ONs containing the analogs and the complementary RNAs was less favorable in terms of enthalpy but more favorable in terms of entropy than that of the corresponding unmodified DNA/RNA duplexes.
= +2.0 ∼ −1.0 kcal mol−1). On comparison of the thermodynamic parameters, it was found that duplex formation between ONs containing the analogs and the complementary RNAs was less favorable in terms of enthalpy but more favorable in terms of entropy than that of the corresponding unmodified DNA/RNA duplexes.
The ΔTm and  values between duplexes 1 and 2 were +3.3 °C and −1.0 kcal mol−1, respectively, while those between duplexes 9 and 10 were −1.8 °C and +0.3 kcal mol−1, respectively. Similarly, the ΔTm and
values between duplexes 1 and 2 were +3.3 °C and −1.0 kcal mol−1, respectively, while those between duplexes 9 and 10 were −1.8 °C and +0.3 kcal mol−1, respectively. Similarly, the ΔTm and 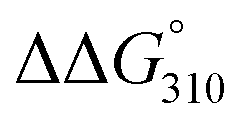 values between duplexes 3 and 4 were −0.1 °C and +2.0 kcal mol−1, respectively, while those between duplexes 11 and 12 were −5.9 °C and +2.0 kcal mol−1, respectively. Thus, incorporation of the analogs into duplexes comprising 13 base pairs destabilized the duplexes more than their incorporation into duplexes composed of 17 base pairs, both thermally and thermodynamically.
values between duplexes 3 and 4 were −0.1 °C and +2.0 kcal mol−1, respectively, while those between duplexes 11 and 12 were −5.9 °C and +2.0 kcal mol−1, respectively. Thus, incorporation of the analogs into duplexes comprising 13 base pairs destabilized the duplexes more than their incorporation into duplexes composed of 17 base pairs, both thermally and thermodynamically.
The base-discriminating abilities of the analogs 1–4 in DNA/RNA duplexes were then assessed (Fig. 2 and Table S1†). The ΔTm (Tm of duplex containing mismatched base pair − Tm of complementary duplex) values for the unmodified duplexes were −0.6 ∼ −12.3 °C, whereas those for the duplexes containing the analogs were −0.3 ∼ −5.2 °C. Thus, the analogs 1–4 have base-discriminating abilities in DNA/RNA duplexes, though weaker than those of the corresponding unmodified nucleosides.
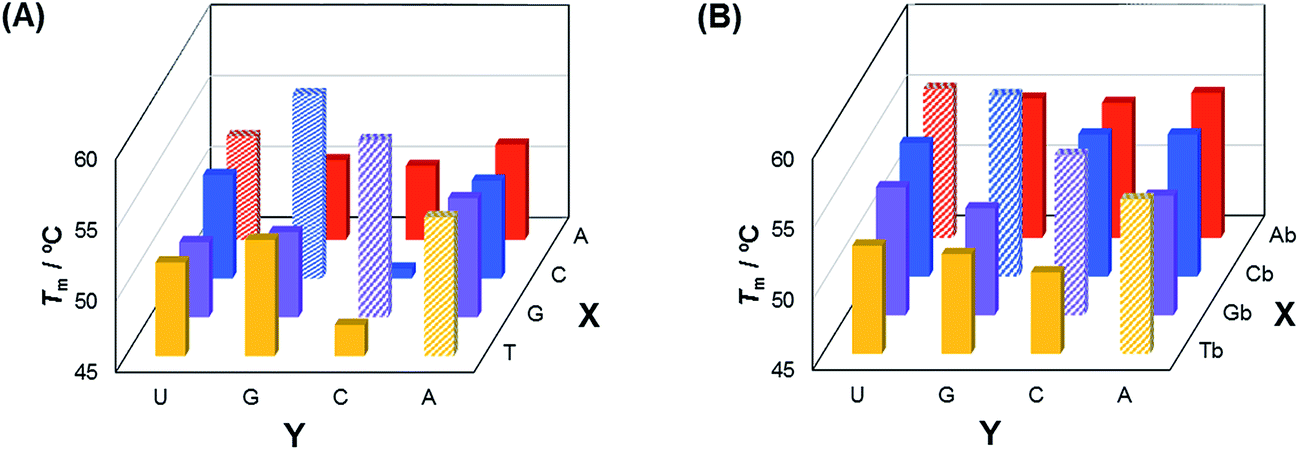 | ||
| Fig. 2 Comparison of the Tm values of the duplexes composed of matched and mismatched sequences. Sequences are shown in Table S1.† | ||
Circular dichroism
To study the global conformations of the duplexes, we analyzed the circular dichroism (CD) spectra of the duplexes composed of ON 1 (17-mer) and of ON 5 (13-mer), containing five analogs (Fig. 3). The ON 1/RNA 1 duplex showed a positive CD band around 270 nm and a negative CD band at 210 nm, which was similar to the spectrum of the unmodified DNA 1/RNA 1 duplex, whereas the shape of the CD spectrum of the ON 5/RNA 5 duplex was different from that of the unmodified DNA 5/RNA 5 duplex. These results suggest that the global conformation of the ON 1/RNA 1 duplex composed of 17 base pairs is similar to that of the unmodified DNA 1/RNA 1 duplex, while the global conformation of the ON 5/RNA 5 duplex comprising 13 base pairs is different from that of the unmodified DNA 5/RNA 5 duplex.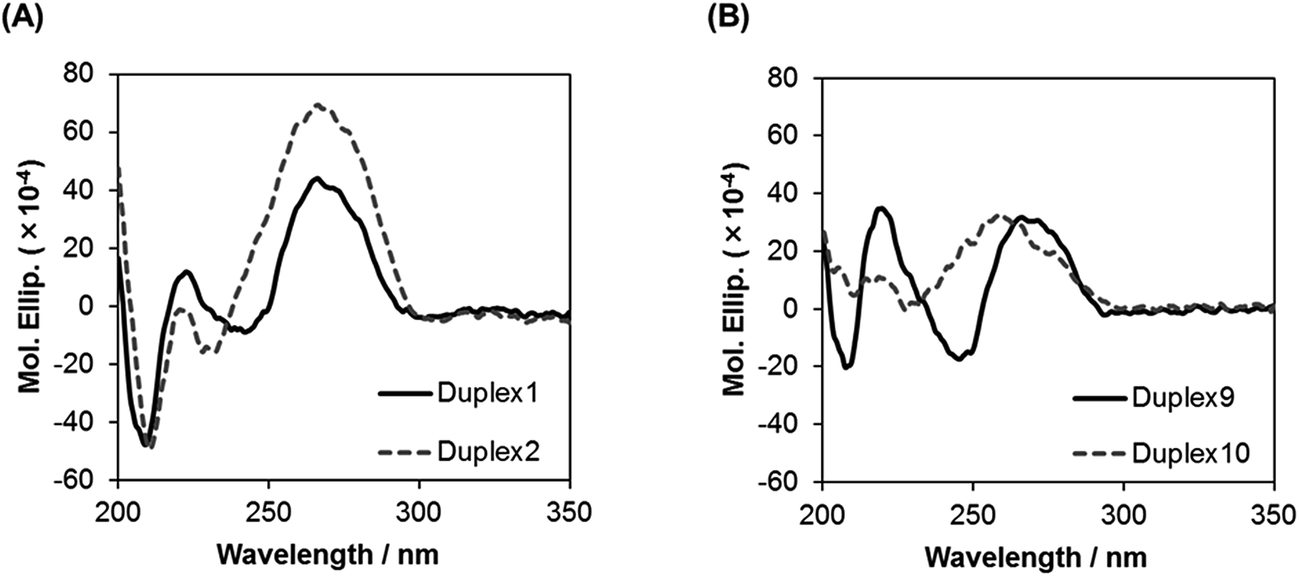 | ||
| Fig. 3 CD spectra of duplexes in a buffer of 10 mM sodium phosphate (pH 7.0) and 0.1 M NaCl at 15 °C. Concentration of duplexes: 4 μM. | ||
Nuclease resistance
Previously, we reported that BGNA–DNA chimeras are more resistant to hydrolysis by a snake venom phosphodiesterase (a 3′-exonuclease) than unmodified DNA.5c To be able to function as drugs, AONs must be stable in blood, which contains various enzymes, including nucleases. Therefore, we investigated the stability of BGNADNA chimeras in bovine serum (BS). DNA 7 or ON 7, labeled with fluorescein at the 5′-end, was incubated in a buffer containing 30% BS and analyzed by polyacrylamide gel electrophoresis under denaturing conditions (Fig. 4). The half-life of DNA 7 was 35 min, while that of ON 7 was 11.7 h. Thus, the BGNA–DNA chimera (ON 7) was approximately 20-fold more stable in the buffer containing 30% BS than the unmodified DNA 7.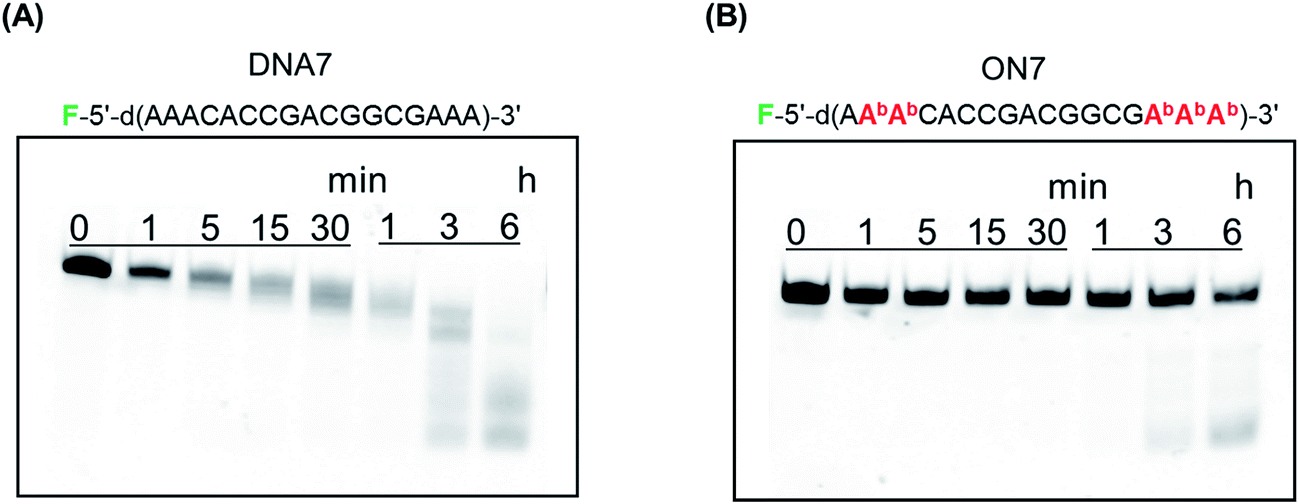 | ||
| Fig. 4 Polyacrylamide gel electrophoresis of (A) DNA 7 and (B) ON 7 treated in a buffer containing 30% BS. F denotes a fluorescein. Experimental conditions are described in the Experimental. | ||
Degradation by human recombinant RNase H1
To function as therapeutics, AONs need to elicit RNase H activity in human cells. We previously reported that a duplex between a BGNA–DNA chimera and a complementary RNA could act as a substrate for Escherichia coli RNase H.5c In this study, we examined cleavage of RNAs by human recombinant RNase H1. A DNA fragment of E. coli mal E gene encoding maltose binding protein (MBP) and a fragment encoding catalytic domain of human RNase H1 (residues 136-286, H-domain) were subcloned into the multiple cloning site of pET15b, resulting in pET-MBP-hRNase H1 (H-domain) vector for expressing MBP-hRNase H1 (H-domain) fusion protein. Human recombinant RNase H1 was expressed from the pET-MBP-hRNase H1 (H-domain) vector in E. coli BL21(DE3) competent cells. The duplex between DNA 1 or ON 1 and RNA 7, labeled with fluorescein at the 5′-end, or DNA 5 or ON 5 and RNA 8, also labeled with fluorescein at the 5′-end, was incubated in a buffer containing human recombinant RNase H, and the products were analyzed by polyacrylamide gel electrophoresis under denaturing conditions (Fig. 5). The initial rates of degradation of the DNA 1/RNA 7 and ON 1/RNA 7 duplexes (17-mers) were 206 pmol min−1 and 215 pmol min−1, respectively, while those of the DNA 5/RNA 8 and ON 5/RNA 8 duplexes (13-mers) were 302 pmol min−1 and 100 pmol min−1, respectively. Thus, the ability of ON 5 (13-mer) containing five analogs to elicit human RNase H activity was found to be weaker than that of the unmodified DNA 5, while the ability of the ON 1 (17-mer) containing five analogs to elicit human RNase H activity was comparable with that of the unmodified DNA 1.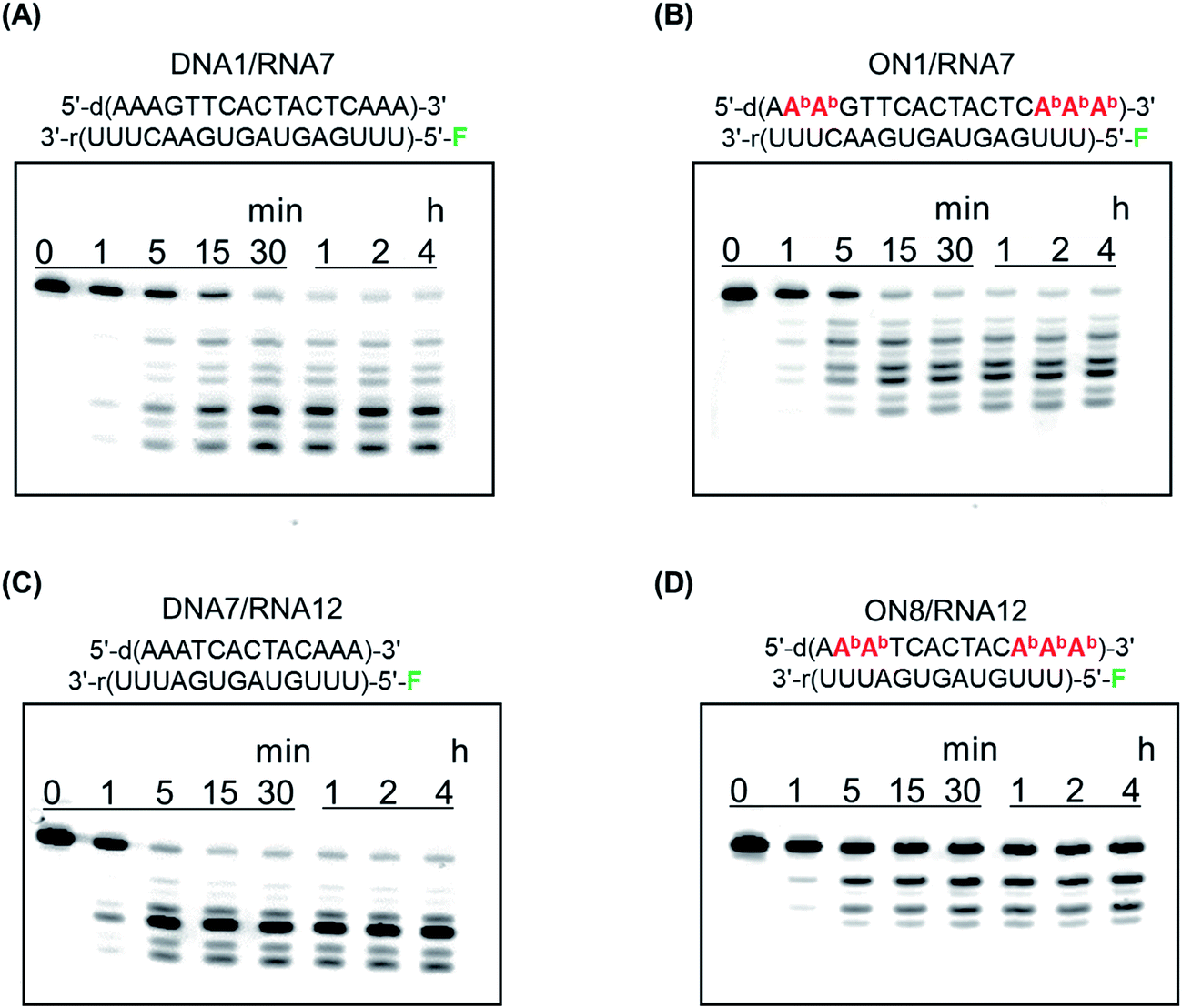 | ||
| Fig. 5 Polyacrylamide gel electrophoresis of (A) DNA 1/RNA 7 duplex, (B) ON 1/RNA 7 duplex, (C) DNA 7/RNA 12 duplex, and (D) ON 8/RNA 12 duplex hydrolyzed by human recombinant RNase H1. F denotes a fluorescein. Experimental conditions are described in the Experimental. | ||
X-ray co-crystal structural analysis has shown that human RNase H recognizes eight base-pairs in a DNA/RNA duplex.8 ON 1 contains eleven contiguous unmodified nucleoside residues, while ON 5 has seven consecutive unmodified nucleoside moieties. This may partially explain why the ON 5/RNA duplex is a poor substrate for human RNase H. Furthermore, CD spectra suggested that the global conformation of the ON 1/RNA duplex was similar to that of the unmodified DNA/RNA duplex, while the global conformation of the ON 5/RNA duplex was different from that of the unmodified DNA/RNA duplex. This is also likely to influence the abilities of the ONs to elicit human RNase H activity.
Conclusions
We have synthesized nucleoside analogs with a thymine- or guanine-benzene-glycol structure. We have synthesized DNAs incorporating four nucleoside analogs – thymidine, cytidine, adenosine, and guanosine – at the 5′- and 3′-regions of the DNAs. We found that the benzene-glycol nucleic acid (BGNA)–DNA chimeras formed thermally and thermodynamically stable duplexes with complementary RNAs, and these are more stable in bovine serum than unmodified DNA. Furthermore, we found that duplexes formed by BGNA–DNA chimeras and complementary RNAs are good substrates for human RNase H. Based on these results, we believe that BGNA–DNA chimeras are good candidates for the development of therapeutic antisense molecules.Experimental
General remarks
The chemicals were purchased from Sigma-Aldrich, Kanto Chemical Co. Inc., Tokyo Chemical Industry Co., Ltd., Nacalai Tesque Inc., Wako Pure Chemical Industries, Ltd., and Glen Research Co. All chemicals were used without further purification. Gibco® bovine serum was purchased from Thermo Fisher Scientific Inc. A pET15b expression vector was purchased from Novagen, Merck Millipore, Japan. E. coli BL21(DE3) competent cells were purchased from NEW ENGLAND BioLabs® Japan Inc. CDCl3 (CIL) or DMSO-d6 (CIL) was used as a solvent for obtaining NMR spectra. Chemical shifts (δ) are given in parts per million (ppm) downfield from (CH3)4Si (δ 0.00 for 1H NMR in CDCl3), or a solvent (for 13C NMR and 1H NMR in DMSO-d6) as an internal reference with coupling constants (J) in Hz. The abbreviations s, d, and q signify singlet, doublet, and quartet, respectively.Oligonucleotide synthesis
The synthesis was carried out with a DNA/RNA synthesizer by the phosphoramidite method. Deprotection of DNAs was performed in concentrated NH4OH at 55 °C for 16 h. Deprotection of RNAs was performed in concentrated NH4OH/EtOH (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at 55 °C for 4 h. 2′-O-TBDMS groups were removed by Et3N·3HF in DMSO at 65 °C for 90 min. The reaction was quenched with 0.1 M TEAA buffer (pH 7.0) and the mixture desalted using a Sep-Pak C18 cartridge. The oligonucleotides were purified by 20% PAGE containing 7 M urea and/or reversed-phase C-18 HPLC using a gradient of solution A (5% MeCN in 0.1 M TEAA buffer, pH 7.0) and solution B (50% MeCN in 0.1 M TEAA buffer, pH 7.0).
:1, v/v) at 55 °C for 4 h. 2′-O-TBDMS groups were removed by Et3N·3HF in DMSO at 65 °C for 90 min. The reaction was quenched with 0.1 M TEAA buffer (pH 7.0) and the mixture desalted using a Sep-Pak C18 cartridge. The oligonucleotides were purified by 20% PAGE containing 7 M urea and/or reversed-phase C-18 HPLC using a gradient of solution A (5% MeCN in 0.1 M TEAA buffer, pH 7.0) and solution B (50% MeCN in 0.1 M TEAA buffer, pH 7.0).
MALDI-TOF/MS analysis of ONs
The spectra were obtained with a time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). A solution of 3-hydroxypicolinic acid (3-HPA) and diammonium hydrogen citrate in H2O was used as the matrix. Data of synthetic ONs: DNA 1: m/z = 5145.40 (calcd for C166H208N65O96P16 [M − H]− 5145.40); DNA 2: m/z = 5098.51 (calcd for C164H208N61O98P16 [M − H]− 5097.35); DNA 3: m/z = 5177.60 (calcd for C166H208N65O98P16 [M − H]− 5177.40); DNA 4: m/z = 5127.60 (calcd for C166H210N59O100P16 [M − H]− 5127.37); DNA 5: m/z = 3919.72 (calcd for C127H158N53O70P12 [M − H]− 3919.62); DNA 6: m/z = 3871.68 (calcd for C125H158N49O72P12 [M − H]− 3870.57); DNA 7: m/z = 5743.48 (calcd for C192H229N76O101P17 [M − H]− 5743.88); ON 1: m/z = 5245.06 (calcd for C181H208N65O91P16 [M − H]− 5245.57); ON 2: m/z = 5198.15 (calcd for C179H208N61O93P16 [M − H]− 5197.53); ON 3: m/z = 5277.04 (calcd for C181H208N65O93P16 [M − H]− 5277.57); ON 4: m/z = 5228.18 (calcd for C181H210N59O95P16 [M − H]− 5227.55); ON 5: m/z = 4018.86 (calcd for C142H158N53O65P12 [M − H]− 4018.79); ON 6: m/z = 3970.97 (calcd for C140H158N49O67P12 [M − H]− 3970.75); ON 7: m/z = 5844.20 (calcd for C207H229N76O96P17 [M − H]− 5844.05); RNA 1: m/z = 5389.64 (calcd for C161H196N59O121P16 [M − H]− 5389.19); RNA 2: m/z = 5467.74 (calcd for C163H198N65O119P16 [M − H]− 5467.27); RNA 3: m/z = 5387.00 (calcd for C161H198N61O119P16 [M − H]− 5387.22); RNA 4: m/z = 5436.10 (calcd for C163H198N65O117P16 [M − H]− 5435.27); RNA 5: m/z = 4080.14 (calcd for C122H148N41O95P12 [M − H]− 4080.39); RNA 6: m/z = 4158.26 (calcd for C124H150N47O93P12 [M − H]− 4158.47); RNA 7: m/z = 5927.30 (calcd for C188H221N60O130P17 [M − H]− 5927.65); RNA 8: m/z = 4618.50 (calcd for C149H173N42O104P13 [M − H]− 4618.85).Thermal denaturation study
The solution containing the duplex in a buffer comprising 10 mM sodium phosphate (pH 7.0) and 100 mM NaCl was heated at 100 °C for 5 min, cooled gradually to an appropriate temperature, and then used for the thermal denaturation study. The thermally induced transition of each mixture was monitored at 260 nm with a UV/vis spectrometer fitted with a temperature controller in quartz cuvettes with a path length of 1.0 cm and a 3.0 μM duplex concentration in a buffer of 10 mM sodium phosphate (pH 7.0) and 0.1 M NaCl. The sample temperature was increased by 0.5 °C min−1. The thermodynamic parameters of the duplexes on duplex formation were determined by calculations based on the slope of a 1/Tm vs. ln(CT/4) plot, where CT (1, 3, 6, 12, 15, 21, 30, and 60 μM) is the total concentration of single strands.Partial hydrolysis of ONs in a buffer containing bovine serum
Each ON (3000 pmol) labeled with fluorescein at the 5′-end was incubated with 30% bovine serum in a buffer (150 μL) comprised of 0.1 M Tris–HCl (pH 8.0) and 20 mM MgCl2 at 37 °C. Aliquots (5 μL) were taken at 0, 0.5, 1, 5, 30, 60, 180, 360 min and mixed with the loading buffer (15 μL) containing Tris–borate–EDTA (TBE) buffer and 20% glycerin on ice. Each sample was analyzed by 20% denaturing PAGE at room temperature at 20 mA for 2 h. The gel was visualized by use of a Luminescent Image analyzer LAS-4000 (Fujifilm).DNA constructs
A DNA fragment of E. coli mal E gene encoding maltose binding protein (MBP) and a fragment encoding catalytic domain of human RNase H1 (residues 136-286, H-domain) were subcloned into the multiple cloning site of pET15b, resulting in pET-MBP-hRNase H1 (H-domain) vector for expressing MBP-hRNase H1 (H-domain) fusion protein. Between the two genes, a specific cleavage site for human Rhinovirus 3C (HRV3C) protease was engineered. All the DNA fragments were prepared by the standard PCR method.Protein expression and purification
The pET-MBP-hRNase H1 (H-domain) vector was transformed into E. coli BL21(DE3). The cells were cultured in Luria–Bertani medium at the 37 °C until OD600 reached to approximately 0.4. Final 0.2 mM of isopropyl-β-D-galactoside was added to induce the recombinant protein expression, and the temperature was shifted to 20 °C. After 20 hours, the cells were harvested. The bacterial cells were suspended to the lysis buffer (pH 7.0) containing 40 mM sodium phosphate, 1 M NaCl, 0.5 mM EDTA and 2 mM dithiothreitol (DTT). Then, the cells were lysed by sonication, and the supernatant was collected by centrifugation. First, a cleared supernatant was passed through DEAE-Sepharose® (GE Healthcare Life Sciences Corp, Piscataway, NJ, USA), and then the fusion protein was captured by Amylose resin (New England Biolabs, Ipswich, MA, USA). From the fusion protein, MBP tag was removed by on-column digestion by adding HRV3C protease at room temperature. The digested hRNase H1 (H-domain) was purified by cation exchange chromatography by using SP-Sepharose® (GE Healthcare Life Sciences) and further purified by gel-filtration by Superdex® 75 pg (GE Healthcare Life Sciences). Finally, the enzyme was dialyzed against 20 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl and 5 mM DTT.Hydrolysis of RNA with human recombinant RNase H1
The solution containing ON (600 pmol) and RNA (3000 pmol) labeled with fluorescein at the 5′-end in a buffer comprised of 50 mM Tris–HCl (pH. 8.0), 75 mM KCl, 3 mM MgCl2 and 10 mM dithiothreitol was heated at 100 °C for 5 min. The reaction mixtures were then cooled gradually to an appropriate temperature. Then human RNase H1 (2 pmol) was added to the solution, and the mixture was incubated at 37 °C. Aliquots (5 μL) were taken at 0, 1, 5, 15, 30, 60, 120 and 240 min, and mixed with formamide (15 μL) on ice. Each sample was analyzed by 20% denaturing PAGE at room temperature at 20 mA for 2 h. The gel was visualized by use of a Luminescent Image analyzer LAS-4000 (Fujifilm).Notes and references
- (a) P. C. Zamecnik and M. L. Stephenson, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 280–284 CrossRef CAS PubMed; (b) M. L. Stephenson and P. C. Zamecnik, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 285–288 CrossRef CAS PubMed.
- (a) M. Muthiah, Antisense Nucleic Acid Drug Dev., 2002, 12, 103–128 CrossRef PubMed; (b) T. Aboul-Fadl, Curr. Med. Chem., 2005, 12, 2193–2214 CrossRef CAS PubMed.
- P. M. D. Moreno and A. P. Pêgo, Front. Chem., 2014, 2, 87, DOI:10.3389/chem.2014.00087.
- Editorial, Nat. Biotechnol., 2016, 34, 1078, DOI:10.1038/nbt.3733.
- (a) Y. Ueno, T. Kato, K. Sato, Y. Ito, M. Yoshida, T. Inoue, A. Shibata, M. Ebihara and Y. Kitade, J. Org. Chem., 2005, 70, 7925–7935 CrossRef CAS PubMed; (b) Y. Ueno, A. Kawamura, K. Takasu, S. Komatsuzaki, T. Kato, S. Kuboe, Y. Kitamura and Y. Kitade, Org. Biomol. Chem., 2009, 7, 2761–2769 RSC; (c) N. Niwa, K. Ueda and Y. Ueno, Eur. J. Org. Chem., 2016, 2435–2443 CrossRef CAS.
- Y. Yue, Z.-G. Zheng, B. Wu, C.-Q. Xia and X.-Q. Yu, Eur. J. Org. Chem., 2005, 5154–5157 CrossRef CAS.
- M. F. Jacobsen, M. M. Knudsen and K. V. Gothelf, J. Org. Chem., 2006, 71, 9183–9190 CrossRef CAS PubMed.
- M. Nowotny, S. A. Gaidamakov, R. Ghirlando, S. M. Cerritelli, R. J. Crouch and W. Yang, Mol. Cell, 2007, 28, 264–276 CrossRef CAS PubMed.
Footnote |
| † Electric supplementary information (ESI) available. See DOI: 10.1039/c7ra03896h |
| This journal is © The Royal Society of Chemistry 2017 |