
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biodegradation of the neonicotinoid insecticide acetamiprid in surface water by the bacterium Variovorax boronicumulans CGMCC 4969 and its enzymatic mechanism†
Shi-Lei Sun a,
Wen-Long Yanga,
Jing-Jing Guoa,
Yi-Ning Zhoua,
Xue Ruia,
Chen Chena,
Feng Ge*b and
Yi-Jun Dai*a
a,
Wen-Long Yanga,
Jing-Jing Guoa,
Yi-Ning Zhoua,
Xue Ruia,
Chen Chena,
Feng Ge*b and
Yi-Jun Dai*a
aJiangsu Key Laboratory for Microbes and Functional Genomics, Jiangsu Engineering and Technology Research Center for Industrialization of Microbial Resources, College of Life Science, Nanjing Normal University, Nanjing 210023, People's Republic of China. E-mail: daiyijun@njnu.edu.cn; Fax: +86-25-85891067; Tel: +86-25-85891731
bNanjing Institute of Environmental Sciences, Ministry of Environmental Protection, Nanjing 210042, People's Republic of China. E-mail: gefeng@nies.org
First published on 11th May 2017
Abstract
The plant growth-promoting rhizobacterium Variovorax boronicumulans CGMCC 4969 was used to degrade the neonicotinoid insecticide, acetamiprid (AAP), in surface water, and the enzymatic mechanisms of AAP degradation in V. boronicumulans CGMCC 4969 were explored. V. boronicumulans CGMCC 4969 degraded 34.7% of 2 mg L−1 AAP over 120 h with a degradation half-life of 182 h in surface water, and the major metabolite was the amide product, (E)-N2-carbamoyl-N1-[(6-chloro-3-pridyl) methyl]-N1-methylacetamidine (IM-1-2). Gene cloning and over-expression studies proved that AAP hydration to IM-1-2 was mediated by a nitrile hydratase (ANHase). Addition of AAP to the mineral salt medium (MSM) broth significantly upregulated the ANHase gene expression by 1.6-fold, when compared with that in the control without AAP. Co-expression of the ANHase gene with its activator gene (anhC) apparently increased ANHase activity 21-fold for AAP hydration compared with the ANHase gene alone. The independent over-expression of anhC gave rise to competitive inhibition on the β-subunit of the ANHase and resulted in decreased ANHase activity. This ANHase is versatile, hydrating aromatic, N-heterocyclic, and aliphatic nitrile compounds. The present study shows the potential of V. boronicumulans CGMCC 4969 in the bioremediation of AAP contaminated water.
1. Introduction
Neonicotinoids are a class of heterocyclic ring-containing systemic pesticides, which are used for the control of sucking insect pests such as aphids, whiteflies, planthoppers, thrips, some micro-Lepidoptera, and coleopteran pests.1 They act as an agonist and selectively bind the nicotinic acetylcholine receptor in the central nervous system of the insect, leading to paralysis and death.2 Recent studies have shown that neonicotinoid insecticides may not only affect pest insects but also non-target organisms such as pollinators and birds.3 Because most neonicotinoids persist in soils for a year or more and are water soluble, more than 80% of neonicotinoid residues remaining in the soil of treated crops eventually enter surface waters or groundwater. Recent surveys in nine countries showed 80% of surface waters were contaminated with neonicotinoids at levels of 0.14 to 18 μg L−1,4 which are sublethal to aquatic arthropods.Among the seven commercial neonicotinoid insecticides, acetamiprid (AAP) belongs to the chloronicotinyl subclass and its widespread use is due to its high stability and insecticidal activity. However, recent studies suggested that AAP causes acute and chronic toxicity in mammals and, moreover, AAP has become the focus of attention because of its apparent toxicity to bees.5,6 In addition, AAP is highly water soluble at 4.25 g L−1, which is 20-fold higher than another neonicotinoid, thiacloprid (THI), containing the same pharmacophore cyanoguanidine moiety. Because of this AAP easily leaches into surface water and groundwater, and therefore AAP residues in aqueous environments have been detected in some recent reports. For example, Leandro et al. detected AAP in shallow groundwater in a cotton-growing region of Mato Grosso, Brazil.7 Anderson et al. reported AAP detection in the playa wetlands of Texas.8 Wu et al. detected AAP at a concentration of 0.2 μg L−1 in the Jingmi River, Beijing, China.9 Struger reported that AAP was detected at concentrations greater than the guideline value in the surface waters of southern Ontario, Canada.10 AAP also persists regardless of bio-treatment and has been found in wastewater treatment plant effluents in concentrations ranging from 50 ng L−1 to 16 μg L−1.11 The AAP residues in water may affect the sensitive aquatic invertebrates living in it and therefore produce risks to the ecosystem.
AAP is known to be stable under alkaline conditions and can persist for more than 30 d.12 Elimination of AAP residues from the environment by chemical and photochemical methods have been largely reported. Khan et al. reported heterogeneous photocatalysed degradation of AAP in aqueous suspensions of a semiconductor.13 Mitsika et al. employed the Fenton reaction to oxidize AAP in water samples.14 However, chemical degradation of AAP is expensive, environmentally harmful, and requires extreme conditions. In contrast, microbial degradation could avoid these disadvantages and is a promising approach for the remediation of AAP contamination. Many microorganisms have been reported to degrade AAP. For example, the yeast Rhodotorula mucilaginosa IM-2 transformed AAP to IM-1-3.15 The bacterium Ensifer meliloti CGMCC 7333 degraded AAP to IM-1-2.16 Pigmentiphaga sp. AAP-1 and D-2 and Stenotrophomonas sp. THZ-XP degraded AAP to form IM-1-4.17–19 The bacterium Rhodococcus sp. BCH2 degraded AAP to IM-1-4, which was further degraded to IC-O.20 The bacterium Stenotrophomonas maltophilia CGMCC 1.1788 degraded AAP via N-demethylation to form IM 2-1.21 In a previous study, we reported that the plant growth-promoting rhizobacterium Variovorax boronicumulans CGMCC 4969 degraded thiacloprid to its amide metabolite and the carcinogen acrylamide to acrylic acid.22,23 Bacteria belonging to the genus Variovorax are common inhabitants of water and soil. Variovorax species display diverse metabolic features and catabolic capabilities making them a promising choice for applications in bioremediation studies. For example, V. paradoxus was used as a bioaugmentation agent for the bioremediation of pesticide linuron-contaminated soils.24 Because there have been no reports on AAP biodegradation in aqueous environments, we primarily investigated the ability of V. boronicumulans CGMCC 4969 to bioremediate AAP contamination in surface water samples. Meanwhile, the genomic DNA of V. boronicumulans CGMCC 4969 has been sequenced to identify the corresponding gene clusters involved in AAP degradation and these genes were functionally over-expressed in Escherichia coli. The characteristics of the CGMCC 4969 enzyme over-expressed in E. coli that was responsible for AAP degradation were studied in detail.
2. Experimental
2.1 Chemicals and media
AAP and THI were obtained from Jiangsu Pesticide Research Institute Company Ltd., Nanjing, China (>97% purity). Indole-3-acetonitrile (IAN), benzonitrile, butyronitrile, 2-cyanopyridine, 3-cyanopyridine, hexanedinitrile, isobutyronitrile, and succinonitrile were purchased from Sigma-Aldrich (Shanghai, China; each of 98% purity). HPLC grade acetonitrile and methanol were purchased from TEDIA (Fairfield, OH, USA). All other reagents were of analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). The broth for cell cultivation was Luria–Bertani medium containing 10.0 g of peptone, 5.0 g of yeast extract, and 10.0 g of NaCl per liter of water. MSM medium contained 1.36 g of KH2PO4, 2.13 g of Na2HPO4, 0.50 g of MgSO4·7H2O, and 10 mL of metal solution in 1.0 L of deionized water (pH 7.5). The metal solution contained 0.40 g of CaCl2·2H2O, 0.30 g of H3BO3, 0.04 g of CuSO4·5H2O, 0.10 g of KI, 0.20 g of FeSO4·7H2O, 0.40 g of MnSO4·7H2O, 0.20 g of NaMoO4·2H2O, and 10.0 mL of concentrated HCl in 1.0 L of deionized water.2.2 Bacterial strains and plasmids
The wild bacterium V. boronicumulans CGMCC 4969 was deposited in the China General Microbiological Culture Collection Center (CGMCC) (Beijing, China). Escherichia coli Rosetta (DE3) was used as a host strain for gene over-expression and stored in our laboratory. The plasmids pET28a (+) and pET21a (+) (Novagen, Inc., Madison, WI, USA) were used as expression vectors.2.3 Biodegradation of AAP by V. boronicumulans CGMCC 4969 in surface water samples
Resting cells of V. boronicumulans CGMCC 4969 were used for AAP degradation and their preparation was described in our previous report.25 These resting cells were used to determine the AAP degradation in surface water samples. The water samples were collected from CaiYue Lake, Nanjing, China. The physicochemical properties of the collected water were: pH 7.0; total phosphorus of 0.08 mg L−1, total Kjeldahl nitrogen of 2.24 mg L−1, and chemical oxygen demand of 4.37 mg L−1. The water samples were filtered through a sterilized 0.22 μm membrane. 20 mL of each water sample were poured into a 100 mL flask and then the resting cells of V. boronicumulans CGMCC 4969 were added to a final concentration of 1.3 × 108 cell per mL while the final AAP concentration was 2 mg L−1. AAP degradation broth excluding bacterial cells was used as a control. The experiments were conducted under the above cultivation conditions. Every 24 h, 1 mL of the broth was collected and centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for 10 min to remove residual cells, and the supernatant was filtered using a 0.22 μm membrane prior to HPLC analysis.
000 g for 10 min to remove residual cells, and the supernatant was filtered using a 0.22 μm membrane prior to HPLC analysis.
2.4 Chemical analysis
The degradation of AAP was analyzed using an Agilent 1200 HPLC system equipped with an HC-C18 column (4.6 × 250 mm, Agilent Technologies, Santa Clara, CA, USA). The mobile phase contained water, acetonitrile, and 0.01% acetic acid (water/acetonitrile, 65:35). The flow rate of the mobile phase was 1 mL min−1. The signal was monitored at a wavelength of 235 nm. LC-MS was conducted using an Agilent 1290 HPLC-DAD and an Agilent 6460 HPLC-q3MS/MS system equipped with an electrospray ion source (Agilent Technologies, Wilmington, DE, USA) that was operated in the positive ion mode. For the other aromatic nitriles, the HPLC conditions were the same as those in a previous report.26 The 1H and 13C NMR spectra for the metabolite were obtained by using a Bruker AV-400 NMR spectrometer (Bruker, Faellanden, Switzerland) operating at 400 and 100 MHz, respectively. The solvent for NMR analysis was dimethylsulfoxide-d6. Chemical shifts were referenced against internal tetramethylsilane. Several NMR techniques were used to assign proton and carbon atom chemical shifts (δ), including distortionless enhancement by polarization transfer, heteronuclear single-quantum correlation spectroscopy, and hetero-nuclear multiple bond correlation.
2.5 RNA extraction and quantitative PCR
| Target genes | Primer | Sequence (5′–3′) | Amplicon size (bp) |
|---|---|---|---|
| NHase | NHa-F | GCCAATACCGACGACCAGCAC | 172 |
| NHa-R | TGGTCTCGGGCAAGGTGGT | ||
| 16S | 16S-F | TACTGGGCGTAAAGCGTGCG | 174 |
| 16S-R | ATTGCCTTCGCCATCGGTGT |
2.6 Construction of plasmids containing different components of the CGMCC 4969 ANHase gene cluster
A MiniBEST bacterial genomic DNA extraction kit (TaKaRa, Dalian, China) was employed for the extraction of the genomic DNA of V. boronicumulans CGMCC 4969. The sequence of the genomic DNA was determined by the Huada Genomics Institute (Shenzhen, China). Plasmids containing different components of the ANHase gene cluster (Fig. 1) were constructed to clarify the functions of the independent genes in the ANHase gene cluster responsible for AAP degradation. The primers are listed in Table 2 and were synthesized by Sangon Biotech (Shanghai) Co., Ltd. | ||
| Fig. 1 Construction of recombinant plasmids and over-expression strains containing different components of the CGMCC 4969 ANHase gene cluster and the compositions of the ANHase gene clusters in V. boronicumulans CGMCC 4969 and E. meliloti CGMCC 7333. | ||
| Primer | Sequence (5′–3′) |
|---|---|
| a Underlined bases indicate the restriction enzyme sites of EcoRI (GAATTC) and XhoI (CTCGAG), respectively. Boldface bases indicate the homologous fragment used in the ligation between the target fragment and the expression vector. | |
| P1-F | ACAGCAAATGGGTCGCGGATCCGAATTCATGACCGGCCATGACCACTCCCAC |
| P1-R | ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGTGCCGCGGGCTCCAGGTAGCTT |
| P2-F | ACAGCAAATGGGTCGCGGATCCGAATTCATGACCGGCCATGACCACTCCCAC |
| P2-R | ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGGCCCGGAAAGTCTTCGCCGC |
| P3-F1 | ACAGCAAATGGGTCGCGGATCCGAATTCATGACCGGCCATGACCACTCC |
| P3-R1 | ATGGTATATCTCCTTCTTAAAGTTCTATGCCGCGGGCTCCAGGTAG |
| P3-F2 | AGAACTTTAAGAAGGAGATATACCATATGAAGGCGAACGACATGCCG |
| P3-R2 | ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGGCCCGGAAAGTCTTCGCCGC |
| P4-F | ACAGCAAATGGGTCGCGGATCCGAATTCATGAAGGCGAACGACATGCCG |
| P4-R | ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGCTAGCCCGGAAAGTCTTCGCC |
| P6-F1 | ACAGCAAATGGGTCGCGGATCCGAATTCATGACCGGCCATGACCACTCCCAC |
| P6-R1 | GTGAGTCGTATTAATTTCGCGGGATCTATGCCGCGGGCTCCAGGTAG |
| P6-F2 | TAGATCCCGCGAAATTAATACGACTC |
| P6-R2 | ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGCTAGCCCGGAAAGTCTTCGC |
To investigate the influence of the downstream hypothetical protein (ANHc) on ANHase activity, two plasmids named pNAB and pNABC containing ANHase structural genes without and with anhC were constructed, respectively (Fig. 1). The ANHase structural α and β genes (anhAB) and anhAB with anhC (anhABC) were amplified by polymerase chain reaction (PCR) using primer pairs P1 (P1-F and P1-R) and P2 (P2-F and P2-R), which have a pET28a homologous base located before the corresponding restriction enzyme sites, EcoRI and XhoI. The PCR system was composed of 0.4 μL of genomic DNA template of CGMCC 4969 (100 ng μL−1), 0.4 μL of each primer (20 mmol L−1), 4 μL of PrimerSTAR buffer (Mg2+ plus, 5×), 0.2 μL of PrimerSTAR DNA polymerase (2.5 U μL−1, Takara), 2 μL of dNTP (2.5 mmol L−1 each), and 12.6 μL of sterilized distilled water. The total volume of the reaction mixture was 20 μL. After an initial pre-denaturation for 5 min at 95 °C, the cycling conditions were as follows: denaturation for 50 s at 95 °C, annealing for 40 s at 60 °C, and extension for 2 min at 72 °C. A final 10 min extension step at 72 °C was performed after 29 cycles. The PCR product was analyzed using 1% agarose gel electrophoresis. The anhAB or anhABC fragment was ligated to the expression plasmid pET28a, which was performed according to the protocol of the ClonExpress II one step cloning kit (Vazyme Biotech, Nanjing, China). The recombinant plasmids pNAB and pNABC were verified using DNA sequencing by Springen Biotech (Nanjing, China).
To examine the influence of the enhanced expression of anhC on the ANHase activity for AAP hydration, the efficient Shine–Dalgarno (SD) sequence of the T7 phage from the pET expression system was introduced to the 5′-terminus of anhC by overlap extension PCR. First, anhAB was amplified by PCR using primers P3-F1 and P3-R1; meanwhile, anhC was amplified using primers P3-F2 and P3-R2. The SD sequence was added to the downstream primer P3-R1 and upstream primer P3-F2 as a homologous fragment. Finally, the two DNA fragments were combined using the outer primers P3-F1 and P3-R1, and the resulting fragment was named anhABDC. In addition, the strong T7 promoter was also introduced to the 5′-terminus of anhC according to the method referred to above, except that the primers P6-F1, P6-R1, P6-F2, and P6-R2 were used and the homologous fragment was the T7 promoter. The resulting DNA fragment was named anhABT7DC. The fragments anhABDC and anhABT7DC were inserted into pET28a according to the above-mentioned protocol, and the obtained plasmids were named pNABDC and pNABT7DC, respectively.
We also constructed a double-plasmids expression system employing plasmids pET28a and pET21a. The anhC fragment was independently inserted into the expression vector pET21a using primers P4-F and P4-R to obtain the plasmid pNC, and then the two plasmids were co-transformed in the host E. coli Rosetta (DE3).
2.7 Expression and purification of recombinant ANHase in E. coli Rosetta (DE3) pLysS
Transformation of the recombinant plasmids to the competent cells of E. coli Rosetta (DE3) pLysS and over-expression of the recombinant ANHase were conducted according to the method described in our previous report.25 To investigate degradation by the E. coli resting cells containing different anhs, the over-expressed strains were washed twice with 0.2 mol L−1 sodium phosphate buffer (pH 7.5) and then resuspended in the same buffer containing 500 mg L−1 AAP, and the optical density of the cells at 600 nm was adjusted to 3. After transformation for 10 min at 30 °C, each sample was analyzed by HPLC. To further explore the expression of the different recombinant anhs, equal amounts of cell pellets were suspended in a 1 mL volume of 0.2 mol L−1 phosphate buffer and disrupted by sonication (10 s) for 3 min at ice temperature. The soluble and insoluble fractions were separated by centrifugation at 12000 g for 10 min at 4 °C prior to recombinant protein detection. The purification of the ANHase by His-tag affinity chromatography was conducted according to the protocol of the manufacturer of the chromatography resin (Novagen, Inc.). Western blot and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) were carried out according to the method described by Yang et al.27
2.8 Enzyme assay
The standard assay was carried out by mixing 500 mg L−1 AAP and an appropriate amount of enzyme in 0.2 mol L−1 sodium phosphate buffer (pH 7.5). The reaction mixtures with total volumes of 1 mL were incubated for 10 min at 37 °C and quenched by addition of 10% (v/v) 2 mol L−1 hydrochloric acid. Then, the samples were centrifuged at 12000 g for 10 min and the supernatants were analyzed by HPLC. The ANHase activity toward aliphatic nitrile compounds was assessed by quantifying the amount of ammonia released during the reaction according to the methods reported by Okamoto and Eltis.28 The ANHase reaction was coupled with an aliphatic amidase from CGMCC 4969 (GenBank accession number AFP67897), which was heterologously produced in E. coli Rosetta with a His-tag at the N terminus and purified using a Ni-NTA resin. One unit (U) of ANHase activity was defined as the amount of enzyme that catalyzed the formation of 1 μmol of product in 1 min.
2.9 Characteristics of the purified recombinant ANHase
The optimal pH was determined by adding AAP to different 0.2 mol L−1 sodium citrate (pH 4–6), phosphate (pH 6–8), and Tris–HCl (pH 8–10) buffers to a final concentration of 500 mg L−1. For pH stability, the enzyme was incubated at 4 °C for 12 h in buffers at different pH values and the residual ANHase activity was determined using the method previously described. Reaction mixtures at temperatures from 20 to 60 °C at the optimal pH were used to determine the optimal temperature of ANHase activity. For thermal stability, the enzyme was pre-incubated at different temperatures for 1 h, and the non-heated ANHase was used as the control and its activity was defined as 100%.The effects of metal ions on ANHase activity were determined by individually adding CoCl2, CaCl2, CuCl2, FeCl2, MnCl2, ZnCl2, MgCl2, and AgNO3 at a final concentration of 1 mmol L−1. Acetone, ethanol, ethyl acetate, dichloromethane, hexane, methanol, and isopropanol (at a volume ratio of 2%) were individually added to the standard reaction mixture to test the effects of organic reagents on ANHase activity. Enzyme activity without any additives was used as the control and its activity for AAP was defined as 100%.
To broaden the application spectrum of the CGMCC 4969 ANHase, THI, IAN, 2-cyanopyridine, 3-cyanopyridine, benzonitrile, and the aliphatic nitriles acetonitrile, butyronitrile, isobutyronitrile, succinonitrile, and hexanedinitrile were used as substrates. Each reaction was conducted in a 1 mL mixture containing 2 mmol L−1 substrate and an appropriate amount of enzyme.
The kinetic parameters for the activity of the ANHase toward AAP were calculated by determining the initial velocity of AAP hydration in the range of from 0.2 to 5 mmol L−1 AAP. The maximal hydration rate (Vmax) and apparent Michaelis–Menten constant (Km) were deduced from Lineweaver–Burk plots.
3. Results and discussion
3.1 Biodegradation of AAP by V. boronicumulans CGMCC 4969 in the culture medium and surface water
The ability of the resting cells of V. boronicumulans CGMCC 4969 to degrade AAP was primarily examined in phosphate buffer. As shown in Fig. 2A–C, after transformation for 48 h, a new polar peak at retention time of 3.91 min appeared, whereas no comparable peak was observed upon analysis of controls containing the bacterium or AAP alone, and 41.6% of AAP was degraded (Fig. 2D). LC-MS analysis indicated that the metabolite displayed a protonated parent ion (M + H) at m/z 241, a sodiated adduct (M + Na) at m/z 263, a fragment ion (M − CHON) at m/z 198, a fragment ion (M − C3H2NO) at m/z 157, a fragment ion (M − C4H8N3O) at m/z 126, and a fragment ion (M − C6H6N2Cl) at m/z 99. The 13C NMR data of the purified metabolite were as follows: 165.4, 160.8, 149.5, 149.4, 139.5, 133.7, 124.5, 49.3, 36.1, and 16.4; The 1H NMR data of the purified metabolite were as follows: 8.33 (s, 1H), 7.76 (d, 1H, J = 7.2 Hz), 7.48 (d, 1H, J = 8.4), 6.29, 6.13 (b, 2H), 4.61 (s, 2H), 2.91 (s, 3H), and 2.12 (s, 3H). The mass and NMR data of the metabolite formed by AAP degradation by V. boronicumulans CGMCC 4969 were found to be identical to those reported for (E)-N2-carbamoyl-N1-[(6-chloro-3-pridyl)methyl]-N1-methylacetamidine (usually denoted as IM-1-2),16 and therefore, the metabolite was identified as IM-1-2.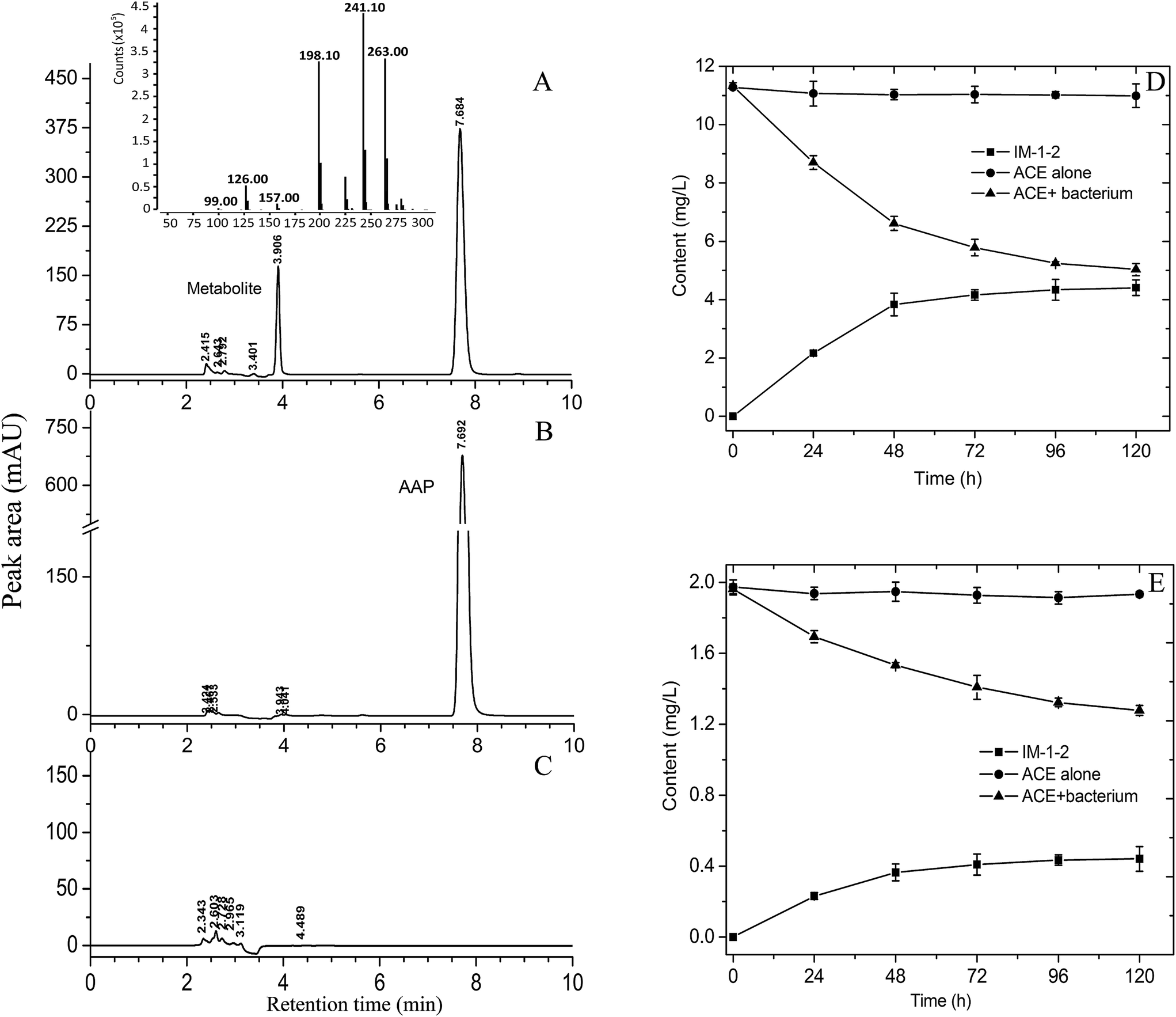 | ||
| Fig. 2 HPLC and LC-MS analyses and the time course of AAP biodegradation by resting cells of V. boronicumulans CGMCC 4969. (A) Transformation broth with inoculation of bacterium and addition of AAP; (B) transformation broth with AAP alone; (C), transformation broth with inoculation of bacterium alone. (D) Resting cells with inoculation of bacterium (2.5 × 109 cell per mL) and AAP (12 mg L−1) in 50 mmol L−1 phosphate buffer. (E) Resting cells with inoculation of bacterium (1.28 × 108 cell per mL) and AAP (2 mg L−1) in water samples. | ||
As shown in Fig. 2E, after inoculating V. boronicumulans CGMCC 4969 in surface water and incubating for 5 days, the AAP content was reduced from 1.96 mg L−1 to 1.28 mg L−1 and the degradation rate was 34.7%. AAP degradation fitted first-order dissipation kinetics (R = 0.99) with a half-life of 182 h. In contrast, AAP was barely degraded in control water without bacterial inoculation. We previously isolated an E. meliloti CGMCC 7333, which could degrade 65.1% of 500 mg L−1 AAP in 96 h with a half-life of 63 h.16 However, this strain showed weak AAP degradation in surface water samples (data not shown). E. meliloti (formerly Sinorhizobium meliloti) is a symbiotic nitrogen-fixing bacterium, and is usually limited by application of microbes–plants combined remediation. In contrast, Variovorax spp. are common inhabitants of soil and water environments, and therefore V. boronicumulans CGMCC 4969 showed potential ability to degrade AAP in water.
The metabolism of AAP in plants, animals, and soils involves three pathways: N-demethylation of AAP to IM-2-1; oxidative cleavage of the cyanoimine group to form IM-1-3, and subsequent N-deacetylation to form IM-1-4; hydration of AAP to the N-carbamoylimine derivative (IM-1-2), which is spontaneously hydrolyzed to IM-1-4.29 As shown in Fig. 2D, 0.44 mg L−1 IM-1-2 was formed and the molar conversion rate (the amount of IM-1-2 formed divided by the amount of AAP reduced) was 66.2%, indicating that AAP hydration to IM-1-2 is the major metabolic pathway in the degradation of AAP in surface water by V. boronicumulans CGMCC 4969. This metabolic pathway is different from that of biodegradation of AAP by Micrococcus luteus SC 1204, which occurs via the formation of the benzothiazole 2-(2-hydroxyethylthio) metabolite.30
3.2 Bioinformatics analysis of the V. boronicumulans CGMCC 4969 ANHase gene cluster
A typical strain of the genus Variovorax, Variovorax paradoxus S110, has two ANHase gene clusters: one involving three genes (Vapar_1665, Vapar_1666, and Vapar_1667) and another with two genes (Vapar_2111 and Vapar_2112). In contrast, V. paradoxus EPS has only one ANHase gene cluster with three genes (Varpa_1815, Varpa_1816, and Varpa_1817). In the present study, genomic DNA sequencing analysis indicated that there is only one ANHase coding gene cluster in the genome of V. boronicumulans CGMCC 4969, which is composed of three genes (Fig. 1), a 642 bp α-subunit coding gene (named anhA), a 666 bp β-subunit coding gene (named anhB), and a 378 bp hypothetical protein coding gene (named anhC) (GenBank accession number KC460345). During the microbial metabolism of nitrile compounds, ANHase converts the nitrile compound to an amide intermediate and an amidase subsequently converts the amide product to an acid metabolite, and therefore the amidase coding gene is usually located at a site close to the ANHase gene. For example, in the low molecular mass ANHase gene cluster of Rhodococcus rhodochrous J1, an amidase encoding gene amdA was located 1.9 kb downstream of the ANHase coding gene nhlBA,31 and an amidase gene was found to be located upstream of the ANHase coding gene in Pseudomonas chlororaphis B23.32 However, as shown in Fig. 2, there is no adjacent amidase gene in the ANHase gene cluster of CGMCC 4969. The genes upstream, anhA, code the enzymes related to folic acid synthesis and the genes downstream, anhC, code an oxidoreductase and an uncharacterized membrane protein (Fig. 1). The same results, namely that no amidase coding gene was located near the ANHase gene cluster, were observed in another AAP-degrading bacterium, Ensifer meliloti 1021 (Fig. 1). Both CGMCC 4969 and CGMCC 7333 have the same rank of anhA (α-subunit), anhB (β-subunit), and anhC (accessory protein) and have the same overlapping sequence “ATGA” between anhA and anhB. However, the overlapping sequence “ATGA” also appeared between anhB and anhC of CGMCC 4969, whereas a 14 bp overlapping sequence “TTGAACCCGCATAG” appeared between anhB and anhC of CGMCC 7333.Protein blast analysis indicated that ANHc exhibits the maximum identity of 95% with the ANHase accessory protein of Variovorax sp. Root473 (GenBank accession number AGI48675), and about 75% identity with the ANHase accessory protein of Bradyrhizobium elkanii (GenBank accession number WP_028165648). However, CGMCC 4969 ANHc only has 41.7% identity with the ANHase accessory protein of E. meliloti CGMCC 7333 (GenBank accession number KF601244), a nitrogen-fixing bacterium shown to be capable of degrading AAP.16
3.3 Transcript analysis of the ANHase gene cluster in V. boronicumulans CGMCC 4969
To further analyze the influence of AAP on the expression of the ANHase gene cluster, MSM medium containing AAP as the only nitrogen source was used to cultivate V. boronicumulans CGMCC 4969 cells. As shown in Fig. 3, AAP treatment significantly upregulated the expression of the ANHase gene by 1.6-fold, when compared with that in the control, suggesting the participation of V. boronicumulans CGMCC 4969 cells in the degradation of AAP. | ||
| Fig. 3 mRNA expression levels of the ANHase gene of V. boronicumulans CGMCC 4969. CK, control; AAP, addition of AAP to the culture medium. Error bars represent the standard deviations of three replicates. | ||
3.4 Effect of co-expression of anhC with anhAB and the effect of the expression level of anhC on ANHase activity for AAP hydration
The control E. coli-pET28a that did not harbor the ANHase gene cluster did not convert AAP to IMI-1-2 (ESI Fig. S1†), while E. coli-pET28a-anhAB (strain P1) could make this conversion, which indicates that CGMCC 4969 ANHase was responsible for AAP degradation to IM-1-2. As shown in Fig. 4, E. coli-pET28a-anhABC (strain P2) showed an 18-fold higher ANHase activity than strain P1. SDS-PAGE analysis (Fig. 5A) indicated that the P1 ANHase was mainly expressed with the inclusion body as compared to the total protein fraction (lane P1T) with the soluble protein fraction (lane P1S). In lane P2T, the over-expressed ANHc with molecular weight of 14 kDa can be clearly observed. The total protein expression level of ANHa was not higher, whereas the soluble fraction (lane P2S) was significantly higher, when compared with P1. These results prove that ANHc plays an important role in the maturation of CGMCC 4969 ANHase by improving its solubility and therefore increasing its enzymatic activity.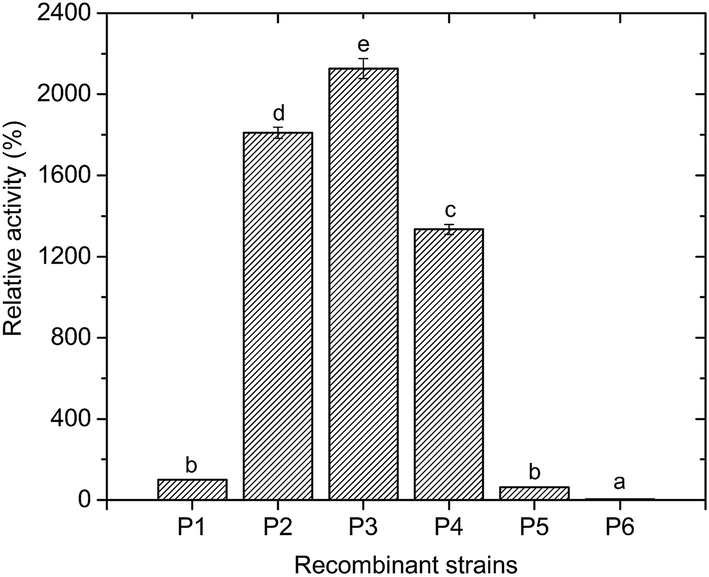 | ||
| Fig. 4 Activity analysis of recombinant E. coli Rosetta strains containing different components of the CGMCC 4969 ANHase gene cluster. The ANHase activity was assayed under the standard assay conditions with AAP as the substrate. Different letters (a–e) above the columns represent significant differences at p ≤ 0.05 according to the Duncan test. | ||
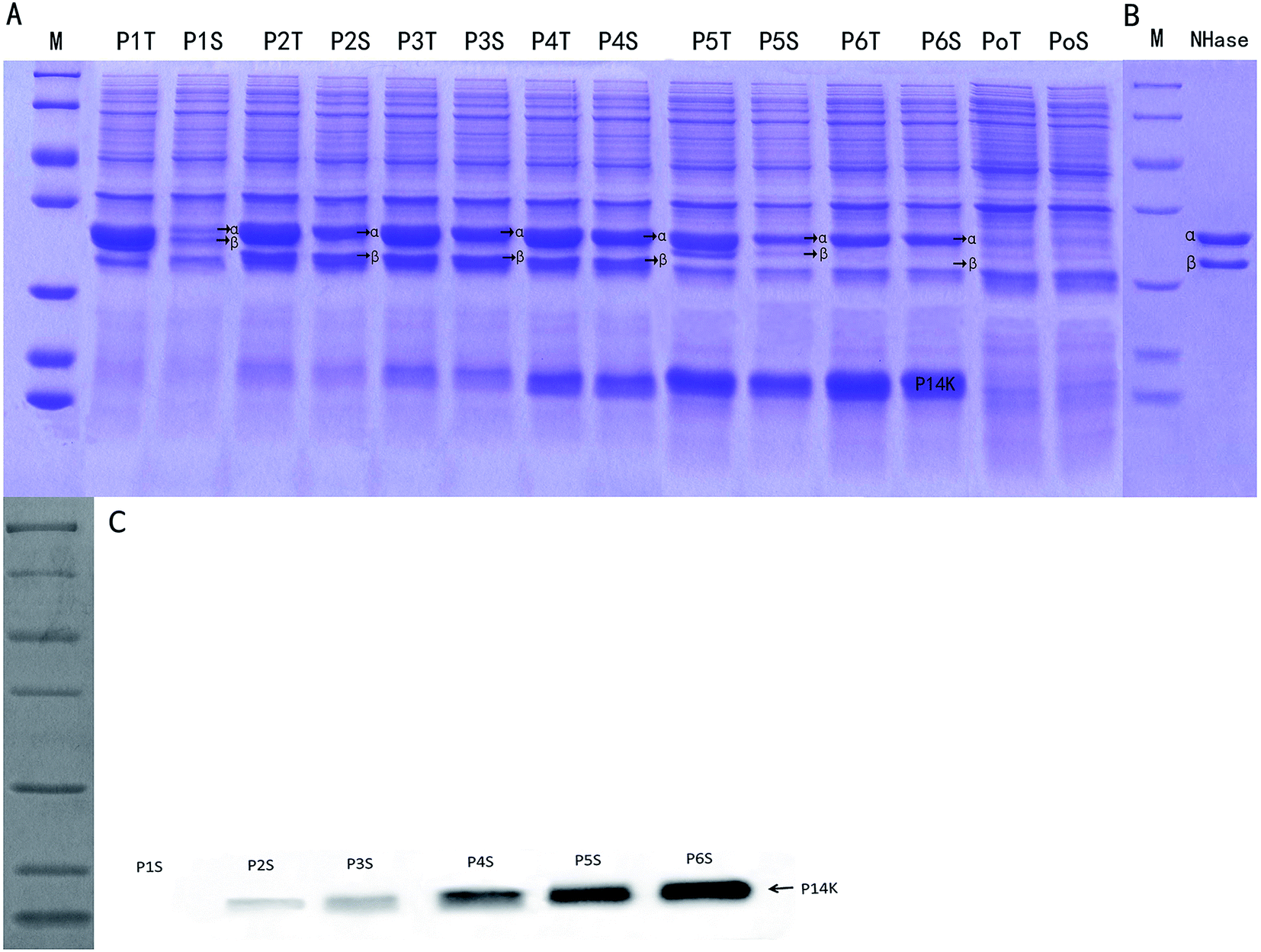 | ||
| Fig. 5 SDS-PAGE and western blotting analyses of different components of the CGMCC 4969 ANHase gene cluster over-expressed in E. coli Rosetta and the purified recombinant ANHase. (A) SDS-PAGE of different components of the CGMCC 4969 ANHase gene cluster; lanes P1T, P2T, P3T, P4T, P5T, P6T represent the total protein of E. coli Rosetta (DE3) P1–P6, and lanes P1S, P2S, P3S, P4S, P5S, P6S represent the soluble protein of E. coli Rosetta (DE3) P1–P6, respectively; lanes PoT and PoS represent the control E. coli Rosetta (DE3) containing the pET28a plasmid alone; lane M represents standard protein markers (116.0, 66.2, 45.0, 35.0, 25.0, 18.4, and 14.4 kDa from top to bottom); (B) purified recombinant ANHase from strain P3; (C) western blotting analysis of the expression levels of the activator. A 6× histidine tag was added to the N-terminus of the α-subunit and C-terminus of the β-subunit of strains P1, P5 and the N-terminus of the α-subunit and C-terminus of the activator for strains P2, P3, P4, P6. | ||
The SD fragment, the RNA polymerase binding site, was introduced between anhB and anhC, and the constructed plasmid pNABDC was over-expressed in E. coli (named strain P3). As shown in Fig. 5, lanes P3T and P3S, the expression level of the activator protein ANHc was further increased compared to P2 as shown by SDS-PAGE (Fig. 5A) and western blotting analysis (Fig. 5C). The ANHase activity of P3 for AAP hydration was the highest and was 21-fold higher than that of P1 and 1.2-fold higher than that of P2. Increasing expression levels of ANHc coincided with promotion of ANHase activity. Therefore, we further engineered the independent over-expression of plasmid pNABT7DC, containing the T7 promoter sequence before the SD sequence, and plasmid pNC with the insertion of only anhC into pET21a. The pNC plasmid was co-expressed in E. coli with pNAB or pNABC. As shown in Fig. 5A, the strains P4, P5, and P6 are associated with gradually rising amounts of ANHc (lanes P4S, P5S, and P6S), and their expression levels were higher than those of P3 according to western blotting analysis (Fig. 5C). However, on the contrary, the ANHase activities of P4, P5, and P6 for AAP hydration were lower than that of strain P3 (Fig. 4). Strains P5 and P6 exhibited even lower ANHase activity than strain P1 without co-expression of ANHc. Comparing the ANHase expression of P5 with P1, the amount of ANHa in P5 (lane 5S) was higher than that in P1, whereas the amount of ANHb in P5 was lower than that in P1. Because the active ANHases exist as α2β2 tetramers and bind one metal atom per αβ unit, the reduced amount of ANHb in P5 decreased its ANHase activity to 62% of the activity of P1. Strain P6 exhibited the highest expression level of ANHc, whereas the expression of ANHb was hardly observed in lane 6S, and HPLC analysis indicated that P6 had very little ANHase activity for AAP hydration (Fig. 4).
We also studied the effect of co-expression of the E. meliloti CGMCC 7333 anhC with anhAB on the ANHase activity for AAP hydration. The results indicate that strain P2 of CGMCC 7333 only had 4-fold higher ANHase activity than strain P1 (data not shown). In the present study, strain P2 with over-expressed CGMCC 4969 anhABC had 18-fold higher ANHase activity than strain P1 with over-expressed anhAB. We speculate that the poor enhancement of ANHase activity by CGMCC 7333 ANHc was due to the longer overlapping sequence between CGMCC 7333 anhB and anhC compared to that of CGMCC 4969. In strain P5, although the expression level of ANHc was much higher than in P2, the enhanced ANHc expression did not increase the ANHase activity, which was 30-fold lower than the activity of P2 and 38% lower than that of P1. As compared with strain P1, harboring pNAB, the independent plasmid pNC (pET21a-anhC) was further introduced to P5. The independent over-expression of ANHc displayed an inhibitory effect on the level of the soluble protein fraction of ANHb but a promoting effect on that of ANHa (Fig. 5 lane 5S). These reverse effects of ANHc on the expressions of ANHa and ANHb have not been reported in previous studies and the mechanism needs further exploration.
3.5 Biochemical properties of the recombinant ANHase
Although the kinetic parameters of the ANHase toward various nitriles have been extensively reported,33 parameters for the neonicotinoid insecticide AAP degradation have rarely been cited. As shown in Fig. 6, the Km and Vmax values of the ANHase for AAP were 21.83 mmol L−1 and 4.84 μmol min−1 mg−1, respectively.
 | ||
| Fig. 6 The kinetic constants of CGMCC 4969 ANHase for the degradation of AAP. The kinetic parameters of the ANHase toward AAP were determined over a range of substrate concentrations (0.2–5 mmol L−1) under standard assay conditions. | ||
| Substrate | Relative activity ± standard deviation (%) |
|---|---|
| a ANHase activity was determined under standard assay conditions. The specific activity of AAP (0.67 U mg−1) was taken as 100%. | |
| ACE | 100 ± 1.61 |
| THI | 230.16 ± 4.24 |
| IAN | 1501.19 ± 39.62 |
| 2-Cyanopyridine | 5068.10 ± 45.77 |
| 3-Cyanopyridine | 7144.42 ± 110.42 |
| Benzonitrile | 5216.56 ± 72.81 |
| Acetonitrile | 35.03 ± 1.72 |
| Butyronitrile | 19.35 ± 0.71 |
| Isobutyronitrile | 3.82 ± 0.12 |
| Succinonitrile | 25.99 ± 2.03 |
| Hexanedinitrile | 1.81 ± 0.22 |
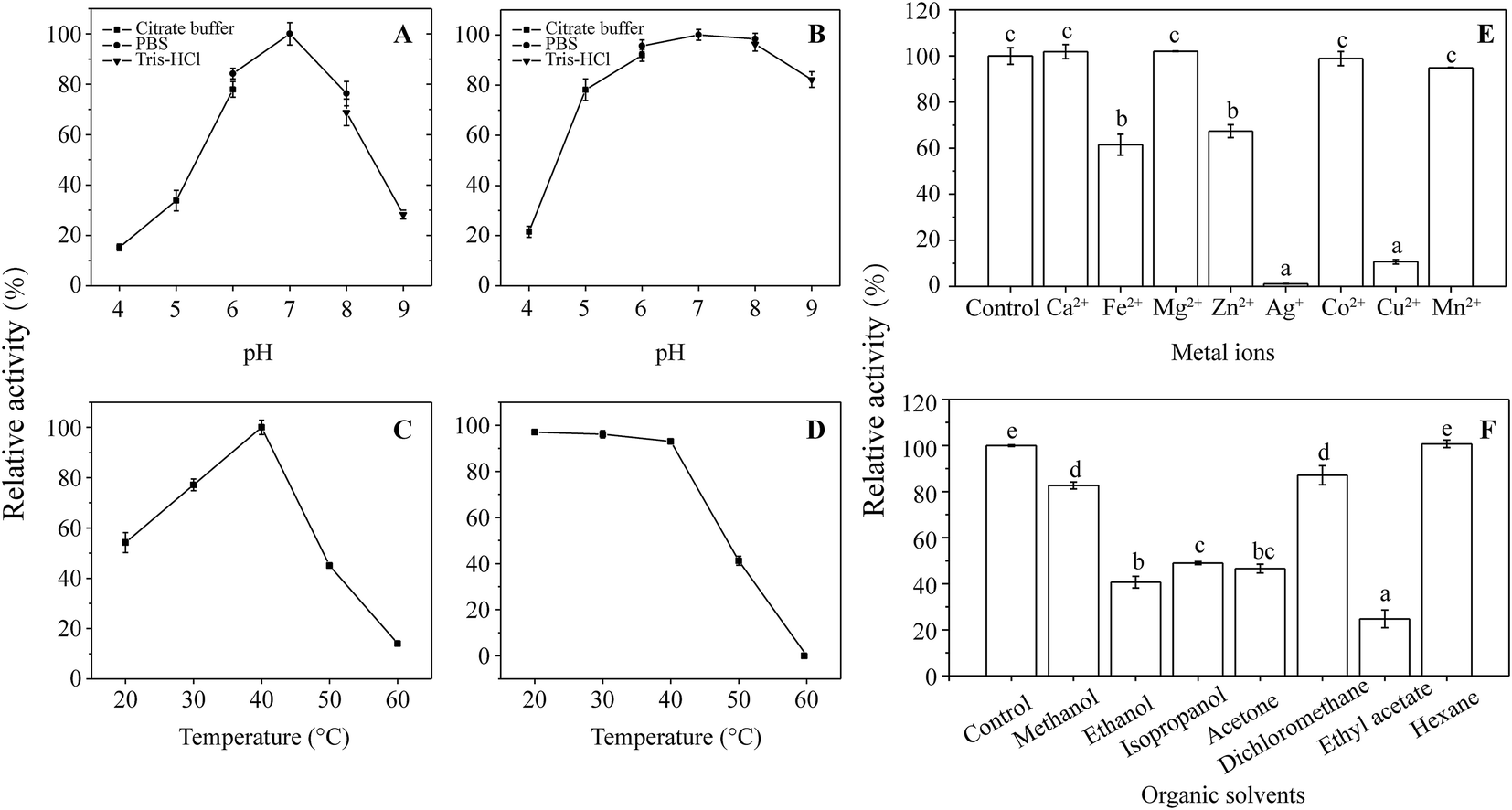 | ||
| Fig. 7 Enzymatic characterization of the purified recombinant ANHase. (A) Effects of pH on the activity of ANHase; (B) effects of pH on the stability of ANHase; (C) effects of temperature on the activity of ANHase; (D) effects of temperature on the stability of ANHase; (E) effects of metal ions on the activity of ANHase; (F) effects of organic solvents on the stability of ANHase. Different letters (a–e) above the columns represent significant differences at p ≤ 0.05 according to the Duncan test. | ||
As shown in Fig. 7F, ethanol, isopropanol, acetone, and ethyl acetate inhibited ANHase activity by more than 50% compared with the control. Methanol and dichloromethane retained >80% activity when 2% (v/v) organic solvent was added to the reaction system. In contrast, hexane, with a logP value of 3.5, displayed no inhibition of ANHase activity, which may because biocatalysis is likely to occur with solvents of higher logP values and inhibition in the presence of hydrophilic solvents (logP < 2.0).41
4. Conclusions
In the present study, we found the neonicotinoid insecticide AAP was degraded to IM-1-2 in surface water by V. boronicumulans CGMCC 4969, and this was mediated by ANHase. Co-expression of downstream anhC apparently improved ANHase activity 21-fold, but independent over-expression of anhC inhibited the expression of the β-subunit, which resulted in decreasing ANHase activity. CGMCC 4969 ANHase is a versatile ANHase that is capable of hydrating aromatic, N-heterocyclic, and aliphatic nitriles.Acknowledgements
This research was financed by the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions, the National Science Foundation of China (grant No. 31570104), and the Academic Natural Science Foundation of Jiangsu Province (grant No. 14KJA180004).References
- P. Jeschke, R. Nauen, M. Schindler and A. Elbert, J. Agric. Food Chem., 2011, 59, 2897–2908 CrossRef CAS PubMed.
- M. Tomizawa, D. L. Lee and J. E. Casida, J. Agric. Food Chem., 2000, 48, 6016–6024 CrossRef CAS PubMed.
- D. Goulson and D. Kleijn, J. Appl. Ecol., 2013, 50, 977–987 CrossRef.
- C. A. Morrissey, P. Mineau, J. H. Devries, F. Sanchez-Bayo, M. Liess, M. C. Cavallaro and K. Liber, Environ. Int., 2015, 74, 291–303 CrossRef CAS PubMed.
- D. Gibbons, C. Morrissey and P. Mineau, Environ. Sci. Pollut. Res. Int., 2015, 22, 103–118 CrossRef CAS PubMed.
- L. W. Pisa, V. Amaral-Rogers, L. P. Belzunces, J. M. Bonmatin, C. A. Downs, D. Goulson, D. P. Kreutzweiser, C. Krupke, M. Liess, M. McField, C. A. Morrissey, D. A. Noome, J. Settele, N. Simon-Delso, J. D. Stark, J. P. Van der Sluijs, H. Van Dyck and M. Wiemers, Environ. Sci. Pollut. Res. Int., 2015, 22, 68–102 CrossRef CAS PubMed.
- C. Leandro, V. Souza, E. F. Dores and M. L. Ribeiro, J. Braz. Chem. Soc., 2008, 19, 1111–1117 CrossRef.
- J. Anderson, C. Dubetz and V. Palace, Sci. Total Environ., 2015, 505, 409–422 CrossRef CAS PubMed.
- X. L. Wu, L. Meng, Y. Wu, Y.-Y. Luk, Y. Ma and Y. Du, J. Braz. Chem. Soc., 2015, 26, 131–139 CAS.
- J. Struger, J. Grabuski, S. Cagampan, E. Sverko, D. McGoldrick and C. H. Marvin, Chemosphere, 2017, 169, 516–523 CrossRef CAS PubMed.
- I. Carra, J. A. Sánchez Pérez, S. Malato, O. Autin, B. Jefferson and P. Jarvis, J. Chem. Technol. Biotechnol., 2016, 91, 72–81 CrossRef CAS.
- V. Guzsvány, J. Csanádi and F. Gaál, Acta Chim. Slov., 2006, 53, 52 Search PubMed.
- A. Khan, M. Haque, N. A. Mir, M. Muneer and C. Boxall, Desalination, 2010, 261, 169–174 CrossRef CAS.
- E. E. Mitsika, C. Christophoridis and K. Fytianos, Chemosphere, 2013, 93, 1818–1825 CrossRef CAS PubMed.
- Y. J. Dai, W. W. Ji, T. Chen, W. J. Zhang, Z. H. Liu, F. Ge and S. Yuan, J. Agric. Food Chem., 2010, 58, 2419–2425 CrossRef CAS PubMed.
- L. Y. Zhou, L. J. Zhang, S. L. Sun, F. Ge, S. Y. Mao, Y. Ma, Z. H. Liu, Y. J. Dai and S. Yuan, J. Agric. Food Chem., 2014, 62, 9957–9964 CrossRef CAS PubMed.
- H. Tang, J. Li, H. Hu and P. Xu, Process Biochem., 2012, 47, 1820–1825 CrossRef CAS.
- G. Wang, W. Yue, Y. Liu, F. Li, M. Xiong and H. Zhang, Bioresour. Technol., 2013, 138, 359–368 CrossRef CAS PubMed.
- H. Yang, X. Wang, J. Zheng, G. Wang, Q. Hong, S. Li, R. Li and J. Jiang, Int. Biodeterior. Biodegrad., 2013, 85, 95–102 CrossRef CAS.
- S. S. Phugare and J. P. Jadhav, Clean: Soil, Air, Water, 2015, 43, 296–304 CrossRef CAS.
- T. Chen, Y. J. Dai, J. F. Ding, S. Yuan and J. P. Ni, Biodegradation, 2008, 19, 651–658 CrossRef CAS PubMed.
- Z. H. Liu, Y. M. Cao, Q. W. Zhou, K. Guo, F. Ge, J. Y. Hou, S. Y. Hu, S. Yuan and Y. J. Dai, Biodegradation, 2013, 24, 855–864 CrossRef CAS PubMed.
- H. J. Zhang, Q. W. Zhou, G. C. Zhou, Y. M. Cao, Y. J. Dai, W. W. Ji, G. D. Shang and S. Yuan, J. Agric. Food Chem., 2012, 60, 153–159 CrossRef CAS PubMed.
- M. Owsianiak, A. Dechesne, P. J. Binning, J. C. Chambon, S. R. Sørensen and B. F. Smets, Environ. Sci. Technol., 2010, 44, 7622–7627 CrossRef CAS PubMed.
- F. Ge, L.-Y. Zhou, Y. Wang, Y. Ma, S. Zhai, Z.-H. Liu, Y.-J. Dai and S. Yuan, Int. Biodeterior. Biodegrad., 2014, 93, 10–17 CrossRef CAS.
- S.-L. Sun, T.-Q. Lu, W.-L. Yang, J.-J. Guo, X. Rui, S.-Y. Mao, L.-Y. Zhou and Y.-J. Dai, RSC Adv., 2016, 6, 15501–15508 RSC.
- Y. Yang, T. Chen, P. Ma, G. Shang, Y. Dai and S. Yuan, Biodegradation, 2010, 21, 593–602 CrossRef CAS PubMed.
- S. Okamoto and L. D. Eltis, Mol. Microbiol., 2007, 65, 828–838 CrossRef CAS PubMed.
- J. E. Casida, J. Agric. Food Chem., 2011, 59, 2923–2931 CrossRef CAS PubMed.
- T. Kanjilal, C. Bhattacharjee and S. Datta, J. Water Process Eng., 2015, 6, 21–31 CrossRef.
- H. Komeda, M. Kobayashi and S. Shimizu, J. Biol. Chem., 1996, 271, 15796–15802 CrossRef CAS PubMed.
- M. Nishiyama, S. Horinouchi, M. Kobayashi, T. Nagasawa, H. Yamada and T. Beppu, J. Bacteriol., 1991, 173, 2465–2472 CrossRef CAS PubMed.
- Y.-S. Feng, P.-C. Chen, F.-S. Wen, W.-Y. Hsiao and C.-M. Lee, Process Biochem., 2008, 43, 1391–1397 CrossRef CAS.
- R. A. Cramp and D. A. Cowan, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1999, 1431, 249–260 CrossRef CAS.
- T. Nagasawa, H. Nanba, K. Ryuno, K. Takeuchi and H. Yamada, Eur. J. Biochem., 1987, 162, 691–698 CrossRef CAS PubMed.
- R. A. Pereira, D. Graham, F. A. Rainey and D. A. Cowan, Extremophiles, 1998, 2, 347–357 CrossRef CAS PubMed.
- A. Banerjee, R. Sharma and U. C. Banerjee, Appl. Microbiol. Biotechnol., 2002, 60, 33–44 CrossRef CAS PubMed.
- M. Odaka, K. Fujii, M. Hoshino, T. Noguchi, M. Tsujimura, S. Nagashima, M. Yohda, T. Nagamune, Y. Inoue and I. Endo, J. Am. Chem. Soc., 1997, 119, 3785–3791 CrossRef CAS.
- Y. Asano, K. Fujishiro, Y. Tani and H. Yamada, Agric. Biol. Chem., 2014, 46, 1165–1174 Search PubMed.
- S. Prasad, J. Raj and T. C. Bhalla, Indian J. Microbiol., 2009, 49, 237–242 CrossRef CAS PubMed.
- C. Laane, S. Boeren, K. Vos and C. Veeger, Biotechnol. Bioeng., 1987, 30, 81–87 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01501a |
| This journal is © The Royal Society of Chemistry 2017 |