DOI:
10.1039/C7RA03872K
(Paper)
RSC Adv., 2017,
7, 30941-30948
Degradation of azo dye with activated peroxygens: when zero-valent iron meets chloride†
Received
5th April 2017
, Accepted 9th June 2017
First published on 15th June 2017
Abstract
Degradation of acid orange 7 (AO7) by Fe0-based Advance Oxidation Process (AOPs) with common peroxygens, persulfate (PS), peroxymonosulfate (PMS) and hydrogen peroxide (H2O2), was investigated, in which sulfate radicals (SO4˙−) and/or hydroxyl radicals (˙OH) are powerful oxidizing species. The effects of Fe0 dosage, peroxygen concentration, initial pH and the presence of chloride on the degradation of AO7 were examined. The AO7 degradation efficiencies by four systems, including Fe0, Fe0/H2O2, Fe0/PMS and Fe0/PS were compared. AO7 degradation rate by Fe0 activated AOPs in descending order is H2O2 ≧ PS > PMS. Increasing acidity and iron dosage favored a rapid degradation of AO7. The presence of chloride greatly inhibited dye removal in Fe0/H2O2 and Fe0/PS systems, whilst accelerated dye degradation was observed in the Fe0/PMS system. In contrast, mineralization of AO7 in the Fe0/PMS/Cl− system was minimal, because of formation of lots of refractory chlorinated phenols as identified by GC-MS. These findings are useful for selecting the most appropriate technology for textile wastewater treatment, depending on the wastewater constituents and pH.
1. Introduction
It is reported that over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kinds of synthetic dyes are widely used in the textile, plastic, paper, food, cosmetic, pharmaceutical and photographic industries. An estimated 10–20% of dyes under production and in the dyeing process are discharged to the environment.1 Therefore, textile wastewater is one of the important industrial pollution sources in developing countries. Azo dyes are of great concern due to their extensive use, carcinogenesis and biorecalcitrance for traditional biological treatment technologies.2 In consideration of the increasingly strict legislations and regulations, it is urgent for the associated industries to develop economically viable technology to remove azo dyes from industrial effluents.
000 kinds of synthetic dyes are widely used in the textile, plastic, paper, food, cosmetic, pharmaceutical and photographic industries. An estimated 10–20% of dyes under production and in the dyeing process are discharged to the environment.1 Therefore, textile wastewater is one of the important industrial pollution sources in developing countries. Azo dyes are of great concern due to their extensive use, carcinogenesis and biorecalcitrance for traditional biological treatment technologies.2 In consideration of the increasingly strict legislations and regulations, it is urgent for the associated industries to develop economically viable technology to remove azo dyes from industrial effluents.
Iron is one of the most abundant metals in the Earth's crust. In recent years, zero-valent iron (ZVI) has been extensively applied to environmental remediation because of its ability to reduce organic pollutants, such as, chlorinated solvents, polychlorobiphenyls (PCBs), pesticides and dyes under anoxic conditions.3–11 More recently, several investigations have reported oxidation of ZVI with oxygen can lead to the formation of reactive oxygen species (ROS) capable of degrading pollutants that cannot be reduced by ZVI. Yields of ROS from oxygen activation with ZVI are significantly affected by pH.12,13 Fe0 or ferrous ions (Fe(II)) released from Fe0 corrosion, react with oxygen to produce H2O2, which further reacts with Fe(II) via a well-known Fenton reaction to produce ˙OH (eqn (1)–(4)).14–16
| | |
Fe0 + 1/2O2 + H2O → Fe2+ + 2OH−
| (1) |
| | |
Fe0 + O2 + 2H+ → Fe2+ + H2O2
| (2) |
| | |
Fe0 + H2O2 → Fe2+ + 2OH−
| (3) |
| | |
Fe2+ + H2O2 → Fe3+ + OH− + ˙OH
| (4) |
In addition to oxygen, ZVI has been successfully used to activate a series of peroxygens like H2O2, persulfate (PS, S2O82−) and peroxymonosulfate (PMS, HSO5−).17–21 Fe0 is a strong electron donor (E0(Fe2+/Fe0) = −0.447 V vs. NHE)19 and is expected to induce reductive decomposition of H2O2 (E0(H2O2/H2O) = 1.77 V vs. NHE),22 PS (E0(S2O82−/SO42−) = 2.01 V vs. NHE) and PMS (E0(HSO5−/HSO4−) = 1.85 V vs. NHE)23 (eqn (5) and (6)). ZVI is thought to act as a continuous slow-release source of Fe(II) during peroxygens activation. The peroxo bond in these peroxygens breaks down by Fe(II) to form highly reactive oxidants, such as ˙OH and SO4˙− (eqn (7) and (8)). Therefore, peroxygens activation with ZVI has been applied to degrade several contaminants such as aniline, bisphenol, trichloroethene, phenol, acid black 24 and pentachlorophenol.17,24–28 To the best of our knowledge, a comparative study on the degradation of the same azo dye by ZVI-based peroxygens activation has not been reported.
| | |
Fe0 + S2O82− → Fe2+ + 2SO42−
| (5) |
| | |
Fe0 + HSO5− → Fe2+ + SO42− + OH−
| (6) |
| | |
Fe2+ + S2O82− → Fe3+ + SO42− + SO4˙−
| (7) |
| | |
Fe2+ + HSO5− → Fe3+ + OH− + SO4˙−
| (8) |
The present study was an attempt to compare the degradation efficiencies of Acid Orange 7 (AO7, a typical azo dye) by ZVI activation with three common peroxygens. The effects of pH, ZVI dosage, chloride ions on dye degradation were investigated. Additionally, TOC and GC-MS measurement were conducted to characterize the extent of mineralization and intermediate products during treatment.
2. Experimental
2.1. Materials
All chemical reagents used in this study were in AR grade and were used without further purification. Hydrogen peroxide (30%, v/v), sodium chloride (NaCl), sodium hydroxide (NaOH), sulfuric acid (H2SO4, 98%), and the Fe0 powder were obtained from Sinopharm. Acid orange 7 (C16H11N2O4SNa) and Oxone® (2KHSO5·KHSO4·K2SO4) were bought from Sigma-Aldrich. Potassium persulfate (K2S2O8) was purchased from Alfa Aesar. All solutions were prepared with deionized water.
2.2. Experimental procedures
The degradation experiments were conducted at room temperature in 250 mL batch reactors, and used the same batch reactors to make all comparisons. For all the experiments, the initial concentration of AO7 was 0.08 mmol L−1. The initial pH values of all solutions were adjusted by 0.1 mol L−1 sulfuric acid or 0.1 mol L−1 sodium hydroxide. At given intervals, the solution was sampled and examined on U-2910 spectrophotometer at 484 nm for testing the degradation kinetics. For the measurement of mineralization and products, samples were quenched by sodium nitrite with a ratio of NaNO2/react solution was 1:1. All experiments were conducted in duplicate.
2.3. Analysis
The analysis of the degradation intermediates was carried out by gas chromatography-mass spectrometry (Agilent 7890A model GC with DB-5 MS. 30 mm × 320 μm × 0.5 μm capillary column coupled with 5975A inert XL MSD model MS). Helium was used as the carrier gas at a rate of 1.0 mL min−1. 1 μL samples were injected at 250 °C in splitless mode. The sequence of GC temperature program was followed as: start at 40 °C, hold for 2 min; 40–100 °C (12 °C min); 100–200 °C (5 °C min); 200–270 °C (20 °C min−1), hold for 5 min. The ion source temperature was maintained at 230 °C. Qualitative detector was used in the electron impact (EI) mode at 70 eV with the mass scanned range (30–400 m/z). The unknown peaks were identified using NIST08 mass spectral library database.
The mineralization of AO7 aqueous solutions was examined by a Shimazu TOC-VCPH analyzer. The sample volumes were 10 mL. The flow rate and pressure of carrying gas (air) was set as 150 mL min−1 and 300 kPa, respectively.
A pseudo first-order kinetic model is used to describe the degradation kinetics in different activation systems. The kinetic expression is represented as eqn (9), where Ct is the residual AO7 concentration at time t (min); C0 is the initial AO7 concentration. k denotes the observed pseudo first-order rate constant (min−1). The constant k is calculated by the slope of a plot of ln(Ct/C0) versus t and is summarized in Table 1.
Table 1 The calculated pseudo first-order rate constant (10−3 min−1) of AO7 oxidation in Fe0-peroxygens systems
| |
Fe |
Fe/H2O2 |
Fe/PMS |
Fe/PS |
| k |
R2 |
k |
R2 |
k |
R2 |
k |
R2 |
| Conditions: m(Fe0) = 0.025 g L−1, c(Ox) = 1.0 mmol L−1, c(AO7) = 0.08 mmol L−1. Conditions: c(Ox) = 1.0 mmol L−1, c(AO7) = 0.08 mmol L−1, pH = 2.5. Conditions: in Fe/PMS, the maximum dosage of Fe0 was 0.05 g L−1. Conditions: m(Fe0) = 0.025 g L−1, c(AO7) = 0.08 mmol L−1, pH = 2.5. Conditions: m(Fe0) = 0.025 g L−1, c(AO7) = 0.08 mmol L−1, c(Ox) = 1.0 mmol L−1, pH = 2.5. |
| pHa |
2.5 |
6.5 |
0.983 |
160 |
0.978 |
120 |
0.930 |
120 |
0.982 |
| 4.0 |
— |
— |
7.2 |
0.997 |
63 |
0.992 |
68 |
0.948 |
| 7.0 |
— |
— |
— |
— |
28 |
0.998 |
5.3 |
0.995 |
| Iron dosage (g L−1)b |
0.005 |
2.1 |
0.913 |
59 |
0.950 |
74 |
0.999 |
26 |
0.998 |
| 0.025 |
8.0 |
0.973 |
91 |
0.900 |
54 |
0.958 |
140 |
0.988 |
| 0.1c |
37 |
0.994 |
220 |
0.942 |
91 |
0.985 |
260 |
0.999 |
| Peroxygens concentration (mmol L−1)d |
0 |
— |
— |
6.4 |
0.984 |
7.6 |
0.988 |
8.2 |
0.999 |
| 0.5 |
— |
— |
55 |
0.980 |
56 |
0.945 |
67 |
0.999 |
| 1.0 |
— |
— |
96 |
0.940 |
81 |
0.906 |
130 |
0.994 |
| 1.5 |
— |
— |
150 |
0.986 |
45 |
0.991 |
160 |
0.969 |
| Cl− concentration (mmol L−1)e |
0 |
7.6 |
0.988 |
120 |
0.929 |
51 |
0.969 |
120 |
0.993 |
| 50 |
7.6 |
0.996 |
100 |
0.992 |
140 |
0.911 |
92 |
0.964 |
| 300 |
6.7 |
0.999 |
110 |
0.969 |
440 |
0.957 |
43 |
0.982 |
| 400 |
5.7 |
0.999 |
150 |
0.986 |
780 |
0.978 |
44 |
0.984 |
3. Results and discussion
3.1. Effectiveness of the various activated peroxygens
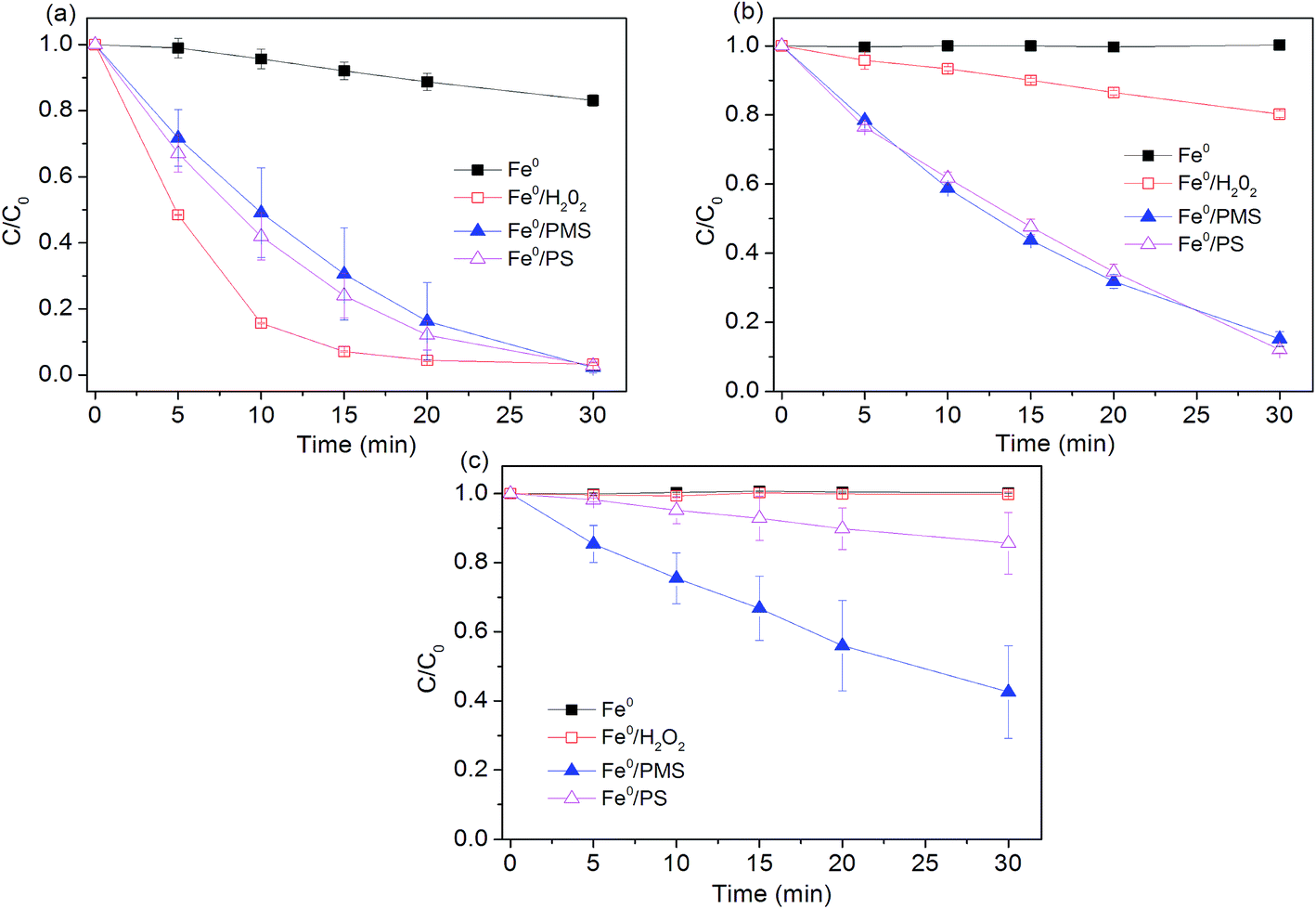
Experiments were conducted to determine effectiveness of Fe0 activated peroxygens on the removal of AO7 by changing initial pH of solution while keeping Fe0 loading and peroxygens concentration constant. As shown in Fig. 1, degradation efficiency of AO7 decreased with the increasing pH value. About 20% of dye was decomposed in Fe0/air system at pH = 2.5, while no measurable degradation was observed at other tested pH. Under acidic conditions, indirect oxidation of dye by strong oxidants generated by Fe0 (eqn (4), (7) and (8)) was responsible for AO7 degradation besides the direct reductive decolorization on iron surface which results in the cleavage of the azo bonds of AO7 (eqn (10)).13| |
–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– + 2H+ + 2e− → –NH + HN– N– + 2H+ + 2e− → –NH + HN–
| (10) |
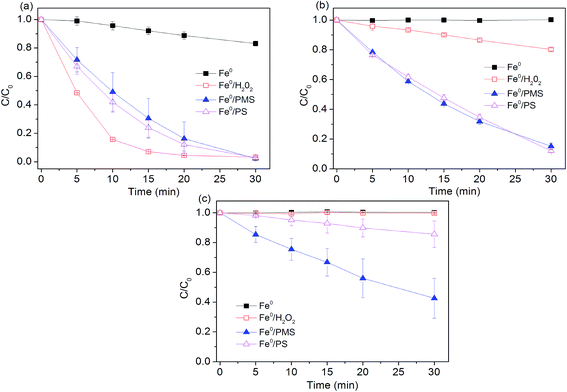 |
| | Fig. 1 Effect of pH value on AO7 degradation rates. (a) pH0 = 2.5; (b) pH0 = 4.0; (c) pH0 = 7.0. Conditions: c(AO7)0 = 0.08 mmol L−1; m(Fe0)0 = 0.025 g L−1; c(Ox)0 = 1 mmol L−1. | |
Mielczarski et al.29 reported that iron oxide/hydroxide on the Fe0 surface was thin at pH = 3.0, but mainly existed in the solution. Accumulation of iron oxide on the iron surface significantly affected the electron transfer between Fe0 and oxygen (or dye) at pH > 4.0,29 thus inhibiting the dye degradation. Among the three tested Fe0-peroxygens systems, Fe0/H2O2 system exhibited a better degradation efficiency at pH = 2.5, with a constant rate of 0.16 min−1, higher than those (0.12 min−1) in two SO4˙−-based systems (Table 1). This can be ascribed to the higher oxidation capacity of ˙OH than SO4˙− at highly acidic pH. However, at higher pH (4.0 and 7.0), Fe0/H2O2 system became inefficient towards dye degradation. At pH 7.0, no measurable degradation of dye occurred. In contrast, despite of reduction in reaction rates at pH 4.0 and 7.0, Fe0/PMS and Fe0/PS systems still led to a considerable degradation of AO7. Some researchers tried to explain it from the perspective of electron transfer mechanism.18 They thought electron transfer from the iron surface to H2O2 undergoes an inner-sphere electron transfer process that is slower than outer-sphere electron transfer mechanism as supposed in Fe0/PMS and Fe0/PS systems. This proposed explanation is supported by Al-Shamsi et al.18 and Rastogi et al.30 who both observed that activated H2O2 system was less efficient to oxidize trichloroethylene (TCE) and polychlorinated biphenyls (PCBs) compared to the activated PS and PMS systems.
3.2. Effect of Fe0 dosage
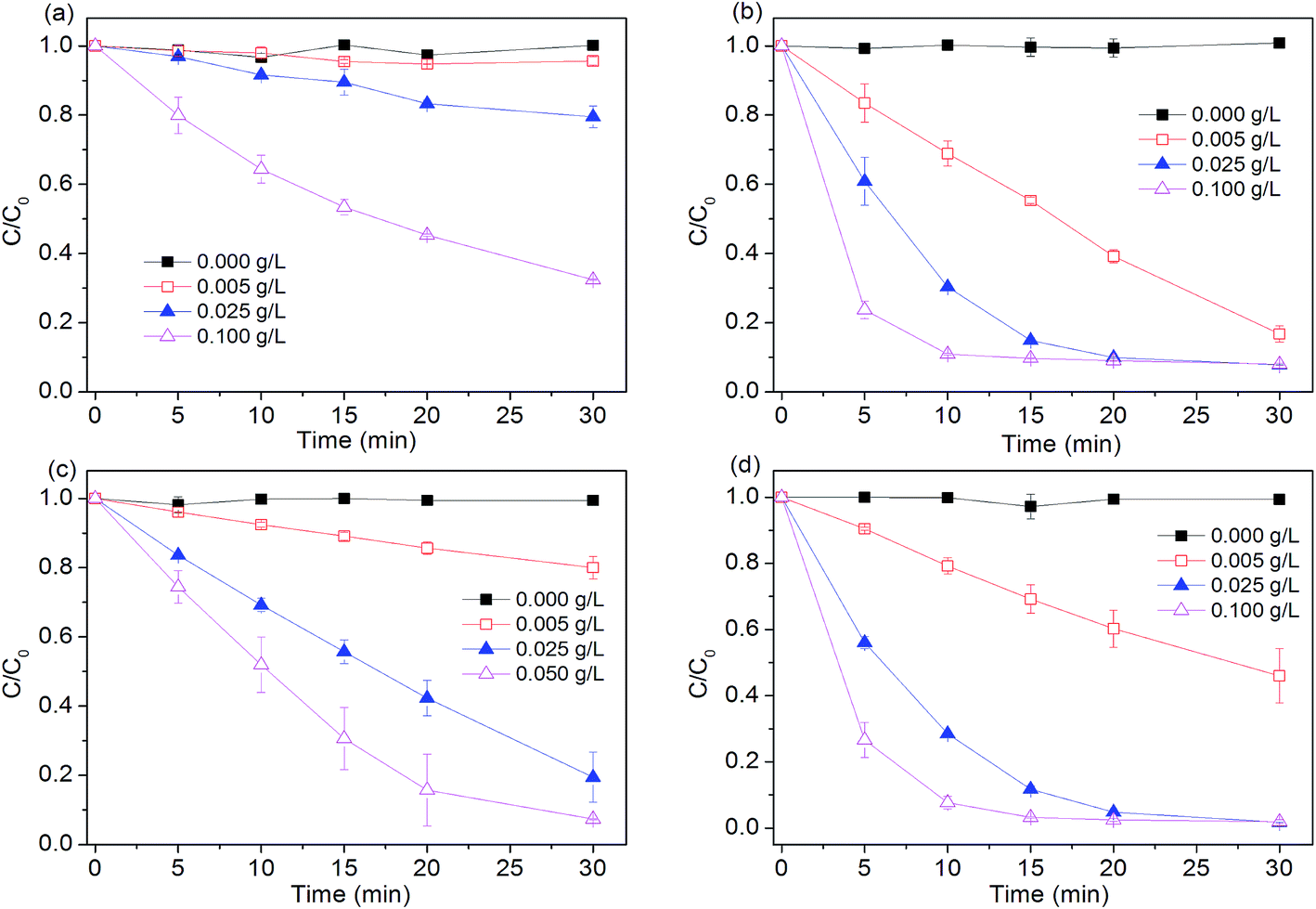
Effects of Fe0 dosage (0.005, 0.025, 0.1 g L−1) on dye degradation by four Fe0-based systems were investigated. As seen in Fig. 2, without Fe0, peroxygen itself can not directly oxidize dye within the examined time. Degradation efficiency of four tested systems all increased with increasing iron dosage. For example, about 90% of dye was degraded in Fe0/H2O2 process at 10 min, while other reactions with relatively lower Fe0 dosage (0.005, 0.025 g L−1) needed longer reaction time of 20 and 30 min to achieve the comparable removal of dye, respectively. Increasing Fe0 dosage from 0.005 to 0.1 g L−1 led to a 17.6, 3.7 and 10-fold increase in pseudo first-order rate constant in Fe0, Fe0/H2O2 and Fe0/PS, respectively. Increase in total surface area and availability of more Fe0 reactive sites should be responsible for the enhanced degradation of dye with increasing iron dosage.
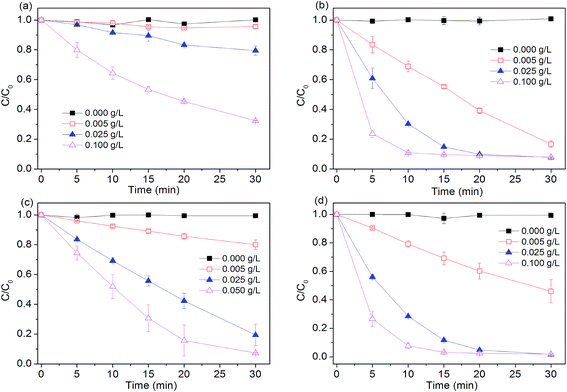 |
| | Fig. 2 Effect of Fe0 dosage on AO7 removal in four systems. (a) Fe0; (b) Fe0/H2O2; (c) Fe0/PMS; (d) Fe0/PS. Conditions: c(AO7)0 = 0.08 mmol L−1; c(Ox)0 = 1 mmol L−1; pH0 = 2.5 ± 0.2. | |
3.3. Effects of peroxygens doses
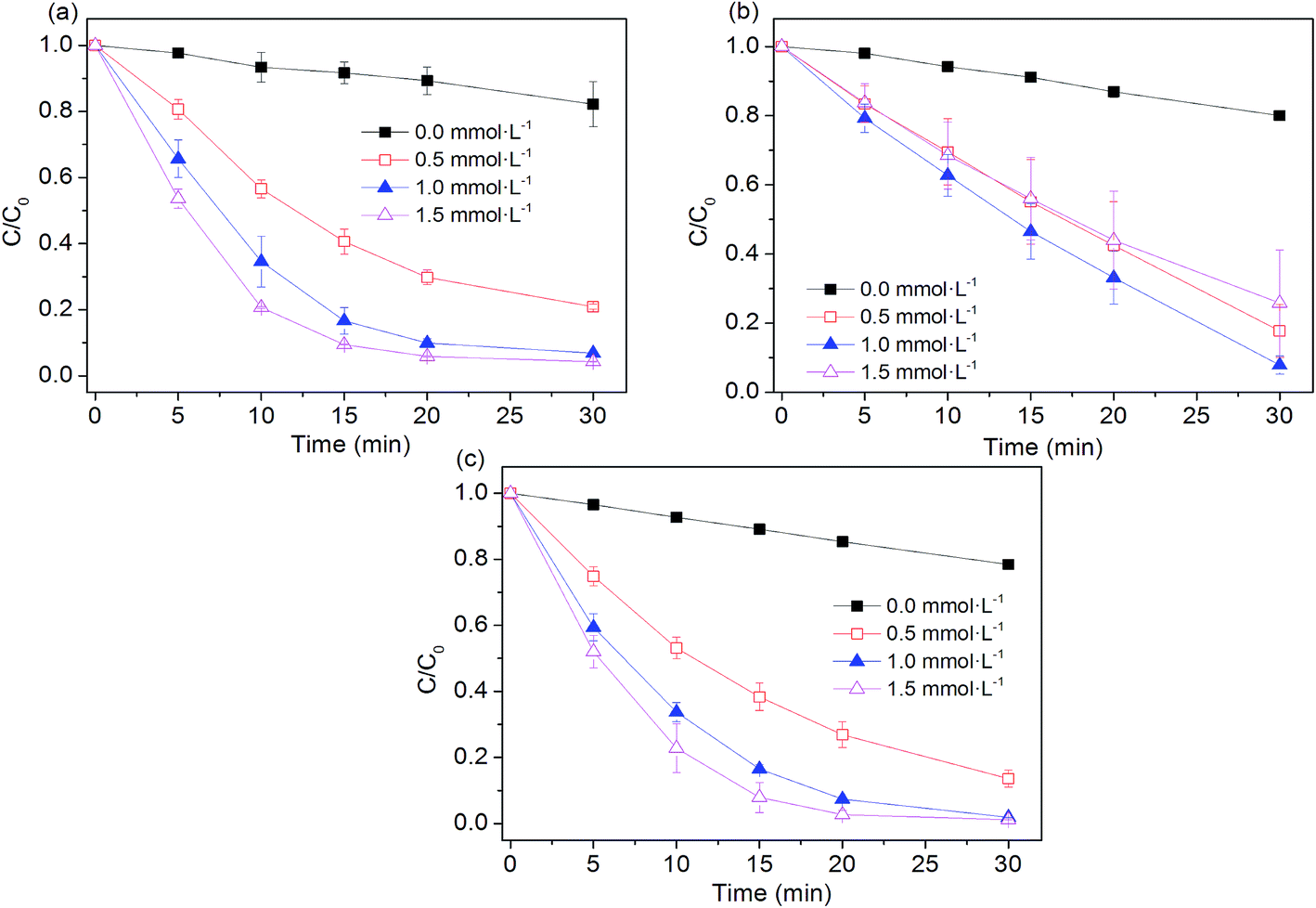
The influence of peroxygens concentrations on the oxidation of AO7 with PS, PMS, H2O2 was studied at the ZVI dosage of 0.005 g. As expected, Fig. 3 shows that degradation rates of AO7 increased as PS and H2O2 dosage increased because of the enhanced generation of hydroxyl radical and sulfate radical as availability of their precursors (i.e. PS and H2O2) was increased. Briefly, the rate constant for dye degradation increased by a factor of 2.7 and 2.4 as concentration of H2O2 and PS increased from 0.5 to 1.5 mmol L−1. As shown in Fig. 3b, the extent of AO7 degradation increased from 20% to 92.1% when PMS concentration increased from 0 to 1 mmol L−1 within 30 min. As PMS concentration was over 1 mmol L−1, the removal of AO7 was decreased to about 70%. A similar trend was also observed during the oxidative degradation of bisphenol A (BPA) by UV activated PMS.31 An excess of HSO5− may scavenge parts of SO4˙− radicals and generate the less reactive SO5˙− radicals (eqn (11)),31,32 thus decreasing the degradation efficiency of dye pollutant.| | |
HSO5− + SO4˙− → HSO4− + SO5˙−
| (11) |
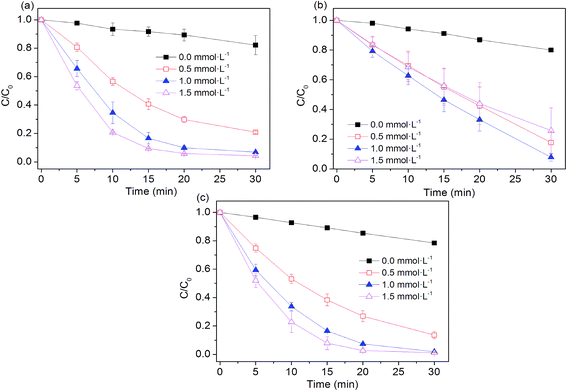 |
| | Fig. 3 Effect of peroxygens concentration on AO7 removal in four systems. (a) Fe0/H2O2; (b) Fe0/PMS; (c) Fe0/PS. Conditions: c(AO7)0 = 0.08 mmol L−1; m(Fe0)0 = 0.025 g L−1; pH0 = 2.5 ± 0.2. | |
3.4. Effect of Cl− concentration
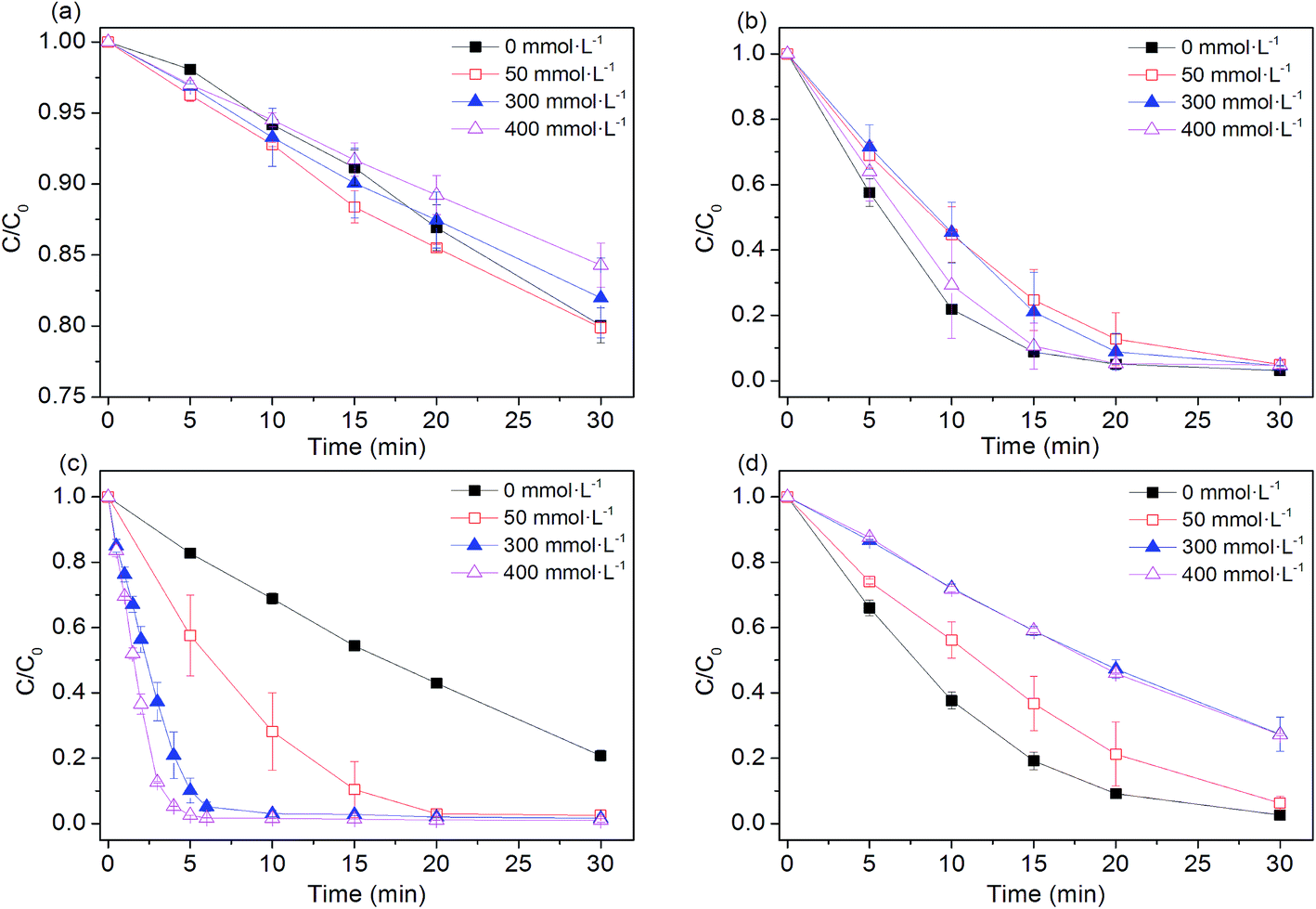
Higher level of NaCl is one of the remarkable feature of textile wastewater, as large amount of NaCl (50–80 g L−1) is frequently applied in dyeing processes in order to improve dye fixation and completion.33 Our previous studies demonstrated that performance of SO4˙−-based advanced oxidation processes (SR-AOPs) is significantly affected by the presence of chloride.34–43 Therefore, it is necessary to examine efficiencies of four Fe0-based processes in the presence of chloride. As shown in Fig. 4, the NaCl in aqueous solution did not obviously affect the dye removal in the Fe0 system. Effects of chloride on Fe0-induced pollutant removal are quite complicated, depending on the nature of pollutants to be treated. For example, the presence of chloride can destroy the passive oxide layers and help to maintain efficiency of Fe0 for nitrobenzene reduction.44 However, Hwang et al.45 reported that a higher concentration of NaCl (>3 g L−1) had a significant inhibitory effect on nitrate reduction by Fe0, although Fe0 is known as a pitting and crevice corrosion promoter.46
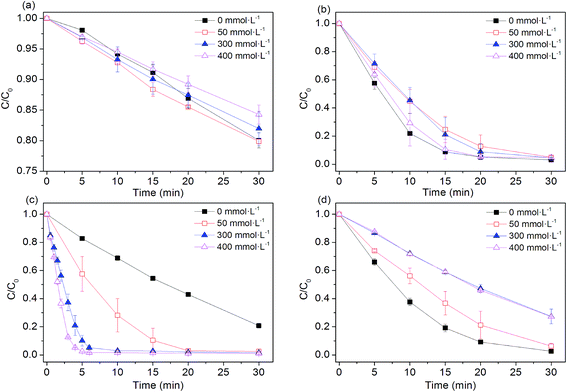 |
| | Fig. 4 Effect of chloride ions on AO7 degradation rates. (a) Fe0; (b) Fe0/H2O2; (c) Fe0/PMS; (d) Fe0/PS. Conditions: c(AO7)0 = 0.08 mmol L−1; m(Fe0)0 = 0.025 g L−1; c(Ox)0 = 1 mmol L−1; pH0 = 2.5 ± 0.2. | |
In the Fe0/H2O2 and Fe0/PS systems, degradation rates of AO7 decreased in the presence of chloride. The negative effects of chloride ion on the efficiency of the Fe0/H2O2 and Fe0/PS systems may be explained by (Laat and Le 2006):47 (1) the formation of Fe(III) or Fe(II)-chlorocomplexes (FeCl+, FeCl2+, FeCl2+) (eqn (12)–(14)) and (2) the scavenging effect of chloride ion for hydroxyl and sulfate radicals (eqn (15)–(18)). When chloride concentrations were less than 50 mmol L−1, degradation efficiency of TCE,48 p-nitrosodimethylaniline49 and AO7 (ref. 50) by PS activation with Fe0 was not significantly affected. In the presence of excess chloride, the reaction of ˙OH or SO4˙− with Cl− leads to the formation of chlorine atoms (Cl˙) (E0(Cl˙/Cl−) = 2.41 V vs. NHE) and of dichloride anion radicals (Cl2˙−) (E0(Cl2˙−/2Cl−) = 2.09 V vs. NHE).51 Cl˙/Cl2˙− can oxidize H2O2 and Fe(II) and but are less reactive with organic solutes than either ˙OH or SO4˙−. It should be noted that dye degradation was significantly accelerated in Fe0/PMS/Cl− systems. For example, at [Cl−] = 400 mmol L−1, k increased 15 times higher than that in the absence of chloride. Our previous investigations found that PMS, rather than H2O2 and PS, could directly react with Cl− to produce HOCl and Cl2 (eqn (19) and (20)),37–43,51 enhancing the dye bleaching rate.
| | |
Fe3+ + 2Cl− ↔ FeCl2+
| (14) |
| | |
˙ClOH− + H+ ↔ H2O + Cl˙
| (16) |
| | |
Cl− + SO4˙− ↔ Cl˙ + SO42−
| (17) |
| | |
Cl− + HSO5− → SO42− + HOCl
| (19) |
| | |
2Cl− + HSO5− + H+ → SO42− + Cl2 + H2O
| (20) |
3.5. AO7 mineralization and byproducts identification
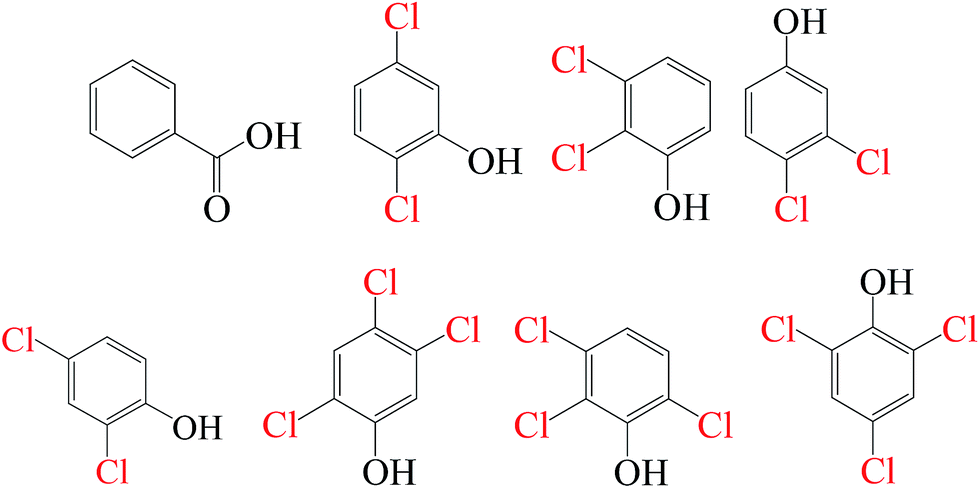
As evidenced in the previous studies, chlorinated organic intermediates would be generated when amounts of chloride are present in SR-AOPs. Therefore, it is necessary to identify the reaction byproducts and evaluate the mineralization, besides testing degradation rates. GC-MS data (Fig. S1–S18,† Table 2) show that some chlorinated compounds were produced in SR-AOPs. Seven chlorinated phenols, including 2,3,6-trichlorophenol, 2,4,5-trichlorophenol, 2,4,6-trichlorophenol, 3,4-dichlorophenol, 2,5-dichlorophenol, 2,3-dichlorophenol, 2,4-dichlorophenol, were identified in Fe0/PMS/Cl− system, whereas only three chlorophenols like 2,4,5-trichlorophenol, 2,4,6-trichlorophenol, 2,3,5-trichlorophenol were detected in Fe0/PS/Cl− system. According to the known Material Safety Data Sheet (MSDS) of pure chemical, acute toxicity, expressed as rat Lethal Dose, 50% (LD50), are 820 mg kg−1 for 2,4,5-trichlorophenol, 820 mg kg−1 for 2,4,6-trichlorophenol, 1685 mg kg−1 for 3,4-dichlorophenol, 2376 mg kg−1 for 2,3-dichlorophenol and 47 mg kg−1 for 2,4-dichlorophenol, respectively, much greater than 3418 mg kg−1 for their parent compound, AO7. This indicates that AO7 was transformed to more toxic and recalcitrant organic byproducts although AO7 itself has been efficiently degraded in Fe0-based oxidation systems.
Table 2 The transformation products during oxidation of AO7 in Fe0-peroxygens systems
| Systems |
Identified by products |
| Fe0/Cl− |
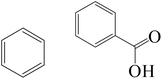 |
| Fe0/H2O2/Cl− |
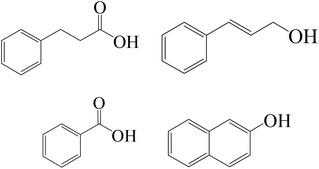 |
| Fe0/PS/Cl− |
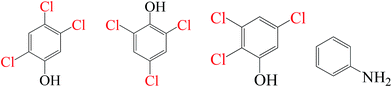 |
| Fe0/PMS/Cl− |
 |
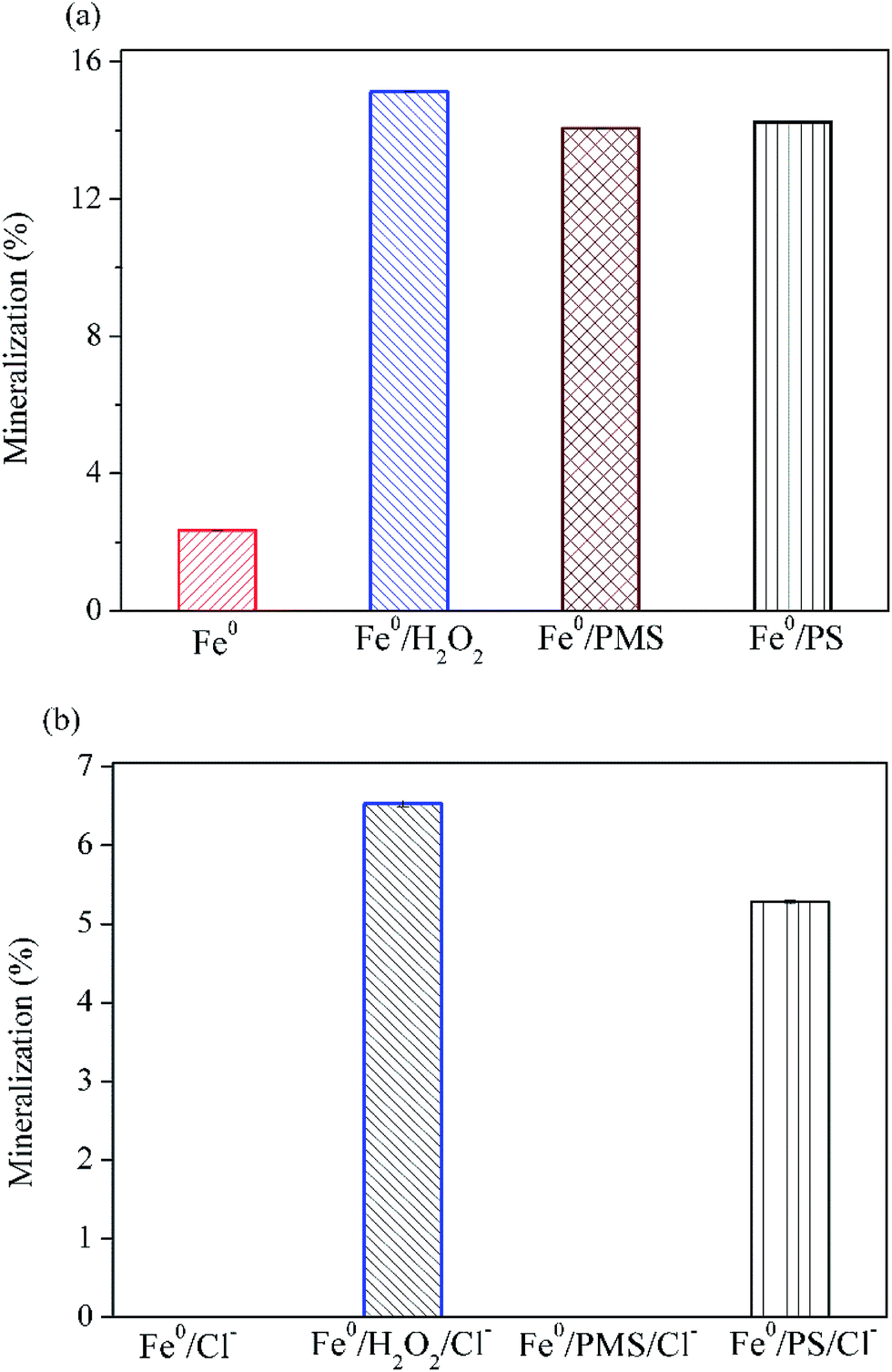
Fig. 5 illustrates that 15.1% of AO7 could be mineralized without the addition of chloride after 30 min oxidation in Fe0/H2O2 system. The extent of mineralization in descending order is Fe0/H2O2 > Fe0/PS > Fe0/PMS > Fe0 (Fig. 5a). In the presence of chloride, TOC removal were dramatically decreased in four systems (Fig. 5b). In particular, no measurable TOC removal were observed in Fe0/PMS system, in sharp contrast to its rapid degradation rate as shown in Fig. 4. In combination with the GC-MS data, it is evident that AO7 in Fe0/PMS/Cl− system was only converted to some chlorinated byproducts, although it was rapidly bleached. In general, extents of mineralization in these tested systems were not satisfactory. It is probably because (1) AO7 is just degraded via a chromophore cleavage, as evidenced by GC-MS; (2) Fe0, as a strong reductant, may continuously consume highly reactive species which are critical for a complete mineralization of organic pollutant. The development of processes combining Fe-based peroxygens oxidation systems with further biological methods is a possible route to improve the removal of recalcitrant degradation intermediates.
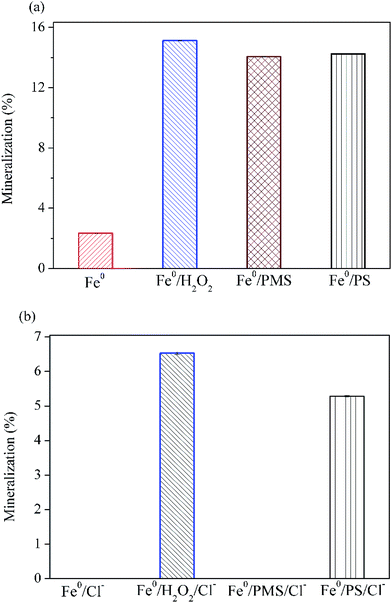 |
| | Fig. 5 Variation of mineralization of AO7 with different peroxygens after 30 min. (a) Without Cl−; (b) with Cl−. Conditions: c(AO7)0 = 0.08 mmol L−1; m(Fe0)0 = 0.025 g L−1; c(Ox)0 = 1 mmol L−1; pH0 = 2.5 ± 0.2; c(NaCl)0 = 50 mmol L−1 (if any). | |
3.6. Mechanism discussion
Under the present experimental conditions, two major mechanisms govern the removal process of dye: (1) reduction and (2) degradation. Fe0 undergoes a couples of corrosion reactions in acidic solution, accompanying with reduction of O2 or pollutants on its surface. Azo dye reduction is thought to involve a two-step process.52 Initially, nascent hydrogen is generated after the oxidation of Fe0. The newly formed Fe2+ on the Fe0 surface enables the azo group of AO7 catalytically hydrogenated to form short-lived hydrazo intermediates, followed by a further hydrogenation of the unstable transition products to form stable aromatic amines.
In addition to dye pollutant, O2 can be directly reduced by Fe0 powder to generate H2O2 (eqn (2)), providing the reactants for the Fenton reaction yielding ˙OH radical. A faster iron redox cycle at the iron surface takes place through a rapid reduction of Fe3+ to Fe2+ by Fe0. The kinetic rates of iron corrosion are dependent upon the intrinsic reactivity of the selected Fe material and other environmental factors, such redox conditions and pH.53 At neutral pH, a thick layer of iron oxides, maghemite or lepidocrocite, is formed as a result of hydrolysis, precipitation and transformation of Fe3+, thereby diminishing the reactivity of Fe0.
In the presence of peroxygens, ˙OH or SO4˙− is produced from the catalytic decomposition of H2O2, PS and PMS by Fe0 and/or Fe2+. Surface-catalyzed Fe2+ oxidation may play an important role in peroxygens activation. Keenan and Sedlak12 found that accelerated Fe2+ oxidation in the presence of Fe surface significantly contributed to the enhanced HCHO yields from oxidation of CH3OH at pH > 6.0. Therefore, it is reasonably expected that surface-catalyzed Fe2+ reactions with peroxygens vary with the homogeneous reactions and deserves in-depth investigations in future.
4. Conclusions
In this work, commercially available Fe0 powder was used to activate three common peroxygens (H2O2, PS and PMS) for the degradation of a model hazardous azo dye (i.e., AO7). Experimental data indicate Fe0 was an effective activator for peroxygens to treat azo dye at acidic pH. The highest AO7 oxidation and mineralization were achieved in Fe0 activated H2O2 system at pH 2.5. However, the ability of Fe0 activated H2O2 system to oxidize AO7 was less than those of Fe0/PS and Fe0/PMS systems at pH ≧ 4.0. Increasing iron dosage and peroxygens concentration favored a rapid degradation of AO7 in Fe0/PS and Fe0/H2O2 systems. Addition of chloride could greatly inhibit dye removal in Fe0/H2O2 and Fe0/PS systems, whereas dye degradation was accelerated in Fe0/PMS system. In contrast, no measurable mineralization of AO7 in Fe0/PMS/Cl− system was observed. Some refractory chlorinated phenols, such as 2,3,6-trichlorophenol, 2,4,5-trichlorophenol, 2,3,5-trichlorophenol, 2,4,6-trichlorophenol, 3,4-dichlorophenol, 2,5-dichlorophenol, 2,3-dichlorophenol and 2,4-dichlorophenol were identified by GC-MS. In conclusion, Fe0/H2O2 system is recommended to treat acidic and saline wastewater, while Fe0/PS and Fe0/PMS processes are more suitable for treatment of low salinity wastewater.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (No. 21677031), National Key Research and Development Program of China (2016YFC0400501/2016YFC0400509), Shanghai Pujiang Program and DHU Distinguished Young Professor Program.
References
- H. Zollinger, Colour Chemistry-Synthesis, 1987 Search PubMed.
- Z. H. Wang, W. H. Ma, C. C. Chen and J. C. Zhao, J. Hazard. Mater., 2009, 168, 1246 CrossRef CAS.
- H. L. Lien and W. X. Zhang, Appl. Catal., B, 2007, 77, 110 CrossRef CAS.
- G. V. Lowry and K. W. Johnson, Environ. Sci. Technol., 2004, 38, 5208 CrossRef CAS.
- S. Choe, Y. Y. Chang, K. Y. Hwang and J. Khim, Chemosphere, 2000, 41, 1307 CrossRef CAS.
- J. Cao, L. P. Wei, Q. G. Huang, L. S. Wang and S. K. Han, Chemosphere, 1999, 38, 565 CrossRef CAS.
- G. Roy, P. D. Donato, T. Görner and O. Barres, Water Res., 2003, 37, 4954 CrossRef CAS.
- S. Nam and P. G. Tratnyek, Water Res., 2000, 34, 1837 CrossRef CAS.
- F. S. Freyria, B. Bonelli, R. Sethi, M. Armandi, E. Belluso and E. Garrone, J. Phys. Chem. C, 2011, 115, 24143 CAS.
- Y. He, J. F. Gao, F. Q. Feng, C. Liu, Y. Z. Peng and S. Y. Wang, Chem. Eng. J., 2012, 179, 8 CrossRef CAS.
- Y. Yuan, H. Q. Li, B. Lai, P. Yang, M. Gou, Y. X. Zhou and G. Z. Sun, Ind. Eng. Chem. Res., 2014, 53, 2605 CrossRef CAS.
- C. R. Keenan and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 1262 CrossRef CAS.
- S. H. Chang, K. S. Wang, S. J. Chao, T. H. Peng and L. C. Huang, J. Hazard. Mater., 2009, 166, 1127 CrossRef CAS.
- O. X. Leupin and S. J. Hug, Water Res., 2005, 39, 1729 CrossRef CAS.
- C. E. Noradoun and I. F. Cheng, Environ. Sci. Technol., 2005, 39, 7158 CrossRef CAS PubMed.
- S. H. Joo, A. J. Feitz, D. L. Sedlak and T. D. Waite, Environ. Sci. Technol., 2005, 39, 1263 CrossRef CAS PubMed.
- C. J. Liao, T. L. Chung, W. L. Chen and S. L. Kuo, J. Mol. Catal. A: Chem., 2007, 265, 189 CrossRef CAS.
- M. A. Al-Shamsi, N. R. Thomson and S. P. Forsey, Chem. Eng. J., 2013, 232, 555 CrossRef CAS.
- J. Y. Zhao, Y. B. Zhang, X. Quan and S. Chen, Sep. Purif. Technol., 2010, 71, 302 CrossRef CAS.
- S. Y. Oh, S. G. Kang and P. C. Chiu, Sci. Total Environ., 2010, 408, 3464 CrossRef CAS.
- C. J. Liang and M. C. Lai, Environ. Eng. Sci., 2008, 25, 1071 CrossRef CAS.
- Z. H. Wang, R. T. Bush, L. A. Sullivan and J. S. Liu, Environ. Sci. Technol., 2013, 47, 6486 CAS.
- Z. H. Wang, R. T. Bush, L. A. Sullivan, C. C. Chen and J. S. Liu, Environ. Sci. Technol., 2014, 48, 3978 CrossRef CAS PubMed.
- I. Hussain, Y. Q. Zhang and S. B. Huang, RSC Adv., 2014, 4, 3502 RSC.
- X. X. Jiang, Y. L. Wu, P. Wang, H. J. Li and W. B. Dong, Environ. Sci. Pollut. Res., 2013, 20, 4947 CrossRef CAS PubMed.
- S. Rodriguez, L. Vasquez, A. Romero and A. Santos, Ind. Eng. Chem.
Res., 2014, 53, 12288 CrossRef CAS.
- Y. X. Wang, H. Q. Sun, X. G. Duan, H. M. Ang, M. O. Tadé and S. B. Wang, Appl. Catal., B, 2015, 172–173, 73 CrossRef CAS.
- H. Y. Shu, M. C. Chang and C. C. Chang, J. Hazard. Mater., 2009, 167, 1178 CrossRef CAS PubMed.
- J. A. Mielczarski, G. M. Atenas and E. Mielcazrski, Appl. Catal., B, 2005, 56, 289 CrossRef CAS.
- A. Rastogi, S. R. Al-Abed and D. D. Dionysiou, Appl. Catal., B, 2009, 85, 171 CrossRef CAS.
- J. Sharma, I. M. Mishra, D. D. Dionysiou and V. Kumar, Chem. Eng. J., 2015, 276, 193 CrossRef CAS.
- P. Maruthamuthu and P. Neta, J. Phys. Chem., 1977, 81, 937 CrossRef CAS.
- Washington and DC, Environmental Protection Agency, 1997.
- S. N. Ramjaun, R. X. Yuan, Z. H. Wang and J. S. Liu, Electrochim. Acta, 2011, 58, 364 CrossRef CAS.
- R. X. Yuan, S. N. Ramjaun, Z. H. Wang and J. S. Liu, Chem. Eng. J., 2012, 192, 171 CrossRef CAS.
- R. X. Yuan, S. N. Ramjaun, Z. H. Wang and J. S. Liu, Chem. Eng. J., 2012, 209, 38 CrossRef CAS.
- C. L. Fang, D. X. Xiao, W. Q. Liu, X. Y. Lou, J. Zhou, Z. H. Wang and J. S. Liu, Chemosphere, 2016, 144, 2415 CrossRef CAS PubMed.
- J. Zhou, J. H. Xiao, D. X. Xiao, Y. G. Guo, C. L. Fang, X. Y. Lou, Z. H. Wang and J. S. Liu, Chemosphere, 2015, 134, 446 CrossRef CAS PubMed.
- R. X. Yuan, Z. H. Wang, Y. Hu, B. H. Wang and S. M. Gao, Chemosphere, 2014, 109, 106 CrossRef CAS PubMed.
- X. Y. Lou, Y. G. Guo, D. X. Xiao, Z. H. Wang and J. S. Liu, Environ. Sci. Pollut. Res., 2013, 20, 6317 CrossRef CAS PubMed.
- L. Xu, R. X. Yuan, Y. G. Guo, D. X. Xiao, Y. Cao, Z. H. Wang and J. S. Liu, Chem. Eng. J., 2013, 217, 169 CrossRef CAS.
- Z. H. Wang, R. X. Yuan, Y. G. Guo, L. Xu and J. S. Liu, J. Hazard. Mater., 2011, 190, 1083 CrossRef CAS PubMed.
- R. X. Yuan, S. N. Ramjaun, Z. H. Wang and J. S. Liu, J. Hazard. Mater., 2011, 196, 173 CrossRef CAS PubMed.
- W. Z. Yin, J. H. Wu, P. Li, X. D. Wang, N. W. Zhu, P. X. Wu and B. Yang, Chem. Eng. J., 2012, 184, 198 CrossRef CAS.
- Y. Hwang, D. Kim and H. S. Shin, Environ. Technol., 2015, 36, 1178 CrossRef CAS.
- T. X. Liu, X. M. Li and T. D. Waite, Environ. Sci. Technol., 2013, 47, 7350 CrossRef CAS.
- J. D. Laat and T. G. Le, Appl. Catal., B, 2006, 66, 137 CrossRef.
- C. J. Liang, Z. Wang and N. Mohanty, Environ. Sci. Technol., 2006, 370, 271 CAS.
- L. R. Bennedsen, J. Muff and E. G. Sogaard, Chemosphere, 2012, 86, 1092 CrossRef CAS PubMed.
- H. Li, J. Wan, Y. Ma, Y. Wang and Z. Guan, RSC
Adv., 2015, 5, 99935 RSC.
- S. N. Ramjaun, Z. H. Wang, R. X. Yuan and J. S. Liu, J. Environ. Chem. Eng., 2015, 3, 1648 CrossRef CAS.
- L. J. Matheson and P. G. Tratnyek, Environ. Sci. Technol., 1994, 28, 2045 CrossRef CAS.
- X. Sun, T. Kurokawa, M. Suzuki, M. Takagi and Y. Kawase, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2015, 50, 1057 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The online version of this article contains supplementary material, which is available to authorized users. See DOI: 10.1039/c7ra03872k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Luoyan Aia,
Ying Huanga,
Juekai Zhangac,
Sitong Liac,
Jiawei Chenac and
Fei Yanga
*ab,
Luoyan Aia,
Ying Huanga,
Juekai Zhangac,
Sitong Liac,
Jiawei Chenac and
Fei Yanga